

 Multicomponent reaction of diazocarbonyl and dibenzenesulfonimide-SCF3 reagents with BAr4 salts in the presence of Zn(NTf2)2 gives α,α′-difunctionalized trifluoromethylthio compounds.
Multicomponent reaction of diazocarbonyl and dibenzenesulfonimide-SCF3 reagents with BAr4 salts in the presence of Zn(NTf2)2 gives α,α′-difunctionalized trifluoromethylthio compounds.
Abstract
A new Zn-mediated trifluoromethylthiolation-based bifunctionalization reaction is developed. In this process, simultaneous C–SCF3 and C–C bond formation takes place in a multicomponent reaction, in which an aryl and a SCF3 group arise from different reagents. Our studies show that the reaction mechanism is similar to the Hooz multicomponent coupling. The process involves in situ generation of BAr3, which reacts with a diazocarbonyl compound, and the reaction is terminated by an electrophilic SCF3 transfer. The reaction can also be extended to fluorination based bifunctionalization which proceeds with somewhat lower yield than the analogous trifluoromethylthiolation reaction.
The Hooz multicomponent reaction is based on coupling of diazocarbonyl compounds with organoboranes and electrophiles (Scheme 1).1 This and related reactions2 involve the formation of an adduct of the diazocarbonyl and the organoboron reagent followed by 1,2-migration of an alkyl substituent from the boron and being terminated by the reaction of an electrophile with the generated boron enolate (Scheme 1). The reaction is very useful for the synthesis of α,α′-bifunctionalized carbonyl compounds with formation of one or two new carbon–carbon bonds. As a part of our research program in organofluorine chemistry, we have developed several bifunctionalization methods3 based on the introduction of F/CF3/SCF3 groups. Recently, our interest4 turned to the synthesis of α,α′-(geminal)bifunctionalized species by using diazocarbonyl compounds1d,5 and electrophilic fluorine (F/CF3/SCF3) transfer reagents in multicomponent reactions.
Scheme 1. The Hooz multicomponent reaction of organoboranes and electrophiles E+.

Development of new methods for the synthesis of SCF3 compounds is particularly important, as functionalized trifluoromethylthiolates are attractive species in pharmaceutical industry, in crop protection and even in Positron Emission Tomography (PET) based medical diagnostics.6 The broad interest in synthesis of structurally diverse SCF3 compounds6a–c,7 is based on the favorable properties of the trifluoromethylthiolation group. Trifluoromethylthiolates can modify the binding properties and lipophilicity (Hansch parameter8π = 1.43) of bioactive small molecules. For instance, the activity of cephalosporin antibiotics can be substantially improved by installing a SCF3 functionality in cefazaflur,9 and tiflorex is a more efficient anorectic drug (Fig. 1) than its –CF3 analog.10
Fig. 1. Examples of SCF3 containing drugs.

Many excellent methods have been reported recently for mono-trifluoromethylthiolation of organic substrates.6a–c,7,11 Our efforts have been focused on the development of trifluoromethylthiolation based bifunctionalization reactions to create structural complexity in a single multicomponent reaction. We have recently reported an efficient Rh-catalyzed procedure for geminal oxy-trifluoromethylthiolation of diazocarbonyl compounds (Scheme 2a).4c In the present study we aimed to develop a trifluoromethylthiolation based bifunctionalization reaction involving carbon–carbon coupling. There are relatively few reactions reported in the literature, in which bifunctionalization of diazoketones involve simultaneous C–SCF3 and C–C bond formation. A recent example is published by Wang and coworkers12 for the asymmetric trifluoromethylthiolation of sulfonium ylides via sigmatropic rearrangement (Scheme 2b), which is based on the Doyle–Kirmse13 reaction. Our idea was to develop a new multicomponent reaction, in which the SCF3 and aryl groups arise from different reagents. Considering the tremendous problems in functional group incompatibility in such multicomponent couplings, we hypothesized that the Hooz reaction (Scheme 1) could be a suitable platform for the realization of this transformation.
Scheme 2. Trifluoromethylthiolation based bifunctionalization of diazocarbonyl compounds (a–d) including reactions with C–C and C–SCF3 coupling (b–d).

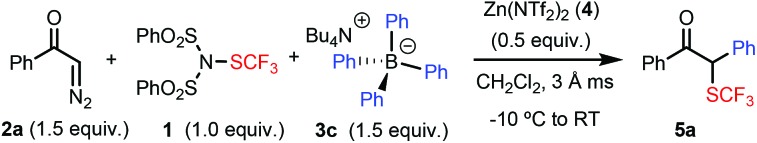
Our initial studies were performed under the typical conditions1 of Hooz multicomponent reactions (Scheme 2c). In this process, diazoketone 2a, dibenzenesulfonimide141 (as electrophile) were reacted with various organoboron species (including phenylboronic acid/boroxine and BPh3). However, the reaction proceeded with low yield (up to 15%) even under strictly inert conditions (see below). When we modified the typical conditions of the Hooz reaction by application of tetraphenylborate derivatives (such as 3c) and Zn(NTf2)2 (4), the SCF3/Ph-bifunctionalized product 5a was obtained in high yield (Scheme 2d).
After careful optimization we found that the reaction proceeds in 81% yield when excess amounts of diazoketone 2a (1.5 equiv.) and phenyl source 3c (1.5 equiv.) were reacted with SCF3 transfer reagent 1 in the presence of (0.5 equiv.) Zn-salt (4) and molecular sieves at –10 °C (Table 1, entry 1). Deviations from these optimal conditions led to decreased yields or the formation of SCF3 product 5a could not be observed. Reduction of the amount of Zn-mediator 4 (entry 2) led to a decrease of the yield of 5a (37%) and without 4, formation of 5a was not observed at all (entry 3). As mentioned above, Rh2(OAc)4 was an excellent catalyst in the geminal oxy-trifluoromethylthiolation of diazocarbonyl compounds4c (Scheme 2a). However, phenyl-trifluoromethylthiolation of 2a did not occur, when Zn-mediator 4 was replaced by a Rh-catalyst (entry 4).
Table 1. Deviation from the optimal reaction conditions for the α,α′-trifluoromethylthiolation–phenylation of diazoketone 2a a .
| ||
| Entry | Deviation from the standard conditions | Yield b (%) |
| 1 | None | 81 |
| 2 | 0.25 equiv. Zn(NTf2)2 (4) | 37 |
| 3 | Without Zn(NTf2)2 (4) | <5 |
| 4 | Rh2(OAc)4 (5 mol%) instead of 4 | 0 |
| 5 | Pd(OAc)2 (15 mol%) instead of 4 | 30 |
| 6 | Zn(OTf)2 instead of 4 | <5 |
| 7 | 0.5 equiv. of 3c | 28 |
| 8 | Na(BPh4) instead of 3c | 52 |
| 9 | (PhBO)3 (3a) instead of 3c, without 4 | 0 |
| 10 | BPh3 (3b) instead of 3c, without 4 | 15 |
| 11 | ZnPh2 instead of 3c | <5 |
| 12 | ZnPh2 instead of 4 and BPh33b instead of 3c | 0 |
| 13 | PhMe as the solvent | 71 |
| 14 | THF as the solvent | 35 |
| 15 | MeCN as the solvent | 0 |
| 16 | Without 3Å ms | 59 |
| 17 | 22 °C instead of –10 °C | 66 |
aTo reagent 1 (0.1 mmol), Bu4N(BPh4) (3c) (0.15 mmol), Zn(NTf2)2 (4) (0.05 mmol) and 80 mg 3Å molecular sieves (ms) was added a solution of diazoketone 2a (0.15 mmol) in CH2Cl2 (1.0 ml) at –10 °C. This mixture was stirred at –10 °C for 2 h before allowing it to warm up to RT overnight.
bIsolated yield.
When Pd(OAc)2 was used instead of 4, a complex reaction mixture was obtained, from which 5a could be isolated in 30% yield (entry 5). According to the analysis of the crude reaction mixture by 19F NMR, in this process a large amount (up to 35%) of Ph–SCF3 was formed indicating that two components (1 and 3c) of the three-component reaction may react directly in a Pd-catalyzed process. Other Zn-salts in place of 4, such as Zn(OTf)2, were not able to mediate the reaction (entry 6). Using Na(BPh4) instead of Bu4N(BPh4) 3c led to a decrease of the yield (52%) probably because of its poor solubility in DCM (entry 8). This gave the idea to study various solubilizing reagents together with Na(BPh4) (see below). As mentioned above, phenylboroxine 3a was inefficient as phenyl source (entry 9) and application of BPh3 (3b) (typical Hooz conditions) instead of 3c/4 led to a poor yield (15%) of 5a (entry 10). A boron-based phenyl source is important for the bifunctionalization reaction, as ZnPh2 instead of 3c gave no product 5a (entry 11). The combination of BPh3 (3b) and phenylzinc reagent ZnPh2 did not result in product 5a formation, indicating that phenyl transfer cannot happen from an external phenylzinc source in the presence of BPh3 (entry 12). We briefly screened the solvent effects as well. The reaction proceeds with good yield (71%) in toluene (entry 13) but in more polar solvent, such as in THF (entry 14) the yield is lower (35%) and we did not observe any formation of 5a in acetonitrile (entry 15). The dry conditions are apparently important for the high yield of 5a, as without molecular sieves the yield dropped to 59% (entry 16). At room temperature instead of –10 °C, the yield was decreased to 66% (entry 17).
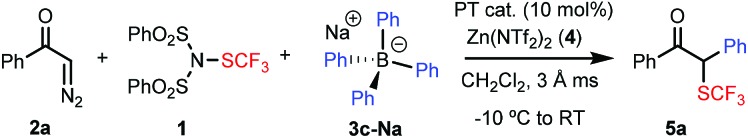
As mentioned above, the reactions proceeded with high yield with Bu4N(BPh4) 3c, which is soluble in DCM, while the yield dropped, when sparingly soluble Na(BPh4) was employed (c.f. entries 1 and 8). Therefore, we attempted to increase the yield (52%) of the reaction with Na(BPh4) using phase transfer (PT) catalysts (Table 2). Using Bu4N(BPh4) 3c and Bu4N(NTf2) in 10 mol% as PT catalyst (Table 2 entries 2 and 3) increased the yield to 66% and 64%, respectively. However, the yields with Na(BPh4) in the presence of PT catalysts were still lower than with Bu4N(BPh4) 3c as the phenyl source.
Table 2. Phase transfer experiments for the 1,1-trifluoromethyl-thiolation-phenylation a .
| ||
| Entry | Phase transfer (PT) catalyst | Yield b of 5a (%) |
| 1 | None | 52 |
| 2 | Bu4N(BPh4) | 66 |
| 3 | Bu4N(NTf2) | 64 |
aStandard conditions according to Table 1, entry 1.
bIsolated yield.
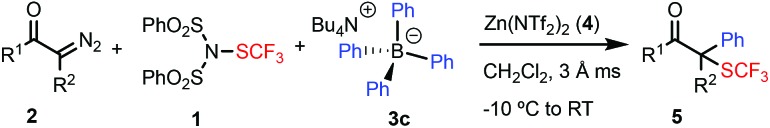
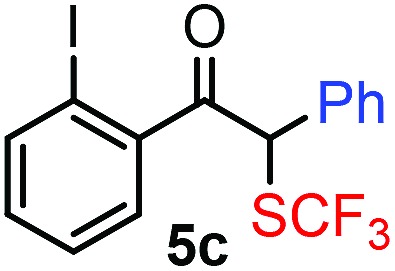
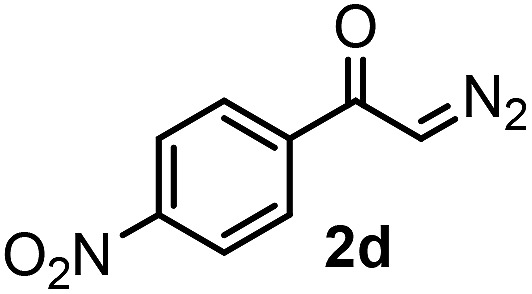
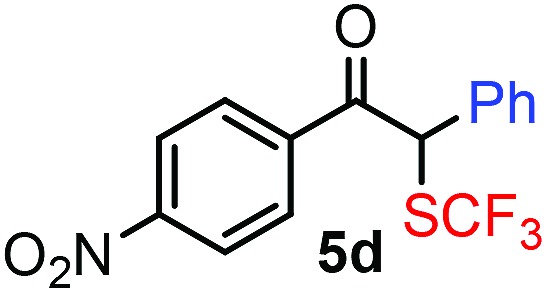
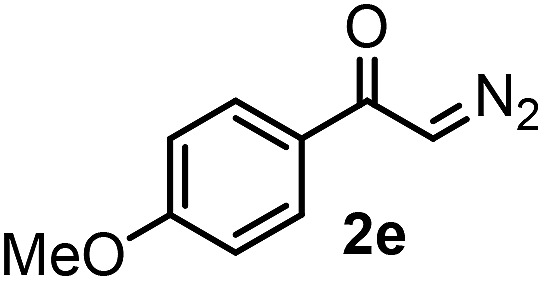
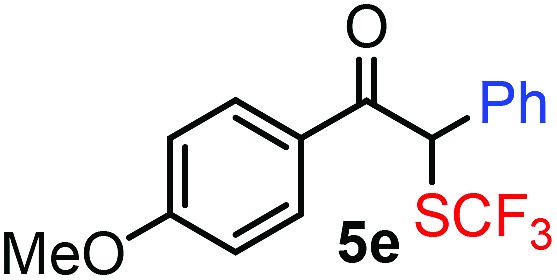
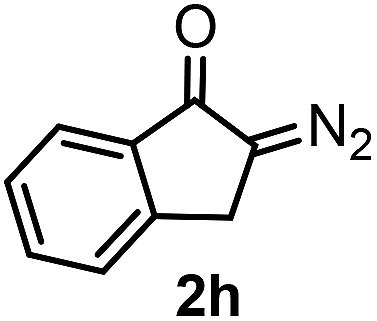
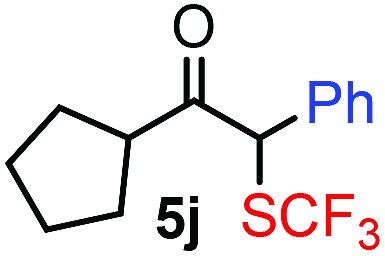
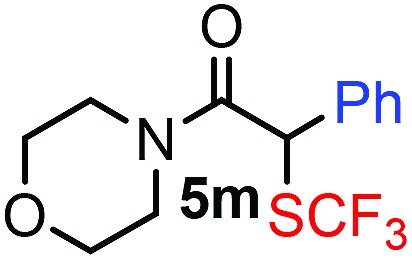
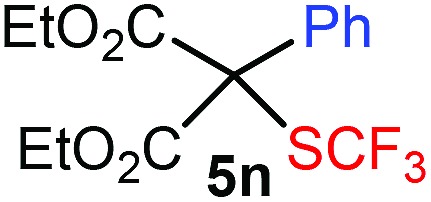
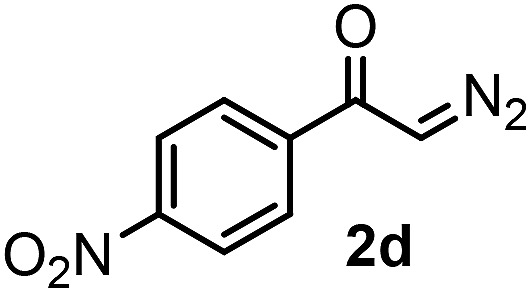
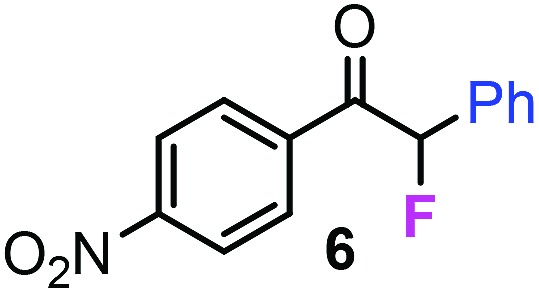
With the optimized reaction conditions in hand we investigated the substrate scope of this reaction by varying the diazocarbonyl and the organoboron reagents. Diazoketones bearing halogen or EWD nitro substituents (Table 3, entries 2–4) on the aryl ring (2b–d) reacted with high yields (73–84%), similarly to phenyl derivative 2a (entry 1), affording the corresponding SCF3 derivatives 5b–d. The aromatic iodo substituent in 2c (entry 3) remained unchanged under the reaction affording 5c, which has a useful handle for subsequent Pd-catalyzed coupling reactions. With the presence of an electron donating methoxy group (2e) in the substrate, the yield (60%) was somewhat lower than for the phenyl derivative 5a. Aryldiazoketone 2f containing a similar tetrazole motif as cefazaflur (Fig. 1) reacted smoothly providing 5f in 68% yield (entry 6). Due to the low solubility of 2f in DCM, this reaction was conducted at room temperature instead of –10 °C. Furane based diazoketone 2g reacted with high yield (84%) affording the SCF3 product 5g (entry 7). Disubstituted diazoketone 2h also underwent the phenylation–trifluoromethylthiolation reaction affording 5h, in which the phenyl and SCF3 groups are attached to a quaternary carbon (entry 8). This reaction occurred with lower yield (30%) compared to the formation of the tertiary substituted trifluoromethylthio derivatives, such as 5a, probably because of steric reasons (see below). Not only aromatic but even aliphatic diazoketones (2i–j) could be used as substrates (entries 9 and 10). The successful bifunctionalization of 2i–j could be due to the mild reaction conditions without added base leaving the α-keto hydrogens unchanged. In particular, nonyl derivative 2i reacted with high yield (83%), while the bifunctionalization of cyclopentyl derivative 2j occurred with a lower yield (48%). Unlike in the Rh-catalyzed oxy-trifluoromethylthiolation reaction,4c diazoester 2k underwent the phenylation–trifluoromethylthiolation reaction smoothly (entry 11) affording 5k in good yield (78%). This result, together with the above mentioned observation (Table 1, entry 4), shows that the trifluoromethylthiolation based bifunctionalization of diazocarbonyl compounds in Rh-catalyzed and in Hooz-type reactions proceed with substantially different mechanisms (see below). A ten-fold scale up of the reaction of diazoester 2k with 1 and 3c (entry 11) could be performed without significant change of the yield (74%). When diazoacetamide 2m was employed, bifunctionalized product 5m was obtained in high yield (entry 12). However, diazomalonate 2n could not be converted to the corresponding product 5n (entry 13), in which the phenyl and SCF3 groups would be attached to a quaternary carbon. We have briefly studied the possibilities for a phenylation–fluorination reaction (entry 14) using NFSI instead of 1 as a fluorine-electrophile source with 2d as the substrate. This reaction occurred with a lower yield (55%) than the analogous process with 1 (entry 4, 84%). In addition, fluoro derivative 6 and (4-nitrophenyl)-2-phenylethan-1-one were formed in a 1 : 1 mixture, indicating that protonation of the key reaction intermediate is about as fast as fluorination. The observation that substantial amounts of protonation product formed in the fluorination (entry 12) compared to the trifluoromethylthiolation (entry 4) reaction under similar conditions confirms our previous conclusion15 that 1 is a more efficient electrophile than NFSI in bifunctionalization reactions (see also the control experiments below). Due to their similar polarity, the separation of the mixture of the fluorinated and protonated products was cumbersome, which somewhat decreases the synthetic utility of this phenylation–fluorination-based bifunctionalization of diazoketones. The yield (55%) of 6 was determined after chromatography using a sample, which was contaminated with the protonated analog [1-(4-nitrophenyl)-2-phenylethan-1-one]. However, 6 could be further purified by selective oxidation of the protonated byproduct (see ESI†).
Table 3. 1,1-Trifluoromethylthiolation–phenylation of diazo compounds 2 with SCF3-source 1 and Bu4N(BPh)4 (3a) a .
| |||
| Entry | Diazocarbonyl compound 2 | Carbonyl compound 5 | Yield b of 5 (%) |
| 1 |
|
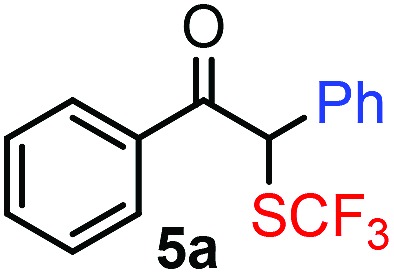
|
81 |
| 2 |
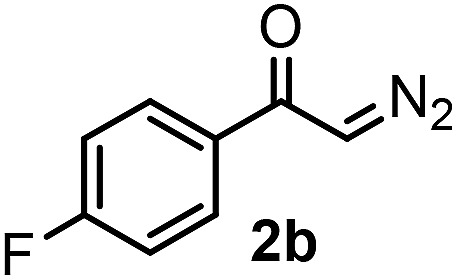
|
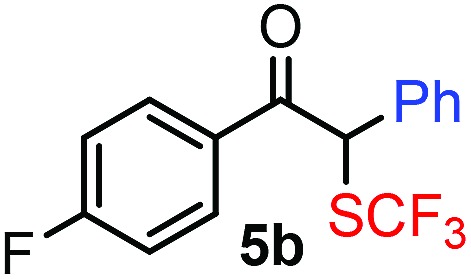
|
80 |
| 3 |
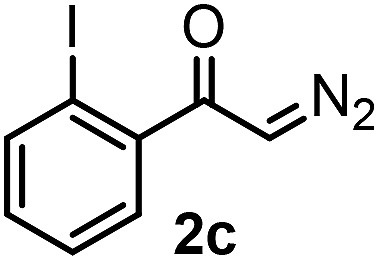
|
|
73 |
| 4 |
|
|
84 |
| 5 |
|
|
60 |
| 6 |
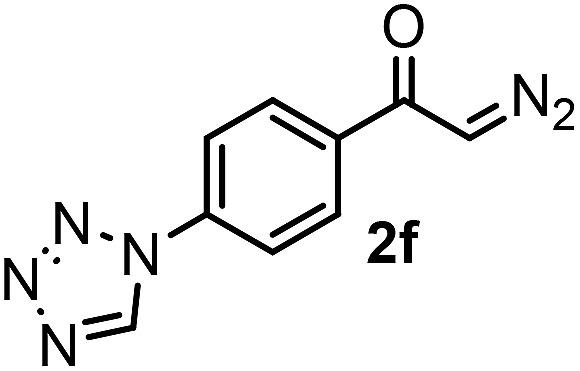
|
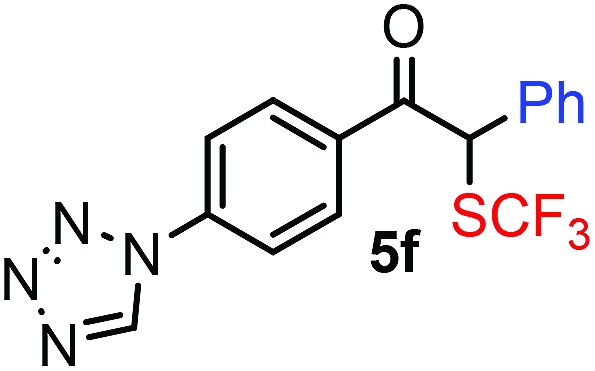
|
68 c |
| 7 |
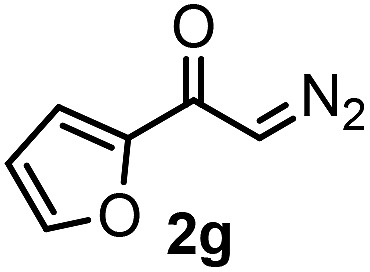
|
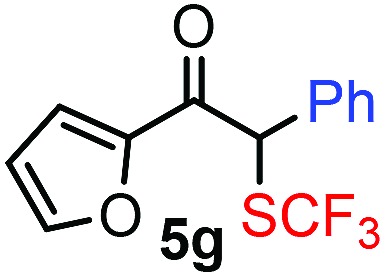
|
84 |
| 8 |
|
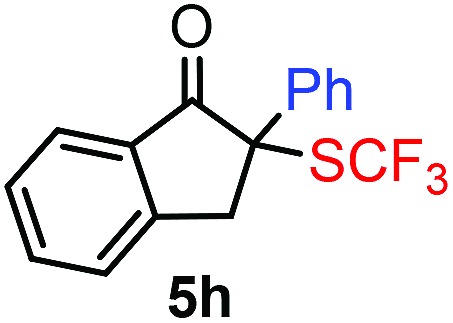
|
30 c |
| 9 |
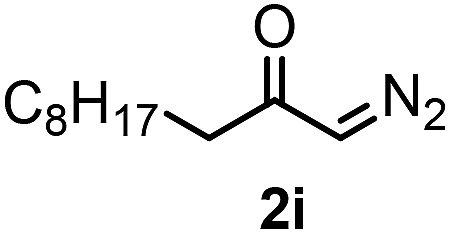
|
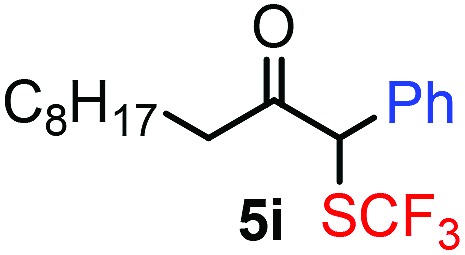
|
83 |
| 10 |
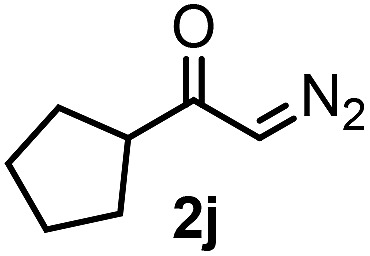
|
|
48 |
| 11 |
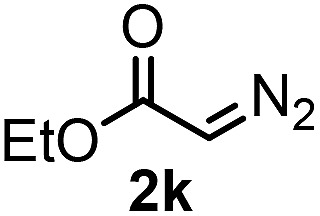
|
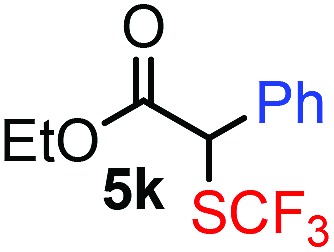
|
78(74) d |
| 12 |
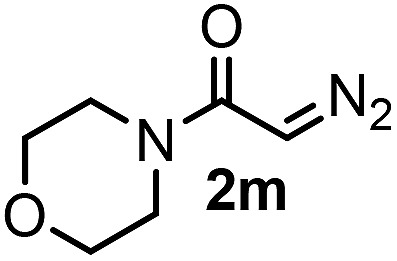
|
|
80 |
| 13 |
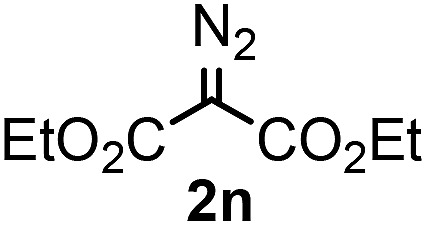
|
|
0 |
| 14 |
|
|
55 c , e , f |
aUnless otherwise stated: to 1 (0.1 mmol), Bu4N(BPh4) (3c) (0.15 mmol), Zn(NTf2)2 (4) (0.05 mmol) and 3Å ms (80 mg) was added a solution of 2 (0.15 mmol) in CH2Cl2 (1.0 ml) at –10 °C, stirred for 2 h before warmed up to RT overnight.
bUnless otherwise stated isolated yield.
cRT overnight.
d1.0 mmol scale.
eInstead of 1, NFSI was used.
fYield of 6 (determined by 1H-NMR analysis) contains side product 1-(4-nitrophenyl)-2-phenylethan-1-one.
We have also varied the aryl source, BAr4, of the bifunctionalization reaction (eqn (1)–(4)). When the phenyl substituent of 3c was replaced by other aryl groups, we had to slightly modify the reactions conditions, such as the counter ion of the BAr4 reagent and/or the reaction temperature. The yields with these reagents were lower (36–48%) than with 3c. When the chloro-phenyl derivative 3d and 1 were reacted with diazoester 2k or diazoketone 2o, the corresponding bifunctionalized products 5o and 5p were formed in 48% and 47% yields, respectively (eqn (1) and (2)). These species have one (5o) or two (5p) aromatic halogenide handles for further functionalization by cross-coupling reactions. The reaction with thiophene transfer reagent 3e was conducted at room temperature affording 5q in 45% yield (eqn (3)). Trifluoromethylthio product 5r with two different heterocyclic rings was obtained in 36% yield by reaction of 2g and 3e with 1 at room temperature (eqn (4)). We also attempted to perform alkylation–trifluoromethylthiolation reactions using Bu4N(BBu4), 3f. However, in this reaction (eqn (5)) formation of 5s was not observed, instead Bu-SCF3 (7) was formed by the rapid reaction of 1 and 3f.
 |
1 |
 |
2 |
 |
3 |
 |
4 |
 |
5 |
In order to get insights into the reaction mechanism, we performed a couple of control experiments (Scheme 3). First, we wanted to determine the sequence of the reactions among the four reaction components, such as 1, 2a, 3c and 4. Therefore, the systematic changes of the 11B NMR spectrum of the reaction of Bu4N(BPh4) 3c and Zn(NTf2)24 was monitored. Pure 3c gave a sharp 11B NMR signal at –6.6 ppm, which immediately disappeared, when an equimolar amount of Zn(NTf2)2 (4) was added (Scheme 3). The reaction of 3c and 4 led to appearance of a new, broad signal at 67.6 ppm. The value of the 11B NMR shift and the broad shape of the signal indicated formation of BPh33b from Bu4N(BPh4) 3c by boron to zinc transmetallation.16 In this process PhZnNTf2 (8) was probably also formed. Phenylzinc derivative 8 rapidly reacted with residual water reversibly adsorbed by the molecular sieves to give benzene (9), which was observed by 1H NMR in the reaction mixture. The other product of the hydrolysis of PhZnNTf2 (8) is probably Zn(OH)NTf2 (10). When the resulted reaction mixture was reacted with diazoketone 2a a new broad peak appeared at 45.6 ppm in the 11B NMR spectrum, which was assigned to vinyloxy-boronate 13.17 Species like 13 are known to form in the Hooz reaction via formation of adduct 11, followed by formation of 12 and a subsequent borotropic shift.1,17b Subsequent addition of dibenzenesulfonimide 1 to the reaction mixture led to the formation of trifluoromethylthiolation product 5a, which could be observed by 19F NMR. In this last step (13 → 5a), the Zn-mediator 4 or its hydroxy derivative 10 may play an important role. For example, the electrophilic SCF3 transfer may be accelerated by coordination of Zn to the oxygen atom of 13, which facilitates the cleavage of the boron–oxygen bond and delivery of 14. As mentioned above, the three-component reaction of 2a, BPh3 (3b) and 1 proceeds with a notoriously low yield (Scheme 2c, and Table 1/entry 10). This yield could not be improved by using strictly dry conditions in the reaction. However, a high yield was observed for a large variety of diazocarbonyl compounds, when 3c/4 conditions were used involving: (a) in situ generation of BPh3 (Scheme 3), (b) in situ removal of H2O and (c) Zn-mediated assistance of the electrophilic attack on 13.
Scheme 3. Suggested mechanism for the 1,1-trifluoro-methylthiolation–arylation of diazocarbonyl compounds. Chemical shifts (δ) are given in ppm.

Further control reactions (Scheme 4) confirmed the above mechanistic suggestions (Scheme 3). As mentioned above, Zn(OTf)2 (16), which is a close analog of Zn(NTf2)2 (4) did not mediate the reaction of 2a, 3a and 1 to form 5a (Table 1, entry 6). This may be explained by the fact that the reaction of 16 and 3c did not lead to an in situ formation of BPh33b (Scheme 4a), which is the prerequisite for the formation of 5avia11 (Scheme 3). A further confirmation of the Hooz-type reaction mechanism via vinyloxy-boronate intermediate 13 arose from the control reaction, when 1 was replaced by another electrophile, such as benzaldehyde derivative 17 (Scheme 4b). The result of this reaction was formation of 18, which most probably formed by reaction of 13 and 17 in the terminating step of the reaction. As mentioned above, the phenylation–fluorination reaction (Table 2, entry 12) with NFSI occurred with much lower yield than the corresponding phenylation–trifluoromethylthiolation with 1 (Table 2, entry 4). In the fluorination reaction, significant amounts of protonation product formed presumably because of competing electrophilic protonation of 13. This led us to the conclusion that 1 is a better electrophile than NFSI in this bifunctionalization reaction (see above). Indeed, when we performed a competitive reaction of 2a, 3a and equimolar amounts of 1 and NFSI, we observed exclusive formation of the trifluoromethylthiolated product 5a without formation of the fluorinated product 19.
Scheme 4. Control experiments to support the mechanism given in Scheme 3.

An alternative to the above Hooz-type mechanism could be an initial reaction of the PhZn species 8 directly with the diazo compound 2a without involvement of BPh3. However, this hypothesis seems to be in conflict with the attempted phenylation–trifluoromethylthiolation reactions with ZnPh2 species without application of Zn(NTf2)24 (Table 1, entries 11 and 12). In these reactions the bifunctionalized product 5a did not form.
In this report, we have described a new arylation–trifluoromethylthiolation reaction for an α,α′-bifunctionalization of diazocarbonyl compounds. This process can be performed as a multicomponent reaction, in which the aryl and SCF3 groups arise from different reagents, from 3 and 1. This Hooz-type coupling is a novel approach for bifunctionalization based trifluoromethylthiolation, as for example the previously reported4c Rh-catalyzed oxy-trifluoromethylthiolation. The arylation–trifluoromethylthiolation reaction is initiated by Zn assisted formation of BAr3 from 3. According to our mechanistic studies the reaction follows a Hooz-type reaction mechanism, which is terminated by electrophilic SCF3 transfer from 1. As far as we know this is the first Hooz-type reaction for the synthesis of organofluorines from electrophilic transfer reagents. The reaction can also be extended to the phenylation–fluorination process, using the fluorine analog (NFSI) of 1.
Conflict of interest
The authors declare no competing financial interest.
Supplementary Material
Acknowledgments
We thank the Knut och Alice Wallenbergs Foundation (Dnr: 2018.0066) and Swedish Research Council (VR) for financial support.
Footnotes
†Electronic supplementary information (ESI) available: Experimental procedures, characterization and NMR spectra of the products. See DOI: 10.1039/c9sc00829b
References
- (a) Hooz J., Linke S. J. Am. Chem. Soc. 1968;90:5936. [Google Scholar]; (b) Hooz J., Linke S. J. Am. Chem. Soc. 1968;90:6891. [Google Scholar]; (c) Sun C.-L., Shi Z.-J. Chem. Rev. 2014;114:9219. doi: 10.1021/cr400274j. [DOI] [PubMed] [Google Scholar]; (d) Li H., Zhang Y., Wang J. Synthesis. 2013;45:3090. [Google Scholar]; (e) Paraja M., Plaza M., Valdés C. Synlett. 2017;28:2373. [Google Scholar]; (f) Barluenga J., Valdés C. Angew. Chem., Int. Ed. 2011;50:7486. doi: 10.1002/anie.201007961. [DOI] [PubMed] [Google Scholar]
- (a) Peng C., Zhang W., Yan G., Wang J. Org. Lett. 2009;11:1667. doi: 10.1021/ol900362d. [DOI] [PubMed] [Google Scholar]; (b) Barluenga J., Tomás-Gamasa M., Aznar F., Valdés C. Nat. Chem. 2009;1:494. doi: 10.1038/nchem.328. [DOI] [PubMed] [Google Scholar]; (c) He Z., Zajdlik A., Yudin A. K. Dalton Trans. 2014;43:11434. doi: 10.1039/c4dt00817k. [DOI] [PubMed] [Google Scholar]
- (a) Janson P. G., Ghoneim I., Ilchenko N. O., Szabó K. J. Org. Lett. 2012;14:2882. doi: 10.1021/ol3011419. [DOI] [PubMed] [Google Scholar]; (b) Ilchenko N. O., Janson P. G., Szabo K. J. J. Org. Chem. 2013;78:11087. doi: 10.1021/jo401831t. [DOI] [PubMed] [Google Scholar]; (c) Ilchenko N. O., Tasch B. O. A., Szabó K. J. Angew. Chem., Int. Ed. 2014;53:12897. doi: 10.1002/anie.201408812. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ilchenko N. O., Cortés M. A., Szabo K. J. ACS Catal. 2016;6:447. [Google Scholar]; (e) Ilchenko N. O., Hedberg M., Szabo K. J. Chem. Sci. 2017;8:1056. doi: 10.1039/c6sc03471c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yuan W., Szabó K. J. Angew. Chem., Int. Ed. 2015;54:8533. doi: 10.1002/anie.201503373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Yuan W., Eriksson L., Szabó K. J. Angew. Chem., Int. Ed. 2016;55:8410. doi: 10.1002/anie.201602137. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yuan W., Szabó K. J. ACS Catal. 2016;6:6687. [Google Scholar]; (c) Lübcke M., Yuan W., Szabó K. J. Org. Lett. 2017;19:4548–4551. doi: 10.1021/acs.orglett.7b02139. [DOI] [PubMed] [Google Scholar]
- (a) Doyle M. P., Duffy R., Ratnikov M., Zhou L. Chem. Rev. 2010;110:704. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]; (b) Ford A., Miel H., Ring A., Slattery C. N., Maguire A. R., McKervey M. A. Chem. Rev. 2015;115:9981. doi: 10.1021/acs.chemrev.5b00121. [DOI] [PubMed] [Google Scholar]; (c) Xia Y., Qiu D., Wang J. Chem. Rev. 2017;117:13810. doi: 10.1021/acs.chemrev.7b00382. [DOI] [PubMed] [Google Scholar]; (d) Xiao Q., Zhang Y., Wang J. Acc. Chem. Res. 2013;46:236. doi: 10.1021/ar300101k. [DOI] [PubMed] [Google Scholar]; (e) Guo X., Hu W. Acc. Chem. Res. 2013;46:2427. doi: 10.1021/ar300340k. [DOI] [PubMed] [Google Scholar]
- (a) Xu X.-H., Matsuzaki K., Shibata N. Chem. Rev. 2015;115:731. doi: 10.1021/cr500193b. [DOI] [PubMed] [Google Scholar]; (b) Shao X., Xu C., Lu L., Shen Q. Acc. Chem. Res. 2015;48:1227. doi: 10.1021/acs.accounts.5b00047. [DOI] [PubMed] [Google Scholar]; (c) Rossi S., Puglisi A., Raimondi L., Benaglia M. ChemCatChem. 2018;10:2717. [Google Scholar]; (d) Zhang J., Yang J.-D., Zheng H., Xue X.-S., Mayr H., Cheng J.-P. Angew. Chem., Int. Ed. 2018;57:12690. doi: 10.1002/anie.201805859. [DOI] [PubMed] [Google Scholar]; (e) Li M., Zheng H., Xue X.-s., Cheng J.-p. Tetrahedron Lett. 2018;59:1278. [Google Scholar]; (f) Verhoog S., Kee C. W., Wang Y., Khotavivattana T., Wilson T. C., Kersemans V., Smart S., Tredwell M., Davis B. G., Gouverneur V. E. J. Am. Chem. Soc. 2018;140:1572. doi: 10.1021/jacs.7b10227. [DOI] [PubMed] [Google Scholar]
- Tlili A., Billard T. Angew. Chem., Int. Ed. 2013;52:6818. doi: 10.1002/anie.201301438. [DOI] [PubMed] [Google Scholar]
- Hansch C., Leo A., Taft R. W. Chem. Rev. 1991;91:165. [Google Scholar]
- DeMarinis R. M., Boehm J. C., Dunn G. L., Hoover J. R. E., Uri J. V., Guarini J. R., Phillips L., Actor P., Weisbach J. A. J. Med. Chem. 1977;20:30. doi: 10.1021/jm00211a006. [DOI] [PubMed] [Google Scholar]
- (a) Silverstone T., Fincham J., Plumley J. Br. J. Clin. Pharmacol. 1979;7:353. doi: 10.1111/j.1365-2125.1979.tb00945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Giudicelli J., Richer C., Berdeaux A. Br. J. Clin. Pharmacol. 1976;3:113. doi: 10.1111/j.1365-2125.1976.tb00578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Guyon H., Chachignon H., Tognetti V., Joubert L., Cahard D. Eur. J. Org. Chem. 2018;2018:3756. [Google Scholar]; (b) Xu C., Ma B., Shen Q. Angew. Chem., Int. Ed. 2014;53:9316. doi: 10.1002/anie.201403983. [DOI] [PubMed] [Google Scholar]; (c) Alazet S., Ismalaj E., Glenadel Q., Le Bars D., Billard T. Eur. J. Org. Chem. 2015;2015:4607. [Google Scholar]
- Zhang Z., Sheng Z., Yu W., Wu G., Zhang R., Chu W.-D., Zhang Y., Wang J. Nat. Chem. 2017;9:970. doi: 10.1038/nchem.2789. [DOI] [PubMed] [Google Scholar]
- Doyle M. P., Tamblyn W. H., Bagheri V. J. Org. Chem. 1981;46:5094. [Google Scholar]
- Zhang P., Li M., Xue X.-S., Xu C., Zhao Q., Liu Y., Wang H., Guo Y., Lu L., Shen Q. J. Org. Chem. 2016;81:7486. doi: 10.1021/acs.joc.6b01178. [DOI] [PubMed] [Google Scholar]
- Mai B. K., Szabó K. J., Himo F. Org. Lett. 2018;20:6646. doi: 10.1021/acs.orglett.8b02628. [DOI] [PubMed] [Google Scholar]
- (a) Boudier A., Darcel C., Flachsmann F., Micouin L., Oestreich M., Knochel P. Chem.–Eur. J. 2000;6:2748. doi: 10.1002/1521-3765(20000804)6:15<2748::aid-chem2748>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]; (b) Hupe E., Knochel P., Szabó K. J. Organometallics. 2002;21:2203. [Google Scholar]; (c) Jimeno C., Sayalero S., Fjermestad T., Colet G., Maseras F., Pericàs M. A. Angew. Chem., Int. Ed. 2008;47:1098. doi: 10.1002/anie.200703103. [DOI] [PubMed] [Google Scholar]; (d) Bedford R. B., Gower N. J., Haddow M. F., Harvey J. N., Nunn J., Okopie R. A., Sankey R. F. Angew. Chem., Int. Ed. 2012;51:5435. doi: 10.1002/anie.201202219. [DOI] [PubMed] [Google Scholar]
- (a) Mukaiyama T., Inomata K., Muraki M. J. Am. Chem. Soc. 1973;95:967. [Google Scholar]; (b) Pasto D. J., Wojtkowski P. W. Tetrahedron Lett. 1970;11:215. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.