

Summary:
Keloid disorder (KD) is a fibroproliferative condition caused by dysregulated wound healing following wounding of the skin. The pathogenesis of KD has not been fully elucidated and current treatment is unsatisfactory. There is increasing evidence of the role of stem cells in KD. This review discusses the role of embryonic stem (ESC)-like cells and mesenchymal stem cells in the pathogenesis of KD. It is proposed that dysfunction of the ESC-like population localized to the endothelium of the microvessels and perivascular cells within the keloid-associated lymphoid tissues may give rise to the aberrant fibroblasts and myofibroblasts via a mesenchymal stem cell intermediate in keloid lesions, by undergoing an endothelial-to-mesenchymal transition. We also discuss the role of the renin-angiotensin system (RAS), the immune system, and the inflammatory response, on stem cell proliferation and differentiation. The understanding of the precise roles of these stem cells and interplay of the associated regulatory pathways could lead to the development of targeted therapy for this enigmatic and challenging condition. The demonstration of the expression of components of the RAS and cathepsins B, D, and G that constitute bypass loops of the RAS, by the ESC-like population, suggests that the primitive population may be a therapeutic target by modulation of the RAS, using existing medications.
INTRODUCTION
Keloid disorder (KD) is a fibroproliferative condition associated with excessive dermal collagen deposition into the extracellular matrix (ECM), in response to wounding of the skin.1,2 In contrast to hypertrophic scars, keloid lesions (KLs) extend beyond the boundaries of the original wound and rarely regress. The most common sites of predilection for KLs are the anterior chest, shoulders, upper back, and earlobes.3 These lesions present as firm, shiny, rubbery lesions and are often associated with pruritus, pain, disfigurement, and joint contracture.4 Disfiguring KLs can lead to profound functional and psychological sequelae, impacting the patient’s quality of life.5,6
KLs can occur in individuals of any ethnicity but it is most prevalent in patients with darker skin pigmentation especially those of African descent with an incidence of 6%–16%.3,7 Conversely, KLs are extremely rare in albinos.8 KD affects men and women equally, with a peak age of onset of 10–30 years.3,9 Several studies investigating patients with a positive family history of KD suggest a predominantly autosomal dominant pattern of inheritance with incomplete penetrance in the predisposition.10,11 Additionally, human leukocyte antigens polymorphisms have been implicated in KD.12 Two genome-wide association studies have also isolated 4 single-nucleotide polymorphisms associated with KD.11,13,14
The mainstay treatment for KLs includes intralesional corticosteroid injections, either as a monotherapy or in combination with other treatment modalities15 including cryotherapy, 5-flurouracil, radiotherapy, laser therapy, surgical excision, or silicon occlusive dressing. Despite a plethora of treatment options, the outcome of the treatment is often unsatisfactory with recurrence rates of 45%–100%.16,17
Stem cells are cells that possess unlimited self-renewal capacity and the ability to give rise to daughter cells capable of undergoing differentiation into specialized differentiated cells.18,19 There is increasing evidence of the involvement of stem cells, the renin-angiotensin system (RAS), and the immune system, in the pathogenesis of KD.20,21
This review outlines the characteristic of the major cell populations within KLs, focuses on the role of stem cells and their potential roles in the generation of the aberrant fibroblasts and myofibroblasts, the RAS, and the immune system, in KD.
FIBROBLASTS AND MYOFIBROBLASTS
Histologically, KLs are composed of thick, hyalinized, eosinophilic bundles of types I and III collagen fibers within a mucinous ECM.1 (Fig. 1). Fibroblasts and myofibroblasts, which are the most dominant cell types within KLs, are responsible for collagen production and wound contraction, respectively. Keloid-derived fibroblasts differ in phenotype from their normal counterparts by being more resistant to apoptosis and express more type I collagen, α-smooth muscle actin (α-SMA), and transforming growth factor-β1 (TGF-β1)-activator thrombospondin.22
Fig. 1.
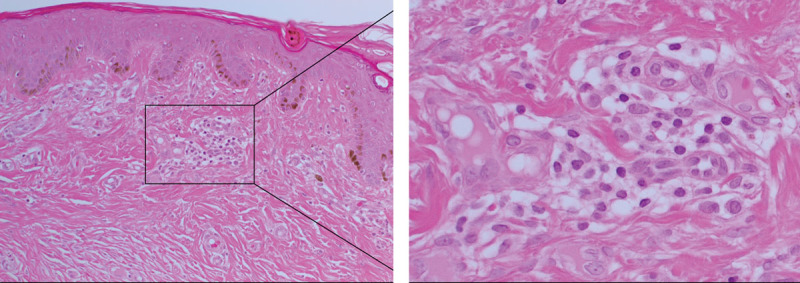
Hematoxylin & eosin staining of keloid tissue demonstrating the abundant collagen deposition within the dermis and the presence of the keloid-associated lymphoid tissues containing microvessels and inflammatory cells (inset), just beneath the epidermis. Original magnification: 200×.
The origin of myofibroblasts and fibroblasts in KLs has not been fully elucidated. Exaggerated release of both TGF-β1 and interleukin-6 has been implicated in the pathogenesis of other types of organ-related fibrotic diseases.23–26 Overproduction of TGF-β1, normally responsible for regulating fibroblasts, results in increased proliferation of fibroblasts and their subsequent differentiation into myofibroblasts, culminating in excessive production of ECM and formation of the persistent pathological scar (Fig. 2). These aberrant fibroblasts and myofibroblasts may originate from mesenchymal stem cells (MSCs) through a process of epithelial-to-mesenchymal transition or endothelial-to-mesenchymal transition (EndoMT), or from bone marrow mesenchymal precursors, as proposed for Dupuytren’s disease.27,28
Fig. 2.
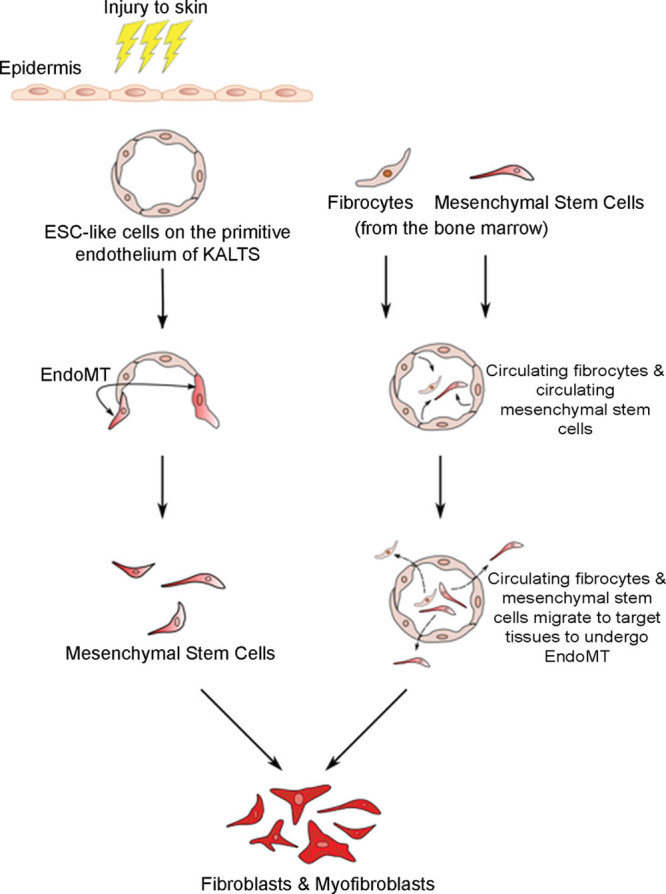
A proposed model of keloid disorder demonstrating the potential source of dysregulated fibroblasts and myofibroblasts, characteristic of keloid lesions. Following an injury, the embryonic stem cells (ESC)-like cells on the endothelium of the microvessels give rise to mesenchymal stem cells (MSCs) through a process of endothelial-to-mesenchymal transition (Endo-MT) induced by cytokines such as transforming growth factor-β1 (TGF-β1). These MSCs undergo differentiation into aberrant fibroblasts and myofibroblasts, characteristic excessive extracellular matrix deposition, and fibrosis seen in keloid lesions. This process is regulated by the renin-angiotensin system (RAS) and the immune system/inflammatory process. Through Endo-MT, these aberrant fibroblasts and myofibroblasts may also be derived from circulating MSCs and fibrocytes from the bone marrow which migrate to the target tissues.
MESENCHYMAL STEM CELLS
MSCs are multipotent stem/progenitor cells with restricted differentiation capability and capacity for self-renewal.29 These cells are defined by their capacity to differentiate down-specific lineages in vitro, into a variety of phenotypically mature and functional cells such as osteoblasts, chondrocytes, fibrocytes, and adipocytes. These processes are influenced by stimuli from their microenvironmental niche. MSCs are characterized by their ability to adhere to plastic, expression of CD73, CD90, and CD105 cell surface markers, and the lack of endothelial or hematopoietic surface markers such as CD34.30
MSCs play a vital role in attenuating inflammation and fibrosis, ultimately promoting wound healing.31 Such properties have been observed in models simulating inflammation-related fibrotic conditions such as renal and lung fibrosis and liver cirrhosis.24–26
MSCs have been isolated from the bone marrow, umbilical cord blood, placenta, and adipose tissues.29 More recently, the origin of MSCs has been linked to embryonic stem cells (ESCs) that are derived from the inner cell mass of blastocyst and they possess pluripotent capabilities.32 Several groups have successfully induced differentiation of human ESCs (hESCs) into MSCs which, in turn, differentiate into adipocytes, osteocytes, chondrocytes, and myocytes.33 Findings of Lee et al.34 suggest that neural crest cells that differentiated from the hESCs at the neural rosette stage are endowed with the plasticity for differentiation to the MSCs precursors.
EMBRYONIC STEM CELL-LIKE CELLS
Bagabir et al.35 report the presence of keloid-associated lymphoid tissues (KALTs) located just deep to the epidermis of KLs. These KALTs consist of aggregations of T cells expressing CD3 and CD4, B cells expressing CD20, macrophages expressing CD68 and CD163, and mast cells expressing c-kit, tryptase, and OX40L-FITC.35 Zhang et al.36 have identified a population of cells expressing elevated levels of the ESC markers Octamer-4 and stage specific embryonic antigen-4 (OCT4) randomly distributed in the dermal layer of keloid tissue suggesting the presence of keloid-derived stem cells in persistent pathological scars. We have recently demonstrated a primitive population of cells that expresses ESC markers OCT4, NANOG, SOX2, and pSTAT3, localized to the endothelium of microvessels and perivascular cells within the KALTs in KLs.20 We propose this putative ESC-like population as a potential source of the aberrant myofibroblasts and fibroblasts20,35,36 through a MSC intermediate.
Elevated levels of the cytokines interleukin-6 and interleukin-17 have been shown to increase the number of benign tumor-like stem cells and have been implicated in the provision of an inflammatory niche within KLs.36 It has been proposed that exposure to the inflammatory microenvironment can trigger the production of a MSC intermediate which is able to differentiate into keloid fibroblasts and myofibroblasts.
ENDOTHELIAL-TO-MESENCHYMAL TRANSITION
EndoMT is a complex process by which endothelial cells lose their unique surface markers, such as CD31 and vascular endothelial cadherin, and express products that are specific to MSCs or myofibroblasts such as α-SMA, vimentin, and type I collagen27,37 (Fig. 2). EndoMT shares many similarities with the traditional epithelial-to-mesenchymal transition. Both processes utilize the same signaling pathway and culminate in the production of cells with mesenchymal phenotypes.38 This phenotypic transformation is induced by a variety of cytokines, including TGF-β, secreted by chronic inflammatory cells such as macrophages and lymphocytes through the upregulation of the transcription factors Snail, Slug, and Twist. After losing their cell-cell adhesion, the newly formed mesenchymal cells are highly migratory and are able to infiltrate surrounding tissues.
EndoMT is essential for the structural development of the valves and septa of the heart during embryonic development.25 Several studies have shown the involvement of EndoMT in pathological tissue fibrosis affecting the kidney, heart, and lung.39,40
Lee et al.27 have demonstrated expression of the endothelial marker CD31 and the mesenchymal marker vimentin, by the endothelial cells of microvessels in the dermis of keloid tissue. Furthermore, they are able to induce EndoMT in human dermal microvascular endothelial cells using wingless protein (Wnt-3a) which suggests that keloid myofibroblasts may be of an endothelial origin. We propose that the differentiation of the ESCs-like cells by an EndoMT process through an MSC intermediate is responsible for the genesis of these aberrant fibroblasts and myofibroblasts (Fig. 2).
THE RENIN-ANGIOTENSIN SYSTEM
The RAS is an endocrine system that physiologically regulates cardiovascular homeostasis and electrolyte balance.41 A decrease in renal perfusion causes the juxtaglomerular cells in the kidneys to release the enzyme renin. Renin then induces the proteolytic cleavage of angiotensinogen to produce angiotensin I which is in turn converted to angiotensin II (ATII) by angiotensin-converting enzyme (ACE). ATII is a potent vasoactive hormone that acts through 2 main receptors namely angiotensin II receptor 1 (ATIIR1) and angiotensin II receptor 2 (ATIIR2). ATIIR1 mediates most of the classical cardiovascular and renal effects of ATII, namely blood pressure regulation and water and sodium homeostasis.42 ATIIR1 modulates the immune and inflammatory response locally resulting in the recruitment of inflammatory cells, cellular proliferation, and accumulation of ECM.42 ATIIR2 has opposing actions to that of ATIIR1.43
ATII is known to induce fibrosis in multiple organ systems through ATIIR1 signaling. This profibrotic process occurs both independently and in the presence of TGF-β, ultimately leading to cardiac, liver, and pulmonary fibrosis and systemic sclerosis.44 Research has established a positive association between increased expression of ATII and the development of pathological scarring.45,46 Stawski et al. investigated the profibrotic potential of ATII using mouse models and demonstrated induction of dermal inflammation and fibrosis in the skin via MCP1 upregulation and the accumulation of fibroblasts.47 Additionally, blockage of ATII receptors by competitive ATIIR1 antagonists has been shown to significantly reduce the pliability and vascularity of KLs and hypertrophic scars.48 Furthermore, the RAS has been implicated in the regulation of stem cell proliferation and differentiation, giving rise to dysregulated fibroblasts.49
We have recently demonstrated the expression of components of the RAS: pro-renin receptor, ACE, ATIIR1, and ATIIR2 by the ESC-like population on the endothelium of the microvessels and perivascular cells within the KALTs in KLs.21 Recent reports documenting improvement of KLs following treatment for mild hypertension with low-dose enalapril, an ACE inhibitor, further support a role for the RAS in KD.50 This suggests that the primitive population could be a novel therapeutic target by modulation of the RAS using existing medications. We have also demonstrated the expression of cathepsins B, D, and G by the ESC-like population within KLs, suggesting the existence of bypass loops for the RAS.51 These findings underscore the potential development of more effective modulation of the RAS, using existing medications.
IMMUNE SYSTEM AND INFLAMMATION
Inflammation is a protective response initiated by the body’s immune system when exposed to pathogens, infection, external injuries, or tissue stress.52 This defence mechanism is mediated by immune cells such as macrophages, mast cells, and T cells through the collaborative effect of both the innate and adaptive immune system. The innate immune system is a nonspecific, immediate, and broad response which acts as the first-line of defence against foreign microorganisms.53 The phagocytic macrophages, in response to bacterial constituents, engulf the bacterium and induce the secretion of cytokines and chemokines such as IL-1, IL-12, IL-23, and tumor necrosis factor-α (TNF-α). These proteins attract neutrophils and monocytes and initiate the inflammation process. Furthermore, the innate system plays a vital role in the activation of the adaptive immunity. Dendritic cells facilitate the adaptive response by endocytosing pathogens and acting as antigen presenting cells for the antigen-specific circulating naive T lymphocytes.54 This results in their clonal expansion and differentiation into effector cells. Some of these cells persist after the sensitizing antigen has been eliminated and are then identified as memory cells. They form the foundation for immunological memory and facilitate a more rapid and effective host response against specific pathogens when they are reencountered. There has also been increasing evidence for an important role for both the innate and adaptive immune system in tissue regeneration and repair.55
Inflammation is known to regulate several stem cell niches. MSCs have been shown to suppress the immune system through interferon-γ and the concomitant presence of the pro-inflammatory cytokines TNF-α, IL-1α, or IL-1β.56 In a rat experimental bowel fibrosis model, treatment with anti-TNF-α demonstrates a reduction in inflammation, and fibrosis and a decreased expression of pro-collagen TGF-β and insulin growth factors suggesting a possible direct pro-fibrotic effect on myofibroblasts by TNF-α.57 Additionally, selective administration of interferon-γ, a hallmark of the Th1 immune response, to myofibroblasts in an experimental renal fibrosis model, results in amelioration of fibrosis with reduced collagen synthesis and α-SMA production.58 This immunosuppressive property allows MSCs to bypass allogenic barriers and regulate hematopoietic homeostasis within the bone marrow. When exposed to the inflammatory milieu, MSCs produce growth factors such as TGF-β, epidermal growth factor, fibroblast growth factor, platelet-derived growth factor, vascular endothelial growth factor, and insulin growth factor. In turn, these compounds encourage tissue repair by stimulating a cascade of molecular events consisting inhibition of inflammatory responses, increased activity of endothelial cells and fibroblasts, and the proliferation and differentiation of tissue progenitor cells.59
Fu et al.60 have demonstrated that activated T cells, in in vitro models, simulate skeletal muscle injury and initiate muscle regeneration, through direct regulation and expansion of skeletal muscle stem cells. This further illustrates the crosstalk interaction between the inflammatory response and stem cells, thus opening possibilities for restoring tissue integrity through the regulation of the inflammatory response and its proposed effects on ESC differentiation.
CONCLUSIONS
Current treatment for KD is unsatisfactory. There is accumulating evidence demonstrating the involvement of an ESC-like population within the KALTs, which gives rise to aberrant fibroblasts and myofibroblasts, via an MSC intermediate, by an endoMT process. The expression of components of the RAS, and cathepsins B, D, and G, by this ESC-like population suggests a critical role for the RAS in regulating this primitive population. There is also increasing evidence of the involvement of the immune system and inflammatory response in stem cell dysregulation. Understanding the complex signaling pathways involved in the regulation of the disease-specific stem cells will lead to novel treatment of this hitherto unsolved medical problem.
Footnotes
Published online 16 May 2019.
Disclosure: Drs. Itinteang, Davis, and Tan are inventors of a provisional patent application Treatment of Fibrotic Conditions (PCT/NZ2016/050187). The authors are otherwise not aware of any commercial associations or financial relationships that might pose or create a conflict of interest with information presented in any submitted manuscript. Mr. Lim has no financial interest to declare in relation to the content of this article.
REFERENCES
- 1.Andrews JP, Marttala J, Macarak E, et al. Keloids: the paradigm of skin fibrosis - Pathomechanisms and treatment. Matrix Biol. 2016;51:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiao H, Zhang T, Fan J, et al. The superficial dermis may initiate keloid formation: histological analysis of the keloid dermis at different depths. Front Physiol. 2017;8:885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gauglitz GG, Korting HC, Pavicic T, et al. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med. 2011;17:113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Attar A, Mess S, Thomassen JM, et al. Keloid pathogenesis and treatment. Plast Reconstr Surg. 2006;117:286–300. [DOI] [PubMed] [Google Scholar]
- 5.Lemonas P, Ahmad Irfan, Falvey H, et al. Keloid scars: the hidden burden of disease. Pigmentary Disorders. 2015;2:231 [Google Scholar]
- 6.Bock O, Schmid-Ott G, Malewski P, et al. Quality of life of patients with keloid and hypertrophic scarring. Arch Dermatol Res. 2006;297:433–438. [DOI] [PubMed] [Google Scholar]
- 7.Chike-Obi CJ, Cole PD, Brissett AE. Keloids: pathogenesis, clinical features, and management. Semin Plast Surg. 2009;23:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang C, Murphy GF, Akaishi S, et al. Keloids and hypertrophic scars: update and future directions. Plast Reconstr Surg Glob Open. 2013;1:e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayat A, Arscott G, Ollier WE, et al. Keloid disease: clinical relevance of single versus multiple site scars. Br J Plast Surg. 2005;58:28–37. [DOI] [PubMed] [Google Scholar]
- 10.Halim AS, Emami A, Salahshourifar I, et al. Keloid scarring: understanding the genetic basis, advances, and prospects. Arch Plast Surg. 2012;39:184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shih B, Bayat A. Genetics of keloid scarring. Arch Dermatol Res. 2010;302:319–339. [DOI] [PubMed] [Google Scholar]
- 12.Brown JJ, Ollier WE, Thomson W, et al. Positive association of HLA-DRB1*15 with keloid disease in Caucasians. Int J Immunogenet. 2008;35:303–307. [DOI] [PubMed] [Google Scholar]
- 13.Nakashima M, Chung S, Takahashi A, et al. A genome-wide association study identifies four susceptibility loci for keloid in the Japanese population. Nat Genet. 2010;42:768–771. [DOI] [PubMed] [Google Scholar]
- 14.Zhu F, Wu B, Li P, et al. Association study confirmed susceptibility loci with keloid in the Chinese Han population. PLoS One. 2013;8:e62377.23667473 [Google Scholar]
- 15.Gauglitz GG. Management of keloids and hypertrophic scars: current and emerging options. Clin Cosmet Investig Dermatol. 2013;6:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arno AI, Gauglitz GG, Barret JP, et al. Up-to-date approach to manage keloids and hypertrophic scars: a useful guide. Burns. 2014;40:1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edriss AS, Mesták J. Management of keloid and hypertrophic scars. Ann Burns Fire Disasters. 2005;18:202–210. [PMC free article] [PubMed] [Google Scholar]
- 18.Watt FM, Driskell RR. The therapeutic potential of stem cells. Philos Trans R Soc Lond B Biol Sci. 2010;365:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biehl JK, Russell B. Introduction to stem cell therapy. J Cardiovasc Nurs. 2009;24:98–103; quiz 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grant C, Chudakova DA, Itinteang T, et al. Expression of embryonic stem cell markers in keloid-associated lymphoid tissue. J Clin Pathol. 2016;69:643–646. [DOI] [PubMed] [Google Scholar]
- 21.Humphries H, Brasch HD, van Schaijik B, et al. Expression of components of the renin-angiotensin system by embryonic stem cell-like population within keloid lesions. Plast Reconstr Surg. 2019. In press. [DOI] [PubMed] [Google Scholar]
- 22.Ehrlich HP, Desmoulière A, Diegelmann RF, et al. Morphological and immunochemical differences between keloid and hypertrophic scar. Am J Pathol. 1994;145:105–113. [PMC free article] [PubMed] [Google Scholar]
- 23.Pohlers D, Brenmoehl J, Löffler I, et al. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim Biophys Acta. 2009;1792:746–756. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goumans MJ, van Zonneveld AJ, ten Dijke P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: a switch to cardiac fibrosis? Trends Cardiovasc Med. 2008;18:293–298. [DOI] [PubMed] [Google Scholar]
- 26.Yue X, Shan B, Lasky JA. TGF-β: Titan of lung fibrogenesis. Curr Enzym Inhib. 2010;6:54–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee WJ, Park JH, Shin JU, et al. Endothelial-to-mesenchymal transition induced by Wnt 3a in keloid pathogenesis. Wound Repair Regen. 2015;23:435–442. [DOI] [PubMed] [Google Scholar]
- 28.Tan K, Withers AHJ, Tan ST, et al. The role of stem cells in Dupuytren’s disease: a review. Plast Reconstr Surg Glob Open. 2018;6:e1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ullah I, Subbarao RB, Rho GJ. Human mesenchymal stem cells – current trends and future prospective. Biosci Rep. 2015;35:e00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seo BF, Jung SN. The immunomodulatory effects of mesenchymal stem cells in prevention or treatment of excessive scars. Stem Cells Int. 2016;2016:6937976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pera MF, Reubinoff B, Trounson A. Human embryonic stem cells. J Cell Sci. 2000;113 (Pt 1):5–10. [DOI] [PubMed] [Google Scholar]
- 33.Barberi T, Willis LM, Socci ND, et al. Derivation of multipotent mesenchymal precursors from human embryonic stem cells. PLoS Med. 2005;2:e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee G, Kim H, Elkabetz Y, et al. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat Biotechnol. 2007;25:1468–1475. [DOI] [PubMed] [Google Scholar]
- 35.Bagabir R, Byers RJ, Chaudhry IH, et al. Site-specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br J Dermatol. 2012;167:1053–1066. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Q, Yamaza T, Kelly AP, et al. Tumor-like stem cells derived from human keloid are governed by the inflammatory niche driven by IL-17/IL-6 axis. PLoS One. 2009;4:e7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho JG, Lee A, Chang W, et al. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. 2018;9:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeisberg EM, Potenta SE, Sugimoto H, et al. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi SH, Hong ZY, Nam JK, et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin Cancer Res. 2015;21:3716–3726. [DOI] [PubMed] [Google Scholar]
- 41.Brewster UC, Setaro JF, Perazella MA. The renin-angiotensin-aldosterone system: cardiorenal effects and implications for renal and cardiovascular disease states. Am J Med Sci. 2003;326:15–24. [DOI] [PubMed] [Google Scholar]
- 42.Audoly LP, Oliverio MI, Coffman TM. Insights into the functions of type 1 (AT1) angiotensin II receptors provided by gene targeting. Trends Endocrinol Metab. 2000;11:263–269. [DOI] [PubMed] [Google Scholar]
- 43.Carey RM, Wang ZQ, Siragy HM. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension. 2000;35:155–163. [DOI] [PubMed] [Google Scholar]
- 44.Murphy AM, Wong AL, Bezuhly M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair. 2015;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.On N, Koh SP, Brasch HD, et al. Embryonic stem cell-like population in Dupuytren’s disease expresses components of the renin-angiotensin system. Plast Reconstr Surg Glob Open. 2017;5:e1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ehanire T, Ren L, Bond J, et al. Angiotensin II stimulates canonical TGF-β signaling pathway through angiotensin type 1 receptor to induce granulation tissue contraction. J Mol Med (Berl). 2015;93:289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stawski L, Han R, Bujor AM, et al. Angiotensin II induces skin fibrosis: a novel mouse model of dermal fibrosis. Arthritis Res Ther. 2012;14:R194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hedayatyanfard K, Ziai SA, Niazi F, et al. Losartan ointment relieves hypertrophic scars and keloid: A pilot study. Wound Repair Regen. 2018;26:340–343. [DOI] [PubMed] [Google Scholar]
- 49.Durik M, Sevá Pessôa B, Roks AJ. The renin-angiotensin system, bone marrow and progenitor cells. Clin Sci (Lond). 2012;123:205–223. [DOI] [PubMed] [Google Scholar]
- 50.Iannello S, Milazzo P, Bordonaro F, et al. Low-dose enalapril in the treatment of surgical cutaneous hypertrophic scar and keloid–two case reports and literature review. MedGenMed. 2006;8:60. [PMC free article] [PubMed] [Google Scholar]
- 51.Paterson C, Lee VM, Brasch HD, de Jongh J, et al. Expression of cathepsins B, D and G by the embryonic stem cell-like population within human keloid tissues and keloid-derived primary cell lines. Plast Reconstr Surg. 2019. In press. [DOI] [PubMed] [Google Scholar]
- 52.Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12:1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. 2010;125:S3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Julier Z, Park AJ, Briquez PS, et al. Promoting tissue regeneration by modulating the immune system. Acta Biomater. 2017;53:13–28. [DOI] [PubMed] [Google Scholar]
- 56.Ren G, Zhang L, Zhao X, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–150. [DOI] [PubMed] [Google Scholar]
- 57.Valatas V, Filidou E, Drygiannakis I, et al. Stromal and immune cells in gut fibrosis: the myofibroblast and the scarface. Ann Gastroenterol. 2017;30:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inagaki Y, Nemoto T, Kushida M, et al. Interferon alfa down-regulates collagen gene transcription and suppresses experimental hepatic fibrosis in mice. Hepatology. 2003;38:890–899. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Chen X, Cao W, et al. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol. 2014;15:1009–1016. [DOI] [PubMed] [Google Scholar]
- 60.Fu X, Xiao J, Wei Y, et al. Combination of inflammation-related cytokines promotes long-term muscle stem cell expansion. Cell Res. 2015;25:655–673. [DOI] [PMC free article] [PubMed] [Google Scholar]