

Abstract
A series of benzimidazole carboxamide derivatives have been synthesized and characterized by 1H-NMR, 13C-NMR and HRMS. PARP inhibition assays and cellular proliferation assays have also been carried out. Compounds 5cj and 5cp exhibited potential anticancer activities with IC50 values of about 4 nM against both PARP-1 and PARP-2, similar to the reference drug veliparib. The two compounds also displayed slightly better in vitro cytotoxicities against MDA-MB-436 and CAPAN-1 cell lines than veliparib and olaparib, with values of 17.4 µM and 11.4 µM, 19.8 µM and 15.5 µM, respectively. The structure-activity relationship based on molecular docking was discussed as well.
Keywords: poly(ADP-ribose) polymerase, PARP enzyme inhibition, benzimidazole carboxamide
1. Introduction
Poly(ADP-ribose) polymerase-1 (PARP-1) is a kind of enzyme closely involved in the DNA damage repair process. The PARP family contains 18 subtypes, while only PARP-1 and PARP-2 contain a DNA binding domain which facilitates the recognition and localization of the DNA damage site [1]. In DNA damage repairing process, PARP-1 and PARP-2 catalyze the degradation of nicotinamide adenine dinucleotide (NAD+) to nicotinamide and ADP-ribose, and the synthesis of poly(ADP-ribose) on the acceptor proteins by the formed ADP-ribose as substrate, which is the essential process in the repair of DNA [2,3,4]. The inhibition of PARP will cause synthetic lethal effects in cells, which makes PARP a hot target in cancer therapy [5].
Because of the similarities in structure to the natural substrate nicotinamide, such heterocyclic derivatives as quinazoline, phenanthridone, phthalazine and benzimidazole were developed as different generations of PARP inhibitor scaffolds [4,6,7]. so far, there are four approved parp inhibitors—olaparib, Niraparib, Rucaparib and Talazoparib (Figure 1)—which are on the market as chemotherapy drugs. Olaparib, based on the 2H-phthalazin-1-one scaffold, was the first FDA-approved oral PARP inhibitor drug for therapy of BRCA-mutated cancer in women with recurrent ovarian cancer [8,9]. The clinical trial of the drug combination of olaparib and taxol for terminal gastric cancer treatment finally failed in 2018. Niraparib, a PARP inhibitor derived from the indazole carboxamide lead developed by Merck, was approved by the FDA in 2017 for treatment of Pt-sensitive recurring (PSR) ovarian and peritoneal cancers without the limitation of BRCA mutations [10,11,12]. Besides competitively inhibiting catalytic active site of PARP-1, niraparib has the strongest ability to capture PARP-1 on DNA chains [13]. Rucaparib, another PARP inhibitor on the market derived from tricyclic indolactam, was the first PARP inhibitor put into clinical trials, and then approved in 2016 by the FDA for the treatment of BRCA-mutated terminal ovarian cancer [14]. Rucaparib was approved for maintenance treatment of PSR ovarian cancer in 2018. Talazoparib, consisting of a 2H-phthalazin-1-one scaffold, like olaparib, was approved for the treatment of patients carrying germ line BRCA-mutated, HER2 negative ovarian cancer in late 2018 [15].
Figure 1.

Structures of approved PARP inhibitors on the market.
The benzimidazole carboxamide scaffold is the basis of an effective series of PARP-1 inhibitors due to its relatively low molecular weight and high intrinsic potency, which made veliparib (ABT-888) a promising PARP inhibitor lead compound for antitumor drugs [14,16,17,18,19]. The amide group acts as an analogue of nicotinamide to bind with the PARP-1 active site. The intermolecular hydrogen bond interactions between the amide bond and Gly-863 and Ser-904 residues in the active site of PARP-1 contribute crucially to the potency, along with the π-π interaction with Tyr-907. However, as shown in the X-ray co-crystal structure of veliparib with PARP-1 (Figure 2), veliparib does not form distinct interactions with the residues in the hydrophobic pocket of PARP-1 [16]. In addition, veliparib exhibits a relatively low ability to trap PARP-1 on DNA and relatively low cellular activity in cancer cells, as compared with other PARP-1 inhibitors [13,20,21].
Figure 2.

X-Ray co-crystal structure of veliparib with PARP-1 [16].
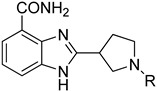
In this report, we designed a series of benzimidazole carboxamide derivatives based on veliparib, in which an aromatic ring substituting alkyl side chain was attached to the nitrogen atom in the five-member cyclic amine, in order to improve the combination of the compound and the active site of PARP. A phenyl group was attached to the terminal of the side chain for the sake of increasing the membrane permeability. Since PARP inhibitors will cause synthetic lethal effect specifically in BRCA-mutated cells, two cell lines, namely, MDA-MB-436 (a BRCA-1-mutated breast cancer cell line) and CAPAN-1 (a BRCA-2-mutated pancreatic cancer cell line) were selected to conduct cell proliferation assay. We discovered two potent PARP-1 and PARP-2 inhibitors exhibiting good potency in these two cell lines, against which veliparib and olaparib exhibited a relatively lower potency.
2. Results and Discussion
2.1. Chemistry
The benzimidazole ring was constructed by a ten-step large scale synthesis procedure as described previously (Scheme 1) [17,22]. Cbz-protected cyclic amine carboxylic ester B1_6 was hydrolyzed to give acid B1. Then B1 was coupled with 2,3-diaminobenzamide dihydrochloride under N,N′-carbonyldimidazole catalysis to give the amide product N1, which was then refluxed in acetic acid to produce Cbz-benzimidazole carboxamide N2. The Cbz protecting group was removed under hydrogenolysis conditions to provide a secondary amine N3. Nucleophilic substitutions using different chloro-substituted aromatic side chains on basic condition gave tertiary amine N4. Based on the structure of N4, we synthesized the target compounds.
Scheme 1.

The synthetic protocol of the compounds.
2.2. PARP Inhibition Assay
In this report, we describe a series of benzimidazole carboxamide-containing PARP inhibitors in which a side chain has been introduced at the point of attachment of five-member cyclic amine expected to improve the activity. Table 1 shows the PARP-1 and PARP-2 inhibition assay results. The inhibition percentages at 10 nM of 16 compounds were measured, followed by IC50 values for the six compounds that showed relatively higher inhibition potency. Compounds 5cc, 5ch, 5ci, 5cj, 5co and 5cp showed relatively good PARP-1 inhibition potency, with IC50 values near or lower than 10 nM. By comparing IC50 of 5cd, 5cp and 5cc, in which side chain contains 2-4 carbon length of phenylketone fragments, 5cp exhibited the much better potency than 5cc and 5cd, indicating that the style/position of carbonyl group, as well as its interactions with residues in the binding pocket may affect the inhibition. The docking study showed that 5cp could bind to three important amino acid residues, Gly-863, Ser-904 and Glu-988, but 5cc and 5cd only interact with two of these residues, lacking the hydrogen bond with Glu-988. However, reduction of carbonyl group on 5cd and 5cp to a hydroxy group in 5cf and 5cg respectively, led to a dramatic decrease of the enzyme inhibition activities. The probable reason is that the binding pocket occupied by five-member ring prefers a lipophilic group (not shown in the docking diagram), so it is easy to understand why compounds 5cl and 5cm containing hydrophilic amine groups displayed lower enzyme potency. Moreover, 5ci, 5cj and 5cp with different substituents in the para position of the benzene ring exhibited much better enzyme potency that all the other compounds, as well as veliparib. Compounds 5ca and 5cb, 5cn and 5co containing a N-phenylamine group or N-benzamide group, all displayed some enzyme inhibition activities, but slight lower than those phenylketone compounds 5cc, 5cd, 5ci, 5cj and 5cp. Compounds 5ce and 5ck both containing phthalimide group, a bulky group with large steric hindrance, displayed different enzyme inhibition activities, whose structure-activity relationship needs to be proved by more experiments.
Table 1.
PARP inhibition assay.
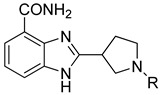
| Compound | R | PARP-1 Inhibition % (10 nM) | PARP-2 Inhibition % (10 nM) | PARP-1 IC50 (nM) | PARP-2 IC50 (nM) |
|---|---|---|---|---|---|
| 5ca |
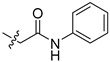
|
17.6 | 34.5 | / | / |
| 5cb |
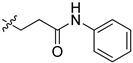
|
19.9 | 41.2 | / | / |
| 5cc |
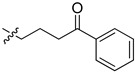
|
42.8 | 62.3 | 12.2 | 5.8 |
| 5cd |
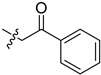
|
−1.1 | 7.2 | / | / |
| 5ce |
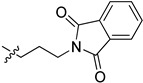
|
22.0 | 74.5 | / | / |
| 5cf |
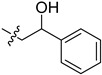
|
5.0 | 9.0 | / | / |
| 5cg |
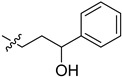
|
−2.1 | 6.5 | / | / |
| 5ch |
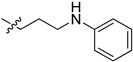
|
47.5 | 66.2 | 7.1 | 3.3 |
| 5ci |
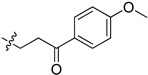
|
59.2 | 62.5 | 5.9 | 4.5 |
| 5cj |
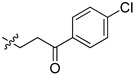
|
65.7 | 65.6 | 3.9 | 4.2 |
| 5ck |
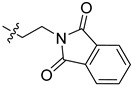
|
6.0 | 37.1 | / | / |
| 5cl |
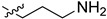
|
26.8 | 46.4 | / | / |
| 5cm |
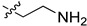
|
24.8 | 32.8 | / | / |
| 5cn |
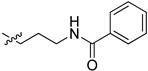
|
19.4 | 39.1 | / | / |
| 5co |
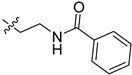
|
38.4 | 66.2 | 11.1 | 5.7 |
| 5cp |
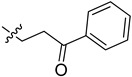
|
68.0 | 76.7 | 3.6 | 3.2 |
| pc1 | Veliparib | 63.7 | 78.3 | 5.3 | 1.6 |
2.3. Cell Proliferation Assay
Table 2 shows the results of cell proliferation assay of the analogues. Compared with compounds 5cd and 5cc, 5cp showed the lowest IC50, which indicated that the side chain with three-carbon alkyl group exhibits the highest activity, while nitrogen atoms in the side chains decreased the potency, especially in CAPAN-1 cell line, as was shown in compounds 5ca, 5cb and 5co. The terminal benzene group also played a vital part in exhibiting activity which facilitates membrane permeability, indicated in compounds 5cc, 5cp, 5cl and 5cm. Meanwhile, by comparing 5cc, 5cp, 5cf and 5cg, it was concluded that carbonyl group facilitated increasing the activity, since the reduction of the carbonyl to a hydroxy group greatly weakened the cellular potency. Compound 5cj showed an even lower IC50 than 5cp, which indicated that electron-withdrawing groups enhanced the activity, while electron-donating group in 5ci impaired the activity, as compared with 5cp.
Table 2.
Cell proliferation assay.
| Compound ID | R | MDA-MB-436 IC50 (μM) | CAPAN-1 IC50 (μM) |
|---|---|---|---|
| 5ca |
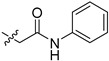
|
22.9 | >100 |
| 5cb |
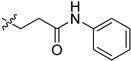
|
90.4 | >100 |
| 5cc |
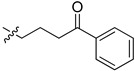
|
31.9 | 20.7 |
| 5cd |
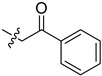
|
55.0 | 82.5 |
| 5ce |
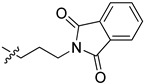
|
>100 | >100 |
| 5cf |
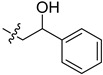
|
61.0 | >100 |
| 5cg |
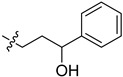
|
74.1 | >100 |
| 5ch |
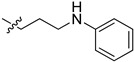
|
>100 | >100 |
| 5ci |
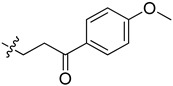
|
38.6 | 48.1 |
| 5cj |
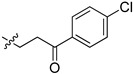
|
17.4 | 11.4 |
| 5ck |
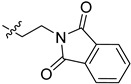
|
>100 | >100 |
| 5cl |

|
>100 | >100 |
| 5cm |
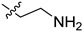
|
>100 | >100 |
| 5cn |
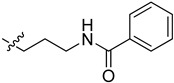
|
>100 | >100 |
| 5co |
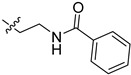
|
>100 | >100 |
| 5cp |
|
19.8 | 15.5 |
| pc1 | Veliparib | >100 | >100 |
| pc2 | Olaparib | 30.2 | >100 |
2.4. Molecular Docking
The two compounds exhibiting the best potency, namely 5cj and 5cp, were chosen for molecular docking. As is shown in the molecular docking diagram of compound 5cj with PARP-1 (Figure 3), the target molecule 5cj was appropriately inserted into the catalytic active site of PARP-1.
Figure 3.

Molecular docking of 5cj with PARP-1.
The amide group acts as hydrogen bond donor and acceptor to form intermolecular hydrogen bonds with Gly-863 and Ser-904, respectively. The electron-rich benzimidazole ring forms π-π stacking interactions with Tyr-907. The nitrogen atom in the meta-position of the amide group serves as a hydrogen bond donor to form a hydrogen bond with Glu-988. Compared with the X-ray co-crystal structure diagram of veliparib with PARP-1, the side chain of 5cj was inserted into the hydrophobic pocket in the active site of PARP-1. Though the docking diagram of 5cj did not show more intermolecular interactions distinctly besides the hydrogen bonding interactions existing in the crystal structure diagram of veliparib, there may exist Van der Waals interactions or hydrophobic interactions between the side chain and the residues in the active site of PARP-1, which facilitated the binding of 5cj with PARP-1, and thus made 5cj a better PARP-1 inhibitor, as compared with veliparib. Docking mode of 5cp is similar to that of 5cj (Figure 4), except the orientation of the side chain, which may be due to the influence of the chlorine atom in the benzene ring.
Figure 4.

Molecular docking of 5cp with PARP-1.
3. Materials and Methods
3.1. Genereral Informations
NMR spectra (1H 400 MHz, 13C 101 MHz) were obtained on Bruker 400 spectrometer (Karlsruhe, Germany) with the indicated solvent and internal standard. Chemical shifts are given in delta (δ) values and coupling constants (J) in Hertz (Hz). The following abbreviations are used for peak multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broadened. Mass spectra were performed on a Waters Micromass Q-TOF Premier Mass Spectrometer (Milford, MA, USA) running as a flow injection acquisition. All solvents and reagents were obtained from commercial sources and used without further purification. Details for the 1H-NMR, 13C-NMR and HRMS of compounds 5ca–5cp are provided in the section “Supplementary Materials”.
3.2. Chemistry
3.2.1. Procedure A: Synthesis of 2-(Pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (N3)
Step 1: Preparation of N-benzyl-1-(trimethylsilyl)-methanamine (B1_2). A solution of B1_1(32 g, 261.1 mmol) and (chloromethyl)trimethylsilane (C1, 84 g, 783.9 mmol) in acetonitrile (500 mL) was stirred and heated at reflux overnight. The mixture was neutralized with 0.6 M NaOH solution (600 mL) and extracted by diethyl ether. The organic phase was dried over Na2SO4 and concentrated. The mixture was separated by column chromatography (silica gel, EtOAc/PE=1:10, Rf value 0.10) to give the title compound (20.62 g, 41%).
Step 2: Preparation of N-benzyl-1-methoxy-N-((trimethylsilyl)methyl)methanamine (B1_3). A solution of methanol (3.99 g, 124.1 mmol) and formalin (10.1 g, 124 mmol) was cooled to 0 °C. B1_2 (20 g, 103.4 mmol) was added and the mixture was stirred for 0.5 h. The mixture was warmed to room temperature and stirred for 3 h. K2CO3 (14.3 g, 103.4 mmol) was added and the mixture was stirred for 1 h. The supernatant was transferred to another reaction flask. K2CO3 (5.0 g, 36.3 mmol) was added. The mixture was filtered, and the filter cake was rinsed by diethyl ether. The organic phase was concentrated to give the title compound (22.08 g, 89.9%).
Step 3: Preparation of methyl 1-benzylpyrrolidine-3-carboxylate (B1_4). A solution of B1_3 (22.06 g, 92.7 mmol) and methyl acrylate (C2, 11.54 g, 139.0 mmol) in DCM (100 mL) was cooled to 0 °C. A solution of trifluoroacetic acid (12.71 g, 111.2 mmol) in DCM (50 mL) was added slowly by drop to the reaction mixture. The solution was warmed to room temperature and stirred for 17 h. NaHCO3 saturated solution (150 mL) was added to the solution and stirred until no gas was produced. The mixture was partitioned between water and DCM. The organic phase was washed with brine, dried over Na2SO4 and concentrated to give the title compound (19.53 g, 96%). 1H-NMR (CDCl3) δ 7.35–7.23 (m, 5H), 3.69 (s, 3H), 3.64 (s, 2H), 3.12–2.99 (m, 1H), 2.98–2.87 (m, 1H), 2.79–2.69 (m, 1H), 2.69–2.57 (m, 1H), 2.58–2.48 (m, 1H), 2.17–2.04 (m, 2H). MS (ESI, pos, ion): 219.9 [M + H]+.
Step 4: Preparation of methylpyrrolidine-3-carboxylate hydrochloride (B1_5). A solution of B1_4 (19.50 g, 88.99 mmol), HCl dioxane solution (22.3 mL, 88.99 mmol) in methanol (150 mL) was treated with 10% Pd/C (1.95 g) and stirred at 50 °C under hydrogen atmosphere for 5 h. The solid was filtered off and the filtrate was concentrated to give the title compound (13.85 g, 94%).
Step 5: Preparation of 1-benzyl-3-methylpyrrolidine-1,3-dicarboxylate (B1_6). The solution of B1_5 (13.85 g, 83.6 mmol) in NaHCO3 saturated aqueous solution (200 mL) was added by toluene (200 mL) and cooled to 0 °C. Benzyl chloroformate (13.83 mL, 83.6 mmol) was added by drop. The mixture was warmed to room temperature and stirred for 6 h, and then partitioned between water and toluene. The organic phase was washed with brine, dried over Na2SO4, and concentrated to give the title compound (20.03 g, 91%).
Step 6: Preparation of 1-((benzyloxy)carbonyl)pyrrolidine-3-carboxylic acid (B1). A solution of B1_6 (20.03 g, 76.1 mmol) in THF (200 mL) was added by water (150 mL) and stirred. A solution of LiOH (3.64 g, 152.2 mmol) in water (100 mL) was added by drop. The mixture was heated and stirred at 60 °C for 5 h and the THF was removed. The mixture was extracted by EtOAc. And the aqueous phase was acidified to pH 2 by 2N HCl and partitioned between EtOAc and water. The organic phase was washed with brine, dried over Na2SO4 and concentrated to give the title compound (16.57 g, 87.4%).
Step 7: Preparation of benzyl 3-((2-amino-3-carbamoylphenyl)carbamoyl)pyrrolidine-1-carboxylate (N1). A solution of B1 (16.57 g, 66.5 mmol) in pyridine (60 mL) and DMF (60 mL) was stirred with N,N′-carbonyldiimidazole (CDI, 11.76 g, 72.5 mmol) at 45 °C for 1 h. 2,3-Diaminobenzamide dihydrochloride (A1, 13.54 g, 60.4 mmol) was added and the mixture stirred at 45 °C temperature for 24 h. After concentration, the residue was partitioned between EtOAc and aqueous NaHCO3. The solid was collected and washed with water and dried to give the titled compound (11.97 g, 52%).
Step 8: Preparation of benzyl 3-(4-carbamoyl-1H-benzo[d]imi-dazol-2-yl)pyrolidine-1-carboxylate (N2). A suspension of N1 (11.97 g, 31.3 mmol) in AcOH (85 mL) was heated at reflux for 2 h. After cooling, the solution was concentrated and the residue partitioned between EtOAc and aqueous NaHCO3. The organic layer was washed with water and concentrated to give the title compound (6.79 g, 59%).
Step 9: Preparation of 2-(pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (N3). A solution of N2 (6.79 g, 40 mmol) in MeOH (250 mL) was treated with 10% Pd/C (0.54 g) and stirred at 50 °C under hydrogen atmosphere for 5 h. The solid was filtered off and the filtrate was concentrated. The residue was stirred in petroleum ether and then filtered to give the titled compound (4.43 g, 100%). 1H NMR (400 MHz, DMSO) δ 7.52–7.30 (m, 2H), 7.13–6.94 (m, 1H), 3.77–3.33 (m, 2.5H), 3.24–3.01 (m, 2.5H), 2.39–2.21 (m, 1H), 2.11–1.96 (m, 1H). MS calcd for C12H15N4O, [M + H]+, 231.1, found 231.1.
3.2.2. Procedure B: Synthesis of 5ca, 5cb, 5cc, 5cd, 5ce, 5ch, 5ci, 5cj, 5ck and 5cp
2-(1-(2-Oxo-2-(phenylamino)ethyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5ca). A solution of N3 (200 mg, 0.87 mmol), 2-chloro-N-phenylacetamide (221 mg, 1.30 mmol) in DMF (5 mL) was treated with K2CO3 (240 mg, 1.74 mmol) and KI (14.4 mg, 0.09 mmol) at 50 °C for 3 h. The solution was concentrated, and the residue was purified by column chromatography (silica gel, DCM/MeOH = 30:1, Rf value 0.22) to give the title compound (111 mg, 35%). 1H-NMR (methanol-d4) δ 7.86 (d, J = 7.6 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.54–7.49 (m, 2H), 7.31–7.24 (m, 3H), 7.11–7.05 (m, 1H), 3.85–3.76 (m, 1H), 3.52–3.39 (m, 2H), 3.17 (dd, J = 6.7, 2.4 Hz, 2H), 3.11–3.03 (m, 1H), 2.93–2.84 (m, 1H), 2.53–2.41 (m, 1H), 2.34 (m 1H). 13C-NMR (MeOD) δ 169.26, 158.72, 137.71, 128.44, 124.09, 122.23, 121.48, 119.98, 58.66, 58.29, 53.45, 37.37, 30.02. HRMS calcd for C20H21N5O2, [M + H]+, 364.1769, found 364.1768.
2-(1-(3-Oxo-3-(Phenylamino)propyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5cb). The title compound was prepared according to procedure B using 4-chloro-1-phenylbutan-1-one in place of 2-chloro-N-phenylacetamide (21%). 1H-NMR (methanol-d4) δ 7.86 (d, J = 7.6 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.53–7.40 (m, 2H), 7.35–7.16 (m, 3H), 7.10–6.97 (m, 1H), 3.79 (dt, J = 13.1, 6.3 Hz, 1H), 3.20 (d, J = 7.4 Hz, 2H), 3.11–2.96 (m, 3H), 2.96–2.84 (m, 1H), 2.67 (t, J = 6.9 Hz, 2H), 2.53–2.28 (m, 2H). 13C-NMR (MeOD) δ 171.18, 169.20, 158.26, 138.29, 128.37, 123.79, 122.27, 121.48, 119.80, 58.22, 53.03, 51.21, 37.01, 34.86, 29.55. HRMS calcd for C21H23N5O2, [M + H]+, 378.1926, found 378.1926.
2-(1-(4-Oxo-4-phenylbutyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5cc). The title compound was prepared according to procedure B using 4-chloro-1-phenylbutan-1-one in place of 2-chloro-N-phenylacetamide (40%). 1H NMR (methanol-d4) δ 8.07–7.97 (m, 2H), 7.88 (d, J = 7.7 Hz, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.65–7.57 (m, 1H), 7.50 (dd, J = 7.6 Hz, 2H), 7.32 (dd, J = 8.9, 6.7 Hz, 1H), 3.81–3.65 (m, 1H), 3.16 (dd, J = 11.1, 7.8 Hz, 2H), 3.04–2.77 (m, 4H), 2.74–2.61 (m, 2H), 2.50–2.37 (m, 1H), 2.35–2.23 (m, 1H), 2.02 (dd, J = 7.2 Hz, 2H). 13C-NMR (MeOD) δ 158.48, 132.79, 128.30, 127.72, 122.16, 121.39, 58.39, 55.02, 53.28, 48.44, 48.23, 36.97, 29.68, 22.85. HRMS calcd for C22H24N4O2, [M + H]+, 377.1973, found 377.1980.
2-(1-(2-Oxo-2-phenylethyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxa-mide (5cd). The title compound was prepared according to procedure B using 2-chloro-1-phenylethan-1-one in place of 2-chloro-N-phenylacetamide in acetone solution (24%). 1H-NMR (DMSO-d6) δ 12.71 (br, 1H), 9.30 (br, 1H), 8.05–7.93 (m, 2H), 7.79 (d, J = 7.6 Hz, 1H), 7.73–7.59 (m, 3H), 7.51 (dd, J = 7.7 Hz, 2H), 7.25 (dd, J = 7.8 Hz, 1H), 4.21–4.03 (m, 2H), 3.79–3.64 (m, 1H), 3.26–3.13 (m, 1H), 3.02–2.85 (m, 2H), 2.79 (td, J = 8.6, 6.0 Hz, 1H), 2.37–2.15 (m, 2H). 13C-NMR (DMSO) δ 197.41, 167.05, 158.73, 136.19, 133.68, 133.25, 129.71, 129.11, 128.43, 122.39, 121.74, 114.79, 61.70, 58.92, 53.65, 40.43, 37.39, 29.47. HRMS calcd for C20H20N4O2, [M + H]+, 349.1660, found 349.1669.
2-(1-(3-(1,3-Dioxoisoindolin-2-yl)propyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5ce). The title compound was prepared according to procedure B using 2-(3-bromopropyl)- isoindoline-1,3-dione in place of 2-chloro-N-phenylacetamide (57%). 1H-NMR (methanol-d4) δ 7.88–7.80 (m, 3H), 7.78–7.73 (m, 2H), 7.66 (dd, J = 8.0, 1.1 Hz, 1H), 7.28 (dd, J = 7.8 Hz, 1H), 3.84–3.76 (m, 2H), 3.70–3.60 (m, 1H), 3.11–3.02 (m, 1H), 2.98 (dd, J = 9.5, 6.5 Hz, 1H), 2.84–2.72 (m, 2H), 2.70–2.60 (m, 2H), 2.40–2.28 (m, 1H), 2.21–2.11 (m, 1H). 13C-NMR (MeOD) δ 169.26, 168.58, 158.51, 133.89, 132.00, 122.64, 122.19, 121.40, 58.28, 53.22, 53.03, 36.94, 35.83, 29.74, 26.83. HRMS calcd for C23H23N5O3, [M + H]+, 418.1875, found 418.1873.
2-(1-(3-(Phenylamino)propyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5ch). The title compound was prepared according to procedure B using N-(3-chloropropyl)aniline in place of 2-chloro-N-phenylacetamide (28%). 1H NMR (400 MHz, DMSO-d6) δ 9.19 (br, 1H), 7.76 (d, J = 7.5 Hz, 1H), 7.62 (d, J = 7.5 Hz, 2H), 7.23 (t, J = 7.8 Hz, 1H), 7.00 (t, J = 7.7 Hz, 2H), 6.47 (dd, J = 16.1, 7.8 Hz, 3H), 5.54 (br, 1H), 3.66 (dd, J = 9.6, 6.9 Hz, 2H), 3.02 (dd, J = 7.0 Hz, 2H), 2.86–2.76 (m, 1H), 2.70 (dd, J = 7.9 Hz, 2H), 2.57 (dd, J = 7.4 Hz, 2H), 2.33–2.13 (m, 2H), 1.72 (p, J = 7.0 Hz, 2H).13C NMR (101 MHz, DMSO) δ 167.12, 159.00, 149.47, 129.29, 122.36, 121.72, 115.87, 112.41, 59.22, 55.32, 53.82, 41.70, 37.19, 29.92, 28.18. HRMS calcd for C21H23N5O2, [M + H]+, 364.2133, found 364.2138.
2-(1-(3-(4-Methoxyphenyl)-3-oxopropyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5ci). The title compound was prepared according to procedure B using 3-chloro-1-(4-methoxy- phenyl)propan-1-one in place of 2-chloro-N-phenylacetamide (33%). 1H-NMR (chloroform-d) δ 11.36 (br, 1H), 9.65 (br, 1H), 8.14–7.93 (m, 3H), 7.64 (d, J = 7.9 Hz, 1H), 7.30 (dd, J = 4.9, 3.8 Hz, 1H), 7.04–6.89 (m, 2H), 3.90–3.83 (m, 3H), 3.80–3.70 (m, 1H), 3.36–3.07 (m, 6H), 2.84–2.71 (m, 1H), 2.55–2.35 (m, 2H), 2.12–2.01 (m, 1H). 13C-NMR (MeOD) δ 197.88, 197.83, 169.21, 163.98, 158.17, 130.20, 129.50, 122.24, 121.47, 113.51, 58.43, 54.67, 53.43, 50.53, 36.97, 36.37, 29.79. HRMS calcd for C22H24N4O3, [M + H]+, 393.1922, found 393.1919.
2-(1-(3-(4-Chloroxyphenyl)-3-oxopropyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5cj). The title compound was prepared according to procedure B using 3-chloro-1-(4-chlorophenyl)- propan-1-one in place of 2-chloro-N-phenylacetamide (20%). 1H-NMR (chloroform-d) δ 11.37 (br, 1H), 9.63 (br, 1H), 8.03–7.84 (m, 3H), 7.63 (d, J = 7.9 Hz, 1H), 7.46–7.40 (m, 2H), 7.30–7.23 (m, 1H), 3.77–3.68 (m, 1H), 3.36–3.00 (m, 6H), 2.77 (dd, J = 9.4, 6.7 Hz, 1H), 2.54–2.35 (m, 2H), 2.11–1.99 (m, 1H). 13C-NMR (MeOD) δ 197.96, 169.22, 158.18, 139.22, 135.19, 129.44, 128.58, 122.24, 121.46, 120.12, 116.39, 58.45, 53.41, 50.09, 37.02, 30.55, 29.35. HRMS calcd for C21H21N4O2Cl, [M + H]+, 397.1427, found 397.1428.
2-(1-(2-(1,3-Dioxoisoindolin-2-yl)ethyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5ck). The title compound was prepared according to procedure B using 2-(2-bromoethyl)- isoindoline-1,3-dione in place of 2-chloro-N-phenylacetamide (72%). 1H-NMR (methanol-d4) δ 7.88–7.78 (m, 3H), 7.78–7.72 (m, 2H), 7.62 (d, J = 8.0 Hz, 1H), 7.27 (dd, J = 7.8 Hz, 1H), 3.95–3.83 (m, 2H), 3.77–3.66 (m, 1H), 3.25–3.11 (m, 2H), 3.03–2.82 (m, 4H), 2.46–2.34 (m, 1H), 2.29–2.17 (m, 1H). 13C-NMR (MeOD) δ 169.19, 168.50, 158.24, 133.87, 132.03, 122.66, 122.21, 121.41, 58.20, 53.51, 53.23, 37.01, 36.13, 29.61. HRMS calcd for C22H21N5O3, [M + H]+, 404.1718, found 404.1719.
2-(1-(3-Oxo-3-phenylpropyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5cp). The title compound was prepared according to procedure B using 3-chloro-1-phenylpropan-1-one in place of 2-chloro-N-phenylacetamide (77%). 1H-NMR (- DMSO-d6) δ 9.10 (br, 1H), 8.05–7.94 (m, 2H), 7.80 (d, J = 7.5 Hz, 1H), 7.77–7.60 (m, 3H), 7.60–7.48 (m, 2H), 7.34–7.19 (m, 1H), 4.08–3.96 (m, 1H), 3.93–3.79 (m, 1H), 3.72–3.60 (m, 3H), 3.60–3.43 (m, 4H), 2.62–2.49 (m, 1H), 2.44–2.27 (m, 1H). 13C-NMR (DMSO) δ 197.43, 166.96, 155.69, 136.48, 134.16, 129.38, 129.31, 129.03, 128.70, 128.53, 122.69, 122.36, 122.13, 56.90, 53.56, 49.87, 36.59, 35.11, 29.83. HRMS calcd for C21H22N4O2, [M + H]+, 363.1817, found 363.1815.
3.2.3. Procedure C: Synthesis of 5cf and 5cg
2-(1-(2-Hydroxy-2-phenylethyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5cf). A solution of 5cd (30 mg, 0.14 mmol) in MeOH (5 mL) was treated by sodium borohydride (10 mg, 0.28 mmol) and stirred at room temperature for 4 h. The solution was concentrated, and the residue was extracted by EtOAc. The organic phase was concentrated to give the title compound (28 mg, 94%). 1H-NMR (methanol-d4) δ 7.87 (d, J = 7.6 Hz, 1H), 7.70–7.63 (m, 1H), 7.46–7.39 (m, 2H), 7.39–7.25 (m, 4H), 4.87–4.82 (m, 1H), 3.81–3.63 (m, 1H), 3.22–3.01 (m, 2.5H), 2.98–2.79 (m, 2.5H), 2.77–2.64 (m, 1H), 2.49–2.36 (m, 1H), 2.29–2.15 (m, 1H). 13C-NMR (MeOD) δ 169.29, 158.90, 143.36, 127.97, 127.16, 125.77, 122.17, 121.38, 100.00, 72.05, 63.33, 58.81, 53.76, 37.17, 29.35. HRMS calcd for C20H22N4O2, [M + H]+, 351.1817, found 351.1821.
2-(1-(3-Hydroxy-3-phenylpropyl)pyrrolidin-3-yl)-1H-benzo[d]imi-dazole-4-carboxamide (5cg). The title compound was prepared according to procedure C using 5cp in place of 5cd (72%). 1H-NMR (methanol-d4) δ 7.86 (dd, J = 7.7, 1.0 Hz, 1H), 7.67 (dd, J = 8.0, 1.1 Hz, 1H), 7.41–7.21 (m, 6H), 4.81–4.76 (m, 1H), 3.87–3.74 (m, 1H), 3.25–3.13 (m, 1H), 3.05–2.98 (m, 2H), 2.96–2.86 (m, 1H), 2.85–2.75 (m, 1H), 2.54–2.43 (m, 1H), 2.39–2.27 (m, 1H), 2.12–1.94 (m, 3H).13C-NMR (MeOD) δ 158.42, 144.70, 127.88, 126.84, 125.50, 122.18, 121.36, 72.80, 58.52, 53.36, 52.76, 37.03, 36.96, 29.28. HRMS calcd for C21H24N4O2, [M + H]+, 365.1973, found 365.1981.
3.2.4. Procedure D: Synthesis of 5cl and 5cm
2-(1-(3-Aminopropyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5cl).A solution of 5ce (500 mg, 1.33 mmol) in EtOH (10 mL) was added by 80% hydrazine hydrate (832 mg, 13.3 mmol), and the solution was heated at reflux for 4 h. The solution was concentrated and the residue was purified by column chromatography (silica gel, MeOH/EtOAc = 1:1, Rf value 0.18) to give the title compound 199 mg (52%). 1H-NMR (methanol-d4) δ 7.86 (dd, J = 7.7, 1.1 Hz, 1H), 7.66 (dd, J = 8.0, 1.0 Hz, 1H), 7.28 (dd, J = 7.8 Hz, 1H), 3.80–3.65 (m, 1H), 3.11 (dd, J = 9.6, 8.0 Hz, 1H), 2.95 (dd, J = 9.6, 6.8 Hz, 1H), 2.88–2.71 (m, 4H), 2.71–2.53 (m, 2H), 2.46–2.35 (m, 1H), 2.33–2.18 (m, 1H), 1.75 (p, J = 7.3 Hz, 2H).13C-NMR (MeOD) δ 169.29, 158.58, 122.16, 121.40, 120.00, 116.49, 58.56, 53.48, 53.44, 39.49, 36.93, 30.50, 29.71. HRMS calcd for C16H21N5O, [M + H]+, 288.1820, found 288.1524.
2-(1-(2-Aminoethyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5cm). The titled compound was prepared according to procedure D using 5ck in place of 5ce (210 mg, 62%). 1H-NMR (methanol-d4) δ 7.88–7.84 (m, 1H), 7.68–7.64 (m, 1H), 7.28 (dd, J = 7.8 Hz, 1H), 3.78–3.67 (m, 1H), 3.08 (dd, J = 9.5, 7.9 Hz, 1H), 2.97 (dd, J = 9.5, 6.5 Hz, 1H), 2.89–2.75 (m, 4H), 2.72–2.61 (m, 2H), 2.47–2.34 (m, 1H), 2.29–2.18 (m, 1H).13C-NMR (MeOD) δ 169.30, 158.75, 121.39, 119.94, 116.55, 58.68, 57.63, 53.36, 39.51, 37.01, 29.83. HRMS calcd for C14H19N5O, [M + H]+, 274.1664, found 274.1674.
3.2.5. Procedure E: Synthesis of 5cn and 5co
2-(1-(3-Benzamidopropyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5cn). A solution of 5cl (150 mg, 0.52 mmol) in DMF (5 mL) was added by benzoyl chloride (95 mg, 0.68 mmol) in ice bath. The solution was stirred for 2 h, and then concentrated. The residue was purified by column chromatography (silica gel, DCM/EtOAc = 1:1, Rf value 0.24) to give the title compound (85 mg, 42%). 1H-NMR (methanol-d4) δ 7.87 (dd, J = 7.7, 1.0 Hz, 1H), 7.85–7.79 (m, 2H), 7.69 (dd, J = 8.0, 1.1 Hz, 1H), 7.56–7.49 (m, 1H), 7.47–7.41 (m, 2H), 7.31 (dd, J = 7.9 Hz, 1H), 3.99–3.88 (m, 1H), 3.60–3.48 (m, 4H), 3.30–3.17 (m, 2H), 3.11–3.01 (m, 2H), 2.65–2.52 (m, 1H), 2.44–2.32 (m, 1H), 2.09–1.97 (m, 2H). 13C-NMR (MeOD) δ 169.27, 156.57, 133.92, 131.42, 128.20, 126.87, 122.48, 121.75, 57.66, 53.56, 53.23, 37.09, 36.61, 29.66, 26.99. HRMS calcd for C22H25N5O2, [M + H]+, 392.2082, found 392.2085.
2-(1-(2-Benzamidoethyl)pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (5co). The title compound was prepared according to procedure E using 5cm in place of 5cl (145 mg, 71%). 1H-NMR (methanol-d4) δ 7.93–7.85 (m, 3H), 7.68 (d, J = 7.9 Hz, 1H), 7.55 (d, J = 7.3 Hz, 1H), 7.47 (dd, J = 7.6 Hz, 2H), 7.31 (dd, J = 7.8 Hz, 1H), 4.12–4.03 (m, 1H), 4.01–3.87 (m, 2H), 3.80 (t, J = 5.9 Hz, 2H), 3.71–3.57 (m, 2H), 3.56–3.47 (m, 2H), 2.74–2.62 (m, 1H), 2.52–2.40 (m, 1H). 13C-NMR (MeOD) δ 169.64, 168.97, 155.15, 133.40, 131.73, 128.26, 127.12, 122.65, 121.95, 57.48, 55.25, 54.12, 36.51, 36.36, 29.49. HRMS calcd for C21H23N5O2, [M + H]+, 378.1926, found 378.1921.
3.3. PARP Inhibition Assay
The PARP-1 and PARP-2 inhibition assays were performed in outsourcing, by a CRO company, Shanghai Medicilon Inc. (Shanghai, China). The PARP-1 and PARP-2 inhibitory activities of the compounds were measured using PARP-1 Chemiluminescent Assay Kit (BPS Bioscience, catalog 80569, San Diego, CA, USA) and PARP-2 Chemiluminescent Assay Kit (BPS Bioscience, catalog 80552), respectively, according to the manufacturer’s instructions. Briefly, PARP-1 or PARP-2 biotinylated substrate was incubated with compounds or solvent control at varying concentrations and an assay buffer containing the PARP-1 or PARP-2 enzyme. After then, the plate was treated with streptavidin-HRP followed by addition of the HRP substrate and the luminescent signal was measured using a chemiluminescence reader (Perkin Elmer Envision 2104 Multi Label Microplate Reader, Waltham, MA, USA).
3.4. Cell Proliferation Assay
The MDA-MB-436 and CAPAN-1 cell lines were provided by Sundia MediTech Company, Ltd., Shanghai, China. A three-day assay was conducted. A cell suspension was prepared. Cell density was counted by an automated cell counter and then diluted to the required density, according to seeding density. 50 μL cells were seeded into 384-well plate in growth medium according to the plate map. The cells were incubated at 37 °C, 5% CO2 overnight.
200× compound solution in DMSO was prepared and diluted with growth medium to 26× final concentration by addition of 26 μL 200× compounds to 174 μL growth medium. Then 2 μL of diluted compound solution was added to cells and the cell was incubated at 37 °C, 5% CO2 for 72 h.
The assay plate was equilibrated to room temperature prior to measurement. 15 μL of CellTiter-Glo Reagent was added into each well and the contents were mixed for 2 min on an orbital shaker to induce cell lysis. The cells were incubated at room temperature for 60 min to stabilize luminescent signal. Luminescence was recorded on the Envision system.
3.5. Molecular Docking
The molecular docking experiment was based on the molecular modeling package SYBYL-X 1.3 (Tripos associate Inc., St. Louis, MO, USA). The 3D structure of PARP-1 receptor for the molecular docking study was downloaded from Protein Data Bank (PDB ID: 2RD6) [16]. The selected ligands were compound 5cj and 5cp in this experiment. The molecular docking experiment was implemented in four steps: (1) The water molecules in the protein crystal structure downloaded from PDB were deleted; (2) The energy of receptor and ligand was optimized; (3) The intrinsic ligand in the binding site of the receptor was deleted, and then the selected target compound was docked into the receptor according to Sybyl-X modules; (4) The molecular docking results obtained by the above steps were handled and analyzed.
4. Conclusions
In summary, a series of benzimidazole carboxamide derivatives have been synthesized and their PARP inhibition activities and cellular activities have been detected. These compounds displayed good PARP inhibition potency, as much as veliparib and olaparib. Compound 5cj exhibited the best activity among 16 synthesized compounds, with IC50 of 3.9 nM against PARP-1 and 4.2 nM against PARP-2. Furthermore, compound 5cj showed significant cytotoxicity against such two BRCA-mutated cell lines. To be specific, as for the cytotoxicity against MDA-MB-436, the IC50 of 5cj is 17.4 μM, much lower than 30.2 μM of olaparib; and especially, as for the cytotoxicity against CAPAN-1, the IC50 of 5cj is 11.4 μM, by contrast, veliparib and olaparib exhibited low potency within the three-day cell proliferation assay. Molecular docking study of 5cj indicated that besides the hydrogen bond interactions formed by the benzimidazole carboxamide scaffold similar to Veliparib, the side chain of 5cj being inserted into the hydrophobic pocket in the active site of PARP-1 leads to a different binding mode.
Supplementary Materials
The following are available online, 1H-NMR, 13C-NMR and HRMS of compounds 5ca–5cp.
Author Contributions
W.W., C.Z. and Y.J. designed the experiments. R.M., M.W., L.T., H.Z. and D.C. performed the experiments. R.M. analyzed the data and wrote the paper. C.Z. and W.W. reviewed and edited the paper. H.Z. acquired funding. All authors have read and approved the manuscript.
Funding
This work was financially supported by Shenzhen Sci. & Tech. Bureau, Project No. JCYJ20170816170342439 and JCYJ20170413113448742.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 5ca–5cp are available from the authors.
References
- 1.Virág L., Szabó C. The therapeutic potential of poly(ADP-ribose)polymerase inhibitors. Pharmacol. Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 2.Jagtap P., Szabó C. Poly(ADP-Ribose)polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 3.Ame´ J.-C., Spenlehauer C., de Murcia G. The PARP superfamily. BioEssays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 4.Burkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. BioEssays. 2001;23:795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- 5.Lord C.J., Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almahli H., Hadchity E., Jaballah M.Y., Daher R., Ghabbour H.A., Kabil M.M., Al-shakliah N.S., Eldehna W.M. Development of novel synthesized phthalazinone-based PARP-1 inhibitors with apoptosis inducing mechanism in lung cancer. Bioorg. Chem. 2018;77:443–456. doi: 10.1016/j.bioorg.2018.01.034. [DOI] [PubMed] [Google Scholar]
- 7.Malyuchenko N.V., Kotova E.Y., Kulaeva O.I., Kirpichnikov M.P., Studitskiy V.M. PARP1 inhibitors: Antitumor drug design. Acta Nat. 2015;7:27–37. [PMC free article] [PubMed] [Google Scholar]
- 8.Menear K.A., Adcock C., Boulter R., Cockcroft X.L., Copsey L., Cranston A., Dillon K.J., Drzewiecki J., Garman S., Gomez S., et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phtha-lazin-1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 2008;51:6581–6591. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 9.Loh V.M., Cockcroft X.L., Dillon K.J., Dixon L., Drzewiecki J., Eversley P.J., Gomez S., Hoare J., Kerrigan F., Matthews I.T.W., et al. Phthalazinones. Part 1: The design and synthesis of a novel series of potent inhibitors of poly(ADP ribose) polymerase. Bioorg. Med. Chem. Lett. 2005;15:2235–2238. doi: 10.1016/j.bmcl.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 10.Scott L.J. Niraparib: First global approval. Drugs. 2017;77:1029–1034. doi: 10.1007/s40265-017-0752-y. [DOI] [PubMed] [Google Scholar]
- 11.Kanjanapan Y., Lheureux S., Oza A.M. Niraparib for the treatment of ovarian cancer. Expert Opin. Pharmaco. 2017;18:631–640. doi: 10.1080/14656566.2017.1297423. [DOI] [PubMed] [Google Scholar]
- 12.Jones P., Altamura S., Boueres J., Ferrigno F., Fonsi M., Giomini C., Lamartina S., Monteagudo E., Ontoria J.M., Orsale M.V., Palumbi M.C., et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): A novel oral poly (ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J. Med. Chem. 2009;52:7170–7185. doi: 10.1021/jm901188v. [DOI] [PubMed] [Google Scholar]
- 13.Murai J., Huang S.Y., Das B.B., Renaud A., Zhang Y., Doroshow J.H., Ji J., Takeda S., Pommier Y. Trapping of PARP-1 and PARP-2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canan Koch S.S., Thoresen L.H., Tikhe J.G., Maegley K.A., Almassy R.J., Li J., Yu X.-H., Zook S.E., Kumpf R.A., et al. Novel tricyclic poly(ADP-ribose) polymerase-1 inhibitors with potent anticancer chemopotentiating activity: Design, synthesis, and X-ray cocrystal structure. J. Med. Chem. 2002;45:4961–4974. doi: 10.1021/jm020259n. [DOI] [PubMed] [Google Scholar]
- 15.Brown J., Kaye S., Yap T. PARP inhibitors: The race is on. Brit. J. Cancer. 2016;114:713–715. doi: 10.1038/bjc.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Penning T.D., Zhu G.-D., Gandhi V.B., Gong J., Liu X., Shi Y., Klinghofer V., Johnson E.F., Donawho C.K., Frost D.J., et al. Discovery of the Poly(ADP-ribose) Polymerase (PARP) Inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the Treatment of Cancer. J. Med. Chem. 2009;52:514–523. doi: 10.1021/jm801171j. [DOI] [PubMed] [Google Scholar]
- 17.Penning T.D., Zhu G.-D., Gandhi V.B., Gong J., Thomas S., Lubisch W., Grandel R., Wernet W., Park C.H., Fry E.H., et al. Discovery and SAR of 2-(1-propylpiperidin-4-yl)-1H-benzimidazole-4-carboxamide: A potent inhibitor of poly (ADP-ribose) polymerase (PARP) for the treatment of cancer. Bioorg. Med. Chem. 2008;16:6965–6975. doi: 10.1016/j.bmc.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 18.Ferraris D., Ficco R.P., Dain D., Ginski M., Lautar S., Lee Wisdom K., Linag S., Lin Q., Lu M.X.-C., Morgan L., et al. Design and synthesis of poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors. Part 4: Biological evaluation of imidazobenzodiazepines as potent PARP-1 inhibitors for treatment of ischemic injuries. Bioorg. Med. Chem. 2003;11:3695–3707. doi: 10.1016/S0968-0896(03)00333-X. [DOI] [PubMed] [Google Scholar]
- 19.Costatino G., Macchiarulo A., Camaioni E., Pellicciari R. Modeling of poly(ADP- ribose)polymerase (PARP) inhibitors. Docking of ligands and quantitative structure- activity relationship analysis. J. Med. Chem. 2001;44:3786–3794. doi: 10.1021/jm010116l. [DOI] [PubMed] [Google Scholar]
- 20.Murai J., Huang S.-Y.N., Renaud A., Zhang Y., Ji J., Takeda S., Morris J., Teicher B., Doroshow J.H., Promier Y. Stereospecific PARP trapping by BMN-673 and comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014;13:433–443. doi: 10.1158/1535-7163.MCT-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pommier Y., O’ Connor M.J., de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016;8:362–368. doi: 10.1126/scitranslmed.aaf9246. [DOI] [PubMed] [Google Scholar]
- 22.Barkalow J.H., Breting J., Gaede B.J., Haight A.R., Henry R., Kotecki B., Mei J., Pearl K.B., Tedrow J.S., Viswanath S.K. Process development for ABT-472, a benzimidazole PARP inhibitor. Org. Process Res. Dev. 2007;11:693–698. doi: 10.1021/op7000194. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.