

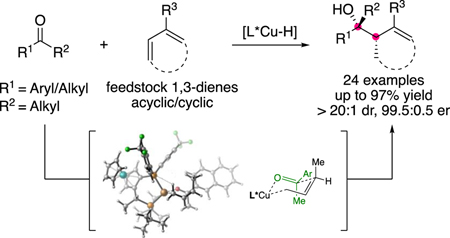
Abstract
Chiral tertiary alcohols are important building blocks for the synthesis of pharmaceutical agents and biologically active natural products. The addition of carbon nucleophiles to ketones is the most common approach to tertiary alcohol synthesis, but traditionally relies on stoichiometric organometallic reagents that are difficult to prepare, sensitive, and uneconomical. We describe a mild and efficient method for the copper-catalyzed allylation of ketones, using widely available 1,3-dienes as allylmetal surrogates. Homoallylic alcohols bearing a wide range of functional groups are obtained in high yield and with good regio-, diastereo-, and enantioselectivity. Mechanistic investigations using density functional theory (DFT) implicate the in situ formation of a rapidly equilibrating mixture of isomeric copper(I) allyl complexes, from which Curtin-Hammett kinetics determine the major isomer of product. A stereochemical model is provided to explain the high diastereo- and enantioselectivity of this process. Finally, this method was applied toward the preparation of an important drug, (R)-Procyclidine, and a key intermediate in the synthesis of several pharmaceuticals.
Graphical Abstract
■ INTRODUCTION
Enantiomerically enriched tertiary alcohols and their derivatives feature prominently in a variety of important pharmaceutical agents and complex natural products.1 Consequently, their efficient synthesis has attracted great attention from synthetic chemists.2 Traditionally, the addition of organomagnesium (Grignard) reagents to ketones has been a popular method to obtain tertiary alcohols in racemic form.3 However, the harsh methods required to prepare these organometallic reagents, as well as their instability and Brønsted basicity, have limited the tolerance of these reagents toward polar functional groups. Furthermore, the necessity to use stoichiometric organometallic reagents, and often, chiral auxiliaries for enantioselective transformations, is intrinsically inefficient and operationally complicating. Thus, the development of highly efficient catalytic, asymmetric strategies for constructing tertiary alcohols remains a goal of high priority in organic synthesis.4
1,3-ienes are important industrial raw materials that are produced on an enormous scale annually (Figure 1a). These chemicals include butadiene5 (about 13 × 106 ton/year production), isoprene6 (about 8 × 105 ton/year) and myrcene7 (about 2500 ton/year). Recently, a number of groups have proposed that these inexpensive and stable compounds could serve as ideal surrogates for stoichiometric organometallic reagents in carbonyl addition reactions. In 2005, a pioneering report by Gendre and Moïse8 demonstrated the first titanium-catalyzed aldehyde allylation using conjugated dienes as reagents (Figure 1b), although due to the highly reactive nature of the titanium-allyl species, the functional group tolerance was limited. Subsequently, Krische5b has developed ruthenium-catalyzed stereoselective aldehyde (or alcohol) allylations with 1,3-butadiene (Figure 1c). Unfortunately, the same method cannot be generally applied to ketones for the synthesis of tertiary alcohols. Despite reports of a number of other transition-metal-catalyzed reductive couplings (Ni,9 Ru,10 Rh11 and Ir12) with conjugated dienes, reactions involving ketones, rather than aldehydes, remain challenging, even in a non-stereoselective manner.
Figure 1.
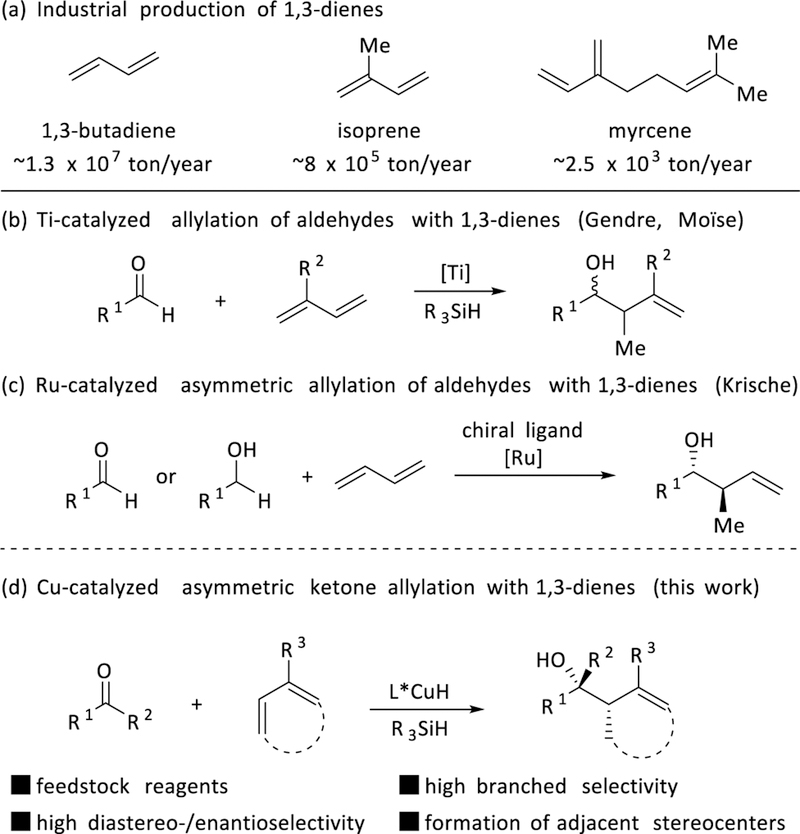
Overview of 1,3-dienes in industry and in catalytic allylation processes.
Over the past several years, a number of research groups, including ours, have reported approaches for the copper-catalyzed hydroamination of unsaturated substrates through the in situ generation of alkylcopper nucleophiles.13 Using this strategy, activated pronucleophiles such as enynes and allenes were successfully engaged in nucleophilic addition reactions with ketones.14 This reactivity pattern has since also been extended to the reductive coupling of olefin pronucleophiles with imines.2e,15
Following this general concept, herein we describe a highly regio- and enantioselective copper-catalyzed method for the allylation of ketones using readily available 1,3-dienes (Figure 1d). Previously, we had reported a single, unoptimized example of this transformation. In addition, we report a computational study of the mechanism of this class of transformations, revealing a complex kinetic basis for diastereo- and enantioselectivity resulting from an equilibrating mixture of allylcopper intermediates of similar energy. Furthermore, we propose a stereochemical model for these allylation processes using non-C2-symmetric JOSIPHOS-derived chiral ligands. Finally, we apply our method toward an efficient and concise synthesis of the pharmaceutical agent (R)-procyclidine and key intermediates in the synthesis of (R)-Oxyphencyclimine, (R)-Oxybutynin and (R)-Oxyphenonium bromide.
■ RESULTS AND DISCUSSION
We began by studying the reaction between 4-methoxyacetophenone (1a) and 1,3-butadiene (1b) under conditions previously reported for Cu-catalyzed reductive coupling reactions (Table 1, entry 1).14 With (R)-DTBM-SEGPHOS (L1) as the ligand, homoallylic alcohol 1 was obtained with 44% yield, 1.2:1 dr and 83.5:16.5 er for the major diastereomer (65.5:34.5 er for the minor). Based on 1H NMR analysis of the crude reaction mixture, the remainder of the ketone underwent direct reduction by copper hydride. When the ligand was exchanged for (S,S)-Ph-BPE (L2), this reduction pathway was suppressed,14a and a 96% yield of 1 was obtained with moderate dr and ee (Table 1, entry 2). Further ligand screening revealed that use of the commercially available JOSIPHOS16 derivative SL-J011–1 further improved the stereoselectivity to 4:1 dr and 97:3 er (96:4 er for the minor diastereomer, Table 1, entry 3).
Table 1.
Evaluation of Reaction Conditions for the CuH-Catalyzed Allylation of 4-Methoxyacetophenone.a
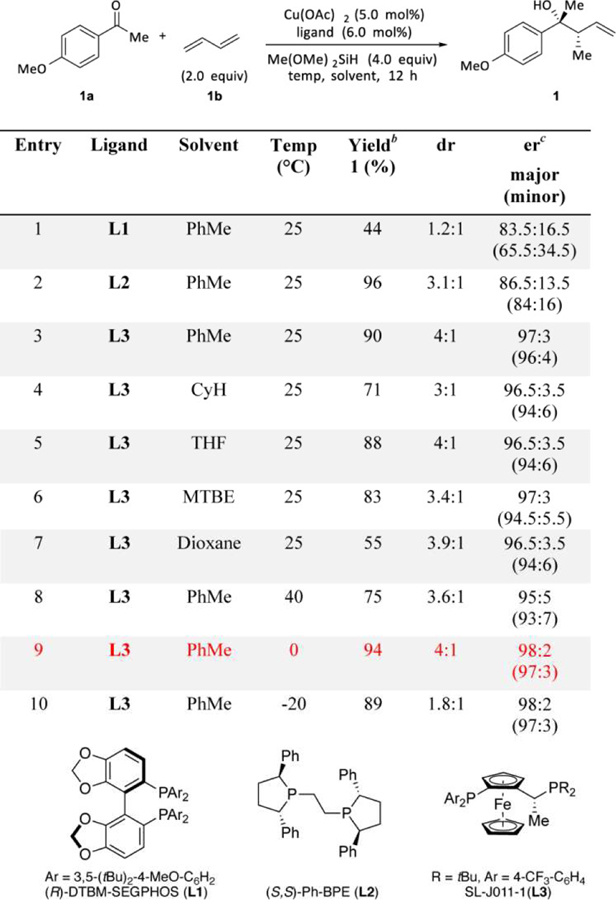 |
Conditions: 0.2 mmol ketone (1 equiv), 1,3-butadiene (2 equiv), copper(II) acetate (0.05 equiv), ligand (0.06 equiv), dimethoxy(methyl)silane (4 equiv) in solvent (0.2 mL), ketone was added slowly by syringe pump; see the Supporting Information for details.
Yield and diastereomeric ratio were determined by 1H NMR spectroscopy of the crude mixture, using dibromomethane as an internal standard.
Enantiomeric ratio was determined by HPLC or SFC analysis on commercial chiral columns, and the relative configuration of 1 was determined by comparing its NMR data with reported data.17
Evaluation of the reaction solvent (Table 1, entry 4–7) indicated that toluene was optimal for this transformation. The results were very sensitive to the reaction temperature: the yield, dr, and er were all diminished at slightly elevated temperatures (40 ˚C, Table 1, entry 8). However, excellent yield (94%), dr (4:1), and er (98:2 and 97:3 respectively for the major and minor diastereomers) were achieved when the reaction was performed at 0 ˚C (Table 1, entry 9). Further lowering of the reaction temperature (−20 ˚C) significantly decreased the dr again (Table 1, entry 10).
Next, the substrate scope of the asymmetric reductive coupling of diverse ketones with acyclic 1,3-dienes was examined (Table 2). A range of chiral homoallylic tertiary alcohols were prepared with excellent yields and enantiomeric purity (>94:6 er). The reaction was compatible with ether (1), alcohol (2), secondary (3) and tertiary amine (4) groups, as well as aromatic heterocycles (5, 6). Cyclic ketones such as 7a reacted with particularly good diastereoselectivity, as well as excellent yield and enantioselectivity. Using acetylferrocene, we obtained enantiomerically enriched ferrocene 8. In addition to butadiene, isoprene was also found to react with good yield and excellent enantioselectivity (9).
Table 2.
Evaluation of the Scope of the Ketone Allylation with Acyclic Dienes.a
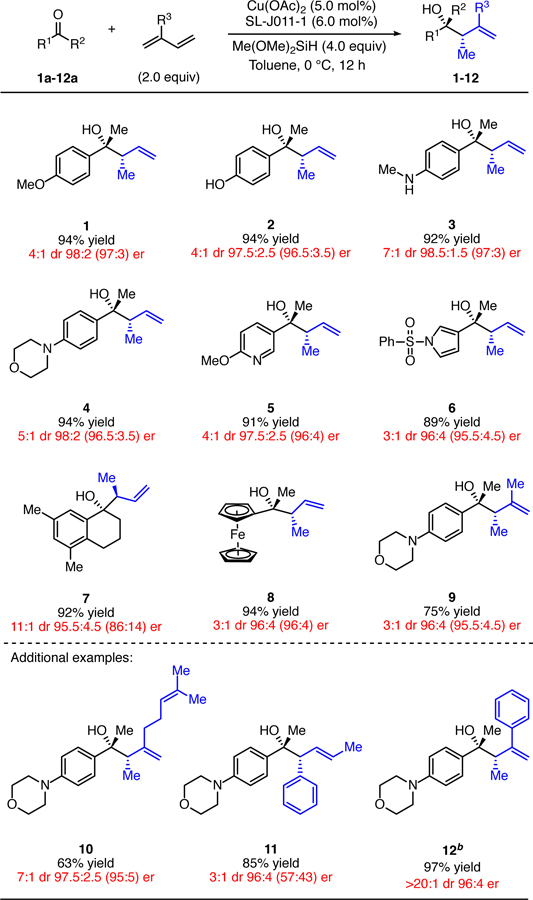 |
Yields indicate the isolated yield of product as a mixture of two diastereomers on a 1.0 mmol scale. Diastereomeric ratios were determined by 1H NMR spectroscopy for both the crude and purified products; enantiomeric ratios were determined by HPLC or SFC analysis on commercial chiral columns; enantiomeric ratios of minor diastereomers are indicated in parentheses after those of the major diastereomers. Yields, diastereomeric ratios, and enantiomeric ratios are the averages for two identical runs. See Supporting Information for full details.
L2 was used instead of L3.
We also surveyed the scope of ketone allylation using cyclic 1,3-dienes. However, under the conditions used for acyclic dienes, the yield of the desired product was unsatisfactory, and direct reduction of the ketone was instead the major reaction that was observed (see Supporting Information for details). We hypothesized that in the case of cyclic dienes, the L3-ligated CuH is unable to react with the diene at a rate competitive with direct ketone reduction. Revisiting our initial ligand evaluation data, we noticed that the use of (S,S)-Ph-BPE (L2) provided less ketone reduction byproduct than with L3 (Table 1, entries 2 and 3).14a Accordingly, we hypothesized that substituting L2 for L3 might be useful in these cases where ketone reduction is a problem: indeed, the catalyst derived from L2 provided greatly improved yields with cyclic diene substrates.
Using L2, several classes of ketones were coupled with cyclic 1,3-dienes in high regio- and enantioselectivity (Table 3). The reaction is most efficient for aryl methyl ketones. A broad range of aromatic carbonyl substituents, including an ortho-substituted arene (11), a pyridine (15), a pyrrole (16), a bromopyrazole (17), and a ferrocene (20) were evaluated, all providing good results. Several additional types of ketones were converted with high yield and stereoselectivity under the same conditions. For instance, a dialkyl ketone (13) and a vinyl methyl ketone (14) underwent allylation with high enantioselectivity. We proposed that the low diastereoselectivity observed in the case of 13 may be due to the minimal steric differentiation between the methyl and methylene groups attached to the carbonyl. Accordingly, we found that our method can be particularly useful on symmetric dialkyl ketones, which react to form homoallylic alcohol products with exceptionally high yield and enantioselectivity (18, 19). Finally, a larger ring diene, 1,3-cycloheptadiene, is also an effective reagent, providing 21 with moderate yield and excellent stereoselectivity.
Table 3.
Scope Evaluation of Ketone Allylation with Cyclic Dienes.a
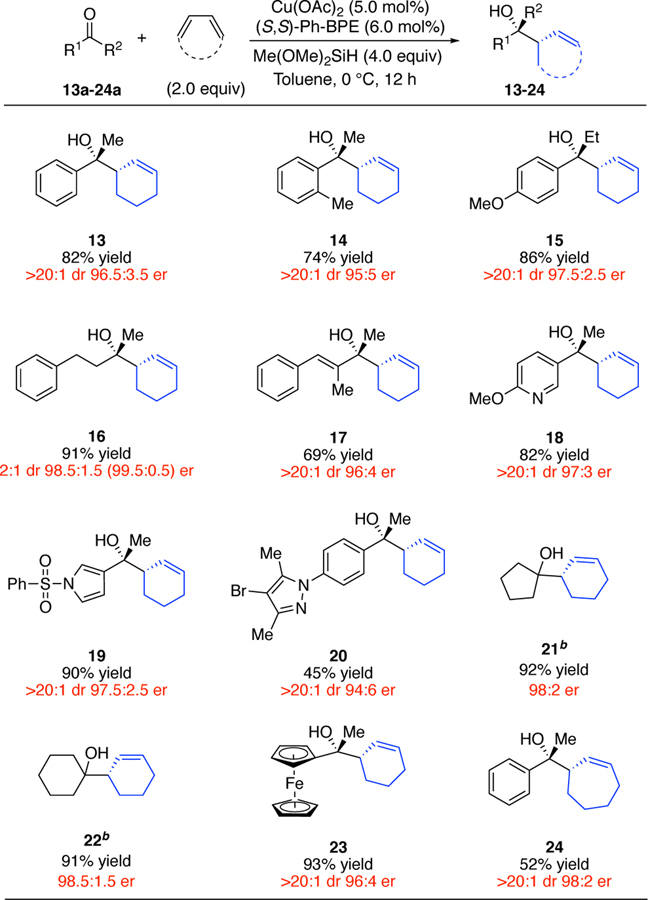 |
Yields indicate the isolated yield of product as a mixture of two diastereomers on a 1.0 mmol scale. Diastereomeric ratios were determined by 1H NMR spectroscopy for both the crude and purified products; enantiomeric ratios were determined by HPLC or SFC analysis on commercial chiral columns; enantiomeric ratios of minor diastereomers are indicated in parentheses after those of the major diastereomers. Yields, diastereomeric ratios, and enantiomeric ratios are the averages for two identical runs. See Supporting Information for full details.
The yield was determined by 1H NMR versus an internal standard due to the volatility of the product.
■ MECHANISTIC STUDIES
The proposed catalytic cycle of this CuH-catalyzed allylation reaction is summarized in Figure 2. We envisioned that a primary allylic copper intermediate (III) might be formed by hydrocupration of a diene. Selectivity-determining nucleophilic addition of III to the ketone would provide copper alkoxide V. Subsequently, σ-bond metathesis with a hydrosilane VI should rapidly regenerate the copper hydride catalyst I, with concomitant formation of the silylated homoallylic alcohol (VII) in a process that is well precedented.14a
Figure 2.
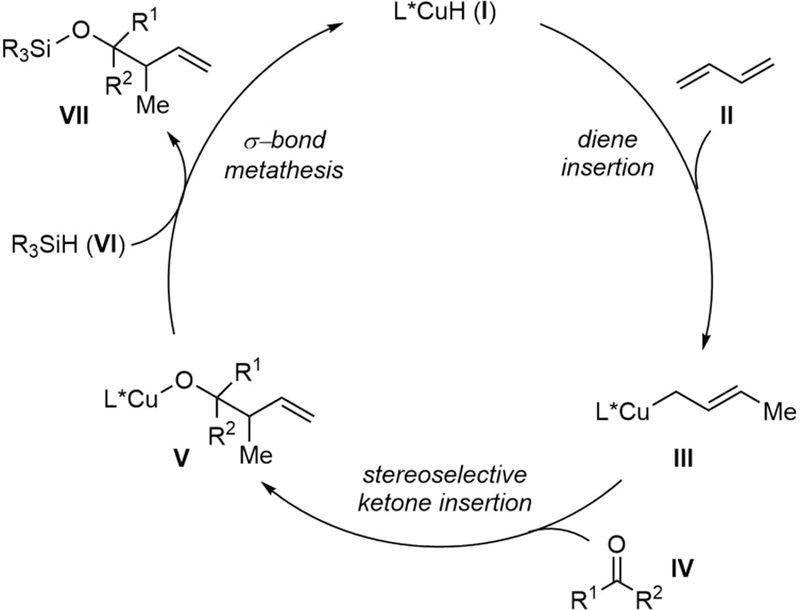
Proposed catalytic cycle.
We performed density functional theory (DFT) calculations to investigate several aspects of this proposed reaction mechanism. First, a comparison of the candidate hydrocupration mechanisms was performed to understand the mechanism of generation of the key allylcopper intermediate. Next, the energies and interconversion barriers of several possible allylic complexes were evaluated. From here, a thorough consideration of possible insertion transition states for the addition of the allylcopper intermediate to ketones was undertaken to reveal the origin the observed diastereoselectivity. Finally, we sought to explain the π-facial selectivity with respect to the ketone. While the mechanism of chiral induction in enantioselective reactions utilizing C2-symmetric ligands such as Ph-BPE has been frequently rationalized using quadrant-diagrams,18a-c analogous intuitive models for less symmetric ligands such as JOSIPHOS derivatives are rare.18d Therefore, we focused on developing an understanding of the high enantioselectivity observed with L3-supported copper catalysts.
We started our computational investigation with a conformational search on the L3-supported CuH catalyst. Two lowest-energy conformers with almost identical energies (22a and 22b, Figure 3) were located. These conformers differ in the arrangement of the six-membered chelate ring. In 22a, the chiral carbon center is puckered out-of-plane, while the two phosphorus atoms and the Cu are nearly co-planar with one of the Cp rings of the ferrocene. In contrast, in 22b, the Cu is puckered out-of-plane, while the two phosphorus atoms and the chiral carbon are nearly co-planar with the ferrocene Cp ring. As a result, the P-tBu and P-Ar substituents in 22a and 22b adopt different orientations, and thus create distinct steric environments around the Cu center. In 22a, the P-tBu group in quadrant IV and the P-aryl group in quadrant II are placed in closer proximity to the Cu center, while the P-tBu and P-aryl groups in quadrants I and III are more distal from the Cu. Therefore, the steric environment of this conformer resembles those of C2-symmetric ligands. In contrast, conformer 22b is pseudo-CS symmetric. The P-tBu and P-aryl groups in quadrants IV and III are placed closer to the Cu center, while quadrants I and II are relatively unoccupied by the ligand as the P-substituents in these quadrants are placed further away from the Cu. Considering the similar stability of 22a and 22b, both ligand conformations were considered when locating the transition states in the proposed catalytic cycle. Our calculations indicated the hydrocupration, 1,3-migration, and ketone addition transition states all are lower in energy when the ligand adopts the conformation in 22a, which has a pseudo-C2-symmetric steric environment (see below). This is consistent with the high efficiency of CuH catalysts with C2-symmetric ligands such as Ph-BPE in promoting similar transformations.
Figure 3.
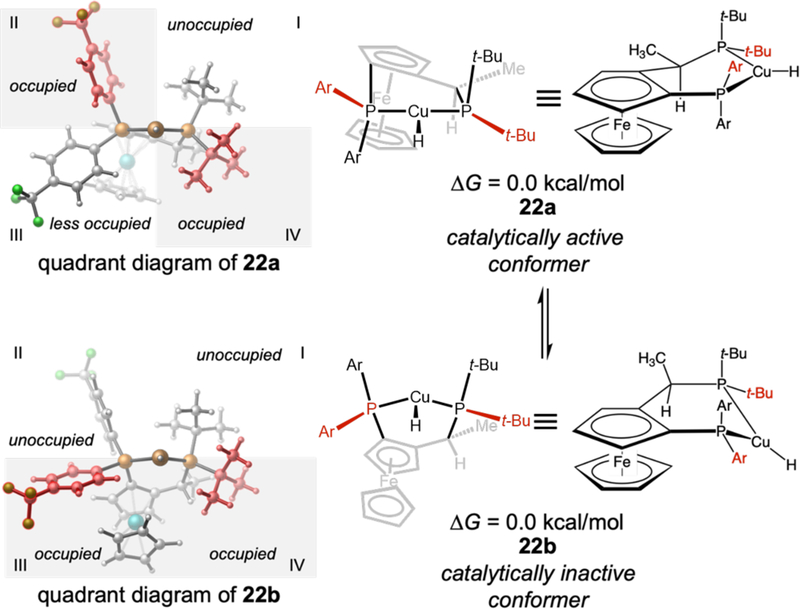
Conformers of the CuH catalyst supported by the SL-J011–1 ligand (L3). The P-Ar and P-tBu groups proximal to the Cu center are highlighted in red.
We selected 2-acetonaphthone and 1,3-butadiene as the model substrates for our computational investigation of the catalytic cycle. Experimentally, this pair of substrates react with 95% yield, 2.5:1 dr, and 93:7 er (for the major diastereomer, 90.5:9.5 er for the minor diastereomer) under the standard reaction conditions (see Supporting Information for details). We hypothesized that, initially, the hydrocupration of 1,3-butadiene might proceed via either direct 1,4-hydrocupration of the diene or via 1,2-hydrocupration followed by a 1,3-migration. Our calculations suggest that this process strongly prefers to occur through the 1,2-addition pathway (TS1a, Figure 4) to form a secondary allylcopper intermediate (23, Figure 5). In comparison, the 1,4-hydrocupration of the diene requires 9.6 kcal/mol higher activation energy (TS1c, Figure 4b). The 1,2-hydrocupration proceeds with moderate π-facial selectivity (∆∆G‡ = 0.8 kcal/mol, Figure 4a) leading initially to an (S)-allylcopper intermediate. However, this stereocenter is rapidly ablated: the secondary allyl complex 23 undergoes facile 1,3-migration via either TS2-cis or TS2-trans to form primary allylcopper intermediates 24-cis and 24-trans, which are similar in energy to each other, and both more stable than 23 (Figure 5). This 1,3-migration step requires a very low barrier and is reversible. Therefore, the cis/trans isomers of the primary allylcopper intermediates exist in equilibrium with each other, and with the branched isomers, prior to the nucleophilic addition to the ketone.
Figure 4.
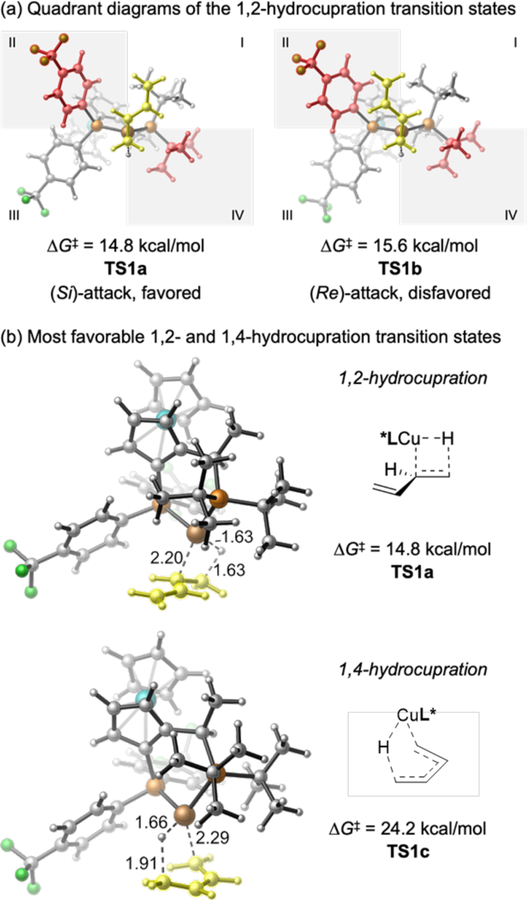
Optimized geometries of the 1,2- and 1,4-hydrocupration transition states. The diene is highlighted in yellow. The P-aryl and P-tBu groups “proximal” to the Cu center are highlighted in red in Figure 4a.
Figure 5.
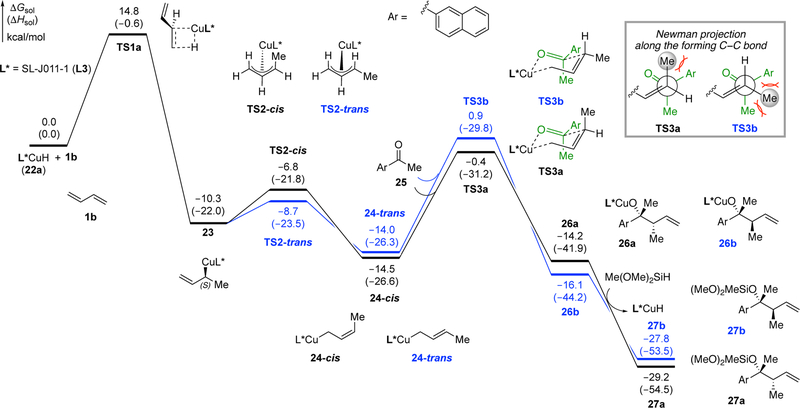
Computed energy profiles of the CuH-catalyzed allylation of 2-acetonaphthone 25. The calculations were performed at the M06–2X/SDD–6–311+G(d,p)/SMD(toluene)//B3LYP/SDD–6–31G(d) level of theory. All energies are with respect to the separate L*CuH catalyst (22a) and reactants (1b and 25).
The enantio- and diastereoselectivity are both determined in the subsequent ketone addition step. We found that the ketone addition occurs through a six-membered Zimmerman-Traxler-type transition state.19 After exhaustive computational investigation of possible transition state isomers, TS3a and TS3b were identified as the most favorable pathways for the ketone additions (see SI for other less favorable TS structures). In both TS3a and TS3b, the bulkier aryl group on the ketone is placed in a pseudo-equatorial orientation, and the methyl substituent is pseudo-axial. Counterintuitively, the preferred pathway for reaction with the ketone takes place from the cis-allylcopper species 24-cis via TS3a, which places the terminal methyl substituent of the allyl group pseudo-axial.19b In comparison, the ketone addition process from 24-trans, which involves a pseudoequatorial methyl substituent, requires an additional 1.3 kcal/mol of activation energy (TS3b). An examination of TS3b reveals the origin of this destabilization: the methyl substituent on the C=C double bond is placed between the aryl and methyl groups of the ketone, and thus induces greater steric repulsions at the forming C–C bond. On the other hand, the 1,3-diaxial repulsions with the same methyl substituent in TS3a are relatively weak because only one H…Me interaction is expected to contribute. The most favorable ketone addition transition state TS3a leads to the alkoxycopper intermediate 26a, from which rapid σ-bond metathesis with a silane generates the observed major product in silyl-protected form (27a). This step is known to be very rapid for copper alkoxides, which renders the ketone addition step effectively irreversible.14a
We next turned our attention to the enantioselectivity of this process, which is determined by the π-facial selectivity of the ketone addition step as dictated by the ligand. Relative to favored transition state structure TS3a, disfavored structure TS3c involves the addition to the opposite face of the ketone (Figure 6). In TS3c, the α-methylene group and the pseudo-axial methyl group of the ketone are both placed in the two quadrants occupied by the “proximal” P-aryl and P-t-Bu groups (highlighted in red in Figure 6). As such, TS3c is destabilized by the steric repulsions with the ligand. In contrast, in the more stable transition state TS3a, the α-methylene group and the pseudo-axial methyl group are both placed in the “unoccupied” quadrants, in which the P-substituents are further away (“distal”) from the Cu center and the substrate. Due to the diminished ligand-substrate steric repulsions, TS3a is 4.0 kcal/mol more stable than TS3c, which is in qualitative agreement with the high levels of enantioselectivity observed in the experiment.
Figure 6.
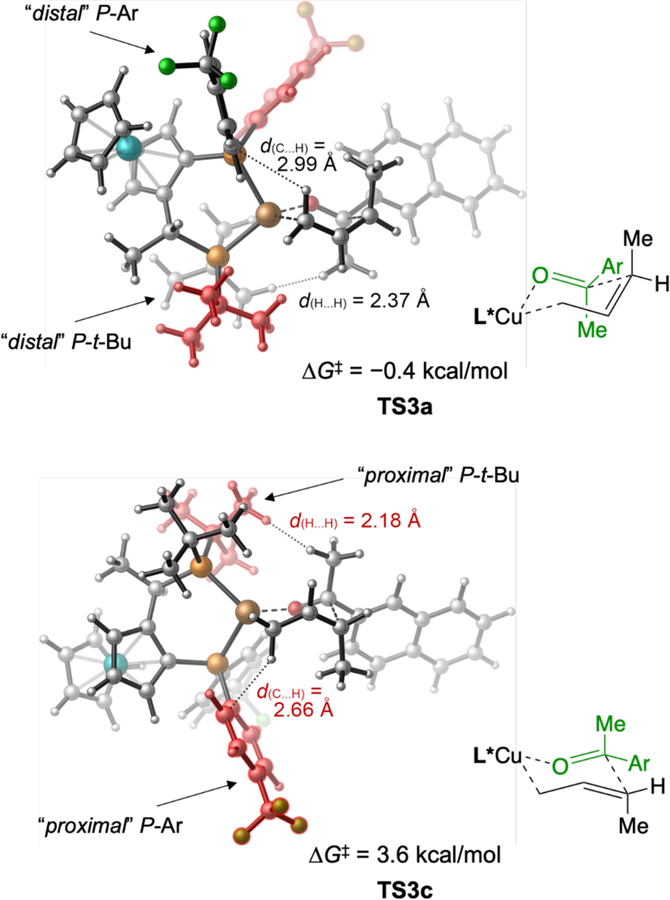
Origin of enantioselectivity.
■ APPLICATIONS
To demonstrate the synthetic utility of this asymmetric transformation, we sought to prepare chiral tertiary alcohol 30, a key intermediate in the synthesis of anticholinergic agents (R)-Oxybutynin,20 (R)-Oxyphenonium bromide21 and (R)-Oxyphencyclimine.22 Currently, these drugs are typically administered in their racemic form, which is synthesized through the addition of a cyclohexyl Grignard reagent to a ketone.23 However, motivated by the decreased side effects24 and higher efficiency22,25 associated with the single enantiomer forms, several groups have developed synthetic routes to enantioenriched key chiral intermediate 30 using chiral auxiliaries,26 chiral pool synthesis,20c an organocatalytic aldol-elimination-hydrogenation-deprotection sequence,20b or palladium-catalyzed asymmetric allylic alkylation followed by functional group interconversions.20d Using our method, we devised an alternative, catalytic, enantioselective synthetic route to this key chiral intermediate (Scheme 1) that yields enantiopure product and does not require the purification of intermediates. From commercially available starting material 28, after CuH-catalyzed coupling with cyclohexadiene, reduction, hydrolysis, and recrystallization, 30 was obtained in 53% overall yield and with over 99.5:0.5 er in a one-pot sequence without the need for chromatography.
Scheme 1.
Synthesis of a Key Tertiary α-Hydroxy Acid Intermediate.
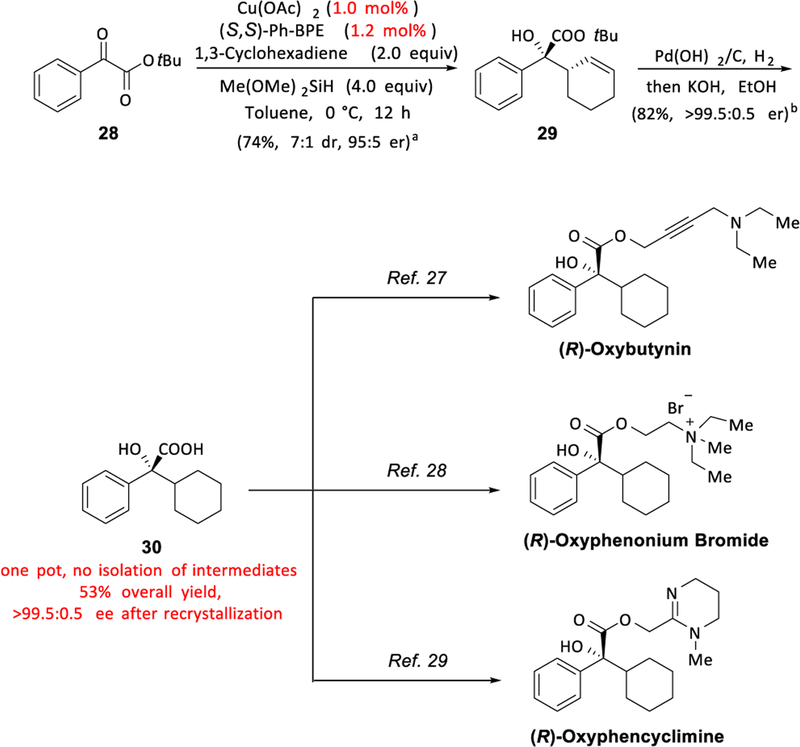
aParenthetical data reflect results of an independent run for which intermediates were isolated, purified, and characterized. See the Supporting Information for details. bAfter recrystallization. See the Supporting Information for details.
We also applied our method toward a new synthetic route to (R)-Procyclidine, a treatment for Parkinson’s disease.30 Biological testing suggests that (R)-Procyclidine has a higher affinity for the relevant muscarinic receptor both in humans and in animal models.22b,30 Thus, an efficient asymmetric synthesis of (R)-Procyclidine would be valuable. We studied the copper-catalyzed allylation of commercially available ketone 31, which provided chiral tertiary alcohol 32. Again, without requiring chromatographic purification, the mixture containing 32 was subjected to simple hydrogenation and direct crystallization to yield (R)-Procyclidine (33) in 58% overall yield and over 99.5:0.5 er.
■ CONCLUSION
In summary, we have developed a highly efficient copper-catalyzed allylation of ketones with feedstock linear and cyclic conjugated dienes. A large variety of chiral tertiary alcohols were prepared in excellent yield, regio-, and enantioselectivity, and with a high level of functional group compatibility. Guided by DFT calculations, a rationale explaining the factors responsible for the enantio- and diastereoselectivity of this transformation was derived. From a mixture of rapidly equilibrating allylcopper intermediates of similar energy, selective reaction of the cis-allyl complex generates the observed diastereomer. Furthermore, a model for the enantioselectivity of the addition of the allylcopper intermediates to ketones was proposed for catalysts bearing the non-C2-symmetric JOSIPHOS ligands. Our method also enabled a new, concise, and enantioselective synthesis of pharmaceutically important drug (R)-Procyclidine and a key intermediate for anticholinergic drugs (R)-Oxyphencyclimine, (R)-Oxybutynin and (R)-Oxyphenonium bromide.
Supplementary Material
Scheme 2.
Synthesis of (R)-Procyclidine.
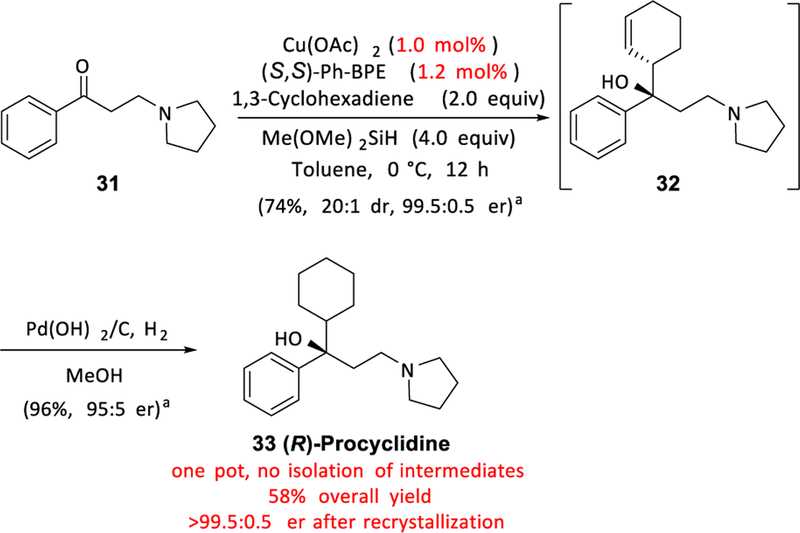
aParenthetical data reflect results of an independent run for which intermediates were isolated, purified, and characterized. See the Supporting Information for details.
ACKNOWLEDGMENT
Research reported in this publication was supported by the National Institutes of Health (GM122483, GM46059, GM058160–17S1, GM128779). The content of this communication solely reflects the research and opinion of the authors and does not necessarily represent the official views of the NIH. Solvias AG is acknowledged for a generous gift of SL-J011–1. R.Y.L. acknowledges Bristol–Myers Squibb for a Fellowship in Synthetic Organic Chemistry. We are grateful to Drs. Scott McCann, Andy Thomas, and Christine Nguyen for advice on the preparation of this manuscript. DFT calculations were performed at the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF.
Footnotes
■ ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.
Experimental procedures and characterization data for all compounds (PDF)
NMR spectra (PDF)
SFC and HPLC traces (PDF)
Computational details and Cartesian coordinates of optimized geometries (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).For selected reviews, see:; (a) Yus M; González-Gómez JC; Foubelo F Diastereoselective Allylation of Carbonyl Compounds and Imines: Application to the Synthesis of Natural Products. Chem. Rev 2013, 113, 5595–5698. [DOI] [PubMed] [Google Scholar]; (b) Ameen D; Snape TJ Chiral 1,1-diaryl compounds as important pharmacophores. MedChemComm 2013, 4, 893–907. [Google Scholar]
- (2).For selected reviews, see:; (a) Yus M; González-Gómez JC; Foubelo F Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev 2011, 111, 7774–7854. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE; Fu J Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev 2003, 103, 2763–2793. [DOI] [PubMed] [Google Scholar]; (c) Pu L; Yu H-B Catalytic Asymmetric Organozinc Additions to Carbonyl Compounds. Chem. Rev 2001, 101, 757–824. [DOI] [PubMed] [Google Scholar]; (d) Riant O; Hannedouche J Asymmetric catalysis for the construction of quaternary carbon centres: nucleophilic addition on ketones and ketimines. Org. Biomol. Chem 2007, 5, 873–888. [DOI] [PubMed] [Google Scholar]; (e) Shibasaki M; Kanai M Asymmetric Synthesis of Tertiary Alcohols and α-Tertiary Amines via Cu-Catalyzed C−C Bond Formation to Ketones and Ketimines. Chem. Rev 2008, 108, 2853–2873. [DOI] [PubMed] [Google Scholar]; (f) Leonori D; Aggarwal VK Lithiation–Borylation Methodology and Its Application in Synthesis. Acc. Chem. Res 2014, 47, 3174–3183. [DOI] [PubMed] [Google Scholar]
- (3).Knochel P; Dohle W; Gommermann N; Kneisel FF; Kopp F; Korn T; Sapountzis I; Vu VA Highly functionalized organomagnesium reagents prepared through halogen-metal exchange. Angew. Chem. Int. Ed 2003, 42, 4302–4320. [DOI] [PubMed] [Google Scholar]
- (4).For selected recent reports of constructing tertiary alcohols with catalytic, asymmetric reactions:; (a) Huang L; Zhu J; Jiao G; Wang Z; Yu X; Deng W; Tang W Highly Enantioselective Rhodium-Catalyzed Addition of Arylboroxines to Simple Aryl Ketones: Efficient Synthesis of Escitalopram. Angew. Chem. Int. Ed 2016, 55, 4527–4531. [DOI] [PubMed] [Google Scholar]; (b) Li K; Shao X; Tseng L; Malcolmson SJ 2-Azadienes as Reagents for Preparing Chiral Amines: Synthesis of 1,2-Amino Tertiary Alcohols by Cu-Catalyzed Enantioselective Reductive Couplings with Ketones. J. Am. Chem. Soc 2018, 140, 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Khan A; Khan S; Khan I; Zhao C; Mao Y; Chen Y; Zhang YJ Enantioselective Construction of Tertiary C-O Bond via Allylic Substitution of Vinylethylene Carbonates with Water and Alcohols. J. Am. Chem. Soc 2017, 139, 10733–10741. [DOI] [PubMed] [Google Scholar]; (d) Brauns M; Mantel M; Schmauck J; Guder M; Breugst M; Pietruszka J Highly Enantioselective Allylation of Ketones: An Efficient Approach to All Stereoisomers of Tertiary Homoallylic Alcohols. Chem. - Eur. J 2017, 23, 12136–12140. [DOI] [PubMed] [Google Scholar]; (e) Robbins DW; Lee K; Silverio DL; Volkov A; Torker S; Hoveyda AH Practical and Broadly Applicable Catalytic Enantioselective Additions of Allyl-B(pin) Compounds to Ketones and α-Ketoesters. Angew. Chem. Int. Ed 2016, 55, 9610–9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Dahlmann M; Grub J; Löser E Butadiene. In Ullmann’s Encyclopedia of Industrial Chemistry Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]; (b) Zbieg JR; Yamaguchi E; McInturff EL; Krische MJ Enantioselective C-H Crotylation of Primary Alcohols via Hydrohydroxyalkylation of Butadiene. Science 2012, 336, 324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Weitz HM; Loser E, Isoprene. In Ullmann’s Encyclopedia of Industrial Chemistry Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- (7).Behr A; Johnen L Myrcene as a Natural Base Chemical in Sustainable Chemistry: A Critical Review. ChemSusChem 2009, 2, 1072–1095. [DOI] [PubMed] [Google Scholar]
- (8).Bareille L; Le Gendre P; Moise C First catalytic allyltitanation reactions. Chem Commun 2005, 775–777. [DOI] [PubMed]
- (9).(a) Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kimura M; Ezoe A; Mori M; Iwata K; Tamaru Y Regio- and Stereoselective Nickel-Catalyzed Homoallylation of Aldehydes with 1,3-Dienes. J. Am. Chem. Soc 2006, 128, 8559–8568. [DOI] [PubMed] [Google Scholar]; (c) Ogoshi S; Tonomori K-I; Oka M-A; Kurosawa H Reversible Carbon−Carbon Bond Formation between 1,3-Dienes and Aldehyde or Ketone on Nickel (0). J. Am. Chem. Soc 2006, 128, 7077–7086. [DOI] [PubMed] [Google Scholar]; (d) Kimura M; Miyachi A; Kojima K; Tanaka S; Tamaru Y Highly Stereo- and Regioselective Ni-Catalyzed Homoallylation of Aldimines with Conjugated Dienes Promoted by Diethylzinc. J. Am. Chem. Soc 2004, 126, 14360–14361. [DOI] [PubMed] [Google Scholar]
- (10).(a) Shibahara F; Bower JF; Krische MJ Ruthenium-Catalyzed C−C Bond Forming Transfer Hydrogenation: Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level Employing Acyclic 1,3-Dienes as Surrogates to Preformed Allyl Metal Reagents. J. Am. Chem. Soc 2008, 130, 6338–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Park BY; Montgomery TP; Garza VJ; Krische MJ Ruthenium Catalyzed Hydrohydroxyalkylation of Isoprene with Heteroaromatic Secondary Alcohols: Isolation and Reversible Formation of the Putative Metallacycle Intermediate. J. Am. Chem. Soc 2013, 135, 16320–16323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jang H-Y; Huddleston RR; Krische MJ A New Catalytic C-C Bond-Forming Hydrogenation: Reductive Coupling of Dienes and Glyoxals under Catalytic Hydrogenation Conditions. Angew. Chem. Int. Ed 2003, 42, 4074–4077. [DOI] [PubMed] [Google Scholar]
- (12).(a) Bower JF; Patman RL; Krische MJ Iridium-Catalyzed C−C Coupling via Transfer Hydrogenation: Carbonyl Addition from the Alcohol or Aldehyde Oxidation Level Employing 1,3-Cyclohexadiene. Org. Lett 2008, 10, 1033–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zbieg JR; Fukuzumi T; Krische MJ Iridium-Catalyzed Hydrohydroxyalkylation of Butadiene: Carbonyl Crotylation. Adv. Synth. Catal 2010, 352, 2416–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For selected reviews, see:; (a) Pirnot MT; Wang Y-M; Buchwald SL Copper Hydride Catalyzed Hydroamination of Alkenes and Alkynes. Angew. Chem. Int. Ed 2015, 55, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mohr J; Oestreich M Balancing C=C Functionalization and C=O Reduction in Cu−H Catalysis. Angew. Chem. Int. Ed 2016, 55, 12148–12149. [DOI] [PubMed] [Google Scholar]; (c) For selected reports, see: Zhu S; Niljianskul N; Buchwald SL Enantio- and regioselective CuH-catalyzed hydroamination of alkenes. J. Am. Chem. Soc 2013, 135, 15746–15749. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yang Y; Shi S-S; Niu D-W; Liu P; Buchwald SL Catalytic asymmetric hydroamination of unactivated internal olefins to aliphatic amines. Science 2015, 349, 62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nishikawa D; Hirano K; Miura M Asymmetric Synthesis of α-Aminoboronic Acid Derivatives by Copper-Catalyzed Enantioselective Hydroamination. J. Am. Chem. Soc 2015, 137, 15620–15623. [DOI] [PubMed] [Google Scholar]; (f) Miki Y; Hirano K; Satoh T; Miura M Copper-Catalyzed Intermolecular Regioselective Hydroamination of Styrenes with Polymethylhydrosiloxane and Hydroxylamines. Angew. Chem. Int. Ed 2013, 52, 10830–10834. [DOI] [PubMed] [Google Scholar]; (g) Xi Y; Butcher TW; Zhang J; Hartwig JF Regioselective, Asymmetric Formal Hydroamination of Unactivated Internal Alkenes. Angew. Chem. Int. Ed 2015, 55, 776–780. [DOI] [PubMed] [Google Scholar]
- (14).(a) Yang Y; Perry IB; Lu G; Liu P; Buchwald SL Copper-catalyzed asymmetric addition of olefin-derived nucleophiles to ketones. Science 2016, 353, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tsai EY; Liu RY; Yang Y; Buchwald SL A Regio- and Enantioselective CuH-Catalyzed Ketone Allylation with Terminal Allenes. J. Am. Chem. Soc 2018, 140, 2007–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For selected reviews, see:; (a) Yamada K-I; Tomioka K Copper-Catalyzed Asymmetric Alkylation of Imines with Dialkylzinc and Related Reactions. Chem. Rev 2008, 108, 2874–2886. [DOI] [PubMed] [Google Scholar]; (b) For selected reports, see: Liu RY; Yang Y; Buchwald SL Regiodivergent and Diastereoselective CuH-Catalyzed Allylation of Imines with Terminal Allenes. Angew. Chem. Int. Ed 2016, 55, 14077–14080. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang Y; Perry IB; Buchwald SL Copper-Catalyzed Enantioselective Addition of Styrene-Derived Nucleophiles to Imines Enabled by Ligand-Controlled Chemoselective Hydrocupration. J. Am. Chem. Soc 2016, 138, 9787–9790. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shao X; Li K; Malcolmson SJ Enantioselective Synthesis of anti-1,2-Diamines by Cu-Catalyzed Reductive Couplings of Azadienes with Aldimines and Ketimines. J. Am. Chem. Soc 2018, 140, 7083–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Colacot TJ A Concise Update on the Applications of Chiral Ferrocenyl Phosphines in Homogeneous Catalysis Leading to Organic Synthesis. Chem. Rev 2003, 103, 3101–3118. [DOI] [PubMed] [Google Scholar]
- (17).Yatsumonji Y; Sugita T; Tsubouchi A; Takeda T Preparation of syn-tertiary homoallylic alcohols utilizing allenyltitanocenes generated by reductive titanation of γ-trimethylsilylpropargylic carbonates. Org. Lett 2010, 12, 1968–1971. [DOI] [PubMed] [Google Scholar]
- (18).(a) Kagan HB; Phat D-T Asymmetric Catalytic Reduction with Transition Metal Complexes. I. Catalytic System of Rhodium(I) with (–)-2,3-O-Isopropylidene2,3-dihydroxy-1,4-bis(diphenylphosphino)butane, a New Chiral Diphosphine. J. Am. Chem. Soc 1972, 94, 6429–6433. [Google Scholar]; (b) Kagan HB In Asymmetric Catalysis; Morrison JD, Ed.; Academic Press: New York, 1985; Vol. 5, pp 1–339. [Google Scholar]; (c) Whitesell JK C2-Symmetry and Asymmetric Induction. Chem. Rev 1989, 89, 1581–1615. [Google Scholar]; (d) Walsh P; Kowzlowski M Fundamentals of Asymmetric Catalysis University Science Books: Sausalito, CA: (2008). [Google Scholar]; (d) Kobayashi K; Yamamoto Y; Miyaura N Pd/Josiphos-Catalyzed Enantioselective alpha-Arylation of Silyl Ketene Acetals and Mechanistic Studies on Transmetalation and Enantioselection. Organometallics 2011, 30, 6323–6327. [Google Scholar]
- (19).(a) Grayson MN; Krische MJ; Houk KN Ruthenium-Catalyzed Asymmetric Hydrohydroxyalkylation of Butadiene: The Role of the Formyl Hydrogen Bond in Stereochemical Control. J. Am. Chem. Soc 2015, 137, 8838–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mejuch T; Gilboa N; Gayon E; Wang H; Houk KN; Marek I Axial Preferences in Allylation Reactions via the Zimmerman–Traxler Transition State Acc. Chem. Res 2013, 46, 1659–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Chapple CR Muscarinic receptor antagonists in the treatment of overactive bladder. Urology 2000, 55, 33–46. [DOI] [PubMed] [Google Scholar]; (b) Tokuda O; Kano T; Gao W-G; Ikemoto T; Maruoka K A Practical Synthesis of (S)-2-Cyclohexyl-2-phenylglycolic Acid via Organocatalytic Asymmetric Construction of a Tetrasubstituted Carbon Center. Org. Lett 2005, 7, 5103–5105. [DOI] [PubMed] [Google Scholar]; (c) Roy S; Sharma A; Chattopadhyay N; Chattopadhyay S An efficient asymmetric synthesis of (S)-2-cyclohexyl-2-phenylglycolic acid, the acid segment of oxybutynin. Tet. Lett 2006, 47, 7067–7069. [Google Scholar]; (d) Trost BM; Xu J; Reichle M Enantioselective Synthesis of α-Tertiary Hydroxyaldehydes by Palladium-Catalyzed Asymmetric Allylic Alkylation of Enolates. J. Am. Chem. Soc 2007, 129, 282–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Hassan WS; Elazazy MS; Elmasry MS Spectroscopic and conductometric assay of oxyphenonium bromide in pure form and in pharmaceuticals. Anal. Bioanal. Electrochem 2014, 6, 28–42. [Google Scholar]; (b) Inhalation preparation for treating asthma Chinese Patent CN102961366A, 2013.
- (22).(a) Schjelderup L; Kozlowski MR; Weissman A; Aasen AJ Antimuscarinic effects of (R)- and (S)-oxyphencyclimine hydrochloride. Pharm. Res 1988, 5, 236–237. [DOI] [PubMed] [Google Scholar]; (b) Waelbroeck M; Camus J; Tastenoy M; Mutschler E; Strohmann C; Tacke R; Schjelderup L; Aasen A; Lambrecht G; Christophe J Stereoselective interaction of procyclidine, hexahydrodifenidol, hexbutinol and oxyphencyclimine, and of related antagonists, with four muscarinic receptors. Eur. J. Pharmacol., Mol. Pharmacol. Sect 1992, 227, 33–42. [DOI] [PubMed] [Google Scholar]
- (23).Ikemoto T; Gao W-G; Takeda M; Igi M Production method of 2-cyclohexyl- 2-hydroxy-2-phenylacetic acid intermediate therefor and production method thereof. US Patent US2003013911 (A1), January 16, 2013.
- (24).Thompson IM; Lauvetz R Oxybutynin in bladder spasm, neurogenic bladder, and enuresis. Urology 1976, 8, 452–454. [DOI] [PubMed] [Google Scholar]
- (25).(a) Feitsma KG; Postma DS; Koeter GH; Nossent GD; Brenth BF; De ZRA Comparative study of the bronchodilating effects of (−)- and (+)-oxyphenonium bromide. Br. J. Clin. Pharmacol 1988, 25, 683–687. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Feitsma KG Enantiomers of oxyphenonium bromide. Analytical and pharmacological aspects. Pharm. Weekbl. Sci 1988, 10, 221–223. [DOI] [PubMed] [Google Scholar]
- (26).(a) Senanayake CH; Fang K; Grover P; Bakale RP; Vandenbossche CP; Wald SA Rigid aminoalcohol backbone as a highly defined chiral template for the preparation of optically active tertiary α-hydroxyl acids. Tetrahedron Lett 1999, 40, 819–822. [Google Scholar]; (b) Grover PT; Bhongle NN; Wald SA; Senanayake CH Chiral Mandelic Acid Template Provides a Highly Practical Solution for (S)-Oxybutynin Synthesis. J. Org. Chem 2000, 65, 6283–6287. [DOI] [PubMed] [Google Scholar]
- (27).Vandenbossche CP; de Croos P; Singh SP; Bakale RP; Wagler TR Formation of (S)-5-Cyclohexyl-5-phenyl-1,3-dioxolane-2,4-dione: A Key Intermediate in the Synthesis of (S)-Oxybutynin Hydrochloride. Org. Process Res. Dev 2010, 14, 921–925. [Google Scholar]
- (28).Vlek JW; Feitsma KG; van der Mark TW; Drenth BFH; Paans AMJ; Vaalburg W Synthesis of d-[11C]oxyphenonium iodide, a potential radioligand for in vivo visualization of human cholinergic muscarinic receptor-sites by positron emission tomography. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1990, 41, 453–456. [DOI] [PubMed] [Google Scholar]
- (29).Schjelderup L; Aasen AJ The absolute configuration of oxyphencyclimine, a parasympatholytic drug. Syntheses of both enantiomers. Acta Chem. Scand., Ser. B 1986, B40, 601–603. [Google Scholar]
- (30).(a) Jevtovic-Todorovic V; Meyenburg AP; Olney JW; Wozniak DF Anti-parkinsonian agents procyclidine and ethopropazine alleviate thermal hyperalgesia in neuropathic rats. Neuropharmacology 2003, 44, 739–748. [DOI] [PubMed] [Google Scholar]; (b) Gao ZG; Liu BY; Cui WY; Li LJ; Fan QH; Liu CG Anti-nicotinic properties of anticholinergic antiparkinson drugs. J. Pharm. Pharmacol 1998, 50, 1299–1305. [DOI] [PubMed] [Google Scholar]; (c) Waelbroeck M; Camus J; Tastenoy M; Lambrecht G; Mutschler E; Tacke R; Christophe J Stereoselectivity of procyclidine binding to muscarinic receptor subtypes M1, M2 and M4. Eur. J. Pharmacol., Mol. Pharmacol. Sect 1990, 189, 135–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.