

Crystal structures of the extracellular domain of rabbit neprilysin and of neprilysin complexed with the inhibitors phosphoramidon or thiorphan are reported.
Keywords: neutral endopeptidase, neprilysin, phosphoramidon, thiorphan
Abstract
Neutral endopeptidase (neprilysin; NEP) is a proteinase that cleaves a wide variety of peptides and has been implicated in Alzheimer’s disease, cardiovascular conditions, arthritis and other inflammatory diseases. The structure of the soluble extracellular domain (residues 55–750) of rabbit neprilysin was solved both in its native form at 2.1 Å resolution, and bound to the inhibitors phosphoramidon and thiorphan at 2.8 and 3.0 Å resolution, respectively. Consistent with the extracellular domain of human neprilysin, the structure reveals a large central cavity which contains the active site and the location for inhibitor binding.
1. Introduction
Neprilysin (EC 3.4.24.11), a peptidase also known as neutral endopeptidase (NEP) or enkephalinase, is a large membrane-bound zinc-dependent ectozyme that cleaves peptide bonds on the amino side of hydrophobic residues (reviewed in Erdös & Skidgel, 1989 ▸; Roques et al., 1993 ▸). Neprilysin was originally extracted and purified from the brush border of rabbit kidney cells (Kerr & Kenny, 1974a ▸,b ▸). It has since been found to be widely distributed, with high concentrations in the central nervous system, intestinal and kidney brush border membranes, lymph nodes and placenta (Roques et al., 1993 ▸).
Neprilysin has important roles in many organs of the body and has attracted attention owing to its role in various human diseases. Its central role in the metabolism of cardiovascular peptides, for example, has made neprilysin the subject of a landmark clinical trial which assessed its inhibition for the treatment of heart failure (McMurray et al., 2014 ▸). Because of its role in the degradation of amyloid β-peptide, the manipulation of neprilysin levels in neuronal tissue has been of therapeutic interest for some time for the treatment of Alzheimer’s disease (Nalivaeva et al., 2014 ▸; Webster et al., 2014 ▸).
The overall structure of this large 94 kDa metalloprotease consists of an N-terminal cytoplasmic region with a short N-terminal cytoplasmic domain (27 residues), a transmembrane region of about 23 hydrophobic residues and a large extracellular domain of about 700 residues (Devault et al., 1987 ▸). Within this domain is found a large cavity that is capable of accommodating peptides with a length of up to 50 residues. The active site contains a single zinc cation coordinated by two histidine residues and a glutamic acid residue.
The structures of several human neprilysin extracellular domains have been published in complex with various inhibitors, including phosphoramidon (Oefner et al., 2000 ▸, 2004 ▸, 2007 ▸; Glossop et al., 2011 ▸; Webster et al., 2014 ▸; Schiering et al., 2016 ▸). Most recently, a human structure free of any bound inhibitor or substrate has been reported (Moss et al., 2018 ▸). The cytoplasmic tail of neprilysin from mouse was also crystallized as part of a complex (Terawaki et al., 2007 ▸). Structural details of a prokaryotic endopeptidase that shares significant homology with human neprilysin, in complex with phosphoramidon, have also been described (Ferraris et al., 2011 ▸). The Mycobacterium tuberculosis zinc-dependent metalloprotease-1 (Zmp1) shares 48% similarity with human neprilysin and is thought to play an important role in pathogenesis, making it a target for the rational design of specific inhibitors (Ferraris et al., 2018 ▸).
The present work represents the first neprilysin structures from a rabbit source, with rabbit having been used as an important model for studying the role of neprilysin in various disease states (Ichiki et al., 2013 ▸; Birner et al., 2012 ▸; Grois et al., 2017 ▸). The rabbit neprilysin sequence is highly similar to that of human neprilysin, with 94% identity and 97% similarity using standard BLAST parameters (Madden, 2002 ▸). Since rabbits are used as an animal model for human diseases related to neprilysin, it is important to structurally compare the enzyme in both species. Indeed, the rabbit model has also been proposed as a good model for the study of M. tuberculosis infection because rabbits in particular display a spectrum of disease states representing specific stages of the disease in humans (Dehnad et al., 2016 ▸; Gupta & Katoch, 2005 ▸; Converse et al., 1996 ▸).
2. Materials and methods
2.1. Macromolecule production
A soluble form of the rabbit enzyme was used in which the cytoplasmic and transmembrane regions had been removed and the soluble portion was expressed in Pichia pastoris strain GS115 (his4 −). The protein could be expressed at levels as high as 20 mg l−1 and purified. Briefly, 200 ml culture medium (concentrated to 50 ml) was loaded onto a Resource Q anion-exchange chromatography column (Pharmacia) and bound protein was eluted from the column with a linear NaCl gradient (20 mM Tris pH 7.0, 0–0.5 M NaCl). The protein was further purified by hydrophobic interaction chromatography (Phenyl Superose HR 10/10, 0.1 M phosphate buffer pH 7.0, 1.5 M ammonium sulfate gradient). Endoglycosidase digestion of glycosylated sec-NEP was next performed with EndoH (New England BioLabs, 50 mM sodium citrate pH 5.5 at 25°C). The protein was then polished by size-exclusion chromatography (Superose 12, 50 mM Tris pH 7.0, 0.1 M NaCl). Macromolecule-production information is summarized in Table 1 ▸.
Table 1. Macromolecule-production information.
| Source organism | Rabbit |
| DNA source | Rabbit kidney cDNA |
| Cloning vector | pSVsec-NEP† |
| Expression vector | pPIC9 |
| Expression host | P. pastoris strain GS115 (his4 −) |
| Complete amino-acid sequence of the construct produced | GICKSSDCIKSAARLIQNMDATAEPCTDFFKYACGGWLKRNVIPETSSRYSNFDILRDELEVILKDVLQEPKTEDIVAVQKAKTLYRSCVNETAIDSRGGQPLLKLLPDVYGWPVATQNWEQTYGTSWSAEKSIAQLNSKYGKKVLINFFVGTDDKNSMNHIIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMIAVAKLIRQEEGLPIDENQISVEMNKVMELEKEIANATTKSEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWSNFTNEIMSTVNINIPNEEDVVVYAPEYLIKLKPILTKYSPRDLQNLMSWRFIMDLVSSLSRTYKDSRNAFRKALYGTTSESATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKKAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWITGAAIVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSANNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGIGQAYRAYQNYVKKNGEEKLLPGIDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGSLQNSVEFSEAFQCPKNSYMNPEKKCRVW |
2.2. Crystallization
The purified protein was concentrated using Centricon 10 dialysis tubes to 20 mg ml−1 in 10 mM MgCl2, 100 mM sodium cacodylate pH 7.0 buffer. Crystals were obtained in hanging drops using crystallization buffer consisting of 18% PEG 4000, 10 mM MgCl2, 100 mM sodium cacodylate pH 7.0. The protein solution (2 µl) was mixed in a 1:1 ratio with 2 µl crystallization buffer and equilibrated against a 1 ml reservoir of crystallization buffer. The crystals were cryoprotected using 25% ethylene glycol. Crystallization information is summarized in Table 2 ▸.
Table 2. Crystallization.
| Method | Hanging-drop vapour diffusion |
| Plate type | 24-well |
| Temperature (K) | 293 |
| Protein concentration (mg ml−1) | 20 |
| Buffer composition of protein solution | 10 mM MgCl2, 100 mM sodium cacodylate pH 7.0 |
| Composition of reservoir solution | 18% PEG 4000, 10 mM MgCl2, 100 mM sodium cacodylate pH 7.0 |
| Volume and ratio of drop | 2 µl protein solution:2 µl reservoir solution |
| Volume of reservoir (ml) | 1 |
2.3. Data collection and processing
Data were collected on beamlines X8C and X12C at the National Synchrotron Light Source (NSLS) as outlined in Table 3 ▸. The images were indexed, integrated and scaled using HKL-2000 and SCALA (Otwinowski & Minor, 1997 ▸; Evans, 2006 ▸).
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Structure (PDB code) | Native (4xbh) | Phosphoramidon complex (4zr5) | Thiorphan complex (5v48) |
|---|---|---|---|
| Diffraction source | X12C, NSLS | X8C, NSLS | X8C, NSLS |
| Wavelength (Å) | 1.1 | 1.1 | 1.1 |
| Temperature (K) | 100 | 100 | 100 |
| Detector | Brandeis B4 | Quantum 4 CCD | Quantum 4 CCD |
| Crystal-to-detector distance (mm) | 262.0 | 279.5 | 280.0 |
| Rotation range per image (°) | 0.5 | 0.66 | 0.66 |
| Total rotation range (°) | 180 | 76.6 | 72.6 |
| Space group | P212121 | P212121 | P212121 |
| a, b, c (Å) | 69.4, 108.1, 212.8 | 68.5, 107.5, 210.6 | 69.0, 108.5, 211.9 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Mosaicity (°) | 0.35 | 0.45 | 0.50 |
| Resolution range (Å) | 39.6–2.11 (2.19–2.11) | 39.2–2.80 (2.90–2.80) | 39.6–3.00 (3.10–3.00) |
| Total No. of reflections | 441341 | 84480 | 69618 |
| No. of unique reflections | 88914 (7408) | 33003 (1755) | 27886 (2245) |
| Completeness (%) | 96.2 (81.3) | 84.4 (45.4) | 85.2 (71.7) |
| Multiplicity | 5.0 | 2.6 | 2.5 |
| 〈I/σ(I)〉 | 21.96 (7.44) | 13.98 (4.86) | 14.16 (4.30) |
| R r.i.m. † | 0.063 | 0.065 | 0.075 |
| CC1/2 | 0.99 (0.95) | 0.98 (0.93) | 0.98 (0.89) |
| Overall B factor from Wilson plot (Å2) | 20.11 | 30.78 | 38.72 |
Estimate based on multiplying R merge by the factor [N/(N − 1)]1/2, where N is the data multiplicity.
2.4. Structure solution and refinement
The 2.1 Å resolution native structure was solved using the Phaser-MR protocol in PHENIX (McCoy et al., 2007 ▸; Adams et al., 2010 ▸) using the structure of human neutral endopeptidase complexed with phosphoramidon (PDB entry 1dmt; Oefner et al., 2000 ▸) as the starting model. This was followed by AutoBuild using residues 55–750 of the rabbit neprilysin sequence NP_001095155 derived from cDNA sequencing (Devault et al., 1987 ▸) and removing the phosphoramidon and glycerol ligands from the starting model. phenix.refine (Afonine et al., 2012 ▸) was used for simulated annealing, and automated water placement was used to aid in assigning the positions of water molecules. This resulted in a model which provided a good overall fit to the electron density. Difference density next to the zinc position matched the shape and size of that expected from the coordination of a phosphate group, which was fitted into the density. It became apparent that four residues in particular could not fit into the experimental electron density: Asn194, Phe363, Phe367 and Phe371. Upon searching the NCBI sequence database, a rabbit sequence predicted from genome sequencing (XP_008264403.1) was found that better matched the densities for these residues. Based on this, residues 194, 363, 367 and 371 were replaced with Lys, Ser, Leu and Met, respectively. Several rounds of refinement using phenix.refine and manual rebuilding confirmed this sequence to fit the data and the final model had an overall R cryst and R free of 0.166 and 0.211, respectively (Table 4 ▸).
Table 4. Structure solution and refinement.
Values in parentheses are for the outer shell.
| Structure (PDB code) | Native (4xbh) | Phosphoramidon complex (4zr5) | Thiorphan complex (5v48) |
|---|---|---|---|
| Resolution range (Å) | 39.60–2.11 (2.19–2.11) | 39.21–2.80 (2.90–2.80) | 39.47–3.00 (3.10–3.00) |
| Completeness (%) | 96.2 (81.3) | 84.4 (45.4) | 85.2 (71.7) |
| σ Cutoff | 1.35 | 1.39 | 1.37 |
| No. of reflections | |||
| Working set | 86914 | 31016 | 25886 |
| Test set | 2000 | 1987 | 2000 |
| Final R cryst | 0.166 (0.175) | 0.192 (0.255) | 0.195 (0.285) |
| Final R free | 0.211 (0.243) | 0.227 (0.297) | 0.245 (0.344) |
| Cruickshank DPI | 0.17 | 0.47 | 0.55 |
| No. of non-H atoms | |||
| Protein | 11182 | 11182 | 11182 |
| Ion | 2 | 2 | 2 |
| Ligand | 164 | 214 | 190 |
| Water | 1506 | 271 | 736 |
| Total | 12854 | 11669 | 12110 |
| R.m.s. deviations | |||
| Bonds (Å) | 0.003 | 0.004 | 0.002 |
| Angles (°) | 0.68 | 0.90 | 0.39 |
| Average B factors (Å2) | |||
| Protein | 24.6 | 33.4 | 45.6 |
| Ion | 19.6 | 24.3 | 37.3 |
| Ligand | 48.2 | 54.5 | 67.0 |
| Water | 32.4 | 24.5 | 31.5 |
| Ramachandran plot | |||
| Most favoured (%) | 97.9 | 97.7 | 97.0 |
| Allowed (%) | 1.95 | 2.16 | 2.90 |
The 2.8 and 3.0 Å resolution structures with bound phosphoramidon or thiorphan, respectively, were refined using the final native structure as the starting model with the bound phosphates removed. Ligands were fitted manually into the difference density, while restraints were prepared using eLBOW in PHENIX with standard settings (Moriarty et al., 2009 ▸). After ligand placement, several rounds of manual adjustment of the model and refinement were carried out using PHENIX until convergence was achieved. Water molecules were either maintained in the locations where they occurred in the native higher resolution structure where this was justified by the experimental maps, or were added manually in a conservative manner. The native structure was used as a reference model during refinement. The atomic coordinates and experimental data have been deposited in the Protein Data Bank (http://www.wwpdb.org; PDB entries 4xbh, 4zr5 and 5v48).
3. Results and discussion
The asymmetric unit in all three structures contained two neprilysin molecules related by pseudo-twofold symmetry (Fig. 1 ▸ a). As in other published structures, each neprilysin molecule has a large central cavity, inside which the zinc active site is found (Fig. 1 ▸ b). It became evident in all three structures that, overall, chain A was more ordered in this packing arrangement than chain B. A relatively disordered loop region between residues 530 and 537 with high B factors is located along one edge of the central cavity, which may provide a clue regarding the mechanism of substrate access. The structures are highly similar to published human neprilysin structures. Differences in nonsimilar residues tend to be located on the outer surface of the enzyme, with the region near the catalytic site being highly conserved (Fig. 1 ▸ a). When the α-carbons of chains A and B from the final native rabbit structure were compared with the published human model used as a starting point for structure solution (PDB entry 1dmt; Oefner et al., 2000 ▸), SUPERPOSE (Maiti et al., 2004 ▸) reported r.m.s.d.s of 0.53 and 0.57 Å, respectively. When the α-carbons of chains A and B were compared with the respective published human native structure with bound phosphate (PDB entry 6gid; Moss et al., 2018 ▸), the r.m.s.d.s were 0.62 and 0.60 Å, respectively.
Figure 1.

(a) Ribbon representation of the asymmetric unit of native rabbit neprilysin. The moiety on the left (light grey) is designated as chain A, while chain B is on the right (dark grey). Red spheres represent the locations of residues that differ from human neprilysin, whereas orange spheres represent residues that are similar but non-identical. The active-site zinc is represented by a silver sphere. (b) Ribbon diagram of chain A coloured according to α-carbon B factors. The large central cavity is depicted using a grey surface representation. The disordered loop region is situated on the front surface of the cavity depiction, from the perspective of the viewer, and represents residues near 530–537. Zinc is represented by a silver sphere at the active site, bound to phosphate. (c) Examples of the quality of 2F o − F c maps (grey) contoured at 1σ around the N-linked acetylglucosamine residues at sites Asn285 (foreground) and Asn311 (background).
In the native structure, the electron density was clear enough to place N-acetylglucosamine (NAG) residues at four distinct sites: Asn145, Asn285, Asn311 and Asn325 (Fig. 1 ▸ c). The positions of the placed NAG residues were retained in the lower resolution structures containing the inhibitors unless the quality of the electron density did not warrant their inclusion in the models. These NAG sites matched the predicted glycosylation sites based on sequence analysis using EnsembleGly (Caragea et al., 2007 ▸) and as noted elsewhere (Koehne et al., 1998 ▸). They are also present in most of the published human neprilysin structures, with the notable exception of the site equivalent to Asn311, where the Asn residue is not modelled as having NAG. An additional site, Asn628, was predicted, and in the case of chain B of the native structure weaker less distinct electron density is observable next to this residue which may indicate the possible presence of NAG. Human neprilysin structures commonly have NAG modelled at this position.
The zinc cation is coordinated by residues His584, His588 and Glu647. The coordination is completed by a phosphate molecule in the native structure (Fig. 2 ▸ a). Residue His712 is very near the ligand site and interacts with the phosphate ion. This residue also remains prominent in the structure of neprilysin bound to phosphoramidon, interacting in a similar manner with the phosphate portion of the ligand (Fig. 2 ▸ b). Residues Arg718 and Asn543, along with portions of the peptide backbones of Ala544 and Val542, stabilize the interaction with the ligand. The hydrophobic portion of phosphoramidon is located within a hydrophobic pocket and Phe107 makes π-stacking interactions with the indole moiety of the inhibitor. Residues Arg718 and Asn543 are also prominent in stabilizing the thiorphan inhibitor (Fig. 2 ▸ c). The aromatic ring of thiorphan is oriented into the hydrophobic pocket, surrounded by Phe107, Ile559, Phe564, Val581 and Trp694. This pocket is occupied by the leucine-like hydrophobic portion of phosphoramidon in its respective structure.
Figure 2.

(a) The active site of native neprilysin, with zinc represented as a silver sphere coordinated to His584, His588, Glu647 and phosphate ion. (b) Active site of neprilysin bound to phosphoramidon. The His584, His588 and Glu647 coordinations of Zn2+ are omitted for clarity. (c) Active site of the thiorphan complex. Ligand-omit F o − F c density maps are contoured at 3σ (green).
When the structures of phosphoramidon- or thiorphan-bound neprilysin were compared with the native phosphate-bound structure, their overlays revealed largely similar structures, with a few notable exceptions (Fig. 3 ▸). To accommodate the aromatic ring of phorphoramidon, the side chains of Arg111 and Arg103 are displaced. Residues Trp694 and Phe107 appear to make adjustments to accommodate the aromatic ring of thiorphan. In the case of residue Trp694 in particular, a difference between the main chain is observed when compared with the native structure, which might be caused by the proximity of the inhibitor. When the phosphate-bound structure is compared with its human counterpart (Moss et al., 2018 ▸), a nearly identical active-site environment is observed (Fig. 4 ▸ a). Similarly, comparing the phosphoramidon-bound rabbit structure with the published human counterpart (Oefner et al., 2000 ▸) reveals a very similar set of interactions, in which the Arg111 and Arg103 side chains are similarly displaced away from the binding site in order to accommodate the hydrophobic aromatic ring structure of the ligand (Fig. 4 ▸ b).
Figure 3.
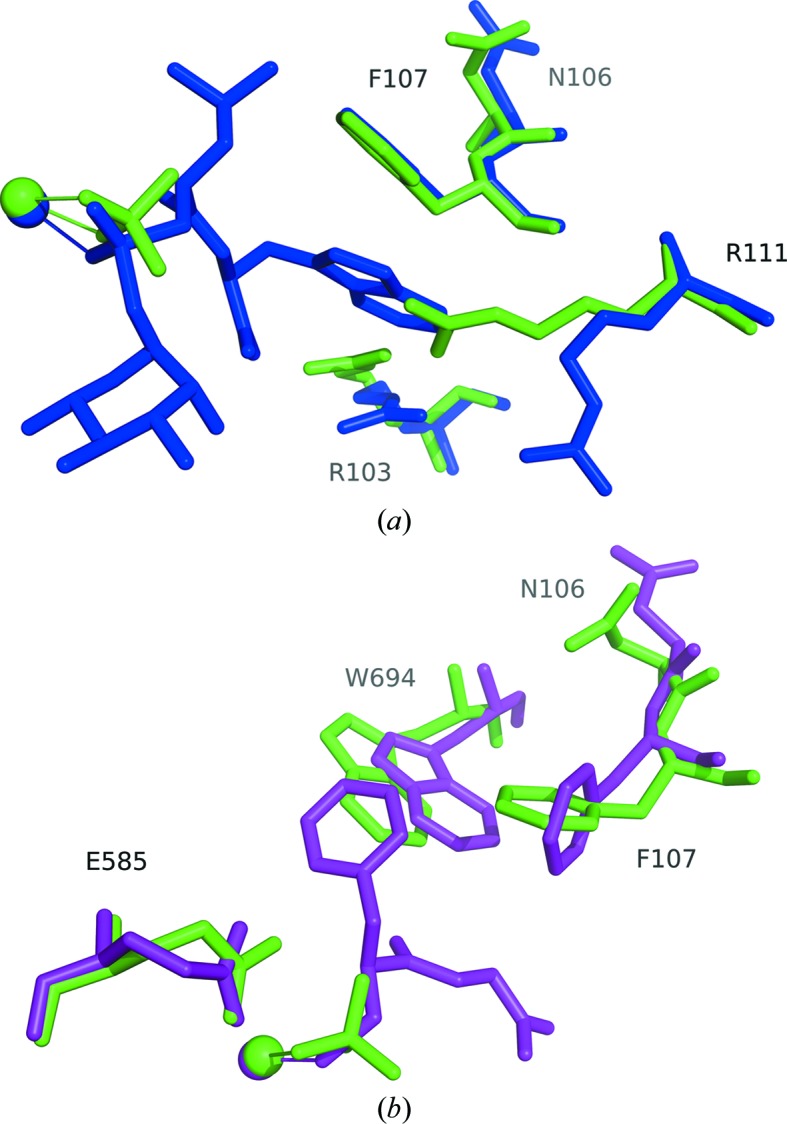
(a) Comparison of the phosphoramidon-bound structure (blue) with the native structure (green). The ligand environment is very similar in both cases, with the notable exceptions of residues Arg111 and Arg103, which are displaced in order to accommodate the large ring of phosphoramidon. (b) Comparison of the thiorphan-bound structure (purple) with the native structure. In this case, there are adjustments in Trp694 and Phe107 to accommodate the aromatic ring of thiorphan. Spheres represent zinc. Alignments were performed against the core amino-acid residues coordinated to the metal ion using the align algorithm in the PyMOL molecular-graphics system (v.1.8; Schrödinger), which was used to produce the figures.
Figure 4.

(a) Overlay of the native rabbit structure (green) with that of its human counterpart (cyan; PDB entry 6gid; Moss et al., 2018 ▸). (b) Overlay of the phosphoramidon-bound rabbit structure (dark blue) with that of its human counterpart (cyan; PDB entry 1dmt; Oefner et al., 2000 ▸). Residues Arg111, Arg103 and His712 from the native phosphate-bound rabbit structure are depicted (green) to highlight the differences in side-chain orientations. (c) Overlay of the phosphoramidon-bound rabbit neprilysin structure (blue) with Zmp1 from M. tuberculosis (red; PDB entry 3zuk; Ferraris et al., 2011 ▸). Phosphoramidon is coloured a darker blue or red, respectively, for clarity. TEA represents N,N′,N′′-triethanolamine in the Zmp1 structure.
Overall, the three rabbit structures share a high similarity with their human neprilysin extracellular domain counterparts, and the active-site zinc is capable of binding a range of ligands. The side chains of Arg103 and Arg111 adopt different conformations in order to accommodate the indole moiety of phosphoramidon, while Phe107 and Trp694 adjust to accommodate the aromatic ring of thiorphan. We would hypothesize that the differences between rabbit and human neprilysin residues that are more distant from the zinc-binding site, but still within the cavity, may contribute to potential differences in longer peptide substrate-binding kinetics, such as were observed between the two species for the amyloid beta 1–40 peptide (Webster et al., 2014 ▸; Leissring et al., 2003 ▸). In this case, k cat for human wild-type neprilysin and the rabbit enzyme were similar, but an approximately eightfold higher K m value was reported in the case of human neprilysin. It is unclear, however, whether these observations result from inherent differences between the enzymes or from protein preparation. Most of the nonsimilar amino-acid substitutions between the two species are found near the outside surface of the protein. However, one residue that stands out as being within the cavity and still relatively near the active site is Ser290. Some 16 Å away from the zinc site, it lines the inner cavity; in the human sequence, a proline residue is found instead. This would be near enough for potentially different interactions with short peptides and, although speculative, it would be interesting to explore whether there is any effect of this residue on the peptide-binding kinetics.
A detailed comparison between human neprilysin and M. tuberculosis Zmp1, both in complex with phosphoramidon, has also been published (Ferraris et al., 2011 ▸). The interactions of Zmp1 with phosphoramidon are very similar to those described here for rabbit neprilysin, with the residues described in Fig. 2 ▸(b) being conserved. In the case of Zmp1, an additional molecule, N,N′,N′′-triethanolamine (TEA), is described in one of the subunits in close proximity to phosphoramidon. We did not observe this in the rabbit neprilysin structure. It should be noted that in the human structure (Oefner et al., 2000 ▸), which is of higher resolution than the rabbit structure presented here, a glycerol molecule is identified near this position. An overlay of the Zmp1 structure with that of rabbit neprilysin is presented in Fig. 4 ▸(c), highlighting the main residue differences surrounding the phosphoramidon binding site. The rhamnose moiety of phosphoramidon is mostly exposed to solvent but lies near Phe545 and Thr537, which are replaced by Tyr454 and Met446, respectively, in Zmp1. Met446 is alternatively found to be a serine residue in the case of human neprilysin. The indole moiety of phosphoramidon is shifted relative to the rest of the inhibitor owing to the proximity of Arg103 and Asp108 in the case of rabbit neprilysin, which are Thr44 and Arg49, respectively, in the case of Zmp1. Additional differences near the indole moiety include Arg111, Gly696 and Tyr698, which are replaced by Phe52, Thr606 and Ser608 in Zmp1. The site that binds TEA in the Zmp1 structure contains differences that are proposed by the authors (Ferraris et al., 2011 ▸) to be promising targets for the structure-based design of specific Zmp1 inhibitors, with Arg615 and Arg616 in Zmp1 replaced by Asn705 and Ser706, respectively, in both human and rabbit neprilysin. Met580 is replaced by Ala489 in the bacterial case, making the region that binds the leucine-like moiety of phosphoramidon more hydrophobic. These differences may be relevant for natural substrate specificity and may be useful for the design of novel specific bacterial inhibitors.
In conclusion, the rabbit neprilysin structures should provide important structural information for inter-species substrate-binding studies going forward. The nonconserved residues between human and rabbit neprilysin, which are relatively distant from the active site, do not appear to show significant effects in the binding mode of the inhibitors phosphoramidon or thiorphan. This is consistent with the rabbit as a model for the study of neprilysin-related disease states. Differences between the active sites of Zmp1 and rabbit neprilysin are consistent with those observed between Zmp1 and human neprilysin, supporting the use of rabbit neprilysin as a good test model for the development of Zmp1 inhibitors in the fight against tuberculosis.
Supplementary Material
PDB reference: soluble rabbit neprilysin, 4xbh
PDB reference: complex with phosphoramidon, 4zr5
PDB reference: complex with thiorphan, 5v48
Acknowledgments
The authors thank Lucie Gonneville for careful technical assistance, and Drs Philippe Crine and Guy Boileau for kindly providing cloning and protein-production resources. Data for this study were measured on beamlines X8C and X12C of the National Synchrotron Light Source. Financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy and from the National Center for Research Resources (P41RR012408) and the National Institute of General Medical Sciences (P41GM103473) of the National Institutes of Health. Computational work was carried out at the Canadian Light Source, which is supported by the Canadian Foundation for Innovation, the Natural Sciences and Engineering Research Council of Canada, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council of Canada and the Canadian Institutes of Health Research.
Funding Statement
This work was funded by Natural Sciences and Engineering Research Council of Canada grant .
References
- Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L.-W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. & Zwart, P. H. (2010). Acta Cryst. D66, 213–221. [DOI] [PMC free article] [PubMed]
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Birner, C., Ulucan, C., Bratfisch, M., Götz, T., Dietl, A., Schweda, F., Riegger, G. A. & Luchner, A. (2012). Naunyn Schmiedebergs Arch. Pharmacol. 385, 1117–1125. [DOI] [PubMed]
- Caragea, C., Sinapov, J., Silvescu, A., Dobbs, D. & Honavar, V. (2007). BMC Bioinformatics, 8, 438. [DOI] [PMC free article] [PubMed]
- Converse, P. J., Dannenberg, A. M. Jr, Estep, J. E., Sugisaki, K., Abe, Y., Schofield, B. H. & Pitt, M. L. (1996). Infect. Immun. 64, 4776–4787. [DOI] [PMC free article] [PubMed]
- Dehnad, A., Ravindran, R., Subbian, S. & Khan, I. H. (2016). Tuberculosis, 101, 1–7. [DOI] [PubMed]
- Devault, A., Lazure, C., Nault, C., Le Moual, H., Seidah, N. G., Chrétien, M., Kahn, P., Powell, J., Mallet, J., Beaumont, A., Roques, B. P., Crine, P. & Boileau, G. (1987). EMBO J. 6, 1317–1322. [DOI] [PMC free article] [PubMed]
- Erdös, E. G. & Skidgel, R. A. (1989). FASEB J. 3, 145–151. [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Ferraris, D. M., Miggiano, R., Rossi, F. & Rizzi, M. (2018). Pathogens 7, 17. [DOI] [PMC free article] [PubMed]
- Ferraris, D. M., Sbardella, D., Petrera, A., Marini, S., Amstutz, B., Coletta, M., Sander, P. & Rizzi, M. (2011). J. Biol. Chem. 286, 32475–32482. [DOI] [PMC free article] [PubMed]
- Fossiez, F., Lemay, G., Labonté, N., Parmentier-Lesage, F., Boileau, G. & Crine, P. (1992). Biochem. J. 284, 53–59. [DOI] [PMC free article] [PubMed]
- Glossop, M. S., Bazin, R. J., Dack, K. N., Fox, D. N., MacDonald, G. A., Mills, M., Owen, D. R., Phillips, C., Reeves, K. A., Ringer, T. J., Strang, R. S. & Watson, C. A. (2011). Bioorg. Med. Chem. Lett. 21, 3404–3406. [DOI] [PubMed]
- Grois, L., Hupf, J., Reinders, J., Schröder, J., Dietl, A., Schmid, P. M., Jungbauer, C., Resch, M., Maier, L. S., Luchner, A. & Birner, C. (2017). PLoS One, 12, e0169743. [DOI] [PMC free article] [PubMed]
- Gupta, U. D. & Katoch, V. M. (2005). Tuberculosis, 85, 277–293. [DOI] [PubMed]
- Ichiki, T., Izumi, R., Cataliotti, A., Larsen, A. M., Sandberg, S. M. & Burnett, J. C. Jr (2013). Peptides, 48, 21–26. [DOI] [PMC free article] [PubMed]
- Kerr, M. A. & Kenny, A. J. (1974a). Biochem. J. 137, 477–488. [DOI] [PMC free article] [PubMed]
- Kerr, M. A. & Kenny, A. J. (1974b). Biochem. J. 137, 489–495. [DOI] [PMC free article] [PubMed]
- Koehne, P., Schäper, C., Graf, K. & Kunkel, G. (1998). Allergy, 53, 1023–1042. [DOI] [PubMed]
- Leissring, M. A., Lu, A., Condron, M. M., Teplow, D. B., Stein, R. L., Farris, W. & Selkoe, D. J. (2003). J. Biol. Chem. 278, 37314–37320. [DOI] [PubMed]
- Lemay, G., Waksman, G., Roques, B. P., Crine, P. & Boileau, G. (1989). J. Biol. Chem. 264, 15620–15623. [PubMed]
- Madden, T. (2002). The NCBI Handbook, 2nd ed. Bethesda: National Center for Biotechnology Information..
- Maiti, R., Van Domselaar, G. H., Zhang, H. & Wishart, D. S. (2004). Nucleic Acids Res. 32, W590–W594. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McMurray, J. J. V., Packer, M., Desai, A. S., Gong, J., Lefkowitz, M. P., Rizkala, A. R., Rouleau, J. L., Shi, V. C., Solomon, S. D., Swedberg, K. & Zile, M. R. (2014). N. Engl. J. Med. 371, 993–1004.
- Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. (2009). Acta Cryst. D65, 1074–1080. [DOI] [PMC free article] [PubMed]
- Moss, S., Subramanian, V. & Acharya, K. R. (2018). J. Struct. Biol. 204, 19–25. [DOI] [PubMed]
- Nalivaeva, N. N., Belyaev, N. D., Kerridge, C. & Turner, A. J. (2014). Front. Aging Neurosci. 6, 235. [DOI] [PMC free article] [PubMed]
- Oefner, C., D’Arcy, A., Hennig, M., Winkler, F. K. & Dale, G. E. (2000). J. Mol. Biol. 296, 341–349. [DOI] [PubMed]
- Oefner, C., Pierau, S., Schulz, H. & Dale, G. E. (2007). Acta Cryst. D63, 975–981. [DOI] [PubMed]
- Oefner, C., Roques, B. P., Fournie-Zaluski, M.-C. & Dale, G. E. (2004). Acta Cryst. D60, 392–396. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Roques, B. P., Noble, F., Daugé, V., Fournié-Zaluski, M.-C. & Beaumont, A. (1993). Pharmacol. Rev. 45, 87–146. [PubMed]
- Schiering, N., D’Arcy, A., Villard, F., Ramage, P., Logel, C., Cumin, F., Ksander, G. M., Wiesmann, C., Karki, R. G. & Mogi, M. (2016). Sci. Rep. 6, 27909. [DOI] [PMC free article] [PubMed]
- Terawaki, S., Kitano, K. & Hakoshima, T. (2007). J. Biol. Chem. 282, 19854–19862. [DOI] [PubMed]
- Webster, C. I., Burrell, M., Olsson, L. L., Fowler, S. B., Digby, S., Sandercock, A., Snijder, A., Tebbe, J., Haupts, U., Grudzinska, J., Jermutus, L. & Andersson, C. (2014). PLoS One, 9, e104001. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: soluble rabbit neprilysin, 4xbh
PDB reference: complex with phosphoramidon, 4zr5
PDB reference: complex with thiorphan, 5v48