

Abstract
Medicarpin is a bioactive pterocarpan that has been attracting increasing attention in recent years. However, its metabolic fate in vivo is still unknown. To clarify its metabolism and the distribution of its metabolites in rats after oral administration, the HPLC-ESI-IT-TOF-MSn technique was used. A total of 165 new metabolites (13 phase I and 152 phase II metabolites) were tentatively identified, and 104, 29, 38, 41, 74, 28, 24, 15, 42, 8, 10, 3, and 17 metabolites were identified in urine, feces, plasma, the colon, intestine, stomach, liver, spleen, kidney, lung, heart, brain, and thymus, respectively. Metabolic reactions included demethylation, hydrogenation, hydroxylation, glucuronidation, sulfation, methylation, glycosylation, and vitamin C conjugation. M1 (medicarpin glucuronide), M5 (vestitol-1’-O-glucuronide) were distributed to 10 organs, and M1 was the most abundant metabolite in seven organs. Moreover, we found that isomerization of medicarpin must occur in vivo. At least 93 metabolites were regarded as potential new compounds by retrieving information from the Scifinder database. This is the first detailed report on the metabolism of ptercarpans in animals, which will help to deepen the understanding of the metabolism characteristics of medicarpin in vivo and provide a solid basis for further studies on the metabolism of other pterocarpans in animals.
Keywords: medicarpin, pterocarpan, metabolites, distribution, in vivo, HPLC-ESI-IT-TOF-MSn
1. Introduction
Pterocarpans are the second largest group of dihydroisoflavonoids and have a special structure skeleton (benzopyran-benzofuran four-ring system). They were originally isolated from plants [1] and then regarded as an important group of phytoalexins, which have antifungal [2] and antibacterial [3] effects. In recent years, many new bioactivities of pterocarpans have been discovered, such as anti-inflammation [4,5,6], antitumor activity [7,8,9], anti-osteoporosis [10,11,12], antimalarial [13], antioxidant [14,15,16], inhibition of neuraminidase [17,18,19] and melanin synthesis [20], estrogenic and anti-estrogenic activity [21], anti-clastogensis [22], immunosuppressive activity [23], and inhibition of acetylcholinesterase [24]. As a result, this group of natural products is getting more and more attention. However, after conducting a systematic literature survey, we surprisingly found that there is only one report on the metabolism of a pure pterocarpan (i.e., trifolirhizin) in animals and only two metabolites (maackiain in feces and maackiain, maackiain-3-O-sulfoacid in urine) were identified [25], though a lot of papers about biosynthesis and the fungal metabolism of pterocarpans have been published. Drug metabolism research has a pivotal role in new drug research and development. In order to effectively utilize pterocarpans, it is important to clarify their metabolism characteristics.
Medicarpin (Figure 1) is a natural pterocarpan-type phytoalexin [26] with a wide variety of biological activities distributed in lots of traditional Chinese medicines (TCM), such as Pueraria Lobata [27], and Hedysari Radix [28]. According to the literature, medicarpin has anti-osteoporotic and bone protection activities even at a low concentration of 10−4 μM on murine bone cells and it can reduce the formation of osteoclasts but increase the formation of osteoprogenitor cells in bone marrow cells (BMCs) in OVx mice at a dose of 10 mg/kg/d for 30 days [10]. In another study, medicarpin treatment (10 mg/kg/d for 30 days) can increase the formation of osteoprogenitor cells in BMCs and osteoid formation in female Sprague-Dawley (SD) rats [11]. It also shows cytotoxic activities in BCA-1 (human breast cancer cells, IC50 = 13.14 μg/mL) and KB (human epidermoid carcinoma of cavity, IC50 = 10.13 μg/mL) cells [29]. In addition, medicarpin can lead to apoptosis and reverse multidrug resistance in P388 leukemia cells (IC50 ≈ 90 μM) [30]. It also has antifungal [31] and antibacterial activities [32]. Additionally, medicarpin can stimulate mesenchymal stem cells to differentiate into brown and beige adipocytes via the adenosine monophosphate-activated protein kinase (AMPK) pathway and boost lipolysis in adipocytes via a protein kinase A (PKA)-dependent pathway, so it is expected to be a potential drug for the treatment of obesity [33,34]. However, the metabolism of medicarpin in vivo and in vitro is still unknown. Therefore, the present study aims to identify its metabolites, clarify its metabolic pathways, and reveal the distribution of medicarpin and its metabolites in rats using high-performance liquid chromatography-electrospray ionization-ion trap-time of flight-multistage mass spectrometry (HPLC-ESI-IT-TOF-MSn).
Figure 1.
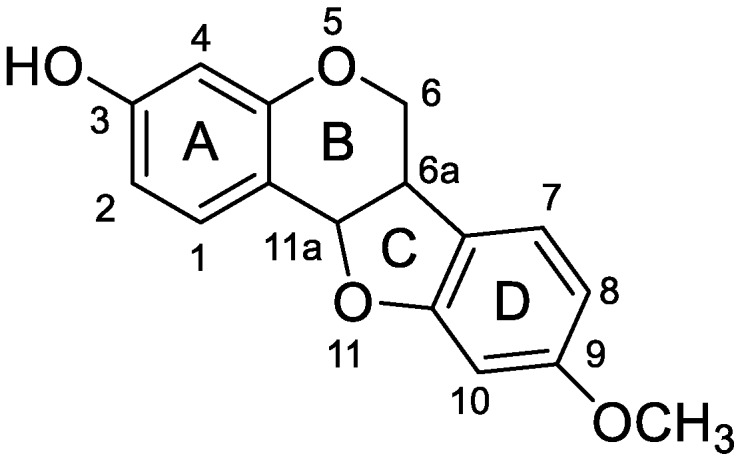
The structure and atom numbering of medicarpin.
2. Results and Discussion
2.1. MS Fragmentation Characteristics of Medicarpin in ESI− Mode and Identification of Medicarpin in Rats
In order to facilitate the description of the ESI− MS characteristics of medicarpin and its metabolites, we proposed a nomenclature for the fragmentation pathways and fragment ions. The four rings were named A, B, C and D, respectively. The cleavable C-C bonds in the skeleton were designated by numbers 1–8, as shown in Figure 2.
Figure 2.

The MS spectra and the proposed fragmentation pathways of medicarpin in ESI− mode.
Medicarpin (C16H14O4) showed [M − H]− at m/z 269.0822 (theoretical mass was 269.0819 Da) in ESI− spectra. The characteristic fragment ions of medicarpin in ESI− mode included m/z 254.0586 ([M − H − CH3•]•−), m/z 161.0173 (C9H5O3, 6,8A− − 2H), m/z 145.0387 (C9H5O2, 6,7A− − 2H), m/z 133.0302 (C8H5O2, 3,4,7A− − H), m/z 132.0270 (C8H4O2, 3,5D− − CH3•), m/z 121.0358 (C7H5O2, 3,5A−) in its MS2 spectra (Figure 2).
2.2. Profiling 165 Metabolites of Medicarpin in Rats
The metabolites of medicarpin were screened by comparing the HPLC chromatograms and MS base peak chromatograms (BPCs) of drug and blank group samples obtained through HPLC-ESI-IT-TOF-MSn analysis, and then confirmed by comparison of extracted ion chromatograms (EICs) between the drug and blank groups. The metabolic reactions were judged by the accurate mass (elemental composition) differences between medicarpin and its metabolites. The accurate mass (elemental composition) differences of −14.01 Da (CH2), +14.01 Da (CH2), +2.01 Da (H2), and +15.99 Da (O) indicated demethylation, methylation, hydrogenation and hydroxylation. The loss of 176.03 Da (C6H8O6) from precursor ion and/or an anion at m/z 175.02 in MS2 spectra indicated that the metabolite was a glucuronide; the loss of 79.95 Da (SO3) from precursor ion indicated that the metabolite was a sulfate; the loss of 162.05 Da (C6H10O5) indicated a hexoside (more likely to be a glucoside); the loss of 158.02 Da (C6H6O5) indicated a vitamin C conjugate [35].
A total of 165 metabolites of medicarpin were confirmed and tentatively identified by the above-mentioned method, and the proposed metabolic pathways of medicarpin are shown in Figure 3. According to the skeleton of molecules, 165 metabolites were classified into 13 categories: (1) metabolites (M1–M3) having the skeleton of medicarpin; (2) metabolites (M4–M10) having the skeleton of hydrogenated medicarpin; (3) metabolites (M11–M26) having the skeleton of demethylated medicarpin; (4) metabolites (M27–M52) having the skeleton of demethylated and hydrogenated medicarpin; (5) metabolites (M53–M75) having the skeleton of demethylated and hydroxylated medicarpin; (6) metabolites (M76–M107) having the skeleton of hydroxylated medicarpin; (7) metabolites (M108–M138) having the skeleton of hydrogenated and hydroxylated medicarpin; (8) metabolites (M139–M149) having the skeleton of demethylated, hydrogenated and hydroxylated medicarpin; (9) metabolites (M150–M156) having the skeleton of demethylated and dihydroxylated medicarpin; (10) metabolites (M157–M161) having the skeleton of dihydroxylated medicarpin; (11) metabolite (M162) having the skeleton of hydrogenated and dihydroxylated medicarpin; (12) metabolites (M163–M164) having the skeleton of hydrogenated and trihydroxylated medicarpin; (13) metabolite (M165) having the skeleton of demethylated, hydrogenated and dihydroxylated medicarpin. The EICs of 165 metabolites are shown in Figures S1–S44.
Figure 3.

The proposed metabolic pathways of medicarpin in rats. Phase I metabolites are in the double line polygon, undetected intermediates are in the dashed line boxes.
2.2.1. Analysis of Metabolites (M1–M3) Having the Skeleton of Medicarpin
M1 and M2 showed [M − H]− at m/z 445.11 and their molecular formulae were predicted to be C22H22O10, so they were isomers. The fragment ions at m/z 269.08 and m/z 175.02 can be detected in both of their MS2 spectra. Therefore, M1 and M2 were determined to be glucuronides of medicarpin. However, medicarpin only has one hydroxyl group, i.e., one glucuronidation site (Figure 4a). Hence, we deduced that isomerization of medicarpin (configuration change or shift of substituent) must occur.
Figure 4.

(a) Extracted ion chromatograms (EIC) of m/z 445.11; (b) EIC of m/z 431.10; (c) EIC of m/z 335.02; (d) EIC of m/z 511.06.
M3 (Figure 5) showed [M − H]− at m/z 427.1049 and its molecular formula was predicted to be C22H20O9. In its MS2 spectra, m/z 269.0792 (C16H13O4, [medicarpin − H]−) formed by losing 158.02 Da (C6H6O5) from [M − H]− was observed, indicating a vitamin C conjugate based on our previous study [35]. Besides, in the ESI+ mode, M3 showed [M + H]+ at m/z 429.1177. In its MS2 spectra, fragment ion at m/z 313.1113 was observed, indicating a neutral loss of 116.01 Da (C4H4O4). Subsequently, m/z 313.1113 yielded fragment ions at m/z 165.0516 (C9H9O3), m/z 161.0577 (C10H9O2) and m/z 137.0600 (C8H9O2). Hence, M3 was medicarpin-3-O-vitamin C.
Figure 5.

The proposed fragmentation pathways of M3 in ESI+ mode.
2.2.2. Analysis of Metabolites (M4–M10) Having the Skeleton of Hydrogenated Medicarpin
M4 showed [M − H]− at m/z 271.0979, and its molecular formula was predicted to be C16H16O4, which had two more hydrogen atoms than that of medicarpin. Therefore, one ring of medicarpin must be cleaved. In its MS2 spectra, the fragment ions at m/z 147.0469 (C9H7O2, 6A− or 3,5B− − 2H ), m/z 135.0466 (C8H7O2, 3,4A− or 3,4B−), m/z 123.0543 (C7H7O2, 6B− + H or 3,5A− + 2H or 2,4A− + 2H), m/z 121.0302 (C7H5O2, 3,5A− or 2,4A−) and m/z 109.0321 (C6H5O2, 2,5A− + 2H) were detected. According to the characteristic fragment ion at m/z 135.0466 (C8H7O2, 3,4A− or 3,4B−), we could deduce that medicarpin had undergone ring cleavage at the bond of C11-C11a as shown in Figure 6a. Hence, M4 was determined to be vestitol. The characteristic fragment ion at m/z 135.05 (C8H7O2) was very useful, because its existence indicated that the C11-C11a bond was cleaved and the hydrogenated medicarpin had the skeleton of a vestitol. The MS2 spectrum of M4 is shown in Figure S45.
Figure 6.

(a) The proposed fragmentation pathways of M4 in ESI− mode; (b) The proposed fragmentation pathways of M10 in ESI− mode.
M5 and M6 showed [M − H]− at m/z 447.13, and their molecular formulae were predicted to be C22H24O10. Moreover, the fragment ions at m/z 271.09, m/z 175.02, m/z 147.05 (C9H7O2, 6A− or 3,5B− − 2H) and m/z 135.05 (C8H7O2, 3,4A− or 3,4B−) could be detected in their MS2 spectra. Based on m/z 135.05 (C8H7O2), we could deduce that the ring-cleavage took place at the bond of C11-C11a. Because a larger CLogP value means a lower polarity and a larger retention time in reversed phase HPLC, so M5 (CLogP = 0.7229, tR = 90.033) was assigned as vestitol-1’-O-glucuronide and M6 (CLogP = 0.9229, tR = 92.050) was vestitol-7-O-glucuronide.
M8 and M9 showed [M − H]− at m/z 429.12 and their molecular formulae were predicted to be C22H22O9, and they yielded a fragment ion at m/z 271.09 by neutral loss of 158.02 Da (C6H6O5). In their MS3 spectra, the characteristic fragment ions at m/z 147.05 (C9H7O2, 6A− or 3,5B− − 2H), m/z 135.05 (C8H7O2, 3,4A− or 3,4B−) and m/z 109.03 (C6H5O2, 2,5A− + 2H) were observed. Thus, M8 and M9 were determined as vestitol vitamin C conjugates.
The molecular formula of M10 (Figure 6b) was calculated as C22H24O13S based on the [M − H]− at m/z 527.0883. The fragment ions at m/z 351.0505 and m/z 271.0944 were observed in MS2 spectra and m/z 147.0546 (C9H7O2), m/z 135.0498 (C8H7O2, 3,4A− or 3,4B−) and m/z 121.0354 (C7H5O2) were detected in MS3 spectra. This indicated that M10 was a hydrogenated medicarpin glucuronide sulfate. Besides, based on m/z 135.0498 (C8H7O2), we could deduce that the bond of C11-C11a in medicarpin was cleaved. Moreover, the fragment ion at m/z 254.9829 (C10H7O6S,), which was derived from precursor ion m/z 527.0901, indicated that the sulfonic group (-SO3H) was linked to the fragment of C10H7O3 (1,5B− − 4H), and the fragment ion at m/z 229.0156 (C9H9O5S), which was obtained from precursor ion m/z 351.0505, indicated that -SO3H was linked to the fragment of C9H9O2 (3,5B−). Hence, we could deduce that the sulfonic group was linked to the hydroxyl group of B ring, while the glucuronyl group was linked to the hydroxyl group of A ring. Therefore, M10 was unambiguously identified as vestitol-7-O-glucuronide-1’-O-sulfate. The MS spectra of M10 are shown in Figure S46.
2.2.3. Analysis of Metabolites (M11–M26) Having the Skeleton of Demethylated Medicarpin
M11–M13 (Figure 4b) showed [M − H]− at m/z 431.10, and their molecular formulae were predicted to be C21H20O10. Compared with [Aglycon − H]− (C15H11O4) at m/z 255.07, the neutral loss was 176.03 Da (C6H8O6), indicating that M11–M13 were glucuronides of demethylated medicarpin.
M14–M20 (Figure 4c) were isomers whose molecular formulae were calculated to be C15H12O7S based on [M − H]− at m/z 335.02. According to the neutral loss 79.95 Da (SO3), we speculated that M14–M20 were sulfates of demethylated medicarpin.
The molecular formulae of M21–M23 (Figure 4d) were predicted to be C21H20O13S based on [M − H]− at m/z 511.06. In their MS2 spectra, fragment ions at m/z 335.02 and m/z 255.07 which were formed by sequential neutral losses of 176.03 Da (C6H8O6) and 79.95 Da (SO3) were observed, demonstrating that M21–M23 were demethylated medicarpin glucuronide sulfates.
Demethylated medicarpin only has two metabolism sites (two free hydroxyl groups), but it has three glucuronidation metabolites (M11–M13), seven sulfation metabolites (M14–M20) and three glucuronidation and sulfation metabolites (M21–M23). Therefore, we can confirm that isomerization of medicarpin (configuration change or shift of substituent) must occur.
2.2.4. Analysis of Metabolites (M27–M52) Having the Skeleton of Demethylated and Hydrogenated Medicarpin
M27 (Figure 7a) showed [M − H]− at m/z 257.0820, and the molecular formula was predicted to be C15H14O4. Compared with C16H14O4 of medicarpin, it has one less carbon atom. In its MS2 spectra, there were a series of fragment ions at m/z 149.0289 (C8H5O3), m/z 147.0481 (C9H7O2), m/z 135.0494 (C8H7O2), m/z 121.0329 (C7H5O2) and m/z 109.0263 (C6H5O2). Based on the fragment ion at 149.0289 (C8H5O3), we could determine that the M27 might be O, Q or R. M37 (Figure 7b) presented [M − H]− at m/z 337.0392, therefore its molecular formula was calculated to be C15H14O7S. We could speculate that it was the sulfate of demethylated and hydrogenated medicarpin based on the neutral loss of 79.95 Da (SO3) and fragment ion at m/z 257.0804 ([Aglycon − H]−). Besides, fragment ions at m/z 151.0443 (C8H7O3), m/z 147.0456 (C9H7O2), m/z 135.0461 (C8H7O2), m/z 121.0267 (C7H5O2), m/z 119.0586 (C8H7O) and m/z 109.0234 (C6H5O2) were observed. Based on fragment ions at m/z 215.0033 (C8H7O5S) and m/z 196.9928 (C8H5O4S), which were yielded from m/z 215.0033 (C8H7O5S) by the loss of 18.01 Da (H2O), we could deduce that the skeleton of C8H7O2 had two hydroxyl groups and the sulfonic group was linked to the B ring. As a result, the aglycon of M37 must be Q. The characteristic fragment ions of M27 and M37 are shown in Figure 7. The MS spectrum of M37 is shown in Figure S47.
Figure 7.

Characteristic fragment ions of M27 (a) and M37 (b) in ESI− mode.
2.2.5. Analysis of Metabolites (M53–M75) Having the Skeleton of Demethylated and Hydroxylated Medicarpin
M53 presented [M − H]− at m/z 271.0612, and the molecular formula was indicated to be C15H12O5. Compared with C16H14O4 of medicarpin, it has a less CH2 and one more oxygen atom. Furthermore, according to the fragment ions at m/z 161.0281 (C9H5O3), m/z 149.0273 (C8H6O3), m/z 137.0283 (C7H5O3), m/z 133.0249 (C8H5O2), m/z 121.0406 (C7H5O2) and m/z 109.0327 (C6H5O2), M53 was determined to be demethylated and hydroxylated medicarpin. However, due to the special structure of the demethylated medicarpin, it is impossible to judge the position of hydroxylation based on the current mass spectrometry data.
2.2.6. Analysis of Metabolites (M76–M107) Having the Skeleton of Hydroxylated Medicarpin
The molecular formulae of M87–M99 were predicted to be C16H14O8S due to the pseudomolecular ion peak [M − H]− at m/z 365.04. In their MS2 spectra, the fragment ion at m/z 285.08 (C16H13O5) formed by the loss of 79.95 Da (SO3) was detected. Therefore, M87–M99 were hydroxylated medicarpin sulfates.
M88 (Figure 8) possessed fragment ions at m/z 285.0727, m/z 270.0564, m/z 161.0254 (C9H5O3, 6,8A− − 2H or 3,5B− − 2H), m/z 139.0337 (C7H7O3, 6,7B− + 2H) and m/z 124.0179 (C6H4O3, 6,7B− + 2H − CH3•). Based on the fragment ion at m/z 124.0179 (C6H4O3, 6,7B− + 2H − CH3•), generated from the fragment ion at m/z 270.0564, we deduced that the newly added hydroxyl group was linked to the D ring of M88.
Figure 8.

The proposed fragmentation pathways of M88 in ESI− mode.
2.2.7. Analysis of Metabolites (M108–M138) Having the Skeleton of Hydrogenated and Hydroxylated Medicarpin
M112 (Figure 9) showed [M − H]− at m/z 287.0943 and the molecular formula was calculated to be C16H16O5, which was two hydrogen atoms and one oxygen atom more than that of medicarpin. Therefore, we speculated that M112 was hydrogenated and hydroxylated medicarpin. In its MS2 spectra, a characteristic fragment ion at m/z 125.0181 (C6H5O3, 2,5A− + 2H) was observed, indicating that the newly added hydroxyl group was linked to the A ring, so it has three possibilities O, P or Q. Besides, based on the fragment ion at m/z 151.0451 (C8H7O3, 3,4A−), we could determine that the bond of C11-C11a was cleaved. Finally, the structure of M112 was determined to be O. Furthermore, the fragment ions of M112 including m/z 163.0388 (C9H7O3, 6A− − H), m/z 149.0532 (C9H9O2, 3,5B−), m/z 145.0348 (C9H5O2, 6A− − H − H2O), m/z 137.0334 (C7H5O3, 2,4A− or 3,5A−) and m/z 134.0482 (C8H6O2, 3,5B− − CH3•) supported our elucidation. The MS2 spectrum of M112 is shown in Figure S48.
Figure 9.

The proposed fragmentation pathways of M112 in ESI− mode.
The molecular formulae of M114–M120 were predicted as C16H16O8S based on their [M − H]− at m/z 367.05. In their MS2 spectra, m/z 287.09 (C16H15O5) was detected, indicating a neutral loss of 79.95 Da (SO3). Therefore, M114–M120 were determined as hydrogenated and hydroxylated medicarpin sulfates.
M114 (Figure 10) showed fragment ions at m/z 287.0921 ([Aglycon − H]−), m/z 272.0681 ([Aglycon − H]− − CH3•), m/z 245.0126 (C9H9O6S), m/z 229.0108 (C9H9O5S), m/z 177.0598 (C10H9O3), m/z 165.0603 (C9H9O3), m/z 163.0388 (C9H7O3), m/z 161.0254 (C9H5O3), m/z 151.0417 (C8H7O3,), m/z 150.0382 (C8H6O3), m/z 149.0653 (C9H9O2), 137.0213 (C7H5O3), m/z 135.0589 (C8H7O2), m/z 135.0094 (C7H3O3), m/z 122.0312 (C7H6O2) and m/z 121.0363 (C7H5O2) in MS2 spectra. According to the fragment ion at m/z 122.0312 (C7H6O2), produced from m/z 272.0681 ([Aglycon − H]− − CH3•), we determined that the aglycon of M114 should be O or P. Moreover, based on the fragment ion at m/z 135.0094 (C7H3O3), we could deduce that the aglycon of M114 was O2 or P. However, because the same fragment ions could be produced from O2 or P, it was impossible to judge the position of hydroxylation based on the current mass spectrometry data. In addition, the elemental composition of m/z 229.0108 (C9H9O5S) suggested that the sulfonic group was connected to the fragment ion of m/z 149.0653 (C9H9O2). Hence, we could determine the sulfate was linked to the hydroxyl group of B ring. The MS2 spectrum of M114 is shown in Figure S49.
Figure 10.

The proposed fragmentation pathways of M114.
M115 exhibited fragment ions at m/z 287.0918 (C16H15O5, [Aglycon − H]−), m/z 230.9941 (C8H7O6S, 3,4B− + SO3), m/z 177.0590 (C10H9O3, 2,5B− − 2H), m/z 165.0631 (C9H9O3, 3,5B− or 2,4B−), m/z 151.0440 (C8H7O3, 3,4B−), m/z 150.0407 (C9H9O3, 3,5B− − CH3• or 2,4B− − CH3•), m/z 147.0412 (C9H7O2, 6A− − H), m/z 139.0448 (C7H7O3, 6B− + H), m/z 136.0182 (C7H4O3, 3,4B− − CH3•), m/z 135.0397 (C8H7O2, 3,4A−) and m/z 121.0325 (C7H5O2, 3,5A− or 2,4A−) in its MS2 spectra. From the characteristic fragment ion at m/z 136.0182 (C7H4O3, 3,4B− − CH3•) yielded from m/z 151.0440 (C8H7O3, 3,4B−), we could identify that the newly added hydroxyl group was linked to the D ring of medicarpin and the C11-C11a bond of medicarpin was cleaved. Moreover, m/z 230.9941 was predicted to be C8H7O6S, which indicated that the sulfonic group was connected to the fragment ion of m/z 151.0440 (C8H7O3, 3,4B−). We therefore confirmed that the sulfonic group was linked to the hydroxyl group of B ring. The probable structure and characteristic fragment ions of M115 are shown in Figure 11. The MS2 spectrum of M115 is shown in Figure S50.
Figure 11.

The probable structure and characteristic fragment ions of M115.
By comparing the fragmentation pathways of M112, M114 and M115, we get the following information: (1) As long as the fragment ion at m/z 125.02 (C6H5O3) is detected, we can deduce that the newly added hydroxyl group is linked to the A ring; (2) If the fragment ions at m/z 151.04 and m/z 136.02 are observed at the same time, we can confirm that the newly added hydroxyl group is attached to the D ring of medicarpin and the ring cleavage is at the bond of C11-C11a.
2.2.8. Analysis of Metabolites (M139–M149) Having the Skeleton of Demethylated, Hydrogenated and Hydroxylated Medicarpin
M140–M146 showed [M − H]− at m/z 353.03, thus their molecular formulae were identified as C15H14O8S. In their MS2 spectra, m/z 273.08 (C15H13O5) generated by a neutral loss 79.95 Da (SO3) was detected. Hence, M140–M146 were demethylated, hydrogenated and hydroxylated medicarpin sulfates.
M144 yielded a series of characteristic fragment ions at m/z 273.0782 ([Aglycon − H]−), m/z 163.0437 (C9H7O3), m/z 161.0315 (C9H5O3), m/z 151.0428 (C8H7O3), m/z 147.0451 (C9H7O2), m/z 137.0308 (C7H5O3), and m/z 121.0276 (C7H5O2). Based on the fragment ion at m/z 151.0428 (C8H7O3), we could deduce that the aglycon of M144 might be O, P, Q, R, S or T. According to the fragment ion at m/z 147.0451 (C9H7O2), we could exclude P, R and T. Besides, based on the fragment ion at m/z 137.0308 (C7H5O3), we could exclude E. Therefore, we could determine that the aglycon of M144 was O or Q. Furthermore, based on the fragment ion at m/z 147.0451 (C9H7O2), we finally deduced that the aglycon of M144 was O1, O2 or Q (Figure 12). The MS2 spectrum of M144 is shown in Figure S51.
Figure 12.

The probable structures (O1, O2, Q) of the aglycon of M144.
The molecular formulae of M147–M149 were predicted as C21H22O11 in line with [M − H]− at m/z 449.11. The fragment ion at m/z 273.08 (C15H13O5) formed by losing 176.03 Da (C6H8O6) from [M − H]− was detected in the MS2 spectra. Hence, M147–M149 were demethylated, hydrogenated and hydroxylated medicarpin glucuronides.
2.2.9. Analysis of Metabolites (M150–M156) Having the Skeleton of Demethylated and Dihydroxylated Medicarpin
M150–M151 showed [M − H]− at m/z 463.09 and the molecular formulae were calculated to be C21H20O10. M152–M156 showed [M − H]− at m/z 367.01, indicating the molecular formulae of C15H12O9S. In their MS2 spectra, the fragment ion at m/z 287.06 (C15H11O6) was detected. Compared with C16H14O4 of medicarpin, it had a less CH2 and two more oxygen atoms. As a result, medicarpin might undergo demethylation and dihydroxylation. Therefore, M150–M151 were glucuronides of demethylated and dehydroxylated medicarpin, while M152–M156 were sulfates of demethylated and dehydroxylated medicarpin.
2.2.10. Analysis of Metabolites (M157–M161) Having the Skeleton of Dihydroxylated Medicarpin
M157–M159 showed [M − H]− at m/z 381.03 and their molecular formulae were determined as C16H14O9S. In MS2 spectra, m/z 381.03 produced [Aglycon − H]− at m/z 301.07 (C16H13O6) by the loss of 79.95 Da (SO3). The elemental composition difference between medicarpin (C16H14O4) and the aglycon of M157–M159 was O2. Therefore, M157–M159 were identified as sulfates of dihydroxylated medicarpin. In MS2 spectra of M159, fragment ions at m/z 366.0054 (C15H10O9S, [M − H]− − CH3•), m/z 301.0711 ([Aglycon − H]−), m/z 286.0477 ([Aglycon − H]− − CH3•) and m/z 203.9750 (C6H4O6S, 6,7D− + 2H + SO3) were detected. Subsequently, m/z 286.0477 yielded a series of fragment ions at m/z 162.0347 (C9H6O3, 2,5D− − CH3•), m/z 161.0269 (C9H5O3, 6,7A− − 2H), m/z 149.0301 (C8H5O3, 3,4,7A− − H), m/z 137.0229 (C7H5O3, 3,5A−), m/z 133.0328 (C8H5O2, 6,7A− − 2H − CO), m/z 124.0254 (C6H4O3, 6,7D− + 2H − CH3•) and m/z 123.0137 (C6H3O3, 2,5A−). Based on the fragment ion at m/z 123.0137 (C6H3O3, 2,5A−), we assured that one hydroxyl group must be linked to the A ring. Besides, the fragment ion at m/z 124.0254 (C6H4O3, 6,7D− + 2H − CH3•) suggested that one hydroxyl group must be linked to the D ring. Furthermore, according to the fragment ion at m/z 203.9750 (C6H4O6S), we could determine that the sulfonic group was linked to the hydroxyl group of D ring. The fragmentation pathways of M159 are shown in Figure 13. The MS spectrum of M159 is shown in Figure S52.
Figure 13.

The proposed fragmentation pathways of M159.
2.2.11. Analysis of Metabolites M162 Having the Skeleton of Hydrogenated and Dihydroxylated Medicarpin
Its molecular formula was calculated to be C16H16O6 based on [M − H]− at m/z 303.0861. Compared with the C16H14O4 of medicarpin, it had two more hydrogen atoms and two more oxygen atoms. Accordingly, M162 was determined as hydrogenated and dihydroxylated medicarpin.
2.2.12. Analysis of Metabolites M163–M164 Having the Skeleton of Hydrogenated and Trihydroxylated Medicarpin
M163 showed [M − H]− at m/z 399.0528 and its molecular formula was predicted to be C16H16O10S. In its MS2 spectra, fragment ions at m/z 381.0296 (C16H13O9S), m/z 301.0664 (C16H13O6) and m/z 286.0504 (C15H10O6) were detected. Therefore, M163 was regarded as hydrogenated and trihydroxylated medicarpin sulfate. The molecular formula of M164 was calculated to be C17H18O10S based on [M − H]− at m/z 413.0521. In its MS2 spectra, fragment ions at m/z 381.0275 (C16H13O9S), m/z 301.0671 (C16H13O6) and m/z 286.0517 (C15H10O6) were detected. Therefore, M164 was determined as hydrogenated, trihydroxylated and methylated medicarpin sulfate.
2.2.13. Analysis of Metabolite M165 Having the Skeleton of Demethylated, Hydrogenated and Dihydroxylated Medicarpin
M165 showed [M − H]− at m/z 369.0297 and its molecular formula was predicted to be C15H14O9S. In its MS2 spectra, fragment ions at m/z 351.0135 (C15H11O8S) and m/z 271.0607 (C15H11O5) were detected, indicating two sequential neutral losses of 18.01 Da (H2O) and 79.95 Da (SO3). Compared with C16H13O4 of medicarpin, m/z 271.0607 had a less CH2 and one more oxygen atom. Therefore, M165 was determined as a demethylated, hydrogenated and dihydroxylated medicarpin sulfate.
2.3. Distribution of 165 Metabolites in Rats
The distribution of 165 metabolites (including 104 metabolites in urine, 29 metabolites in feces, 38 metabolites in plasma, 41 metabolites in the colon, 74 metabolites in the intestine, 28 metabolites in the stomach, 24 metabolites in the liver, 15 metabolites in the spleen, 42 metabolites in the kidney, 8 metabolites in the lung, 10 metabolites in the heart, 3 metabolites in the brain and 17 metabolites in the thymus) of medicarpin and their relative contents in each biological sample are shown in Figure 14. The relative content of a metabolite in each biosample was calculated by (peak area of a metabolite in the sample/total peak area of all metabolites detected in the sample) × 100%. The peak area of a metabolite was calculated from its extracted ion chromatogram.
Figure 14.






The distribution of 165 metabolites of medicarpin and their relative contents in 13 biosamples (a, new compounds). The relative content of a metabolite in each biosample was calculated by (peak area of a metabolite/total peak area of all detected metabolites) × 100%.
From Figure 14, we could find that: (1) in urine, M159 (12.68%, dihydroxylated medicarpin sulfate), M63 (11.86%, demethylated and hydroxylated medicarpin sulfate), M17 (7.18%, demethylated medicarpin sulfate), and M64 (5.34%) were four major metabolites and all of them were sulfates; it is an interesting phenomenon that sulfates other than glucuronides are major metabolites in urine, and the cause is unclear; (2) in feces, M18 (20.34%), M43 (10.35%), M34 (9.89%), M40 (8.86%), M3 (7.57%, Vc conjugate), and M27 (7.25%) were six major metabolites with a relative content higher than 5%, and three of them (M43, M34, M40) were demethylated and hydrogenated medicarpin sulfates; (3) in plasma, M1 (36.15%, medicarpin glucuronide), M5 (12.82%, vestitol-1’-O-glucuronide), M18 (10.81%), M65 (8.00%),and M40 (5.58%) were major metabolites; (4) M1 was the most abundant metabolite in the stomach, liver, spleen, kidney, lung, heart, and thymus. M5, M14 (demethylated medicarpin) were the most abundant metabolites in intestine and liver, respectively. M17 (demethylated medicarpin sulfate) was the most abundant metabolite in the colon and brain; (5) M1, M5 were detected in all 10 organs; M30 (demethylated and hydrogenated medicarpin glucuronide) was detected in nine organs (except the brain); M12 and M27 were detected in eight organs; therefore, these five metabolites were widely distributed in vivo. Considering the high abundance and wide distribution, we suggest that the bioactivities of these metabolites deserve further research in order to fully explain the pharmacological action mechanism of medicarpin. In addition, the reason for specific distribution of some metabolites (e.g., M2, M14, M20, M35, M40, M54, M109) also need further investigation.
The metabolic reactions of medicarpin included demethylation, hydrogenation, hydroxylation, glucuronidation, sulfation, glycosylation, methylation, and conjunction of vitamin C. The relative contents of metabolic reactions and phase I metabolites in each biosample are shown in Table 1, which were calculated by summing the relative contents of all metabolites generated through it and all phase I metabolites, respectively. In urine, the contents of sulfation metabolites, glucuronidation metabolites and vitamin C conjugates were 74.60%, 25.83% and 1.08%, respectively. In feces, the contents of sulfation metabolites, glucuronidation metabolites and vitamin C conjugates were 73.53%, 1.62% and 15.54%, respectively. This indicated that medicarpin was mainly excreted in the form of sulfates. In plasma, the major metabolic reaction was glucuronidation (relative content: 67.87%). The glycosylation metabolite (M50) was only found in urine. Furthermore, the contents of phase I metabolites of medicarpin in most biosamples were very low (<10%), except in the colon (17.57%), which suggested that phase II metabolites are major metabolites.
Table 1.
Relative contents of metabolic reactions and phase I metabolites of medicarpin in 13 biological samples (%).
| Biosample | Relative Contents (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Phase I Reaction | Phase II Reaction | Phase I Metabolites | |||||||
| -CH3 | +2H | +OH | +SO3 | +GlcUA | +Vc | +Glc | +CH3 | ||
| urine | 55.14 | 37.01 | 57.79 | 74.60 | 25.83 | 1.08 | 0.23 | 3.93 | 1.78 |
| feces | 71.51 | 44.43 | 21.55 | 73.53 | 1.62 | 15.54 | — | — 1 | 9.30 |
| plasma | 36.58 | 25.72 | 22.78 | 36.17 | 67.87 | 0.44 | — | 3.16 | — |
| colon | 57.95 | 58.48 | 28.30 | 55.81 | 16.61 | 9.23 | — | 0.78 | 17.57 |
| intestine | 39.56 | 41.46 | 36.17 | 15.62 | 78.04 | 7.60 | — | 11.23 | 0.99 |
| stomach | 39.46 | 22.38 | 26.67 | 35.00 | 54.57 | 1.23 | — | 2.50 | 9.20 |
| liver | 62.62 | 31.43 | 13.33 | 57.11 | 42.09 | — | — | 3.55 | 0.80 |
| spleen | 33.84 | 23.45 | 8.74 | 27.79 | 71.33 | — | — | 1.03 | 0.88 |
| kidney | 60.90 | 30.46 | 34.93 | 63.43 | 36.30 | 0.10 | — | 1.67 | 0.17 |
| lung | 34.25 | 22.91 | 5.29 | 28.22 | 71.78 | — | — | — | — |
| heart | 33.74 | 22.74 | 6.19 | 27.76 | 71.70 | — | — | — | 0.54 |
| brain | 57.11 | 20.86 | — | 57.11 | 42.89 | — | — | — | — |
| thymus | 25.25 | 15.66 | 9.12 | 18.96 | 80.54 | — | — | 2.01 | 0.49 |
1 —, undetected.
3. Materials and Methods
3.1. Chemicals and Reagents
Medicarpin (Molecular formula: C16H14O4, exact mass: 270.0892 Da) was isolated from the Maackia amurensis Radix by chromatographic methods, including 200–300 mesh normal phase silica gel (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) column chromatography and reversed phase C18 silica gel (YMC, YMC Co., Ltd., Kyoto, Japan) column chromatography. The optical rotation angle was = −19.92 (c = 5, CH3OH). Hence, its structure was determined to be (−)-3-hydroxy-9-methoxypterocarpan or (−)-medicarpin on the basis of UV, NMR, and MS data. Its purity was greater than 98% by the peak area normalization method using HPLC-UV analysis at 280 nm. Formic acid (Fisher scientific, Fair lawn, NJ, USA), acetonitrile (Fisher scientific), and methanol (Tianjin Damao Chemicals, Tianjin, China) were of HPLC grade. Ultrapure water was prepared using a Milli-Q water purification system (Millipore, Billerica, MA, USA). Analytical grade sodium carboxymethyl cellulose (CMC-Na) was purchased from Tianjin Guangfu Fine Chemical Research Institute (Tianjin, China). All other chemicals and reagents were of analytical grade.
3.2. Animals and Drug Administration
Eight male Sprague-Dawley rats (220–250 g) were obtained from the Experimental center of Peking University Health Science Center (Beijing, China). All rats were housed in a controlled animal room. Before oral gavage, the rats were randomly divided into two groups (a drug group and a blank group, four rats per group). Each rat was housed in a metabolic cage with water and food ad libitum for three days. Medicarpin was suspended in 0.5% CMC-Na solution and administrated at a dose of 100 mg/kg (body weight) to the drug group rats once a day for four days, while the blank group were given the same volume of 0.5% CMC-Na. All animal treatments were approved by the Biomedical Ethical Committee of Peking University (approval No. LA2015134).
3.3. Sample Collection and Preparation
Urine and feces samples were collected for the first three days after drug administration; plasma samples and organ samples were collected on the last day one hour after drug administration.
Urine samples: All urine samples from the same group were merged and evaporated to dryness at 40 °C using a Laborota 4001 rotator evaporator (Heidolph Instruments GmbH & Co., Schwabach, Germany) under vacuum, respectively. Then, each group added 10 volumes of methanol (10 mL methanol/g residue) and extracted ultrasonically for 30 min. The extracts were filtered and evaporated to dryness at 40 °C. Finally, the residues were dissolved in 2 volumes of methanol (2 mL methanol/g residue), filtered through 0.22 μm membranes, and stored at −80 °C before further analysis.
Feces samples: All feces samples from the same group were dried at 50 °C and crushed into powder, respectively. Then, each group was ultrasonically extracted with 10 volumes (mL/g) of methanol for 30 min and filtered, after that the filtrate was evaporated to dryness at 40 °C. The residues were dissolved in 4 volumes of methanol (4 mL methanol/g residue), filtered through 0.22 μm membranes before being stored at −80 °C.
Plasma samples: An hour after the last drug administration, the blood was collected in heparin tubes by cardiac puncture technique in groups through anesthetizing with an intraperitoneal injection of chloral hydrate. Plasma was obtained after centrifugation at 2292× g (5000 rpm), 4 °C for 15 min using a 3–30 K refrigerated centrifuge (Sigma Laborzentrifugen GmbH, Osterode am Harz Germany). Afterwards, each plasma sample was ultrasonically extracted with 10 volumes (mL/mL) of methanol for 30 min in order to remove the protein content and then centrifuged for 15 min at 2292× g (5000 rpm), 4 °C. The supernatant of each group was evaporated to dryness under vacuum by a rotator evaporator. Then the residue was dissolved in 1 volume of methanol (1 mL methanol/g residue) and filtered through 0.22 μm membranes before stored at −80 °C.
Organ samples: After collecting blood via cardiac puncture, the heart, liver, spleen, lungs, kidneys, colon, intestine, stomach, brain, thymus were rapidly removed and washed three times with cold saline to flush away the surface blood and other residual substances. Later, they were preserved at −80 °C after drying with filter papers. Then, each kind of the organs from the same group were combined and weighed, as well as added 5 times volumes of methanol (5 mL methanol/g tissue). The mixture was homogenized by a T10 homogenizer (IKA Co., Ltd., Staufen, Germany) under low temperature conditions, and ultrasonically extracted by a KQ-2200B ultrasonic cleaning machine (Kunshan Ultrasonic Instruments Co., Ltd., Jiangsu, China) for an hour and then centrifuged at 2292× g (5000 rpm) at 4 °C for 15 min. Additionally, the supernatant was concentrated and dried in a vacuum at 40 °C, reconstituted with 10 times volume of methanol, and maintained at −80 °C. And, before LC-MS analysis they shall be filtered through 0.22 μm membranes.
3.4. Instrumentation and Analytical Conditions
The chromatographic separation was performed on the Agilent HPLC instrument using a Phenomenex Gemini C18 column (250 mm × 4.60 mm, 5 μm). The column oven temperature was maintained at 35 °C and the volume injected was 10 μL. The mobile phases were water (contained 0.1% formic acid, v/v) (A) and acetonitrile (B). The gradient elution program was as follows: 0–10 min, 3% B; 10–130 min, 3–40% B; 130–140 min, 40–100% B; 140–155 min, 100% B. The flow rate was 1.0000 mL/min.
HPLC-ESI-IT-TOF-MSn analysis was performed on a Shimadzu LC-MS-IT-TOF instrument, which consists of a CBM-20A system controller, two LC-20AD pumps, an SIL-20AC autosampler, a CTO-20A column oven, an SPD-M20A PDA detector, an ESI ion source, and an IT-TOF mass spectrometer using the same separation conditions. The mass spectrometer flow rate was 0.2000 mL/min that was split from HPLC effluent and the detection mode included positive ion (ESI+) and negative ion (ESI−) mode with a full-scan covering m/z 100–1000 (MS) and m/z 50–1000 (MS2 and MS3). The other optimal conditions were set as follows: heat block and curved desolvation line temperature was 250 °C and nebulizing nitrogen gas flow was 1.5 mL/min; the interface voltage was (+)-4.5 kV and (−)-5.5 kV and the detector voltage was 1.7 kV. In addition, the ion accumulation time was 30 ms and the relative collision-induced dissociation energy was 70%. The mass range m/z 50–3000 was calibrated by a trifluoroacetic acid sodium solution (2.5 mM). All data were acquired and analyzed by Shimadzu LCMS solution Version 3.60, Formula Predictor Version 1.2, and Accurate Mass Calculator (Shimadzu, Kyoto, Japan).
4. Conclusions
In this paper, the metabolites of medicarpin and their distributions in rats were systematically studied for the first time, and 165 new metabolites (13 phase I metabolites and 152 phase II metabolites) were tentatively identified by HPLC-ESI-IT-TOF-MSn, including 104 metabolites in urine, 29 in feces, 38 in plasma, 41 in the colon, 74 in the intestine, 28 in the stomach, 24 in the liver, 15 in the spleen, 42 in the kidney, eight in the lung, 10 in the heart, three in the brain and 17 in the thymus. Eighty-six sulfate metabolites, five Vitamin C conjugates (M3, M8, M9, M24, M25) and two hydroxylated medicarpin glucuronides (M5 and M6) were regarded as potential new compounds by retrieving information from the Scifinder database. Besides, based on the structure of these metabolites, the metabolic pathways of medicarpin were proposed. The metabolic reactions of medicarpin included demethylation, hydrogenation, hydroxylation, glucuronidation, sulfation, glycosylation, methylation and conjunction of vitamin C, among which sulfation and glucuronidation were the major phase II metabolic reactions. Five metabolites (M1, M5, M30, M12, M27) were widely distributed to nine or ten organs, and the specific distribution of lots of metabolites (e.g., M2, M14, M20, M35, M40, M54, M109) were also discovered. Furthermore, we found that isomerization of medicarpin must occur in vivo. This study is the first comprehensive report on the metabolism of pterocarpans in animals. The results will facilitate the understanding of the metabolism of medicarpin, and will provide a scientific basis for further pharmacological studies on medicarpin and for metabolism research of other pterocarpans in animals.
Supplementary Materials
Figures S1–S52 are available online.
Author Contributions
Conceptualization, F.X.; methodology, F.X.; validation, F.X., T.L.; formal analysis, H.-Y.W.; investigation, H.-Y.W., T.L., R.J.; writing—original draft preparation, H.-Y.W.; writing—review and editing, H.-Y.W., F.X., M.-Y.S., G.-X.L., Y.-L.L., S.-Q.C.; supervision, F.X., M.-Y.S.; project administration, F.X.; funding acquisition, F.X.
Funding
This study was financially supported by the National Natural Science Foundation of China (No. 81573593).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Not available.
References
- 1.Harper S.H., Kemp A.D., Underwood W.G.E. Heartwood constituents of Stoarfzia madagascariensis. Chem. Comm. 1965;14:309–310. [Google Scholar]
- 2.Belofsky G., Kolaczkowski M., Adams E., Schreiber J., Eisenberg V., Coleman C.M., Zou Y., Ferreira D. Fungal ABC tansporter-associated activity of isoflavonoids from the root extract of Dalea formosa. J. Nat. Prod. 2013;76:915–925. doi: 10.1021/np4000763. [DOI] [PubMed] [Google Scholar]
- 3.Rukachaisirikul T., Innok P., Aroonrerk N., Boonamnuaylap W., Limrangsun S., Boonyon C., Woonjina U., Suksamrarn A. Antibacterial pterocarpans from Erythrina subumbrans. J. Ethnopharmacol. 2007;110:171–175. doi: 10.1016/j.jep.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 4.Weng J., Tsao L., Yen M., Wang J., Lin C. Anti-inflammatory constituents and new pterocarpanoid of Crotalaria pallida. J. Nat. Prod. 2003;66:404–407. doi: 10.1021/np020135d. [DOI] [PubMed] [Google Scholar]
- 5.Zhou H., Lutterodt H., Cheng Z., Yu L.L. Anti-inflammatory and antiproliferative activities of trifolirhizin, a flavonoid from Sophora flavescens Roots. J. Agric. Food Chem. 2009;57:4580–4585. doi: 10.1021/jf900340b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L., Liu H., Hsiao P., Kuo L.Y., Lee I., Wu T., Chiou W., Kuo Y. New isoflavonoid glycosides and related constituents from Astragali Radix (Astragalus membranaceus) and their inhibitory activity on nitric oxide production. J. Agric. Food Chem. 2011;59:1131–1137. doi: 10.1021/jf103610j. [DOI] [PubMed] [Google Scholar]
- 7.Kuete V., Sandjo L.P., Djeussi D.E., Zeino M., Kwamou G.M.N., Ngadjui B., Efferth T. Cytotoxic flavonoids and isoflavonoids from Erythrina sigmoidea towards multi-factorial drug resistant cancer cells. Invest. New Drugs. 2014;32:1053–1062. doi: 10.1007/s10637-014-0137-y. [DOI] [PubMed] [Google Scholar]
- 8.Mahajan P., Gnana O.R., Jachak S.M., Bharate S.B., Chaudhuri B. Antioxidant and antiproliferative activity of indigocarpan, a pterocarpan from Indigofera aspalathoides. J. Pharm. Pharmacol. 2016;68:1331–1339. doi: 10.1111/jphp.12609. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen P.H., Le T.V.T., Thuong P.T., Dao T.T., Ndinteh D.T., Mbafor J.T., Kang K.W., Oh W.K. Cytotoxic and PTP1B inhibitory activities from Erythrina abyssinica. Bioorg. Med. Chem. Lett. 2009;19:6745–6749. doi: 10.1016/j.bmcl.2009.09.108. [DOI] [PubMed] [Google Scholar]
- 10.Bhargavan B., Singh D., Gautam A.K., Mishra J.S., Kumar A., Goel A., Dixit M., Pandey R., Manickavasagam L., Dwivedi S.D., et al. Medicarpin, a legume phytoalexin, stimulates osteoblast differentiation and promotes peak bone mass achievement in rats: Evidence for estrogen receptor β-mediated osteogenic action of medicarpin. J. Nutr. Biochem. 2012;23:27–38. doi: 10.1016/j.jnutbio.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Kureel J., Dixit M., Tyagi A.M., Mansoori M.N., Srivastava K., Raghuvanshi A., Maurya R., Trivedi R., Goel A., Singh D. miR-542-3p suppresses osteoblast cell proliferation and differentiation, targets BMP-7 signaling and inhibits bone formation. Cell Death Dis. 2014;5:e1050. doi: 10.1038/cddis.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tyagi A.M., Gautam A.K., Kumar A., Srivastava K., Bhargavan B., Trivedi R., Saravanan S., Yadav D.K., Singh N., Pollet C., et al. Medicarpin inhibits osteoclastogenesis and has nonestrogenic bone conserving effect in ovariectomized mice. Mol. Cell Endocrinol. 2010;325:101–109. doi: 10.1016/j.mce.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 13.Vieira N.C., Espíndola L.S., Santana J.M., Veras M.L., Pessoa O.D., Pinheiro S.M., de Araujo R.M., Lima M.A., Silveira E.R. Trypanocidal activity of a new pterocarpan and other secondary metabolites of plants from Northeastern Brazil flora. Bioorg. Med. Chem. 2008;16:1676–1682. doi: 10.1016/j.bmc.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 14.Rosa A., Deiana M., Corona G., Atzeri A., Incani A., Appendino G., Dessì M.A. Antioxidant properties of extracts and compounds from Psoralea morisiana. Eur. J. Lipid Sci. Technol. 2005;107:521–529. doi: 10.1002/ejlt.200501183. [DOI] [Google Scholar]
- 15.Fotso G.W., Maher F.A., Ngnintedo D., Ango P.Y., Kapche D.G.F.W., Ngameni B., Ngwenya B., Yeboah S.O., Ngadjui B.T., Andrae-Marobela K. Three new isoflavonoids with antioxidant properties from Ptycholobium contortum (N.E.Br.) Brummitt (Leguminosae) Phytochem. Lett. 2015;14:254–259. doi: 10.1016/j.phytol.2015.10.015. [DOI] [Google Scholar]
- 16.Selvam C., Jachak S.M., Gnana Oli R., Thilagavathi R., Chakraborti A.K., Bhutani K.K. A new cyclooxygenase (COX) inhibitory pterocarpan from Indigofera aspalathoides: Structure elucidation and determination of binding orientations in the active sites of the enzyme by molecular docking. Tetrahedron Lett. 2004;45:4311–4314. doi: 10.1016/j.tetlet.2004.04.010. [DOI] [Google Scholar]
- 17.Yuk H.J., Curtis-Long M.J., Ryu H.W., Jang K.C., Seo W.D., Kim J.Y., Kang K.Y., Park K.H. Pterocarpan profiles for soybean leaves at different growth stages and investigation of their glycosidase inhibitions. J. Agric. Food Chem. 2011;59:12683–12690. doi: 10.1021/jf203326c. [DOI] [PubMed] [Google Scholar]
- 18.Ryu Y.B., Curtis-Long M.J., Kim J.H., Jeong S.H., Yang M.S., Lee K.W., Lee W.S., Park K.H. Pterocarpans and flavanones from Sophora flavescens displaying potent neuraminidase inhibition. Bioorg. Med. Chem. Lett. 2008;18:6046–6049. doi: 10.1016/j.bmcl.2008.10.033. [DOI] [PubMed] [Google Scholar]
- 19.Woo H.S., Kim D.W., Curtis-Long M.J., Lee B.W., Lee J.H., Kim J.Y., Kang J.E., Park K.H. Potent inhibition of bacterial neuraminidase activity by pterocarpans isolated from the roots of Lespedeza bicolor. Bioorg. Med. Chem. Lett. 2011;21:6100–6103. doi: 10.1016/j.bmcl.2011.08.046. [DOI] [PubMed] [Google Scholar]
- 20.Mori-Hongo M., Takimoto H., Katagiri T., Kimura M., Ikeda Y., Miyase T. Melanin synthesis inhibitors from Lespedeza floribunda. J. Nat. Prod. 2009;72:194–203. doi: 10.1021/np800395j. [DOI] [PubMed] [Google Scholar]
- 21.Boué S.M., Burow M.E., Wiese T.E., Shih B.Y., Elliott S., Carter-Wientjes C.H., McLachlan J.A., Bhatnagar D. Estrogenic and antiestrogenic activities of phytoalexins from red kidney bean (Phaseolus vulgaris L.) J. Agric. Food. Chem. 2011;59:112–120. doi: 10.1021/jf102255u. [DOI] [PubMed] [Google Scholar]
- 22.Maurich T., Pistelli L., Turchi G. Anti-clastogenic activity of two structurally related pterocarpans purified from Bituminaria bituminosa in cultured human lymphocytes. Mutat. Res. 2004;561:75–81. doi: 10.1016/j.mrgentox.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Zhang W., Xu F., Wang D., Ye J., Cai Q. Buyang Huanwu Decoction ameliorates ischemic stroke by modulating multiple targets with multiple components: In vitro evidences. Chin. J. Nat. Med. 2018;16:0194–0202. doi: 10.1016/S1875-5364(18)30047-5. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad V.U., Iqbal S., Nawaz S.A., Choudhary M.I., Farooq U., Ali S.T., Ahmad A., Bader S., Kousar F., Arshad S., et al. Isolation of four new pterocarpans from Zygophyllum eurypterum (Syn. Z. atriplicoides) with enzyme-inhibition properties. Chem. Biodivers. 2006;3:996–1003. doi: 10.1002/cbdv.200690109. [DOI] [PubMed] [Google Scholar]
- 25.Chen L., Liu Y., Zhang H., Liang S. In-vivo study of metabolism of trifolirhizin in rats. Trad. Chin. Drug Res. Clin. Pharmacol. 2012;23:174–177. [Google Scholar]
- 26.Hargreaves J.A., Mansfield J.W., Coxon D.T. Identification of medicarpin as a phytoalexin in the broad bean plant (Vicia Jaba L.) Nature. 1976;262:318–319. doi: 10.1038/262318a0. [DOI] [Google Scholar]
- 27.Kim J.M., Lee Y.M., Lee G.Y., Jang D.S., Bae K.H., Kim J.S. Constituents of the roots of Pueraria lobata inhibit formation of advanced glycation end products (AGEs) Arch. Pharm. Res. 2006;29:821–825. doi: 10.1007/BF02973900. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y., Chen H., Zhao Y., Wang B., Zhang Q., Zhang L., Tu P. Quantification and stability studies on the flavonoids of Radix hedysari. J. Agric. Food Chem. 2006;54:6634–6639. doi: 10.1021/jf061335o. [DOI] [PubMed] [Google Scholar]
- 29.Rayanil K., Bunchornmaspan P., Tuntiwachwuttikul P. A new phenolic compound with anticancer activity from the wood of Millettia leucantha. Arch. Pharm. Res. 2011;34:881–886. doi: 10.1007/s12272-011-0603-4. [DOI] [PubMed] [Google Scholar]
- 30.Gatouillat G., Magid A.A., Bertin E., El Btaouri H., Morjani H., Lavaud C., Madoulet C. Medicarpin and millepurpan, two flavonoids isolated from Medicago sativa, induce apoptosis and overcome multidrug resistance in leukemia P388 cells. Phytomedicine. 2015;22:1186–1194. doi: 10.1016/j.phymed.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Martínez-Sotres C., López-Albarrán P., Cruz-de-León J., García-Moreno T., Rutiaga-Quiñones J.G., Vázquez-Marrufo G., Tamariz-Mascarúa J., Herrera-Bucio R. Medicarpin, an antifungal compound identified in hexane extract of Dalbergia congestiflora Pittier heartwood. Int. Biodeterior. Biodegrad. 2012;69:38–40. doi: 10.1016/j.ibiod.2011.11.016. [DOI] [Google Scholar]
- 32.Promchai T., Janhom P., Maneerat W., Rattanajak R., Kamchonwongpaisan S., Pyne S.G., Limtharakul T. Antibacterial and cytotoxic activities of phenolic constituents from the stem extracts of Spatholobus parviflorus. Nat. Prod. Res. 2018:1–5. doi: 10.1080/14786419.2018.1512990. [DOI] [PubMed] [Google Scholar]
- 33.Imran K.M., Yoon D., Kim Y. A pivotal role of AMPK signaling in medicarpin-mediated formation of brown and beige. BioFactors. 2018;44:168–179. doi: 10.1002/biof.1392. [DOI] [PubMed] [Google Scholar]
- 34.Imran K.M., Yoon D., Kim Y.S. Medicarpin induces lipolysis via activation of protein kinase A in brown adipocytes. BMB Rep. 2018;51:249–254. doi: 10.5483/BMBRep.2018.51.5.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma L., Xu F., Li F., Wang J., Shang M., Liu G., Cai S. The profiling and identification of the metabolites of 8-prenylkaempferol and a study on their distribution in rats by high-performance liquid chromatography with diode array detection combined with electrospray ionization ion trap time-of-flight multista. Biomed. Chromatogr. 2016;30:175–190. doi: 10.1002/bmc.3534. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.