

Abstract
Motivation:
As the size of high-throughput DNA sequence datasets continues to grow, the cost of transferring and storing the datasets may prevent their processing in all but the largest data centers or commercial cloud providers. To lower this cost, it should be possible to process only a subset of the original data while still preserving the biological information of interest.
Results:
Using 4 high-throughput DNA sequence datasets of differing sequencing depth from 2 species as use cases, we demonstrate the effect of processing partial datasets on the number of detected RNA transcripts using an RNA-Seq workflow. We used transcript detection to decide on a cutoff point. We then physically transferred the minimal partial dataset and compared with the transfer of the full dataset, which showed a reduction of approximately 25% in the total transfer time. These results suggest that as sequencing datasets get larger, one way to speed up analysis is to simply transfer the minimal amount of data that still sufficiently detects biological signal.
Availability:
All results were generated using public datasets from NCBI and publicly available open source software.
Keywords: RNA-Seq, FASTQ, data transfers, high-throughput DNA sequencing
Introduction
The advent of high-throughput DNA sequencing (HTS) in the last decade provides high resolution quantification of individual DNA molecules at the nucleotide level. One can literally count the occurrence of molecules in a biological specimen and determine each molecule’s exact sequence. The utility of measuring complex biological systems with HTS drives the expansion of DNA sequence archives. For example, the National Center for Biotechnology Information’s Sequence Read Archive (NCBI SRA) now contains more than 27 quadrillion base pairs (≈27 petabytes) from more than 4.4 million experiments.1 Given advances in DNA sequencing technology and falling price points, the exponential trend of data accumulation is not likely to end any time soon.
One application of HTS is the quantification of RNA molecules by deep sequencing after conversion of RNA into cDNA, a technique termed RNA-Seq. Evidence suggests that sampling 20 to 25 million RNA molecules with RNA-Seq provides sufficient resolution to capture medium to highly expressed genes, whereas even deeper sequencing to 100 to 200 million reads is likely to detect rare RNA transcripts.2 The depth of sequencing performed on a sample is often a function of a researcher’s sequencing budget which is a real constraint to the quantification of rare molecules. However, as the cost of HTS technology continues to decline, it should be possible to sequence deeper for almost any RNA-Seq application. For example, the Illumina Genome Analyzer released in 2006 was capable of generating 1 gigabase of sequence data, whereas the NextSeq platform in 2017 can produce 120 gigabases (400 million reads) in a single run.3 The more bases a sequencer can read, the deeper a researcher can peer into the molecular landscape of a biological system.
Even if HTS becomes cheap enough for routine deep sequencing of rare transcripts, the larger datasets will still need to be processed with bioinformatics workflows. Currently, a typical RNA-Seq workflow ingests data in FASTQ format, cleans it by trimming unwanted reads, aligns to a reference genome, and quantifies the alignments as RNA transcript counts.4 Transcript counts from multiple biological samples can be combined into a gene expression matrix (GEM),5 where the matrix value GEMi,j is the normalized count (eg, Fragments Per Kilobase of transcript per Million mapped reads; FPKM)6,7 of transcript i in sample j. Among other downstream applications of the GEM is to identify differentially expressed genes8 and generate gene co-expression networks.9-11
The advent of higher molecular resolution into biological systems via improved HTS technology must be coupled with computational advances that can process more and more DNA sequence data. Deep HTS datasets can quickly fill up storage systems, and transferring datasets between workflow execution CPUs can saturate network bandwidth within and between data centers. Furthermore, actual storage space requirements are several times greater than the size of a single dataset due to the large intermediate files created during the workflow. Thus, it will become increasingly important to consider storage and transfer costs into an experiment as dataset generation costs decline.
One way to reduce both storage and network input/output (I/O) costs is to process a reduced amount of DNA sequence data instead of moving the full dataset into a workflow. If the researcher decides that there is sufficient sequencing depth in the subsample, then there would be no need to pay the cost of moving and processing the full dataset. In this study, we explore the effect of transferring and processing partial RNA-Seq datasets using transcript detection as a simple metric. In a proof of concept, we show that our method significantly reduces the total transfer time of a dataset. We predict that partial analysis of datasets will become an important trade-off as researchers sequence deeper into biological samples.
Materials and Methods
To intelligently transfer partial datasets, we require a metric to measure and select a cutoff point and the means to transfer partial files. We chose to run full RNA-Seq workflows on partial datasets, select a cutoff point based on the number of detected transcripts per million mapped reads, and transfer partial datasets over the Internet between cloud computing sites.
In subsequent sections, this article uses the term DNA records or records to mean “the smallest unit of DNA sequence data that can be transferred and processed indivisibly.” In the context of the FASTQ files used as experimental input, a record would mean the 4 adjacent lines in an uncompressed file containing the sequence identifier, bases, duplicate sequence identifier, and quality scores.12 The results in this article were generated from paired-end reads, and for this reason, a logical record includes the corresponding forward and reverse reads.
Experimental setup
To clean the FASTQ files, we used Trimmomatic 0.36.13 To align the reads, we used HISAT2 2.0.5.14 To sort the SAM file, we used SAMtools 1.3.1.15 To map the alignments, we used StringTie 1.3.1c.16 During read alignment, novel splice junction discovery was disabled and only abundances of known reference transcripts were quantified. To load the counts and plot the results, we used R 3.3.2,17 Ballgown 2.6.0,18 ggplot2 2.2.1,19 plyr 1.8.4,20 and reshape2 1.4.2.21
Dataset transfers were performed between clouds in 2 locations. The node in the CloudLab (http://www.cloudlab.us/) cluster at Clemson University had 2 Intel E5-2683 v3 14-core CPUs, 256 GB of ECC RAM, two 1 TB SATA 3G hard disk drives, and a dual-port 10 Gigabit Ethernet adapter.22 The node in the Chameleon (http://www.chameleoncloud.org/) cluster at the University of Chicago had 2 Intel E5-2650 v3 10-core CPUs, 64 GB of ECC RAM, 16 2 TB 12 Gb/s SAS hard disk drives, and a 10 Gigabit Ethernet adapter.23 The software used to perform the transfers was FDT 0.25.1 with the OpenJDK 1.8.0 Java VM running on CentOS 7.4.1708.
Input data
To test the concept of partial dataset processing, we selected 3 human input datasets and 1 pig dataset of varied sequencing depth (ie, DNA sequence records): the human datasets hypoxia (45-55 million records; read length 100), bladder (85-87 million records; read length 76), and nisc2 (189-259 million records; read length 101), and the pig dataset oncopig (55-85 million records; read length 100). All datasets were generated using Illumina HiSeq sequencing systems with paired-end reads.
Our first dataset which we refer to as bladder comes from the project at NCBI with accession PRJNA358425 and includes the runs with accessions SRR5124442, SRR5124443, SRR5124447, SRR5124452, SRR5124453, and SRR5124455.24 Our second dataset which we refer to as hypoxia comes from PRJEB14955 and includes ERR1551404, ERR1551405, ERR1551408, and ERR1551409.25 Our third dataset which we refer to as nisc2 comes from PRJNA231202 and includes the 6 runs SRR1047863 to SRR1047865 and SRR1047869 to SRR1047871.26 Our last dataset which we refer to as oncopig comes from PRJEB8735 and includes the 7 runs ERR777781 to ERR777787.27
Auxiliary input data include the FASTA adapter sequences for the Illumina TruSeq Library Prep Kit. For human runs, we use the Release 26 GRCh38.p10 genome sequence and comprehensive gene annotation for all regions from the Genome Reference Consortium.28 For pig runs, we use the sequence and annotations from Ensemble Release 91.29
Scientific workflow
Before the workflow begins, a FASTQ dataset file is subdivided into a dataset partition factor (DPF) between 1% and 100% of the possible sequence records. Next, Trimmomatic is used to remove adapter sequences and short reads. Then, HISAT2 is run on the trimmed FASTQ file along with the index generated previously using hisat2-build and a file containing known splice sites. The output from HISAT2 is sorted with samtools sort and then processed using StringTie to generate counts in FPKM. We did not account for strand specificity.
The output from StringTie is loaded into R using the Ballgown package.18 For every run at every percent, the number of transcripts with FPKM greater than zero is calculated. The percent values are converted to records and the results are plotted.
Transfer of partial datasets
At the source side in Clemson, the files are stored in a logical volume striped across both disk drives. The 12 FASTQ files from SRR1047863 to SRR1047865 and SRR1047869 to SRR1047871 were transferred using FDT (http://github.com/fast-data-transfer/fdt) to the destination in Chicago. On the Chicago, side files were stored on a single drive in the 16-drive storage array. Both sides use XFS as the file system. The transfer was repeated 5 times.
When the full files were transferred, it was possible to measure the number of detected transcripts at different numbers of records with multiple runs of the RNA-Seq workflow. The slope between successive measurements was calculated for each dataset and expressed as the number of detected transcripts per million records. An arbitrary cutoff of 100 detected transcripts per million records was selected, and the smallest processed record count greater than or equal to the cutoff was chosen for each dataset.
Given that cutoff point, fastq-dump was used to only dump the selected number of records from each SRA file. The resulting partial dataset was again transferred using FDT. These smaller files were transferred in the same manner as before with 5 repetitions.
Results
To simulate a partial RNA-Seq data transfer and processing, we reduced the original datasets from NCBI into 18 subsets of records at these depths: 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, and 90%. For each subset of input data, we ran the RNA-Seq workflow described in the Scientific workflow section and then post-processed the StringTie output to generate our detection measurement defined as the number of transcripts with FPKM > 0. For every dataset in Figure 1, the number of detected transcripts increased with the record count. Within each dataset, there was variability between the runs, but the results tended to cluster together with a similar shape.
Figure 1.

Detected transcripts by number of records for 4 datasets. Each point indicates the number of transcripts with FPKM > 0 measured at the given number of records. All runs were identically analyzed using the workflow of the Scientific workflow section. FASTQ files were sampled at 1% to 100% of the records of the original dataset. Dashed lines at the top of each plot indicate the theoretical maximum number of detected transcripts (217 857 for human and 49 558 for pig). Species for bladder, hypoxia, and nisc2 is Homo sapiens. Species for oncopig is Sus scrofa. FPKM indicates Fragments Per Kilobase of transcript per Million mapped reads.
Figure 2 shows the timing results of full and partial transfers of the nisc2 dataset from Clemson to Chicago. First, the full dataset was transferred and the total transfer time was measured for 5 trials. Then, a subset of each file in the dataset was transferred 5 times. Both the full and partial datasets were transferred over the commodity Internet with the same configuration settings. The total time to transfer all partial datasets was 75% of the time to transfer the full datasets (1.5 hours vs 2 hours). These aggregate times are shown as the rightmost pair of bars in Figure 2.
Figure 2.
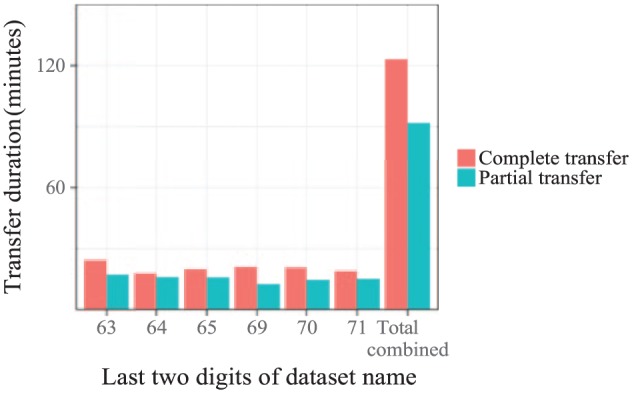
Transfer times of full and partial FASTQ files from nisc2. FASTQ files were transferred between Clemson and Chicago over the public Internet using FDT. The time to transfer a complete dataset is shown with the bars labeled “complete transfer.” The time to transfer a partial dataset satisfying the criteria in the transfer of partial datasets section is shown with the bars labeled “partial transfer.” Reported times within a group are the average of 5 trials. Error bars are too small to be visible. The x-axis gives the last 2 digits of the dataset name, where each dataset name begins with the string SRR10748. The rightmost pair of bars plots the sum total of the times for all datasets within both groups.
Discussion
A primary constraint when sequencing a sample is balancing sequence depth against cost. However, for the experimenter using data that have already been sequenced and stored in a central repository, the primary consideration will be the time and resources required to transfer and process the dataset. In our concept, the data mining experimenter has the option of processing only a subset of the original dataset, thereby reducing computational resources that are becomingly increasingly expensive as HTS dataset sizes swell into hundreds of millions of reads and analysis is increasingly performed in billable cloud compute environments.
A key issue is determining the smallest number of records required to produce the same scientific result as the full dataset, and we point to a simple saturation point as determined by transcript detection. Once a saturation point has been reached, one could pause and examine the results. If there is interesting signal, then there is nothing preventing the user from processing more sequence records. However, if there is no signal, one could drop the experiment and move on to other datasets.
The primary output for our RNA-Seq workflow is count data measured in FPKM for each feature (gene or transcript) in each dataset. We would like to ensure that our partial dataset is able to detect all the features of interest in the full dataset. In our pilot use case, we define a feature is detected when the FPKM measurement for that feature is greater than zero. By continuously processing increasingly larger subsets, it should be possible to detect the threshold at which the number of features with FPKM > 0 is constant; that is, when no new features are detected. However, in the results of Figure 1 across all 4 datasets, we never saw transcript detection saturation as the number of records increased to the maximum. We note that we have tested this proof of principle with a single representative workflow.
Even with the nisc2 dataset having more than 258 million records, there was no saturation, suggesting that either 258 million records is not enough or that some noise is being introduced that is causing the count of detected transcripts to continuously increase. At the time of this writing, 258 million records are in the 99th percentile of public paired-end RNA-Seq runs available at NCBI. It would be unreasonable to expect that any of the 99% of datasets in NCBI smaller than the one we tested would reach a point where the slope was flat. As there was no saturation seen in the nisc2 dataset, we chose a cutoff of 100 detected transcripts per million mapped reads. Although our choice of cutoff was arbitrary, the cutoff points of 133 to 181 million records correspond nicely to the predictions of the literature of 100 to 200 million reads.11 The choice of cutoff value will need to be one of the parameters decided by the experimenter.
It appears that the noise that causes the count of detected transcripts to continuously increase is confined to the low expression transcripts. As seen in Figure 3(A), the number of detected transcripts at different percent records transferred for the largest nisc2 dataset only increases if the detection threshold is FPKM > 0. Ratcheting up the thresholds from >1 to >4 does not detect more transcripts. Furthermore, the same genes are being detected at each transfer (Figure 3(B)). These data suggest that if one is looking at even moderately transcribed genes, these can be effectively captured at low numbers of sequence records.
Figure 3.

Only low-level transcripts accumulate with more sequence records. (A) The number of genes that were detected at 6 FPKM expression thresholds are shown for the 6 nisc2 datasets at each percent transfer. (B) The amount of gene overlap at each transfer level is shown for a representative nisc2 dataset SRR1047863.
In many cases, it may be possible to transfer much fewer than the 133 to 181 million records transferred in our nisc2 experiment, because even though the slope of the number of detected transcripts in Figure 1 never flattens completely, for each dataset around 30 million records the slope of the number of detected transcripts decreases greatly. In Table 1, we estimate the number of transcripts that would have been detected at exactly 30 million records. These predictions for the number of detected transcripts are then compared with the actual number of detected transcripts in the full dataset, yielding a range of percent values which represent the predicted portion of transcripts detected at 30 million records. The minimum value of 71% for SRR1047863 in nisc2 means that even in the worst case, a much smaller cutoff of 30 million records would detect up to 71% of the transcripts detected in the full dataset. While we tested 4 RNA-Seq datasets and saw similar saturation behavior, it is likely that other datasets of variable quality (eg, low RNA quality, rRNA contamination, low quality genome assembly) might exhibit different sensitivities and saturation points. Thus, a saturation curve might need to be generated if the workflow and/or data are very different from the representative workflow we examined.
Table 1.
Estimation of the number of detected transcripts at 30 million records.
| Dataset | Predicted FPKM > 0 | Percent of whole |
|---|---|---|
| Bladder | 97 872-105 433 | 83%-84% |
| Hypoxia | 100 613-102 279 | 90%-93% |
| nisc2 | 101 490-110 594 | 71%-76% |
| Oncopig | 26 865-30 658 | 87%-92% |
Abbreviation: FPKM, Fragments Per Kilobase of transcript per Million mapped reads.
For each run of Figure 1, a linear model of the number of detected transcripts was created using the formula detected ~log(records) (R2 = 0.988-1.0). These models were then used to predict the number of detected transcripts at 30 million records for each run. As each dataset contains between 4 and 7 runs, this table lists the range of predicted transcripts for each dataset. The values for percent of whole were calculated by dividing the predicted number of transcripts at 30 million records by the number of detected transcripts measured in the full dataset as plotted in Figure 1.
At the time of this writing, the mean sequencing depth of public Illumina paired-end RNA-Seq runs was ≈16.5 million records. However, the mean size of all studies (ie, collections of related runs) was 374 million records. In performing certain types of analysis such as the search for differentially expressed genes, it will be necessary to transfer all of the datasets within a related study. Thus, even though the size of individual datasets may currently be small, the aggregate size of the whole study is large enough to benefit from an optimization of the data transfer method.
Likewise, while a 25% reduction in transfer time may not seem significant in the context of a single dataset, a similar reduction applied to all the datasets from an entire study may produce noticeable computational savings. As an example, the full study containing nisc2 consists of more than 3.8 billion records. At the previously measured throughput of 116 million bytes/s, the time to transfer the full study would decrease by 1.25 hours from 5 to 3.75 hours. This reduction in transfer time frees up more network bandwidth and lessens the workflow resource requirements while still capturing transcriptional signals.
Conclusions
For our use case, partial data transfer reduced total transfer time by 25%. Processing smaller datasets given the same amount of time opens the possibility to processing more datasets. For example, more replicates could be incorporated into the experiment leading to better confidence with lowly expressed genes.30 In the end, it will be up to the individual experimenter to decide when signal is captured for their experiment.
Acknowledgments
Results were obtained using CloudLab and Chameleon testbeds supported by the National Science Foundation (award numbers 1743363, 1743363, and 1743358) and the Clemson Palmetto cluster.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Science Foundation (award numbers 1443040 and 1659300).
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: FAF conceived the study. NM and EMB designed and performed the computational experiments. WLP and FAF designed biological experiments. NM, EMB, WLP, WBL, and FAF wrote the manuscript.
ORCID iD: F Alex Feltus  https://orcid.org/0000-0002-2123-6114
https://orcid.org/0000-0002-2123-6114
References
- 1. National Center for Biotechnology Information. SRA database growth. 2019. https://www.ncbi.nlm.nih.gov/sra/docs/sragrowth/
- 2. ENCODE. Standards, guidelines and best practices for RNA-Seq tech. rep. The ENCODE Consortium. 2011. https://genome.ucsc.edu/encode/protocols/dataStandards/ENCODE_RNAseq_Standards_V1.0.pdf
- 3. Illumina. NextSeq series specifications. 2018. https://www.illumina.com/systems/sequencing-platforms/nextseq/specifications.html
- 4. Mihaela P, Daehwan K, Pertea Geo M, Leek Jeffrey T, Salzberg Steven L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Poehlman William L, Mats R, Chris B, Balamurugan D, Feltus Frank A. OSG-GEM: gene expression matrix construction using the open science grid. Bioinform Biol Insights. 2016;10:BBIS38193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621-628. [DOI] [PubMed] [Google Scholar]
- 7. Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA- seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Simon A, Wolfgang H. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ficklin SP, Dunwoodie LJ, Poehlman WL, Christopher W, Roche KE, Feltus FA. Discovering condition-specific gene co-expression patterns using Gaussian mixture models: a cancer case study. Sci Report. 2017;7:8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Butte AJ, Tamayo P, Slonim D, Golub TR, Kohane IS. Discovering functional relationships between RNA expression and chemotherapeutic susceptibility using relevance networks. Proc Natl Acad Sci U.S.A. 2000;97:12182-12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bolger Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature Meth. 2015;12:357-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heng L, Bob H, Alec W, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mihaela P, Pertea Geo M, Antonescue Corina M, Tsung-Cheng C, Mendell Joshua T, Salzberg Steven L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotech. 2015;33:290-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. R Core Team: Vienna; 2016. [Google Scholar]
- 18. Jack F, Frazee Alyssa C, Leonardo CT, Jaffe Andrew E, Leek Jeffrey T. ballgown: Flexible, isoform-level differential expression analysis 2016. R package version 2.6.0. https://bioconductor.riken.jp/packages/3.4/bioc/html/ballgown.html
- 19. Hadley W. Ggplot2: Elegant Graphics for Data Analysis. New York: Springer; 2009. [Google Scholar]
- 20. Hadley W. The split-apply-combine strategy for data analysis. J Stat Softw. 2011;40:1-29. [Google Scholar]
- 21. Hadley W. Reshaping data with the reshape package. J Stat Softw. 2007;21:1-20. [Google Scholar]
- 22. Robert R, Eric E; The CloudLab Team. Introducing CloudLab: scientific infrastructure for advancing cloud architectures and applications. USENIX; login 2014;39:132. [Google Scholar]
- 23. Mambretti J, Chen J, Yeh F. Next generation clouds, the chameleon cloud testbed, and software defined networking (SDN). 2015 International Conference on Cloud Computing Research and Innovation (ICCCRI); October 26-27, 2015; Singapore: 73-79. [Google Scholar]
- 24. Dee LL, Sujoy G, Xiaoran C, et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci Transl Med. 2017;9:eaai8312. [DOI] [PubMed] [Google Scholar]
- 25. Chiang CM, Ilott Nicholas E, Johannes S, et al. Tuning the transcriptional response to hypoxia by inhibiting hypoxia-inducible factor (HIF) prolyl and asparaginyl hydroxylases. J Biol Chem. 2016;291:2066120673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Akula N, Barb J, Jiang X, et al. RNA-sequencing of the brain transcriptome implicates dysregulation of neuroplasticity, circadian rhythms and GTPase binding in bipolar disorder. Mol Psychiatry. 2014;19:1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schook LB, Collares TV, Hu W, et al. A genetic porcine model of cancer. PLoS ONE. 2015;10:1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931-945. [DOI] [PubMed] [Google Scholar]
- 29. Zerbino Daniel R, Premanand A, Akanni Wasiu, et al. Ensembl 2018. Nucleic Acid Res. 2017;46:D754-D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robles José A, Qureshi Sumaira E, Stephen Stuart J, Wilson Susan R, Burden Conrad J, Taylor Jennifer M. Efficient experimental design and analysis strategies for the detection of differential expression using RNA sequencing. BMC Genom. 2012;13:484. [DOI] [PMC free article] [PubMed] [Google Scholar]