

Abstract
A series of 3-amino-5-benzylphenol derivatives were designed and synthesized. Among them, (3-benzyl-5-hydroxyphenyl)carbamates were found to exert good inhibitory activity against M. tuberculosis H37Ra, H37Rv and clinically isolated multidrug-resistant M. tuberculosis strains (MIC = 0.625–6.25 μg/mL). The privileged compounds 3i and 3l showed moderate cytotoxicity against cell line A549. Compound 3l also exhibited potent in vivo inhibitory activity on a mouse infection model via the oral administration. The results demonstrated 3-hydroxyphenylcarbamates as a class of new antitubercular agents with good potential.
Keywords: (3-benzyl-5-hydroxyphenyl)carbamate, Mycobacterium tuberculosis, antitubercular activity
1. Introduction
Tuberculosis (TB) is a dangerous disease caused by M. tuberculosis (Mtb). Today it still poses the serious threat to the human health. According to the report of World Health Organization (WHO), in 2018 there were more than 10.0 million people newly infected with Mtb worldwide [1] and the number of deaths among TB patients was greater than that of HIV patients. Efficient antitubercular agents were developed to fight TB. Typically, the treatment includes a combination of 2–4 antitubercular agents for 6–24 months. The long therapeutic period led to a poor compliance, which readily induced the drug resistance of Mtb. Multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant TB (XDR-TB) were reported to spread over the world and brought new challenges for TB treatment [2,3,4]. In 2018, 560,000 people were infected with rifampin-resistant or MDR-TB. Among them, 230,000 patients died from the disease. In addition, over 30% patients infected with XDR-TB were not curable [5]. In the past 30 years, only bedaquiline [6] and delamanid [7] were marketed for the treatment of MDR-TB. Several candidates including pretomanid [8] and SQ109 [9], are still in the clinical study. Bedaquiline is toxic, mainly for causing irregular heart rhythm [10]. Furthermore, Mtb strains resistant to these new drugs have also been found [11,12]. The situation poses an urgent need for antitubercular agents with new structures and new mechanisms of action [13].
In the previous study, we found that m-amidophenol derivatives YZ-6 and YZ-7 showed potent in vitro inhibitory activity against Mtb H37Ra, H37Rv and clinically isolated MDR-Mtb strains (MIC = 0.39–3.125 μg/mL) (Scheme 1) [14,15]. However, the two compounds did not exert in vivo efficacy on a mouse infection model. Recently, we designed and synthesized a series of new m-amidophenol derivatives with 5-heteroatomic substitutents (OR, SR, NRR′) [16]. One privileged compound YQ-14 showed potent in vitro antitubercular activity and weak in vivo efficacy (Scheme 1). Because the clearance rates of YZ-6 and YQ-14 in mouse liver microsome were high (T1/2 = 6.60 and 5.43 min, respectively), further structural modifications to improve the metabolic stability and in vivo potency are desirable. We designed and synthesized new derivatives of YZ-6 and YZ-7 via the replacement of 1-amido group with sulfamide, urea and carbamate moieties. Such replacements were expected to provide a higher metabolic stability towards enzymatic hydrolysis. In addition, binding with the target may be enhanced considering the introduction of more oxygen and nitrogen atoms. Among the new derivatives, 3-hydroxylphenylcarbamates were found to exhibit potent in vitro and in vivo antitubercular efficacy. The results are reported in this paper.
Scheme 1.

Design of new 3-amino-5-benzylphenol derivatives.
2. Results and Discussion
2.1. Chemistry
The syntheses of 3-amino-5-benzylphenol derived sulfonamides 1a–1b, ureas 2a–2b and carbamates 3a–3p are outlined in Scheme 2. 3-Amino-5-benzylphenol 8 was prepared via the reported procedure and used as the key intermediate [14]. Briefly, 3,5-dinitrobenzoic acid 4 was transformed to the methyl ester via the reflux in methanol with small amount of thionyl chloride. The subsequent substitution of nitro group with lithium methoxide afforded methyl 3-methoxy-5-nitro-benzoate 5. The hydrolysis in aqueous sodium hydroxide and the reduction with NaBH4/BF3·OEt2 provided (3-methoxy-5-nitrophenyl)methanol 6. Friedel-Crafts alkylation of compound 6 with benzene gave 1-benzyl-3-methoxy-5-nitrobenzene 7. The subsequent demethylation and catalytic hydrogenation afforded compound 8. The reaction of 8 with sulfonyl chlorides provided the products 1a–1b. The treatment of 8 with isocyanates afforded the products 2a–2b. The reaction of 8 with chloroformates provided the products 3a–3p in good yields.
Scheme 2.

Synthesis of products 1a–1b, 2a–2b and 3a–3p. Reagents and conditions: (a) SOCl2, CH3OH, reflux, 4 h, 60%; (b) MeOLi, MeOH, reflux, 12 h, 35%; (c) NaOH, CH2Cl2, CH3OH, rt, 3 h, 98%; (d) NaBH4, BF3.OEt2, THF, 0 °C-rt, 1 h, 99%; (e) benzene, CH2Cl2, AlCl3, reflux, 5 h, 53%; (f) BBr3, CH2Cl2, −40 °C–0 °C, 12 h, 60%; (g) H2, 10% Pd/C, CH2Cl2/CH3OH, 4 h, 99%; (h) R1SO2Cl, Et3N, CH2Cl2, 0 °C-rt, 50–75%; (i) R2NCO, Et3N, CH2Cl2, rt, 50–75%; (j) R3OH, triphosgene, Et3N, CH2Cl2, 0 °C-rt, 20–50%.
2.2. Antitubercular Activity against Mtb H37Ra
Antitubercular activity of 1a–1b, 2a–2b and 3a–3p was assessed against avirulent autoluminescent Mtb H37Ra and the results are listed in Table 1 and Table 2. The growth of the bacteria was monitored by the bioluminescence intensity [17]. The minimal inhibitory concentrations (MIC) were determined as the lowest concentration at which the RLU was 90% lower than the drug-free control. Isoniazid (INH) was used as the positive control.
Table 1.
Inhibitory activity of 1a–1b and 2a–2b against Mtb H37Ra.
| Compound | MIC (µg/mL) | CLogP a | |
|---|---|---|---|
| 1a |
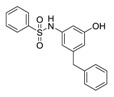
|
50 | 3.98 |
| 1b |
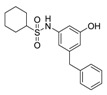
|
50 | 4.38 |
| 2a |
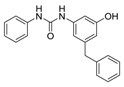
|
12.5 | 4.41 |
| 2b |
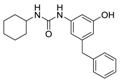
|
6.25 | 4.53 |
| INH | 0.1 | / |
a CLogP was calculated with ChemBioDraw Ultra 12.0.
Table 2.
Inhibitory activity of 3a–3p against Mtb H37Ra.
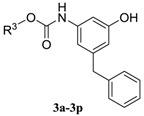
| Compound | R3 | MIC (µg/mL) | CLogP a |
|---|---|---|---|
| 3a |
|
50 | 4.67 |
| 3b |
|
100 | 5.52 |
| 3c |
|
100 | 5.68 |
| 3d |
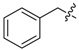
|
10 | 3.63 |
| 3e |
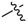
|
25 | 3.01 |
| 3f |
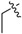
|
5 | 3.54 |
| 3g |
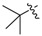
|
5 | 4.24 |
| 3h |
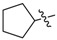
|
2.5 | 4.48 |
| 3i |
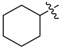
|
1.25 | 5.04 |
| 3j |
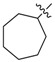
|
1.25 | 5.60 |
| 3k b |
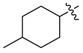
|
1.25 | 5.56 |
| 3l |
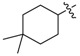
|
0.625 | 6.08 |
| 3m b |
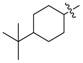
|
25 | 6.88 |
| 3n b |
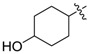
|
25 | 2.95 |
| 3o |
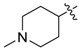
|
100 | 3.06 |
| 3p |
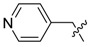
|
100 | 3.33 |
| INH | 0.1 |
a CLogP was calculated with ChemBioDraw Ultra 12.0. b The compound was obtained as a mixture of trans- and cis-isomers.
Benzenesulfonamide 1a and cyclohexanesulfonamide 1b almost did not show inhibitory activity against H37Ra (MIC = 50 μg/mL). However, phenylurea 2a and cyclohexylurea 2b showed higher inhibitory activity (MIC = 12.5 μg/mL and 6.25 μg/mL respectively).
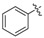
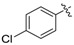
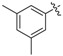
The phenyl carbamate 3a is much less active than its benzamide analog YZ-6 (MIC = 50 μg/mL vs. 5 μg/mL). The substitution on the phenyl ring (3b, 3c) resulted in the loss of the inhibitory activity (MIC = 100 μg/mL). The benzyl carbamate 3d showed better activity (MIC = 10 μg/mL). Furthermore, alkyl carbamates 3e–3m were examined. The methyl carbamate 3e had a low inhibitory activity (MIC = 25 μg/mL), but ethyl and t-butyl carbamates 3f–3g showed improved activity (MIC = 5 μg/mL). The cycloalkyl carbamates (3h–3j) were found to be more potent. The cyclo-hexyl and cyclo-heptyl carbamates 3i and 3j exhibited good inhibitory activity (MIC = 1.25 μg/mL); 4-Methyl-cyclo-hexyl carbamate 3k displayed a similar activity (MIC = 1.25 μg/mL), but 4,4-dimethyl-cyclo-hexyl carbamate 3l afforded better inhibitory activity (MIC = 0.625 μg/mL). The substitution at cyclo-hexyl with bulkier 4-tert-butyl group (3m) decreased the inhibitory activity (MIC = 25 μg/mL). The substitution with hydrophilic group such as hydroxyl (3n) also decreased the inhibitory activity (MIC = 25 μg/mL). N-Methyl-piperidinyl and 4-pyridinylmethyl carbamates 3o and 3p lost the activity (MIC = 100 μg/mL). The results implicated that a hydrophobic moiety is important for the antitubercular activity. However, no general correlation between the MICs and CLogP values was observed.
In order to identify the key pharmacophores, we kept 4,4-dimethyl-cyclo-hexyl carbamate scaffold and replaced 1-hydroxyl with other functional groups. The inhibitory activity of compounds 3q and 3r against Mtb H37Ra are examined and the results are summarized in Table 3. The 1-carboxylic analog 3q is inactive (MIC = 100 μg/mL), however the methoxy analog 3r showed good inhibitory activity (MIC = 2.5 μg/mL).
Table 3.
In vitro inhibitory activity of 3q–3r against Mtb H37Ra.
| Compound | MIC (μg/mL) | CLogP a | |
|---|---|---|---|
| 3q |
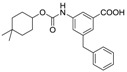
|
100 | 6.73 |
| 3r |
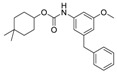
|
2.5 | 6.79 |
| INH | 0.1 |
a CLogP was calculated with ChemBioDraw Ultra 12.0.
2.3. In Vitro Inhibitory Activity against Mtb H37Rv and MDR-Mtb Strains
The compounds 3i and 3l were selected for the further evaluation of inhibitory activity against Mtb H37Rv and clinically isolated multidrug-resistant Mtb strains (MDR-Mtb). The results are summarized in Table 4. Good inhibitory activities (MIC = 2–8 µg/mL) were observed against Mtb H37Rv and MDR-Mtb strains K12, K16, K18, V4.
Table 4.
Inhibitory activity (MIC, µg/mL) of compounds 3i and 3l against Mtb H37Rv and MDR-Mtb strains.
| Mtb Strain | 3i | 3l | RIF | INH |
|---|---|---|---|---|
| H37Rv | 4 | 4 | 0.03 | 0.41 |
| K12 | 2 | 4 | >10 | >10 |
| K16 | 2 | 4 | >10 | >10 |
| K18 | 2 | 4 | >10 | >10 |
| V4 | 4 | 8 | >1 | >10 |
RIF: rifampicin; INH: isoniazid.
2.4. Evaluation of Cytotoxicity and Metabolic Stability
The cytotoxicity of 3i and 3l against human lung cancer cell line A549 was evaluated via MTT method and the results are summarized in Table 5. Moderate cytotoxicities were observed for compounds 3i and 3l (IC50 = 15.8 and 22.6 µg/mL). The selection indices (SI = IC50/MIC) are 12.6 and 36.2 respectively for 3i and 3l. The result demonstrated compound 3l as a more suitable candidate for further evaluation.
Table 5.
Cytotoxicity of compounds 3i and 3l against cell line A549.
| Compounds | IC50 (µg/mL) | MIC (µg/mL, H37Ra) | SI |
|---|---|---|---|
| 3i | 15.8 | 1.25 | 12.6 |
| 3l | 22.6 | 0.625 | 36.2 |
The metabolic stability of 3l in mouse liver microsome was examined. A quick clearance was observed (T1/2 = 1.76 min, CL = 1440 mL/min/gprot). The result indicated that the replacement of 1-amido group in the lead compounds YZ-6 and YZ-7 with carbamate moiety cannot improve the metabolic stability. The reason remains to be investigated.
2.5. In Vivo Inhibitory Activity against Mtb H37Ra
The in vivo efficacy of compound 3l was evaluated in a mouse model infected with autoluminescent Mtb H37Ra. The compound 3l was administered orally (100 mg/kg/day). The untreated control group was administered with sodium carboxymethyl cellulose (CMC-Na) solution. Rifampicin was used as the positive control. After five days treatment, the burdens of the bacteria in lungs and spleens of the mice were determined by the bioluminescence intensity detection. The results are summarized in Figure 1. A significant reduction (1.3 log) of relative light unit (RLU) in the lung was achieved compared with the untreated control group. A small reduction of RLU (0.4 log) was observed in the spleen.
Figure 1.

In vivo inhibitory activity of 3l on a mouse model infected with Mtb H37Ra. Four mice per group were treated for 6 days. Data were presented as mean ± SD. *, p < 0.05; **, p < 0.001.
3. Experimental
3.1. Chemistry
The NMR spectra of 1H and 13C were recorded on Bruker AVANCE 400 or 500 spectrometer (Karlsruhe, Germany). Chemical shifts of protons are reported in parts per million downfield from tetramethylsilane. Peaks are labeled as singlet (s), broad singlet (br), doublet (d), triplet (t), double doublet (dd), doublet of triplets (dt), multiplet (m). The high-resolution mass spectra were analyzed on a SHIMADZU LCMS-IT-TOF mass spectrometer (Tokyo, Japan). Melting points were determined in open capillary tubes on a MPA100 Optimelt automated melting point system. All chemicals were purchased from Sigma-Aldrich and Alfa Aesar chemical companies and were used without further purification. Compounds 4–8 were prepared according to the reported procedures [14].
3.1.1. Synthesis of N-(3-benzyl-5-hydroxyphenyl)benzenesulfonamide (1a)
To a solution of 3-amino-5-benzylphenol 8 (119 mg, 0.6 mmol), DBU (9 mg, 0.06 mmol) in pyridine (2 mL) was added dropwise benzenesulfonyl chloride (211 mg, 1.2 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 5 h. After water (30 mL) was added, the reaction mixture was extracted with EtOAc (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography (petroleum ether/EtOAc = 4:1) to afford 1a as an orange solid (117 mg, 54% yield). M.p. 142–143 °C. 1H-NMR (400 MHz, DMSO) δ 10.10 (s, 1H), 9.37 (s, 1H), 7.75–7.66 (m, 2H), 7.64–7.58 (m, 1H), 7.52 (t, J = 7.5 Hz, 2H), 7.26 (t, J = 7.3 Hz, 2H), 7.21–7.15 (m, 1H), 7.12–7.00 (m, 2H), 6.41 (s, 1H), 6.38 (s, 1H), 6.24 (s, 1H), 3.71 (s, 2H). 13C-NMR (101 MHz, DMSO) δ 158.26, 143.53, 141.25, 140.02, 139.13, 133.23, 129.59, 129.07, 128.80, 127.14, 126.42, 112.05, 111.38, 105.19, 41.50. HRMS (ESI) calculated for C19H17NNaO3S+ [M + Na]+: 362.0788, found: 362.0821. (see Supplementary Materials)
3.1.2. N-(3-Benzyl-5-hydroxyphenyl)cyclohexanesulfonamide (1b)
The compound was synthesized via a similar procedure of 1a, using cyclohexanesulfonyl chloride (236.4 mg, 1.2 mmol) to replace benzenesulfonyl chloride. Orange solid (86 mg, 44% yield). M.p. 140–141 °C. 1H-NMR (400 MHz, DMSO) 1H-NMR (400 MHz, DMSO) δ 9.53 (s, 1H), 9.38 (s, 1H), 7.28 (t, J = 7.2 Hz, 2H), 7.23–7.12 (m, 3H), 6.54 (s, 1H), 6.52 (s, 1H), 6.30 (s, 1H), 3.79 (s, 2H), 2.97–2.84 (m, 1H), 2.05–1.89 (m, 2H), 1.80–1.67 (m, 2H), 1.64–1.52 (m, 1H), 1.45–1.31 (m, 2H), 1.22–1.05 (m, 3H). 13C-NMR (101 MHz, DMSO) δ 158.42, 143.71, 141.43, 140.06, 129.13,128.82, 126.44, 111.62, 110.91, 104.55, 59.30, 41.58, 26.42, 25.22, 24.89. HRMS (ESI) calculated for C19H23NO3S+ [M + H]+: 346.1471, found: 346.1473.
3.1.3. Synthesis of 1-(3-benzyl-5-hydroxyphenyl)-3-phenylurea (2a)
To a solution of 3-amino-5-benzylphenol 8 (119 mg, 0.6 mmol), triethylamine (72 mg, 0.72 mmol) in THF (10 mL) was added dropwise phenylisocyanate (86 mg, 0.72 mmol) at 0 °C. The reaction mixture was stirred at room temperature overnight. After the reaction was quenched with water (30 mL), the mixture was extracted with EtOAc (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography (petroleum ether/EtOAc = 4:1) to afford 2a as a white solid (103 mg, 54% yield). M.p. 178–179 °C. 1H-NMR (400 MHz, DMSO) δ 9.27 (s, 1H), 8.53 (s, 1H), 8.51 (s, 1H), 7.47–7.38 (m, 2H), 7.33–7.16 (m, 7H), 6.95 (t, J = 7.3 Hz, 1H), 6.90 (t, J = 2.0 Hz, 1H), 6.64 (s, 1H), 6.24 (s, 1H), 3.81 (s, 2H). 13C-NMR (101 MHz, DMSO) δ 158.22, 152.81, 143.23, 141.62, 141.21, 140.18, 129.25, 128.84, 126.41, 122.22, 118.55, 109.99, 109.76, 103.46, 41.74. HRMS (ESI) calculated for C20H18N2O2+ [M + H]+: 319.1441, found: 319.1441.
3.1.4. 1-(3-Benzyl-5-hydroxyphenyl)-3-cyclohexylurea (2b)
The compound was synthesized via a similar procedure of 2a, using cyclohexanylisocyanate (90.1 mg, 0.72 mmol) to replace phenylisocyanate. White solid, (74 mg, 38% yield). M.p. 193–194 °C. 1H-NMR (400 MHz, DMSO) δ 9.13 (s, 1H), 8.12 (s, 1H), 7.34–7.24 (m, 2H), 7.22–7.13 (m, 3H), 6.79 (s, 1H), 6.53 (s, 1H), 6.14 (s, 1H), 5.94 (d, J = 7.8 Hz, 1H), 3.75 (s, 2H), 3.49–3.37 (m, 1H), 1.86–1.71 (m, 2H), 1.68–1.57 (m, 2H), 1.37–1.21 (m, 3H), 1.19–1.06 (m, 3H). 13C-NMR (400 MHz, DMSO) δ 158.11, 154.69, 142.95, 141.96, 141.69, 129.19, 128.78, 126.35, 109.21, 109.13, 102.85, 47.89, 41.78, 33.42, 25.71, 24.77. HRMS (ESI) calculated for C20H25N2O2+ [M + H]+: 325.1911, found:325.1891.
3.1.5. Synthesis of Phenyl (3-benzyl-5-hydroxyphenyl)carbamate (3a)
To a solution of phenol (112 mg, 1.2 mmol), triphosgene (178 mg, 0.6 mmol) in CH2Cl2 (10 mL) was added dropwise triethylamine (121 mg, 1.2 mmol) at 0 °C. The reaction mixture was stirred for 0.5 h and then 3-amino-5-benzylphenol 8 (119 mg, 0.6 mmol) was added. The mixture was stirred at room temperature for 5 h. After the reaction was quenched with water (30 mL), the mixture was extracted with EtOAc (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography (petroleum ether/EtOAc = 4:1) to afford 3a as a white solid (61 mg, 32% yield). M.p. 175–176 °C. 1H-NMR (500 MHz, DMSO) δ 10.06 (s, 1H), 9.38 (s, 1H), 7.42 (t, J = 7.9 Hz, 2H), 7.32–7.25 (m, 3H), 7.22–7.17 (m, 5H), 6.87 (s, 1H), 6.84 (s, 1H) 6.31 (s, 1H), 3.80 (s, 2H). 13C-NMR (126 MHz, DMSO) δ 158.20, 152.01, 150.99, 143.30, 141.51, 140.04, 129.87, 129.17, 128.85, 126.43, 125.85, 122.44, 111.15, 110.27, 103.94, 41.77. HRMS (ESI) calculated for C20H18NO3+ [M + H]+: 320.1261, found: 320.1281.
3.1.6. 4-Chlorophenyl (3-benzyl-5-hydroxyphenyl)carbamate (3b)
The compound was synthesized via a similar procedure of 3a, using 4-chlorine phenol (154.3 mg, 1.2 mmol) to replace phenol. White solid (74 mg, 35% yield). M.p. 144–145 °C. 1H-NMR (400 MHz, DMSO) δ 10.10 (s, 1H), 9.37 (s, 1H), 7.50–7.43 (m, 2H), 7.31–7.26 (m, 2H), 7.26–7.22 (m, 2H), 7.22–7.16 (m, 3H), 6.84 (s, 1H), 6.81 (s, 1H), 6.31 (s, 1H), 3.78 (s, 2H). 13C-NMR (101 MHz, DMSO) δ 158.25, 151.69, 149.83, 143.25, 141.49, 139.87, 129.94, 129.75, 129.17, 128.84, 126.43, 124.32, 111.19, 110.29, 104.17, 41.77. HRMS (ESI) calculated for C20H17ClNO3+ [M + H]+: 354.0854, found: 354.0891.
3.1.7. 3,5-Dimethylphenyl (3-benzyl-5-hydroxyphenyl)carbamate (3c)
The compound was synthesized via a similar procedure of 3a, using 3, 5-dimethylphenol (146.5 mg, 1.2 mmol) to replace phenol. White solid (42 mg, 20% yield). M.p. 136–138 °C. 1H-NMR (400 MHz, DMSO) δ 9.95 (s, 1H), 9.31 (s, 1H),7.28 (t, J = 7.4 Hz, 2H), 7.22–7.15 (m, 3H), 6.87 (s, 1H), 6.83 (d, 2H), 6.78 (s, 2H), 6.29 (s, 1H), 3.79 (s, 2H), 2.27 (s, 6H). 13C-NMR (101 MHz, DMSO) δ 158.20, 152.11, 150.89, 143.30, 141.52, 140.05, 139.23, 129.16, 128.83, 127.24, 126.42, 119.89, 111.12, 110.28, 103.99, 41.80, 21.28. HRMS (ESI) calculated for C22H22NO3+ [M + H]+: 348.1568, found: 348.1594.
3.1.8. Benzyl (3-benzyl-5-hydroxyphenyl)carbamate (3d)
The compound was synthesized via a similar procedure of 3a, using benzyl alcohol (129.7 mg, 1.2 mmol) to replace phenol. White solid (94 mg, 47% yield). M.p. 124–125 °C. 1H-NMR (400 MHz, DMSO) δ 9.57 (s, 1H), 9.26 (s, 1H), 7.45–7.32 (m, 5H), 7.27 (t, J = 7.4 Hz, 2H), 7.22–7.14 (m, 3H), 6.83 (s, 1H), 6.78 (s, 1H), 6.24 (s, 1H), 5.11 (s, 2H), 3.77 (s, 2H). 13C-NMR (101 MHz, DMSO) δ 158.14, 153.71, 143.11, 141.55, 140.50, 137.20, 129.15, 128.89, 128.82, 128.45, 126.39, 103.77, 66.02, 41.81. HRMS (ESI) calculated for C21H20NO3+ [M + H]+: 334.1424, found: 334.1438.
3.1.9. Methyl (3-benzyl-5-hydroxyphenyl)carbamate (3e)
The compound was synthesized via a similar procedure of 3a, using methanol (38.5 mg, 1.2 mmol) to replace phenol. White solid (80 mg, 52% yield). M.p. 120–121 °C. 1H-NMR (400 MHz, DMSO) δ 9.42 (s, 1H), 9.24 (s, 1H), 7.28 (t, J = 7.4 Hz, 2H), 7.18 (m, 3H), 6.84 (s, 1H), 6.76 (s, 1H), 6.25 (s, 1H), 3.79 (s, 2H), 3.63 (s, 3H). 13C-NMR (101 MHz, DMSO) δ 158.13, 154.31, 143.05, 141.56, 140.57, 129.15, 128.81, 126.39, 110.58, 110.08, 103.73, 51.92, 41.80. HRMS (ESI) calculated for C15H16NO3+ [M + H]+: 258.1108, found: 258.1125.
3.1.10. Ethyl (3-benzyl-5-hydroxyphenyl)carbamate (3f)
The compound was synthesized via a similar procedure of 3a, using ethanol (55.5 mg, 1.2 mmol) to replace phenol. White solid (80 mg, 49% yield). M.p. 167–168 °C. 1H-NMR (400 MHz, DMSO) δ 9.40 (s, 1H), 9.23 (s, 1H), 7.27 (t, J = 7.4 Hz, 2H), 7.23–7.11 (m, 3H), 6.81 (s, 1H), 6.77 (s, 1H), 6.22 (s, 1H), 4.08 (q, J = 7.0 Hz, 2H), 3.76 (s, 2H), 1.21 (t, J = 7.2 Hz, 3H). 13C-NMR (101 MHz, DMSO) δ 158.09, 153.89, 143.02, 141.57, 140.64, 129.14, 128.81, 126.38, 110.52, 110.12, 103.76, 60.43, 41.81, 14.99. HRMS (ESI) calculated for C16H18NO3+ [M + H]+: 272.1263, found: 272.1281.
3.1.11. tert-Butyl (3-benzyl-5-hydroxyphenyl)carbamate (3g)
The compound was synthesized via a similar procedure of 3a, using tert-butanol (88.9 mg, 1.2 mmol) to replace phenol. White solid (72 mg, 38% yield). M.p. 158–159 °C. 1H-NMR (400 MHz, DMSO) δ 9.19 (s, 1H), 9.15 (s, 1H), 7.27 (t, J = 7.4 Hz, 2H), 7.20–7.15 (m, 3H), 6.80 (s, 1H), 6.78 (s, 1H), 6.18 (s, 1H), 3.76 (s, 2H), 1.44 (s, 9H). 13C-NMR (101 MHz, DMSO) δ 158.01, 153.12, 142.89, 141.65, 140.94, 129.13, 128.80, 126.36, 110.24, 110.01, 103.62, 79.23, 41.83, 28.60. HRMS (ESI) calculated for C18H21NNaO3+ [M + Na]+: 322.1394, found: 322.1414.
3.1.12. Cyclopentyl (3-benzyl-5-hydroxyphenyl)carbamate (3h)
The compound was synthesized via a similar procedure of 3a, using cyclopentanol (103.5 mg, 1.2 mmol) to replace phenol. White solid (80 mg, 43% yield). M.p. 122–123 °C. 1H-NMR (400 MHz, DMSO) δ 9.31 (s, 1H), 9.20 (s, 1H), 7.28 (t, J = 7.4 Hz, 2H), 7.23–7.14 (m, 3H), 6.81 (s, 1H), 6.78 (s, 1H), 6.22 (s, 1H), 5.11–4.98 (m, 1H), 3.76 (s, 2H), 1.92–1.78 (m, 2H), 1.74–1.61 (m, 4H), 1.61–1.51 (m, 2H). 13C-NMR (101 MHz, DMSO) δ 158.07, 153.70, 142.96, 141.60, 140.73, 129.14, 128.80, 126.37, 110.43, 110.07, 103.70, 76.95, 41.83, 32.77, 23.72. HRMS (ESI) calculated for C19H22NO3+ [M + H]+: 312.1573, found: 312.1594.
3.1.13. Cyclohexyl (3-benzyl-5-hydroxyphenyl)carbamate (3i)
The compound was synthesized via a similar procedure of 3a, using cyclohexanol (120.1 mg, 1.2 mmol) to replace phenol. White solid (74 mg, 38% yield). M.p. 156–157 °C. 1H-NMR (400 MHz, DMSO) δ 9.33 (s, 1H), 9.22 (s, 1H), 7.28 (t, J = 7.4 Hz, 2H), 7.22–7.14 (m, 3H), 6.82 (s, 1H), 6.79 (s, 1H), 6.23 (s, 1H), 4.66–4.53 (m, 1H), 3.76 (s, 2H), 1.91–1.80 (m, 2H), 1.77–1.64 (m, 2H), 1.57–1.46 (m, 1H), 1.44–1.23 (m, 5H). 13C-NMR (101 MHz, CDCl3) δ 162.82, 158.19, 147.73, 146.34, 145.48, 133.89, 133.55, 131.12, 115.18, 114.83, 108.44, 77.30, 46.58, 36.84, 30.15, 28.64. HRMS (ESI) calculated for C20H24NO3+ [M + H]+: 326.1737, found: 326.1751.
3.1.14. Cycloheptyl (3-benzyl-5-hydroxyphenyl)carbamate (3j)
The compound was synthesized via a similar procedure of 3a, using cycloheptanol (137.1 mg, 1.2 mmol) to replace phenol. White solid (83 mg, 41% yield). M.p. 133–134 °C. 1H-NMR (400 MHz, DMSO) δ 9.31 (s, 1H),7.28 (t, J = 7.4 Hz, 2H), 7.22–7.13 (m, 3H), 6.81 (s, 1H), 6.79 (s, 1H), 6.23 (s, 1H), 4.90–4.65 (m, 1H), 3.76 (s, 2H), 1.94–1.83 (m, 2H), 1.72–1.58 (m, 4H), 1.53 (s, 4H), 1.48–1.37 (m, 2H). 13C-NMR (101 MHz, DMSO) δ 158.07, 153.42, 142.96, 141.60, 140.78, 129.14, 128.80, 126.37, 110.41, 110.08, 103.70, 74.95, 41.85, 34.11, 28.22, 22.83. HRMS (ESI) calculated for C21H26NO3+ [M + H]+: 340.1937, found: 340.1907.
3.1.15. 4-Methyl-cyclohexyl (3-benzyl-5-hydroxyphenyl)carbamate (3k)
The compound was synthesized via a similar procedure of 3a, using 4-Methylcyclohexanol (137.1 mg, 1.2 mmol) to replace phenol. White solid (89 mg, 44% yield). M.p. 113–114 °C. 1H-NMR (400 MHz, DMSO) δ 9.34 (s, 1H), 9.28 (s, 1H), 7.28 (t, J = 7.4 Hz, 2H), 7.23–7.15 (m, 3H), 6.95–6.72 (m, 2H), 6.24 (s, 1H), 4.81 (s, 1H), 4.49 (s, 1H), 3.77 (d, J = 3.0 Hz, 2H), 1.99–1.88 (m, 1H), 1.82–1.67 (m, 2H), 1.60–1.42 (m, 3H), 1.35–1.26 (m, 2H), 1.09–0.96 (m, 1H), 0.94–0.85 (m, 3H). 13C-NMR (101 MHz, DMSO) δ 158.07, 153.55, 153.44, 142.99, 141.59, 140.77, 140.70, 129.13, 128.80, 126.38, 110.41, 110.13, 103.74, 73.37, 69.68, 41.85, 33.07, 32.10, 31.65, 31.08, 29.63, 29.57, 22.34, 22.18. HRMS (ESI) calculated for C21H25NNaO3+ [M + Na]+: 362.1713, found: 362.1727.
3.1.16. 4,4-Dimethyl-cyclohexyl (3-benzyl-5-hydroxyphenyl)carbamate (3l)
The compound was synthesized via a similar procedure of 3a, using 4,4-Dimethylcyclohexanol (153.8 mg, 1.2 mmol) to replace phenol. White solid (104 mg, 49% yield). M.p. 129–130 °C. 1H-NMR (400 MHz, DMSO) δ 9.34 (s, 1H), 9.23 (s, 1H), 7.28 (t, J = 7.4 Hz, 2H), 7.22–7.14 (m, 3H), 6.81 (s, 1H), 6.79 (s, 1H), 6.23 (s, 1H), 4.68–4.49 (m, 1H), 3.76 (s, 2H), 1.84–1.67 (m, 2H), 1.59–1.47 (m, 2H), 1.47–1.38 (m, 2H), 1.32–1.19 (m, 2H), 0.92 (s, 3H), 0.91 (s, 3H). 13C-NMR (101 MHz, DMSO) δ 157.98, 153.44, 142.91, 141.59, 140.74, 129.14, 128.79, 126.37, 110.45, 110.11, 103.86, 72.42, 41.85, 36.27, 29.75, 27.67. HRMS (ESI) calculated for C22H28NO3+ [M + H]+: 354.2050, found: 354.2064.
3.1.17. 4-(tert-Butyl)-cyclohexyl (3-benzyl-5-hydroxyphenyl)carbamate (3m)
The compound was synthesized via a similar procedure of 3a, using 4-tert-Butylcyclohexanol (187.5 mg, 1.2 mmol) to replace phenol. White solid (84 mg, 37% yield). M.p. 136–137 °C. 1H-NMR (600 MHz, DMSO) δ 9.38 (s, 1H), 9.24 (s, 1H), 7.28 (t, J = 7.5 Hz, 2H), 7.20–7.16 (m, 3H), 6.81 (s, 1H), 6.79 (s, 1H), 6.22 (s, 1H), 4.51–4.43 (m, 1H), 3.76 (s, 2H), 2.06–1.97 (m, 2H), 1.82–1.70 (m, 2H), 1.33–1.24 (m, 2H), 1.12–1.05 (m, 2H), 1.02–0.99 (m, 1H), 0.85 (s, 9H). 13C-NMR (101 MHz, DMSO) δ 158.07, 153.42, 142.95, 141.59, 140.72, 129.14, 128.79, 126.37, 110.44, 110.07, 103.70, 73.59, 46.99, 41.83, 32.60, 32.51, 27.94, 25.55. HRMS (ESI) calculated for C24H32NO3+ [M + H]+: 382.2365, found: 382.2377.
3.1.18. 4-Hydroxycyclohexyl (3-benzyl-5-hydroxyphenyl)carbamate (3n)
The compound was synthesized via a similar procedure to 3a, using 1,4-Cyclohexanediol (139.4 mg, 1.2 mmol) to replace phenol. Sticky liquid (67 mg, 33% yield). 1H-NMR (400 MHz, DMSO) δ 9.34 (s, 1H), 9.23 (s, 1H), 7.28 (t, J = 7.4 Hz, 2H), 7.23–7.14 (m, 3H), 6.88–6.76 (m, 2H), 6.24 (s, 1H), 4.73–4.45 (m, 2H), 3.77 (s, 2H), 3.69–3.45 (m, 1H), 2.03–1.89 (m, 1H), 1.88–1.71 (m, 2H), 1.58 (t, J = 8.6 Hz, 3H), 1.45–1.21 (m, 2H). 13C-NMR (101 MHz, CDCl3) δ 162.81, 158.17, 147.76, 146.35, 145.50, 145.43, 133.90, 133.56, 131.13, 115.14, 114.77, 108.37, 77.11, 75.65, 72.49, 70.43, 46.57, 37.51, 35.61, 34.21, 32.30. HRMS (ESI) calculated for C20H23NNaO4+[M + Na]+: 364.1519, found: 364.1512.
3.1.19. 1-Methylpiperidin-4-yl (3-benzyl-5-hydroxyphenyl)carbamate (3o)
The compound was synthesized via a similar procedure of 3a, using N-Methyl-4-piperidinol (138.2 mg, 1.2 mmol) to replace phenol. Sticky liquid (38 mg, 19% yield). 1H-NMR (400 MHz, DMSO) δ 9.41 (s, 1H), 9.23 (s, 1H), 7.28 (t, J = 7.4 Hz, 2H), 7.23–7.12 (m, 3H), 6.82 (s, 1H), 6.79 (s, 1H), 6.24 (s, 1H), 4.68–4.53 (m, 1H), 3.76 (s, 2H), 2.70–2.57 (m, 2H), 2.23–2.11 (m, 5H), 1.93–1.81 (m, 2H), 1.68–1.53 (m, 2H). 13C-NMR (101 MHz, DMSO) δ 158.09, 153.30, 143.01, 141.58, 140.64, 129.14, 128.81, 126.38, 110.51, 110.08, 103.72, 70.24, 53.05, 46.16, 41.83, 31.30. HRMS (ESI) calculated for C20H24N2O3+ [M + H]+: 341.1860, found: 341.1866.
3.1.20. Pyridin-4-ylmethyl (3-benzyl-5-hydroxyphenyl)carbamate (3p)
The compound was synthesized via a similar procedure of 3a, using 4-Pyridylcarbinol (130.9 mg, 1.2 mmol) to replace phenol. White solid (72 mg, 36% yield). M.p. 164–165 °C. 1H-NMR (400 MHz, DMSO) δ 9.70 (s, 1H), 9.28 (s, 1H), 8.57 (d, J = 5.6 Hz, 2H), 7.38 (d, J = 5.4 Hz, 2H), 7.28 (t, J = 7.4 Hz 2H), 7.25–7.15 (m, 3H), 6.85 (s, 1H), 6.78 (s, 1H), 6.27 (s, 1H), 5.17 (s, 2H), 3.81 (s, 2H). 13C-NMR (101 MHz, DMSO) δ 158.10, 153.40, 150.12, 146.43, 143.18, 141.53, 140.30, 129.17, 128.82, 126.41, 122.06, 110.85, 110.09, 103.75, 64.24, 41.78. HRMS (ESI) calculated for C20H19N2O3+ [M + H]+: 335.1367, found: 335.1390.
3.1.21. 3-Benzyl-5-((((4,4-dimethylcyclohexyl)oxy)carbonyl)amino)benzoic acid (3q)
To a solution of 4,4-dimethylcyclohexan-1-ol (154 mg, 1.2 mmol), triphosgene (178 mg, 0.6 mmol) in CH2Cl2 (10 mL) was added dropwise triethylamine (121 mg, 1.2 mmol) at 0 °C. The reaction mixture was stirred for 0.5 h and then methyl 3-amino-5-benzylbenzoate (144 mg, 0.6 mmol) was added. The mixture was stirred at room temperature for 5 h. After the reaction was quenched with water (30 mL), the mixture was extracted with EtOAc (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography (petroleumether/EtOAc = 4:1) to afford methyl 3-benzyl-5-((((4,4-dimethylcyclohexyl)oxy)carbonyl)amino)benzoate (59 mg, 25% yield).
To a solution of methyl 3-benzyl-5-((((4,4-dimethylcyclohexyl)oxy)carbonyl)amino)benzoate (59 mg, 0.15 mmol) in a mixed solvent (CH2Cl2/CH3OH = 9:1(v/v), 2 mL), was added sodium hydroxide (20 mg, 0.5 mmol). The reaction mixture was stirred at room temperature for 3 h. After the solvent was removed in vacuo, the residue was diluted with water (10 mL). The solution was extracted with diethyl ether (10 mL). The aqueous phase was cooled in an ice-bath, acidified to pH 2–3 with 1M HCl and extracted with AcOEt (10 mL × 2). The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to afford 3q as a white solid (51 mg, 89% yield). M.p. 193–194 °C. 1H-NMR (500 MHz, DMSO) δ 12.89 (s, 1H), 9.71 (s, 1H), 7.95 (s, 1H), 7.60 (s, 1H), 7.43 (s, 1H), 7.33–7.27 (m, 2H), 7.24–7.18 (m, 3H), 4.62 (s, 1H), 3.95 (s, 2H), 1.76 (s, 2H), 1.59–1.50 (m, 2H), 1.46–1.39 (m, 2H), 1.29–1.22 (m, 2H), 0.91 (d, J = 7.4 Hz, 6H). 13C-NMR (126 MHz, DMSO) δ 167.77, 153.57, 142.60, 141.17, 140.12, 132.12, 129.21, 128.97, 128.77, 126.59, 123.95, 123.00, 117.38, 72.78, 41.43, 36.20, 29.77, 27.66. HRMS (ESI) calculated for C23H28NO4+ [M + H]+: 382.2013, found: 382.2011.
3.1.22. 4,4-Dimethylcyclohexyl (3-benzyl-5-methoxyphenyl)carbamate (3r)
A mixture of 3l (35 mg, 0.1 mmol), iodomethane (9.3 µL, 0.15 mmol) and anhydrous K2CO3 (27 mg, 0.2 mmol) in acetone (2 mL) was refluxed for 12 h. The mixture was cooled to room temperature and the solvent was removed in vacuo. The residue was purified by column chromatography over silica gel (hexane/EtOAc = 9:1) to give 3r as a white solid (26 mg, 71% yield). M.p. 193–194 °C. 1H-NMR (400 MHz, DMSO) δ 9.46 (s, 1H), 7.28 (t, J = 7.4 Hz, 2H), 7.22–7.15 (m, 3H), 6.96 (s, 1H), 6.91 (s, 1H), 6.45 (s, 1H), 4.65–4.52 (m, 1H), 3.84 (s, 2H), 3.67 (s, 3H), 1.80–1.71 (m, 2H), 1.57–1.47 (m, 2H), 1.46–1.38 (m, 2H), 1.27–1.22 (m, 2H), 0.91 (d, J = 4.2 Hz, 6H). 13C-NMR (101 MHz, DMSO) δ 160.07, 153.45, 143.25, 141.47, 140.91, 129.10, 128.84, 126.44, 111.43, 108.90, 102.13, 72.54, 55.34, 41.86, 36.22, 29.78, 27.68. HRMS (ESI) calculated for C23H30NO3+ [M + H]+: 368.2222, found: 368.2220.
3.2. Biological Assays
3.2.1. Determination of Minimum Inhibitory Concentration (MIC)
Minimum inhibitory concentration against Mtb H37Ra, H37Rv, and MDR-Mtb strains (provided by Guangzhou Chest Hospital) were determined via previously reported procedures [14].
3.2.2. Evaluation of Cytotoxicity
The cytotoxicities of compounds 3i and 3l were assayed against human lung cancer cell line A549 at concentrations from 100 to 6.25 μg/mL. Approximately 4 × 104 cells, suspended in culture medium, were seeded in the 96-well plate and then allowed to recover for 24 h at 37 °C under 5% CO2. The solution of tested compound was added to the plate and the final concentrations were 100 mg/mL, 50 mg/mL, 25 mg/mL, 12.5 mg/mL, and 6.25 mg/mL, respectively. Each experiment was repeated three times. After being incubated for 72 h, Fresh MTT was added to each well at a final concentration of 0.5 mg/mL and incubated with cells at 37 °C for 4 h. The formazan crystals were dissolved in 150 µL DMSO per each well. The absorbance at 490 nm was measured by microplate microscopy, and the survival rate was calculated by the formula (OD in the experimental group − blank group)/(OD in the cell group − blank group) × 100%. The cytotoxicity is shown as IC50 value, which was calculated by GraphPad Prism Software version 5 [18].
3.2.3. Determination of the Clearance Rate in Mouse Liver Microsome
The mouse liver microsome was purchased from Xenotech company. The liver microsome was incubated in a 96-well plate. The final incubation volume was set at 45 μL, contained NADPH (2 mM), microsomes (0.5 mg/mL), the compound 3l (1 μM), MgCl2 (5.0 mM), DMSO (0.01%) and BSA (0.005%). The plate was incubated at 37 °C for ten minutes, and then was added NADPH to trigger the reaction. The incubation times were 0, 5, 15, 30, 45 and 60 min. Ice-cold acetonitrile was added to terminate the reaction. The mixture was centrifuged at 4 °C for 15 min at 4000 rpm. The concentration of the compound 3l in supernatant was determined by LC-MS/MS. The incubation time was plotted with the logarithm of the drug residual rate in the incubation system. k was calculated by linear regression, and the clearance rate was calculated based the formula [19].
3.2.4. In Vivo Inhibitory Activity against Mtb H37a
The experimental program involving animal feeding and use was submitted to, and approved by, the Animal Welfare Department of Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences.
Mtb H37Ra isolated on plates were homogenized with sterile glass beads in a 250 mL flask containing 50 mL 7H9 with tween80. When RLU reached 2 million/mL, the broth culture was used to infect 4–6-week-old male BALB/c mice by tail vein injection. The day after infection (day 0), RLU counts were determined. The mice were first anesthetized by isoflurane inhalation and the RLU count was determined by laying the breast of the mouse on the detection hole of the luminometer and measuring light production twice for 3 s. Mice with similar RLU readings were randomly allocated to treatment groups (4 mice/group) and individually marked. The treatment groups received: 0.5% CMC-Na alone as negative control; RIF (10 mg/kg) as positive control; 3l (100 mg/kg). The treatment was administered once daily by oral gavage for six days. On the final day of treatment, animals were sacrificed by cervical dislocation. The removed lungs and spleens were homogenized under sterile conditions in a 2 mL volume of PBS. Finally, RLU was detected for each homogenate.
4. Conclusions
In summary, we designed and synthesized a series of new 3-amino-5-benzylphenol derivatives. Several (3-benzyl-5-hydroxyphenyl)carbamates exerted potent in vitro inhibitory activity against Mtb H37Ra, H37Rv and clinically isolated multidrug-resistant Mtb strains. The introduction of hydrophobic carbocycles in the carbamate unit improved the antitubercular activity. The selected compounds 3i and 3l exhibited moderate cytotoxicities against tested cell line. The compound 3l showed good oral efficacy on a mouse infection model. The significant reduction of the bacteria count in the lung was achieved. The further structural optimizations toward the increase of the metabolic stability and in vivo efficacy are currently under investigation.
Supplementary Materials
The spectra of 1H-NMR and 13C-NMR for the target compounds are available online.
Author Contributions
Project administration, Y.-J.C., Z.-Y.L., H.-J.L., C.-T.F. and N.-N.Z.; Supervision, T.-Y.Z. and M.Y.; Writing—original draft, Y.-J.C. and M.Y.; Writing—review & editing, T.-Y.Z. and M.Y.
Funding
This research was funded by the National Natural Science Foundation of China (21772240), the Chinese Academy of Science (YJKYYQ20170036) and Guangzhou Science Technology and Innovation Commission (201707010210, 201604020019). Tian-Yu Zhang received the Science and Technology Innovation Leader of Guangdong Province (2016TX03R095).
Conflicts of Interest
There are no conflicts to declare.
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References
- 1.World Health Organization . WHO Global Tuberculosis Report 2018. World Health Organization; Geneva, Switzerland: 2018. [Google Scholar]
- 2.Zumla A., Raviglione M., Hafner R., Fordham V.R.C. Tuberculosis. N. Engl. J. Med. 2013;368:745–755. doi: 10.1056/NEJMra1200894. [DOI] [PubMed] [Google Scholar]
- 3.Mishra R., Shukla P., Huang W., Hu N. Gene mutations in Mycobacterium tuberculosis: Multidrug-resistant TB as an emerging global public health crisis. Tuberculosis. 2015;95:1–5. doi: 10.1016/j.tube.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Millard J., Ugarte-Gil C., Moore D.A.J. Multidrug resistant tuberculosis. Br. Med. J. 2015;350:h882. doi: 10.1136/bmj.h882. [DOI] [PubMed] [Google Scholar]
- 5.Giffin R., Robinson S., Giffin R. Workshop Summary of Institute of Medicine of the National Academies. The National Academies Press; Washington, DC, USA: 2009. Addressing the Threat of Drug-Resistant Tuberculosis: A Realistic Assessment of the Challenge. [PubMed] [Google Scholar]
- 6.Andries K., Verhasselt P., Guillemont J., Göhlmann W.H., Neefs J.M., Winkler H., Gestel J.V., Timmerman P., Zhu M., Lee E., et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto M., Hashizume H., Tomishige T., Kawasaki M., Tsubouchi H., Sasaki H., Shimokawa Y., Komatsu M. OPC-67683, a nitro-dihydroimidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS. Med. 2006;3:e466. doi: 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stover C.K., Warrener P., VanDevanter D.R., Sherman D.R., Arain T.M., Langhorne M.H., Anderson S.W., Towell J.A., Yuan Y., McMurray D.N., et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 9.Sacksteder K.A., Protopopova M., Barry C.E., Andries K., Nacy C.A. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012;7:823–837. doi: 10.2217/fmb.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox E., Laessig K. FDA approval of bedaquiline —the benefit—risk balance for drug-resistant tuberculosis. N. Engl. J. Med. 2014;371:689–691. doi: 10.1056/NEJMp1314385. [DOI] [PubMed] [Google Scholar]
- 11.Bloemberg G.V., Keller P.M., Stucki D., Trauner A., Borrell S., Latshang T., Coscolla M., Rothe T., Homke R., Ritter C., et al. Acquired Resistance to Bedaquiline and Delamanid in Therapy for Tuberculosis. N. Engl. J. Med. 2015;373:1986–1988. doi: 10.1056/NEJMc1505196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S., Chen J.Z., Cui P., Shi W.L., Shi X.H., Niu H.X., Chan D., Yew W.W., Zhang W.H., Zhang Y. Mycobacterium Tuberculosis Mutations Associated with Reduced Susceptibility to Linezolid. Antimicrob. Agents Chemother. 2016;60:2542–2544. doi: 10.1128/AAC.02941-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dartois V., Barry C.E., 3rd A medicinal chemists’ guide to the unique difficulties of lead optimization for tuberculosis. Bioorg. Med. Chem. Lett. 2013;23:4741–4750. doi: 10.1016/j.bmcl.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang N.N., Liu Z.Y., Liang J., Tang Y.X., Qian L., Gao Y.M., Zhang T.Y., Yan M. Discovery of m-Amidophenol Derivatives as A New Class of Antitubercular Agents. Med. Chem. Commun. 2018;9:1293–1304. doi: 10.1039/C8MD00212F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan M., Zhang T.Y., Zhang N.N., Liu Z.Y., Qian L., Tang Y.X., Cheng Y.J., Zhang X.J. m-Disubstituted phenol derivatives, their preparative methods and antitubercular applications. 107973727A. Patent CN. 2018 May 1;
- 16.Zhang N.N., Tang Y.X., Qian L., Gao Y.M., Liu Z.Y., Zou Z.L., Zhang T.Y., Yan M. Design, synthesis and antitubercular activity of 3-amidophenols with 5-heteroatomic substitutions. Arch Pharm. Chem. Life Sci. 2019;352:e1800277. doi: 10.1002/ardp.201800277. [DOI] [PubMed] [Google Scholar]
- 17.Zhang T.Y., Li S.Y., Nuermberger E.L. Autoluminescent Mycobacterium tuberculosis for rapid, real-time, non-invasive assessment of drug and vaccine efficacy. PLoS ONE. 2012;7:e29774. doi: 10.1371/journal.pone.0029774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mustafa J., Khan S.I., Ma G., Walker L.A., Khan I.A. Synthesis and anticancer activities of fatty acid analogs of podophyllotoxin. Lipids. 2004;39:167–172. doi: 10.1007/s11745-004-1215-5. [DOI] [PubMed] [Google Scholar]
- 19.Bi H.C., Zuo Z., Chen X., Xu C.S., Wen Y.Y., Sun H.Y., Zhao L.Z., Pan Y., Deng Y., Liu P.Q., et al. Preclinical factors affecting the pharmacokinetic behaviour of tanshinone IIA, an investigational new drug isolated from Salvia miltiorrhiza for the treatment of ischaemic heart diseases. Xenobiotica. 2008;38:185–222. doi: 10.1080/00498250701767675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.