

Abstract
Fungi are ubiquitous in our environment, and a healthy immune system is essential to maintain adequate protection from fungal infections. When this protection breaks down, superficial and invasive fungal infections cause diseases that range from irritating to life-threatening. Millions of people worldwide will develop invasive infections during their lives, and mortality for these infections often exceeds 50%. Nevertheless, we are normally colonized with many of the same disease-causing fungi (e.g. on the skin or in the gut). Recent research is dramatically expanding our understanding of the mechanisms by which our immune systems interact with these organisms in health and disease. In this review we will discuss what is currently known about where and how the immune system interacts with common fungi.
Keywords: Candida, Malassezia, Aspergillus, microbiome, Dectin-1, innate immunity
Introduction
Fungi are ubiquitous in the environment, being found on every continent, in the deep sea, in the air, and arctic soil. Mammals have evolved in the presence of diverse fungi, and our immune systems are adapted to their presence. We have innate pattern recognition receptors in our genomes that provide defense against fungal infection (1, 2). We make natural (generated without the requirement for exogenous antigenic stimulation) antibodies that recognize fungi (3). We have harvested fungi for food, fuel, and medicines. Curiously, studies on the microbiota associated with our bodies in health and disease have focused largely on bacteria. Recently, investigators are becoming aware that fungi are a normal part of the microbiota associated with our bodies, be it on the oily skin or the anaerobic gut (4, 5). Interactions between the immune system and these fungi (the “mycobiome”) are being recognized as important for immune homeostasis and for pathology during disease. In this review, we will focus our discussion on host interactions with the fungi that are commonly and naturally part of our microbiota, be it in the context of health or disease. For discussion of immune responses to more overtly pathogenic fungi such as Cryptococcus, Histoplasma, Coccidioides or Mucorales, we direct the reader to other excellent reviews (6–9).
Who are commensal fungi?
The types of fungi that are reported to be associated with the body depend on the site and the methods of detection, although it is clear that representatives of the major fungal phyla, Ascomycota and Basidiomycota, are widely present (Figure 1). Early studies relied on culturing fungi from various anatomical sites, but such methods made little progress in really characterizing the net complexity of commensal fungal communities. DNA-based methods have enabled culture-independent detection and identification of fungi. Approaches such as restriction fragment length polymorphism (RFLP) analysis, oligonucleotide fingerprinting of rRNA genes (OFRG), and denaturing gradient gel electrophoresis (DGGE), were good for revealing more complexity than culturing methods, but were relatively poor at identifying specific fungi. PCR-based methods can be highly specific, but are restricted in what they can identify. High-throughput sequencing technologies can be more comprehensive, but can be challenged by efficiencies of DNA purification and amplification as well as bioinformatics and taxonomical challenges associated with identifying DNA fragments (10).
Figure 1. Distribution of common commensal fungi.

Fungal representation at various sites and sources are indicated. Relative prevalence across multiple studies at each site are represented by the size of the fungal genera name.
The most common targets sequenced for identifying fungi are the internal transcribed spacer regions (ITS) of ribosomal RNA genes. The first culture-independent ITS sequencing-based study to evaluate the oral mycobiome identified >80 genera of fungi, and found that Candida and Cladosporium were most abundantly detected in healthy individuals (11). Another analysis of these data identified Malassezia and Epicoccum to be more abundant (12) and highlights the situation that tools and approaches for fungal rDNA sequence analysis are still being developed and validated.
Fungi are thought to be normal commensals of the mammalian intestines. The mouse gut is home to several common fungi including Candida, Saccharomyces and Cladosporium species, and >50 less commonly detected genera (13, 14). Fungal populations in the mouse gut have been observed to be variable over several months, whereas the intestinal bacterial community remains relatively stable (13). In humans, sequencing approaches commonly detect Saccharomyces and Candida, with other related yeasts as well as some molds including Penicillium being commonly detected (15–17).
Comparing the amount of fungi to, for example, bacteria has proven challenging as it is often difficult to determine how to normalize data appropriately. 16S/ITS sequencing efforts are difficult to compare directly, especially since different types of bacteria and fungi respectively have widely different numbers of repeats of these genomic areas. Metagenomic approaches relying on assembling and identifying genomic DNA from all components of a sample may do better; in the gut, for example, shotgun sequencing efforts have suggested that fungi make up approximately 0.1% of the microorganisms (18, 19). However, fungi are much larger than bacteria, DNA recovery efficiencies are likely very different, and many fungal reference genomes are yet to be produced.
While a comprehensive catalogue of all types of fungi making up the human mycobiota would be enormous, below is a short description of some of the major types of fungi that are currently being reported:
Saccharomycetaceae
Saccharomycetaceae is a family of Ascomyctes that includes common yeasts such as Saccharomyces and Candida. Saccharomyces cerevisiae, baker’s yeast, is found in many foods and is commonly detected in diverse sites of the body. Candida species including C. albicans, C. tropicalis, and C. dubliniensis are dimorphic, meaning that they are yeasts that can also grow in filamentous form (20). These Candida spp. can cause minor skin and mucosal infections in healthy individuals and can cause life-threatening mucosal and blood infections in immunocompromised people. Taxonomically, Candida is a complicated genus with many organisms named Candida not being particularly closely related to these pathogenic organisms. For example, Candida glabrata, a commonly-occurring disease-causing commensal yeast, is more closely related to S. cerevisiae than to C. albicans, is not dimorphic, and is more accurately classified as a Nakaseomyces (21, 22). Other Saccharomycetaceae commonly found in studies of commensal fungi in this group include Pichia, Kazachstania, Debaryomyces, Kluyveromyces and Meyerozyma.
Aspergillus
Aspergillus is a genus of common molds found world-wide that are both harmful and beneficial to people (23). For example, A. niger is used commercially to produce citric acid and A. oryzae is used to ferment starches such as in the production of Sake. In contrast, A. flavus is a plant and animal pathogen that produces the potent carcinogen aflatoxin, and A. fumigatus is a medically important opportunistic pathogen that commonly causes lung infections in immunocompromised patients. A. fumigatus’ natural ecological niche is soil; it grows as a filamentous mass and releases ubiquitous spores into the air. Environmental studies estimate that humans commonly inhale at least several hundred A. fumigatus spores per day (23). These spores are small enough (2-3 μm) to reach the alveoli of the lung. Aspergillus are also major cause of blinding corneal ulcers, and in contrast to most other common fungal infections, occur in immunocompetent individuals (24). Like many other common fungi, nomenclature of Aspergillus spp. can be complicated by the existance of sexual and asexual forms of the organisms that have historically been assigned different names. There are at least eight different teleomorph (sexual form) genera associated with Aspergillus (anamorph, asexual form) including Emericella and Eurotium (25). Various Aspergillus species are commonly detected in microbiota studies of the lung and gut, but as is the case with many of the fungi discussed here, whether these are truly commensal or are simply representative of the enviroment is difficult to establish.
Fusarium
Fusarium is a genus of filamentous mold found ubiquitously in soil, air, and on plants. Many Fusarium species possess the ability to produce mycotoxins and may cause mycotoxicosis following ingestion of food that has been colonized. Fusarium spp. may cause local skin infections that may become invasive or disseminated in immunocompromised individuals (26). Like Aspergillus, Fusarium are also a major cause of blinding corneal ulcers in immunocompetent individuals (24). Commonly detected commensal and infection-causing Fusarium include F. solani, F. oxysporum, and F. moniliforme.
Cladosporium
Cladosporium is another genus of filamentous, spore-forming black molds that are ubiquitous in the soil and on live or dead plant material and are among the most common fungi isolated from air (27). There are over 700 described species, many of which are plant pathogens. Unlike Fusarium, Cladosporium spp. do not produce toxins and are rarely pathogenic in humans, although they have been occasionally found associated with infections of the skin, eyes, brain, sinuses and lung (28). They are also commonly associated with allergies (29).
Malassezia
Fungi of the genus Malassezia are lipid-dependent and lipophilic yeast species that are part of the normal skin microbiota. There are 14 described species, and although most well-known as skin commensals, they are widely distributed, being found at deep sea vents, Antarctic soils, and plant roots (30). Malassezia can be involved in skin disorders including pityriasis versicolor, seborrheic dermatitis, atopic eczema, folliculitis, and dandruff. Rare invasive infections can occur, primarily in neonates or in immunocompromised patients being given intravenous lipid-rich nutrition (31). The three sequenced genomes, M. globosa, M. restricta, and M. sympodialis, reveal less than 5,000 genes (among the smallest of free-living fungi), and a key feature of these genomes are their incomplete fatty acids synthesis metabolic pathways. The organisms rely on secreted lipases, phospholipases, and acid sphingomyelinases to process extracellular lipids. This reliance on often diverse fatty acids for growth can make some Malassezia a challenge to culture (30).
Immune homeostasis with fungi
The microorganisms that occupy mucosal surfaces of the body are critical for appropriately tuning immune responses to ensure efficient responses to pathogens while limiting responses directed towards host tissues and innocuous agents such as allergens (32). Research over the last several decades has provided compelling evidence supporting a role for bacteria in maintaining immune homeostasis, however it is now becoming increasingly clear that the host immune system is also in a dynamic relationship with fungal populations that reside on mucosal surfaces, and disturbance of this relationship can also have serious immune and non-immune consequences for the host (5). Below we will detail what is currently known about the role commensal fungal communities play in modulating immune function.
Probiotic fungi
One of the first indications that fungi could play a role in modulating immune homeostasis comes from the long history of using the yeast Saccharomyces boulardii as a probiotic to alleviate diarrhea and prevent intestinal colonization with Clostridium difficile following broad-spectrum antibiotic therapy (33, 34). S. boulardii was originally isolated by French microbiologist Henri Boulard after a visit to Southeast Asia in the early 1900’s where he observed individuals drinking tea made from skins of tropical lychee and mangosteen fruits to alleviate symptoms of cholera. He isolated the heat-resistant yeast and named it after himself. It is now generally described as a subspecies of S. cerevisiae (Saccharomyces cerevisiae var. boulardii) (35).
S. boulardii is thought to mediate its effects through direct interaction with the microbiota as well as via mechanisms that engage the immune system. The yeast secretes proteases and phosphatases that inactivate toxins produced by intestinal pathogens such as C. difficile and E. coli (36, 37), and can also directly inhibit the growth and invasion of a number of intestinal pathogens including C. albicans, Salmonella typhimurium, and Yersinia enterocolitica (38, 39). More recent work has shown that S. boulardii has immunomodulatory functions that boost immunity to intestinal pathogens and limit pathological inflammation in diseases such as ulcerative colitis, Crohn’s disease, and C. difficile colitis. S. boulardii boosts total intestinal IgA production as well as IgA specific for C. difficile toxin A, although how is still unclear (40). Supernatants from S. boulardii cultures can directly inhibit Toll-like receptor (TLR)-induced inflammatory responses in myeloid dendritic cells isolated from healthy and Crohn’s disease patients (41). S. boulardii supplementation has been documented to have efficacy in maintenance of Crohn’s disease remission (42, 43), as well as for symptomatic improvement in a subset of ulcerative colitis patients (44), which is consistent with the protective effects of S. boulardii administration in experimental mouse models of colitis (45–47). Together, this suggests that the organism likely exerts both pro- and anti-inflammatory effects (Figure 2A).
Figure 2. Interaction of commensal fungi with the mucosal immune system during homeostasis and disseminated fungal infection.

Top panel depicts normal homeostatic conditions. (A) Probiotic Saccharomyces boosts intestinal IgA, directly inhibits intestinal pathogen infection, and also limits pathological inflammatory responses. (B) Commensal Saccharomyces induces monocyte training that limits pathological Candida infection. (C) Commensal Candida being sampled by Langerin+ intestinal dendritic cells and RALDH+ dendritic cells traffic to the peripheral lymph nodes (pLN) in response to commensal Candida to induce lymphocyte homing molecules on pLN high endothelial venules (HEV). (D) Recognition of Candida by intestinal dendritic cells induces a tolerogenic phenotype characterized by IDO expression and induction of Candida-specific regulatory T cells. Bottom panel depicts situation where antibiotic treatment-induced dysbiosis combined with immunosuppression leads to invasive candidiasis. (E) Antibiotic-mediated disruption of commensal bacteria promotes intestinal overgrowth of commensal Candida. (F) Mucosal disruption (for example following chemotherapy-induced mucositis) allows for bloodstream invasion by intestinal commensal Candida and dissemination to peripheral organs. Candida activates complement in the bloodstream which promotes neutrophil and monocyte phagocytosis and activation. (G) DC-derived IL-23p19 activates NK cells to produce GM-CSF needed for neutrophil antimicrobial activity. Neutropenia as well as polymorphisms which impair PMN and monocyte function promote full development of invasive candidiasis.
It will be important to determine if supplementation with probiotic fungi has systemic effects on the immune system as has been demonstrated with numerous members of the bacterial community. Along these lines, a recent study demonstrated that oral administration of the dietary yeast Candida kefyr was not only protective in experimental colitis, but also protected mice from development of experimental autoimmune encephalomyelitis (EAE) (48). This was accompanied by alterations in the bacterial microflora, an increase in mesenteric lymph node regulatory T cells and reduction in lamina propria Th17 cells. Candida kefyr (also known as Candida pseudotropicalis, Kluyveromyces marxianus) is one of those Candida spp. discussed above that is not particularly closely related to C. albicans and other pathogenic Candida.
Immune training by commensal fungi
Although S. boulardii is not a natural resident of the intestinal microflora, other strains of S. cerevesiae are common residents of human skin and gastrointestinal tract (49, 50). Recent work has suggested that the presence of non-pathogenic S. cerevisiae as a commensal (51), and potentially C. albicans (52), might have the ability to “train” the immune system to mount more efficient responses against pathogens. The cell wall of S. cerevisiae, similar to many fungi, consists of an outer layer of mannoproteins that encapsulate a network of β−1,3 and 1,6-glucans, and chitin. These polysaccharides are among targets for innate immune receptors that activate pro- and anti-inflammatory responses (Table 1). Cavalieri and co-workers observed that pre-exposure of human monocytes to particular strains of S. cerevisiae promoted more efficient pro-inflammatory cytokine production (IL-6 and TNF-α) and intracellular killing of bacteria and fungi upon secondary stimulation with microbes (51). This state of “trained immunity” was due to differing amounts of chitin in the yeast strains. Pre-exposure of naïve mice to purified preparations of S. cerevisiae chitin induced efficient protection against a normally lethal systemic infection with C. albicans (Figure 2B). Interestingly, the authors also demonstrated that clinical isolates of S. cerevisiae from stools of healthy and Crohn’s disease patients had significantly elevated levels of chitin compared to laboratory and environmentally isolated strains.
Table 1.
Innate immune receptors recognizing fungi
| Receptor | Predominant immune cell expressiona | Fungal Ligands | Genetic Evidenceb | References |
|---|---|---|---|---|
| Dectin-1 | DCs (diverse populations), Langerhans cells, macrophages, monocytes, neutrophils, mast cells | β-glucan |
C. albicans A. fumigatus P. carnii |
(152–155) |
| Dectin-2 | DCs (lung), Langerhans cells, macrophages, monocytes, neutrophils, γδ T cells | α-mannan |
C. albicans C. glabrata C. neoformans |
(152, 156–158) |
| Dectin-3 (MCL) | Langerhans cells, macrophages, monocytes, neutrophils | α-mannan | C. albicans | (152, 158) |
| Mincle | DCs (lung and adipose tissue), Langerhans cells, macrophages, monocytes, neutrophils, eosinophils, basophils | glyceroglycolipid & mannosyl fatty acids |
C. albicans M. pachydermatis F. pedrosoi |
(116, 152, 159, 160) |
| DC-SIGN | DCs (diverse populations), Langerhans cells, plasmacytoid DCs, macrophages, monocytes | mannan | A. fumigatus | (152, 161, 162) |
| Langerin | DCs (lung and thymus), Langerhans cells, liver macrophages | mannose, fucose, N-acetyl-glucosamine, β-glucan, mannan | N/A | (162) |
| Mannose receptor | DCs (lung, adipose, liver), macrophages, monocytes, γδ T cells | N-mannan | C. neoformans | (152, 163) |
| TLR1/2, 2/6 | Diverse cell types including myeloid immune cells and T and B lymphocytes | phospho-lipomannan (PLM)/glucuronoxylomannan (GXM) |
C. albicans A. fumigatus |
(152, 164–167) |
| TLR4 | Diverse cell types including myeloid immune cells and B cells | O-mannan |
C. albicans A. fumigatus |
(152, 164, 167, 168) |
| Galectin-3 | DCs (diverse populations), Langerhans cells, macrophages, monocytes, neutrophils, activated T cells, γδ T cells, basophils, mast cells, eosinophils | β-mannosides | C. albicans | (152, 169, 170) |
| CD36 | DCs (diverse populations), macrophages, monocytes | β-glucan | C. neoformans | (152, 171) |
| Complement receptor 3 (CR3) | DCs (diverse populations), Langerhans cells, macrophages, monocytes, neutrophils, eosinophils, basophils, NK cells, activated T cells, peritoneal B cells | β-glucan | C. albicans | (80, 152, 172) |
| Mannose-binding lectin (MBL) | Hepatocytes (serum factor) | mannan |
C. albicans A. fumigatus |
(152, 173, 174) |
| Pentraxin-3 | DCs, macrophages, neutrophils (secreted factor) | galactomannan | A. fumigatus | (175–177) |
mRNA expression (mouse) according to ImmGen database (normalized expression cutoff ≥200)
Fungal infections (in vivo evidence) associated with genetic deficiency/mutation in mice or humans
It is intriguing to speculate that the elevated chitin content of commensal S. cerevisiae is important for immune system training, or that strains of S. cerevisiae with elevated chitin content have been selected as a part of the normal intestinal microflora for their beneficial immunomodulatory properties. Another study showed that chitin isolated from Candida albicans can dampen inflammation through the preferential induction of IL-10 by macrophages (53). Whether this anti-inflammatory property of chitin allows for evasion of the immune response or serves as a mechanism to promote commensalism of normal members of the fungal mycobiota remains to be determined. Furthermore, it will be important to determine if chitin recognition by the immune system plays a role in the gastrointestinal protective effects associated with S. boulardii supplementation.
Regulation of lymphocyte recirculation by commensal fungi
The above studies provide evidence that exogenous administration of known species of commensal fungi can modulate immune function, however there is currently limited evidence exploring the idea that indigenous populations of commensal fungi regulate the immune system under normal conditions. One important recent study reported that a unique subset of RALDH+ (retinol dehydrogenase enzyme; necessary for retinoic acid production) dendritic cells is recruited from the intestinal lamina propria to the mesenteric and peripheral lymph nodes (pLNs) shortly after birth, and that their arrival is dependent on the presence of commensal fungi (54) (Figure 2C). These dendritic cells were shown to be required for induction of lymphocyte adhesion molecules MAdCAM-1 and PNAd, necessary for entry of recirculating lymphocytes into lymph nodes. These dendritic cells are not recruited to lymph nodes in germ-free mice, and this causes the abnormal structure and reduced cellularity of secondary lymphoid organs that is observed when mice lack microbial colonization (55). The authors observed that treatment of specific pathogen-free (SPF) mice with antifungal drugs resulted in significant reduction in this dendritic cell population and that exogenous administration of a known mouse commensal fungi, Candida tropicalis (13, 14), was sufficient to increase this population in pLNs. Together the data support the conclusion that the normal presence of commensal fungi has a significant role in maintaining immune homeostasis. Whether these dendritic cells are directly interacting with commensal fungi to promote pLN recruitment is currently unclear, however it was recently shown that a population of Peyer’s patch-associated Langerin-positive dendritic cells in the small intestine are able to sample orally administered C. albicans and C. tropicalis (56) (Figure 2C). It will be interesting to determine whether RALDH+ dendritic cells directly contact commensal fungi to promote their subsequent recruitment to the periphery or if they indirectly facilitate this process.
In related work, Wheeler et al. observed that oral treatment of mice with antifungal drugs resulted in increased disease severity in acute and chronic models of colitis, and exacerbated the development of allergic airway disease (57). Microbiota profiling revealed that the drugs induced restructuring of fungal communities, characterized by reduced representation of Candida spp. and a commensurate increase in Aspergillus, Wallemia, and Epicoccum spp. Furthermore, oral supplementation with a mixture of three fungi found to expand during oral antifungal treatment (Aspergillus amstelodami, Epicoccum nigrum, and Wallemia sebi) was sufficient to recapitulate the exacerbating effects of antifungal drugs on allergic airway disease. Interestingly, these fungi did not induce the changes necessary to recapitulate the increase in disease severity in the acute model of colitis. Together these results indicate that disruption of commensal fungal populations can influence local and peripheral immune responses, although whether a “healthy” commensal fungal population is necessary to balance or modulate bacterial populations or is directly necessary to interact with immune cells (or both) is not yet clear.
Immune tolerance induction by commensal fungi
The interaction between commensal and/or environmental microorganisms and the immune system in normal healthy individuals is thought to promote a “peaceful coexistence” between host and microbe. It is therefore logical to suppose that some commensal fungi would evolve mechanisms to promote their tolerance by the host immune system in order to establish colonization. In the case of Candida, investigators have noted that peripheral blood from healthy human donors contains an expanded population of regulatory T cells specific for the commensal C. albicans, which outnumber C. albicans-specific T effector cells by 5-10 fold (58). Recognition of C. albicans by mouse intestinal Peyer’s patch dendritic cells induces a tolerogenic phenotype in the dendritic cells that is dependent on fungal induction of 2,3 indoleamine dioxygenase (IDO) (59). IDO expression by tolerogenic dendritic cells is important for metabolizing the amino acid tryptophan into substrates that activate the aryl-hydrocarbon receptor (AhR) in T cells to promote regulatory T cell generation (60, 61) (Figure 2D). Further, investigators observed that C. albicans-induced tolerogenic dendritic cells were protective during experimental colitis in mice (59).
Sensing of immune status by commensal fungi
While we typically think of commensal microbe-immune interactions in terms of immune cells responding to microbial stimuli, there is also evidence that commensal fungi respond to the immune status of the host and that this is an important determinant of commensalism versus pathological fungal invasion. During the course of intestinal colonization, C. albicans modulate expression of a transcription factor (Efg1p) that normally regulates expression of a number of cell wall components as well as genes important for yeast-hyphal transition . Upon colonization of the intestines in wild type mice, C. albicans mRNA levels of EFG1 were higher than in C. albicans colonizing T cell-deficient nude mice, suggesting that immunological factors influence the expression of EFG1 (62, 63). Yeast lacking EFG1 could colonize wild type mice initially, but are rapidly lost, suggesting that upregulation of EFG1 is important for adaptation to an immunocompetent environment.
While it is not yet clear how T cell-derived factor(s) influence commensal mycobiota, at least one report suggests that mammalian IL-17a binds directly to C. albicans and promotes fungal adhesion, aggregation, and filamentation in vitro (64). Furthermore, freshly isolated vaginal C. albicans stain positively for IL-17a indicating normal binding in vivo, and pre-exposure to IL-17a ex vivo makes C. albicans more virulent in mouse models of orogastric as well as oropharengeal candidiasis. These findings provide evidence that C. albicans, and potentially other commensal fungi, have evolved to sense host immunity and respond in ways that help ensure their persistence at mucosal surfaces.
Commensal fungi as sources of disease
When the healthy balance between the immune system and commensal fungi breaks down, fungi can become pathogenic. While superficial fungal infections (e.g. or the skin or nails) are extremely common and tend to resolve naturally or with minimal intervention, invasive fungal diseases, especially common in immunocompromised individuals, are hard to treat and can have greater than 50% mortality even in the presence of modern care and antifungal drugs. Worldwide, Candida and Aspergillus are responsible for more than 800,000 life-threatening infections every year (65). Many fungal species found as part of the normal mammalian microbiome can be associated with diseases; however, establishing whether the pathogenic fungi are truly derived from commensal strains is often difficult to establish.
Overgrowth of Candida is associated with skin infections, vaginal infections, oropharyngeal infection, gastrointestinal, and blood infections. After some common bacteria, Candida are the fourth most frequent cause of hospital-acquired blood infections (66). In one recent prospective study of over 100 patients admitted to an intensive care unit, investigators noted a strong association between commensal Candida strains (identified by microsatellite profiling) and strains eventually causing invasive candidiasis (67). Similar previous studies have noted that disease causing strains match commensal strains carried by patients (68–70). Invasive candidiasis is associated with the presence of catheters and recent surgery, procedures involving breaching of the barrier between blood and Candida-colonized surfaces, as well as administration of broad-spectrum antibiotic therapy, which allows for overgrowth of commensal fungi (71).
Although Candida are a normal part of the vaginal microbiome, 5–8% of adult women will experience recurrent vulvovaginal candidiasis (72). The recurrent nature of such infections strongly suggests that the commensal strains cause disease, not consistent re-introduction of foreign strains. Further, vulvovaginitis due to Candida is often associated with use of antibiotics which deplete competing bacteria and presumably allows overgrowth of commensal organisms.
As noted above, Malassezia is a normal resident of the skin and has been implicated in diverse skin disorders (31). Requiring lipids for growth, Malassezia are more associated with oily areas of the skin (Figure 3A). While it is presumed that Malassezia strains found associated with skin disease are derived from the normal commensal strains, few studies have actually tried to establish this. Numerous studies have failed to associate specific species with disease; there is substantial geographic variability in which species are associated with human skin, but diseases seem to be associated with increased prevalence or density of these same species (31). In a recent study of patients with seborrheic dermatitis, investigators established using PCR-based detection that Malassezia restricta and Malassezia globosa are more prevalent in patients with disease compared to controls, although they are found commonly on skin of healthy people (73). More careful sequencing-based analysis of rRNA gene regions revealed that specific strains of the yeasts associated with disease were distinct from common healthy strains, suggesting that disease might be associated with colonization by new strains. Still, whether disease is associated with Malassezia growth/colonization or caused by it remains unclear.
Figure 3. Interactions between fungi and the immune system of the skin.
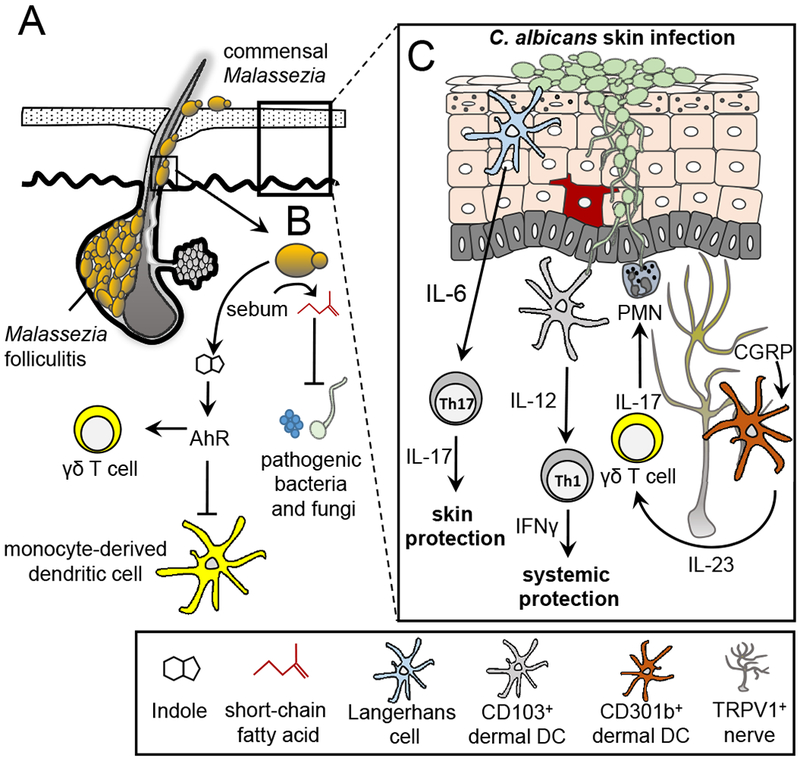
(A) Malassezia species are associated with areas of the skin where sebaceous secretions are present. These fungi utilize long chain fatty acids such as sebum as required carbon sources. Malassezia yeasts have been associated with dermatologic conditions such as Malassezia folliculitis. Normally confined to the infundibulum region of the skin, Malassezia yeasts can invade into the hair follicle causing damage and inflammation. (B) Metabolic byproducts of Malassezia yeasts are important in skin homeostasis. Short chain fatty acids produced from the metabolic breakdown of sebum have dual anti-bacterial and anti-mycotic properties. Potent Ahr indolic ligands produced by Malassezia can inhibit phagocyte responses to TLR stimulation and contribute to cutaneous invariant γδ T cell homeostasis in the skin. (C) Immune responses to Candida albicans skin infection are compartmentalized. In the epidermis, Dectin-1 engagement on LC by C. albicans yeasts stimulates IL-6 production leading to the induction of Th17 CD4+ T cell differentiation and skin immunity. CD103+ dermal DCs detect hyphae C. albicans and produce IL-12 thus priming Th1 CD4+ T cell differentiation and providing systemic protection. Skin TRPV1+ nociceptive nerves are able to detect C. albicans and respond by secreting CGRP which acts on nearby CD301b+ dermal DCs to produce IL-23. γδ T cells respond to IL-23 stimulation by producing IL-17 which activates neutrophils and stimulates anti-microbial peptide production by keratinocytes.
Association of Malassezia with disease is not restricted to the skin. Systemic disease is a consequence of catheter-associated fungal growth that typically develops in catheters used to deliver lipid-rich emulsions as in neonates receiving parenteral nutrition (74). While it is tempting to postulate that the disease-causing fungi originate in the skin, such blood stream infections seem to be largely restricted to Malassezia furfur, a species that is relatively rare on the skin (50, 75). Using analysis of rRNA gene regions, Kaneko et al. noted that M. furfur strains found on the skin around the infected catheter entrance sites matched the strains isolated from blood (74). However, whether this really means that the infection originated from the skin is not clear.
Immunity to fungal infections in the blood
Systemic candidiasis is the most common type of fungal bloodstream infection documented in the developed world and is thought to originate commonly from translocation of gut fungi into the blood (71). More than a dozen species of Candida have been documented to cause bloodstream infections in humans of which 95% are caused by C. albicans, C. glabrata, C. paripsilolis, C. tropicalis, and C. krusei (76). The mechanism through which Candida disseminates from the intestines to the blood is thought to require three initiating events involving 1) intestinal colonization with Candida 2) defects in phagocytic cell function and/or numbers, and 3) intestinal barrier disruption (77) (Figure 2E–G). One of the most common examples where this occurs is in cancer patients receiving chemotherapy. In many cases these patients are treated with antibiotic cocktails which can induce dysbiosis of the normal intestinal microbiota, and pathological overgrowth of commensal Candida spp. (hit 1). The combination of Candida overgrowth with chemotherapy-induced neutropenia (hit 2) and mucositis (hit 3) results in a highly permissible environment through which Candida can disseminate to the bloodstream. Other cases are thought to arise from contamination of intravenous catheters (which may or may not come from skin resident commensals). In these situations, patients usually also present with defects in phagocytic cells, however access to the blood stream is provided directly from contaminated catheters, and likely does not require additional disruption of mucosal barriers (71).
It is well-accepted that the innate immune system is essential to control systemic Candida infections. There is growing evidence that the adaptive immune system plays an important role in controlling systemic infections, although patients with defects in adaptive immunity are not necessarily more susceptible to invasive disease. Below we will review what is known about the innate and adaptive immune components necessary for limiting systemic fungal infections.
First line of defense: Serum Factors
Activation of the complement system by Candida spp. entering the bloodstream is a critical early factor in limiting dissemination and for priming the innate immune system for fungal phagocytosis and antimicrobial activity (78). Although humans with genetic deficiencies in components of the complement system are not more susceptible to invasive Candida infections (79), there is considerable evidence from mouse studies and in vitro studies with human cells that complement does play an important role antifungal immunity. Candida spp. are known to trigger all three pathways of complement activation which promote innate immunity against fungi through opsonization to promote phagocytosis, induction of pro-inflammatory cytokine production, and directly enhancing antimicrobial activity of recruited phagocytes. Activation of the C3 convertase gives rise to the C3b cleavage product that opsonizes and facilitates phagocytosis of fungi by neutrophils and macrophages via the complement receptor CR3 (78). Furthermore, the cleavage products C3a and C5a function as anaphalotoxins that induce pro-inflammatory cytokine production, inflammatory cell recruitment, and enhance antimicrobial activity of phagocytes (78). Importantly, mice deficient in C3 or C5 (precursors for C3a/b and C5a respectively) are highly susceptible to systemic Candida infections, as are mice deficient in the phagocytic receptor CR3. CR3 is unique because in addition to recognizing complement opsonized fungi, it also recognizes Pra1 and β-glucans on the surface of C. albicans to facilitate internalization (80, 81).
Innate immune cells that limit systemic Candida infections:
Neutrophils.
Neutrophils are widely considered the most important innate immune effector cell needed for early control of bloodstream Candida infections. Neutropenia is a major risk factor for development of invasive Candida infection in both mice and humans (77, 82). Furthermore, mutations in genes associated with various aspects of neutrophil recruitment and antimicrobial activity including CXCR1, myeloperoxidase (MPO), and NADPH oxidase are associated with increased risk of systemic Candida infections in humans (83–85). The ability of neutrophils to phagocytose fungi, undergo respiratory burst, secrete antimicrobial granules, and release neutrophil extracellular traps (NETs; decondensed DNA complexed with antimicrobial peptides released to trap and kill pathogens) make them highly toxic to fungi (86). Human neutrophils are also able to prevent yeast to hyphal transition, which is a major virulence factor during infections with C. albicans (87, 88).
In mouse models of disseminated candidiasis, the kidney is the major target organ affected. Neutrophils are rapidly recruited in large numbers to other peripheral organs such as the spleen and liver within 24 hours of infection, which limits fungal growth at these sites. In contrast, for reasons that are not clear, neutrophil recruitment to the kidney is significantly delayed, which can lead to uncontrolled fungal proliferation in this tissue (89). Regardless, the presence of neutrophils in the kidney is essential for controlling Candida infection. A recent study showed that expression of the chemokine receptor Cxcr1, while not required for neutrophil recruitment to the kidney, was essential for neutrophil degranulation and fungal clearance at this site (85). Consistent with this observation, the authors observed that humans with a mutation in CXCR1 (CXCR1-T276) have impaired neutrophil degranulation and are more susceptible to disseminated candidiasis.
Despite their essential anti-fungal properties, excessive neutrophil recruitment and/or activation can also have pathological consequences. Sustained recruitment of neutrophils to the kidney, due to organ-specific production of CCR1 ligands at later times after infection, was shown to be pathogenic in mice, and this contributed to mortality in response to disseminated Candida infection (90). Furthermore, this study showed that CCR1-deficient mice have impaired late recruitment of neutrophils to the kidney and are actually protected from disseminated Candida infection due to less severe immunopathology.
Monocytes/macrophages.
Compared to neutrophils, the role of monocytes and macrophages at early times during bloodstream Candida infection is less clear. Monocytopenia is not a risk factor for invasive Candida infection in humans (91). Also, although monocytes are able to internalize and kill Candida spp., they are less effective than neutrophils (92). However, early work demonstrating that monocyte/macrophage depleted mice are more suceptible to disseminated candidiasis suggests an important role for these cells in anti-fungal host defense (93). The role for monocytes in killing Candida in the bloodstream is likely secondary to neutrophils; however, in peripheral organs such as the kidney where fungi frequently disseminate, monocytes and macrophage appear to play an important protective role. In a detailed analysis of the different leukocyte populations that accumulate in peripheral organs during invasive candidiasis in mice, Ngo et al. observed that inflammatory monocytes rapidly accumulate in in the kidney, liver, spleen, and brain within 24 hours of infection (94). Furthermore, the investigators found that diphtheria toxin-mediated depletion of CCR2+Ly6C+ inflammatory monocytes in mice impairs fungal clearance in the brain and kidney, and leads to decreased survival following invasive Candida infection.
Tissue resident monocytes/macrophages have also recently been shown to play an important role during the early stages of systemic C. albicans infection. Deficiency in the chemokine receptor CX3CR1, which is highly expressed on kidney resident macrophages, resulted in impaired fungal clearance and increased mortality following systemic C. albicans infection (95). This was due largely to reduced macrophage survival in the kidney. This study further demonstrated that an inactivating mutation in Cx3cr1 (Cx3cr1-M280) is associated with increased susceptibility to systemic candidiasis in humans.
Similar to what has been observed with neutrophils, monocytes can also contribute to immunopathology if their recruitment and/or activation is not appropriately controlled. Majer et al. observed that over-production of type 1 interferons (IFN-1) during systemic C. albicans infection promotes excessive monocyte activation and recruitment to the kidney. This initiates an inflammatory cascade that ultimately leads to excessive neutrophil accumulation and lethal kidney immunopathology in mice with disseminated candidiasis. Consistent with this, mice deficient in the type 1 IFN-1 receptor were also highly resistant to systemic C. albicans infection as a result of less severe immunopathology (96).
Dendritic cell-NK cell-neutrophil crosstalk.
Recent studies have shed light on an unexpected relationship between dendritic cells (DCs) and natural killer (NK) cells in limiting disseminated candidiasis in mice. Mice deficient in IL-17a are susceptible to systemic candidiasis, which was initially thought to be due to defective Th17-mediated adaptive immunity (97). However, further investigation revealed that the heightened susceptibility of these mice was not dependent on T cells, but was rather due to a cell-intrinsic defect in NK cell function. The investigators noted that NK cells lacking the IL-17a receptor were functionally impaired (reduced target cell lysis and IFNγ production), and also failed to produce the GM-CSF that is needed to promote neutrophil antimicrobial activity against C. albicans infection. In a subsequent study investigators found that mice with DC-specific deficiency in the kinase Syk, required for signaling downstream of multiple antifungal C-type lectin receptors (Dectin-1, Dectin-2, MCL, and Mincle), were highly susceptible to systemic C. albicans infection (98). In this case, Syk-dependent production of IL-23p19 by dendritic cells in response to C. albicans was necessary to stimulate NK cells to promote GM-CSF production and enhance neutrophil antimicrobial activity (Figure 2G). In contrast to the previous study, however, investigators found that neither IL-17a nor IL-17f was able to directly induce NK cell production of GM-CSF. The reason for this discrepancy is unclear; however, both studies shed light on new and underappreciated roles for DC-NK cell-neutrophil dialogue in coordinating innate control of systemic C. albicans infection. In humans, NK cells have been reported to have both direct (perforin-mediated) and indirect (neutrophil priming-mediated) anti-Candida effector functions (99). Whether a similar crosstalk with dendritic cells exists in humans remains to be determined.
Role of adaptive immunity in systemic Candida infection
While there is considerable evidence pointing to a protective role for the adaptive immune system during mucosal fungal infection, such as in patients with chronic mucocutaneous candidiasis (CMC, detailed below), the role of adaptive immunity in bloodstream Candida infections is less clear. This is highlighted by the fact that individuals with defects in T and B lymphocyte function are not more susceptible to invasive Candida infections (100). However, given that one of the primary risk factors for systemic Candida infection is fungal colonization of mucosal surfaces, defects in adaptive immunity that promote mucosal infection with Candida could predispose these individuals to developing disseminated disease. Notably, mice deficient in critical components of Th1 immunity such as IFN-γ and IL-18 are more susceptible to disseminated candidiasis (101–103). In addition, a recent study found that IL-6 deficient mice, which have expanded Th1 differentiation during C. albicans skin infection, were protected from a secondary systemic C. albicans infection, indicating a potentially important role for Th1 immunity in memory responses to systemic Candida challenge (104, 105). Humans with polymorphisms in IL-12b develop more persistent fungemia during invasive candidiasis (105). Therefore, while not necessarily an initiating risk factor, defects in adaptive immunity to Candida can potentially influence the progression of disease in patients with invasive candidiasis.
Immunity to fungal infections of the skin
The skin is the largest organ of the human body and plays a multifaceted role in providing barrier protection against environmental insult and microbial invasion. It is a compartmentalized organ comprised of the epidermis, dermis and hypodermis layers populated with both hematopoietic and non-hematopoietic cells. The skin harbors a microbiome composed of bacteria, fungi, viruses and parasitic mites with the highest diversity of bacteria and fungi found on drier and exposed sites (50, 106, 107). Microbial communities of the skin contribute to the orchestration of local conditions that establish a symbiotic relationship with the host (108). For example, germ free mice fail to mount a T cell-mediated adaptive response against Leishmania major which was reversed by colonization with the bacterial skin commensal Staphylococcus epidermidis (109). Below we will focus on the interaction between Malassezia and Candida spp. with the host cutaneous immune system.
Malassezia and host defense
As noted above, Malassezia spp. are the dominant fungal member of the skin microbiota and are dependent on processing of external lipids for growth. As such, they thrive on sebaceous gland secretions (Figure 3A). The yeast secrete enzymes necessary for fatty acid metabolism, and a consequence of this is the production of short chain fatty acids that have direct anti-microbial activity in-vitro against bacteria and fungi found on the skin (Figure 3B). One such example is azelaic acid that exhibits antimicrobial activity at lower pH conditions consistent with the pH of normal skin (110). Similarly, ethyl ester derivatives produced by the esterification of medium chain fatty acids by Malassezia have in-vitro anti-mycotic activity (111). In the gastrointestinal tract, short chain fatty acids produced by commensal microbes, as a by-product of carbohydrate fermentation, or by dietary supplementation promotes production of colonic T-regulatory cells (112–114). It is interesting to hypothesize that such regulatory mechanisms could also be induced as a consequence of Malassezia fatty acid metabolism in the skin.
The carbohydrate composition of the cell wall of Malassezia is different from other common fungi in that the main cell wall component is β-(1,6)-glucans instead of the Dectin-1 ligand, β-(1,3)-glucan (115). C-type lectin receptors are still involved in immune responses to Malassezia, but this is largely via Dectin-2 and Mincle (116). Dectin-2 recognizes hydrophilic O-linked mannobiose-rich glycoproteins, while Mincle recognizes hydrophobic glucosyl- and mannosyl-glycolipids (116). Similarly, Malassezia has been observed to activate Toll-like receptor 2 (TLR2) in mast cells (117). Still, the relative roles that these (and other pattern recognition receptors) play in Malassezia-induced commensalism and disease processes are not yet well understood.
Malassezia species have been reported to produce potent aryl hydrocarbon receptor (Ahr) ligands namely indirubin, malassezin, formylindolo[3,2-b] carbazole, indolo[3,2-b]carbazole, pityriazepin, pityriacitrin and tryptanthrin (118–121) (Figure 3B). The aryl hydrocarbon receptor (Ahr) is an orphan nuclear receptor that belongs to the family of basic helix-loop-helix transcription factors and has been shown to positively influence wound healing and contribute to overall skin homeostasis (122–124). It is expressed by diverse cell types in the epidermis leading to the hypothesis that Malassezia-induced Ahr stimulation may promote epithelial cell health and protection from ultraviolet radiation (31). Among CD4+ T cells, Ahr expression in Th17 cells has been shown to increase the production of the proinflammatory cytokines IL-17 and IL-22 in this T cell subset (125). Also, TLR-induced responses in phagocytes are inhibited by Ahr ligands (126), and Ahr plays a crucial role in the homeostasis of cutaneous invariant γδ T cells (127). Thus it is also interesting to hypothesize that Malassezia-induced Ahr stimulation in the skin may directly modulate immune activation in health and disease (Figure 3B). In patients with atopic dermatitis, the diversity of Malassezia species is less than in healthy controls (128). The same observation holds true for non-Malassezia yeasts and filamentous fungi (128), which suggests that a decrease in diversity may alter the niche in a way that is conducive to disease.
Cutaneous Candida albicans infections
As previously mentioned, Candida albicans is a dimorphic and opportunistic fungus capable of colonizing healthy human skin. In the stratum corneum, the upper most cornified and avascular layer of the epidermis, Candida albicans exists mainly in its yeast phase (Figure 3C). In the stratum corneum Candida is exposed to antimicrobial peptides such as β-defensins and LL-37 that have anti-fungal activity (129). At least one fungal defense against these peptides is the observation that secreted aspartic proteases (SAPS) released by Candida can cleave and inactivate LL-37 (130). Candida albicans binds keratinocytes (HaCaT cell line) at least in part through interaction between Gpm1(a fungal Factor H/FHL1-binding protein) and host vitronectin, and binding is associated with induction of proinflammatory cytokine production (131, 132). However, while keratinocytes express PRRs, in vivo evidence of their significance remains to be reported. Recently work by Kaplan et al. has uncovered a role for keratinocytes in licensing epidermal residency to Langerhans cells and tissue resident memory CD8+ T cells through the activation of latent TGFβ (133).
Transition of Candida from the yeast into a pseudo-hyphal form confers the capacity to invade deeper sites such as the dermis and internal organs where they encounter diverse immune cells. Langerhans cells (LCs) reside in the epidermal region while dermal dendritic cells (dDCs), expressing either CD103 or CD11b among other cell surface markers, reside in the dermal layer (134). LCs are capable of inducing CD4+ Th17 cell differentiation through the production of IL-23, IL-6 and IL-1β (135). LCs drive CD4+ Th17 T cell differentiation in response to budding yeasts by a mechanism involving ligation of Dectin-1 and production of IL-6 but not IL-1β and TGFβ (104). Protection for secondary fungal challenge in the skin requires Th17 but not Th1 immunity (104).
Penetrating Candida hyphae are morphologically altered to conceal cell wall β-glucan that could be recognized by Dectin-1, and they encounter dDCs in the dermal layer that consequently induce Th1 and CTL differentiation by producing higher levels of IL-12 and IL-27 without any IL-23 production and only very low levels of IL-1β and IL-6 (135). Protection from secondary systemic challenge with Candida requires Th1 but not Th17 immunity (104). CD11b+ dDCs in the skin are found in close proximity to nerve fibers, and nociceptive neurons expressing transient potential cation channel subfamily V member 1 (TRPV1) are able to sense Candida albicans and stimulate release of CGRP, a neuropeptide with a role in the transmission of pain (136). In this case, CGRP appears to target CD11b+ dDCs and induce production of IL-23, promoting expansion of IL-17 producing γδ T cells which help recruit neutrophils and induce antimicrobial peptide production by keratinocytes (Figure 3C). How this sensing by nociceptors occurs is not clear, although von Andrian and coworkers recently showed the same role for these nerves in an imiquimod-stimulated model of psoriasis-like inflammation in the skin, suggesting that the fibers respond either directly to TLR agonists or to inflammatory mediators produced by other cells in response to TLR agonists (137).
Immunity to fungal infections of the gastrointestinal tract
Infections of the oral cavity
The oral cavity, much like the architecture of the skin, is composed of stratified squamous epithelium with a tough keratinized layer lining the masticatory mucosa and non-keratinized squamous epithelium lining the rest of the oral cavity. Under a normal physiological state, the oral cavity harbors a diverse mycobiota with Candida species found in up to 75% of healthy people surveyed (11). Development of oral Candida infections, such as oral pharyngeal candidiasis (OPC), is often manifested in HIV+ individuals with marked reductions in CD4+ Th17 cell numbers (138). This suggests that normal interactions between commensal fungi and the host immune system are essential to maintain the balance between commensalism and disease. Further, Ghannoum et al. noted an inverse relationship between Candida colonization and abundance of Pichia yeasts in the oral cavity of HIV+ patients, suggesting that fungal competition might also be promoted in different immune environments (139). Pichia was able to inhibit Candida by limiting availability of nutrients, fungal growth and expression of virulence factors (139).
Naglik and coworkers have recently identified a novel virulence factor that aids Candida albicans invasion during oral infection (140). They reported that Candida albicans strains lacking the cell elongation 1 gene (ECE1), a secreted protein expressed during filamentation, form hyphae normally, but fail to penetrate epithelial layers. They determined that this protein is proteolytically processed to generate a pore-forming toxin that is able to kill epithelial cells and that this activity is necessary for tissue penetration. This is the first report of a pore-forming yeast toxin which the investigators named candidalysin (140). It will be interesting in coming years to see what, if any, role this toxin plays in other kinds of infection or whether it participates in competition with other microbes as well.
Chronic mucocutaneous candidiasis
Chronic mucocutaneous candidiasis (CMC) is a variegated group of conditions characterized by persistent or recalcitrant superficial infections caused mainly by Candida albicans. Development of systemic dissemination is uncommon. These infections typically occur in the oral and genital mucosae, skin and nails (141), and the underlying mechanisms that drive these disorders are generally linked with impaired Th17 immunity (142). In humans CMC can arise from mutations in IL-17RA, IL-17RC and IL-17F and downstream IL-17R signaling adaptor Act 1 (142, 143). Patients that have mutations in STAT3 afflicted with autosomal dominant hyper IgE syndrome (AD-HIES) have defects in T cell IL-17 and IL-22 secretion and are affected with CMC (144). Additionally, CMC has also been observed in patients with mutations to Dectin-1 and CARD9, both important for innate anti-fungal immune defense as discussed above (145, 146).
Fungi and inflammatory bowel disease
Inflammatory bowel disease (IBD) is characterized by chronic intestinal inflammation that develops due to a dysregulated immune response to commensal microbes in genetically-susceptible individuals (147). IBD is a group of diseases primarily comprising ulcerative colitis (UC) and Crohn’s disease (CD). Although the exact role that the fungal microbiota plays in IBD pathogenesis is not clear, Iliev et al. have reported that a polymorphic variant of the Dectin-1 gene is associated with disease severity in patients with ulcerative colitis and that mice lacking Dectin-1 are highly susceptible to models of colitis (14). Further, antibodies to yeast (called anti-Saccharomyces cerevisiae antibodies, ASCA) are directed against yeast cell wall mannan and have been identified as a biomarker capable of identifying the majority of patients with Crohn’s disease (CD) (148). Also, genome-wide association studies have revealed potential links between IBD and host anti-fungal immunity including polymorphisms in CARD9 and IL-23R that are associated with susceptibility to disease (149, 150). Together, the data suggest that altered immune interactions with fungi may play a significant role in at least some patients with IBD. However, whether specific types of fungi are involved and how altered immune interactions may contribute to disease initiation or persistence will require further investigation.
Several studies have begun to explore whether IBD is associated with changes in the types of fungi found in fecal samples. An early culture-based study reported enrichment of Candida species (specifically C. dubliniensis and C. parapsilosis) in IBD patients (151). More recent high throughput DNA sequencing-based approaches have noted reduced fungal diversity and prevalence of specific Candida spp. (including C. parapsilosis) in pediatric IBD patients (17), and Candida albicans in adult IBD patients (15). In the latter study, investigators noted an inverse correlation between C. albicans and S. cerevisiae which could be related to a potential homeostatic role of S. cerevisiae as discussed above. These investigators also noted that the presence of Malassezia sympodialis in stool samples was associated with polymorphisms in Dectin-1 and that an IBD-linked CARD9 polymorphism was associated with increases in S. cerevisiae (15). However, how or whether these changes directly affect disease and how or whether these stool-associated fungi interact with immune cells will require deeper investigation.
Future Directions
In this review, we have reflected on recent work investigating interactions between fungi and the immune system in defining healthy and disease states. In the context of health, we are only just beginning to recognize immune mechanisms involved in continuous interaction with environmental and commensal fungal communities. These interactions contribute to maintenance of a healthy immune system that is ideally balanced to allow colonization by commensal organisms but poised to induce effective host defense when challenged with infection. Future studies will provide insights into whether commensal fungal populations and immune responses to them can be manipulated to promote health or ameliorate disease. In the context of disease, effective host immune responses are essential to protect against serious life-threatening fungal infections. Given the prevalence of serious fungal infections, and the associated mortalities, much more effort needs to be applied to understanding how the immune system optimally copes with infectious challenge and how rational strategies may be devised to strengthen these defenses.
References
- 1.Drummond RA, Gaffen SL, Hise AG, Brown GD. 2015. Innate Defense against Fungal Pathogens. Cold Spring Harb. Perspect. Med 5: a019620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Netea MG, Joosten LA, van der Meer JW, Kullberg BJ, van de Veerdonk FL. 2015. Immune defence against Candida fungal infections. Nat. Rev. Immunol 15: 630–42 [DOI] [PubMed] [Google Scholar]
- 3.Rapaka RR, Ricks DM, Alcorn JF, Chen K, Khader SA, et al. 2010. Conserved natural IgM antibodies mediate innate and adaptive immunity against the opportunistic fungus Pneumocystis murina. J. Exp. Med 207: 2907–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seed PC. 2015. The human mycobiome. Cold Spring Harb. Perspect. Med 5: a019810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Underhill DM, Iliev ID. 2014. The mycobiota: interactions between commensal fungi and the host immune system. Nat. Rev. Immunol 14: 405–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibrahim AS, Kontoyiannis DP. 2013. Update on mucormycosis pathogenesis. Curr. Opin. Infect. Dis 26: 508–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson L, Gaab EM, Sanchez J, Bui PQ, Nobile CJ, et al. 2014. Valley fever: danger lurking in a dust cloud. Microbes Infect 16: 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horwath MC, Fecher RA, Deepe GS Jr. 2015. Histoplasma capsulatum, lung infection and immunity. Future Microbiol 10: 967–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rohatgi S, Pirofski LA. 2015. Host immunity to Cryptococcus neoformans. Future Microbiol 10: 565–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang J, Iliev ID, Brown J, Underhill DM, Funari VA. 2015. Mycobiome: Approaches to analysis of intestinal fungi. J. Immunol. Methods 421: 112–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, et al. 2010. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog 6: e1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dupuy AK, David MS, Li L, Heider TN, Peterson JD, et al. 2014. Redefining the human oral mycobiome with improved practices in amplicon-based taxonomy: discovery of Malassezia as a prominent commensal. PLoS One 9: e90899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dollive S, Chen YY, Grunberg S, Bittinger K, Hoffmann C, et al. 2013. Fungi of the murine gut: episodic variation and proliferation during antibiotic treatment. PLoS One 8: e71806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, et al. 2012. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336: 1314–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, et al. 2016. Fungal microbiota dysbiosis in IBD. Gut [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, et al. 2015. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 18: 489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chehoud C, Albenberg LG, Judge C, Hoffmann C, Grunberg S, et al. 2015. Fungal Signature in the Gut Microbiota of Pediatric Patients With Inflammatory Bowel Disease. Inflamm. Bowel Dis 21: 1948–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, et al. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, et al. 2011. Enterotypes of the human gut microbiome. Nature 473: 174–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kullberg BJ, Verweij PE, Akova M, Arendrup MC, Bille J, et al. 2011. European expert opinion on the management of invasive candidiasis in adults. Clin. Microbiol. Infect 17 Suppl 5: 1–12 [DOI] [PubMed] [Google Scholar]
- 21.Gabaldon T, Martin T, Marcet-Houben M, Durrens P, Bolotin-Fukuhara M, et al. 2013. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics 14: 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angoulvant A, Guitard J, Hennequin C. 2016. Old and new pathogenic Nakaseomyces species: epidemiology, biology, identification, pathogenicity and antifungal resistance. FEMS Yeast Res 16 [DOI] [PubMed] [Google Scholar]
- 23.Latge JP. 1999. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev 12: 310–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas PA, Kaliamurthy J. 2013. Mycotic keratitis: epidemiology, diagnosis and management. Clin. Microbiol. Infect 19: 210–20 [DOI] [PubMed] [Google Scholar]
- 25.Pitt JI, Samson RA. 2007. Nomenclatural considerations in naming species of Aspergillus and its teleomorphs. Stud. Mycol 59: 67–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nucci M, Anaissie E. 2007. Fusarium infections in immunocompromised patients. Clin. Microbiol. Rev 20: 695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bensch K, Braun U, Groenewald JZ, Crous PW. 2012. The genus Cladosporium. Stud. Mycol 72: 1–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandoval-Denis M, Sutton DA, Martin-Vicente A, Cano-Lira JF, Wiederhold N, et al. 2015. Cladosporium Species Recovered from Clinical Samples in the United States. J. Clin. Microbiol 53: 2990–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vacher G, Niculita-Hirzel H, Roger T. 2015. Immune responses to airborne fungi and non-invasive airway diseases. Semin. Immunopathol 37: 83–96 [DOI] [PubMed] [Google Scholar]
- 30.Amend A 2014. From dandruff to deep-sea vents: Malassezia-like fungi are ecologically hyper-diverse. PLoS Pathog 10: e1004277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Velegraki A, Cafarchia C, Gaitanis G, Iatta R, Boekhout T. 2015. Malassezia infections in humans and animals: pathophysiology, detection, and treatment. PLoS Pathog 11: e1004523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hooper LV, Littman DR, Macpherson AJ. 2012. Interactions between the microbiota and the immune system. Science 336: 1268–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McFarland LV. 2010. Systematic review and meta-analysis of Saccharomyces boulardii in adult patients. World J. Gastroenterol 16: 2202–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McFarland LV, Surawicz CM, Greenberg RN, Fekety R, Elmer GW, et al. 1994. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 271: 1913–8 [PubMed] [Google Scholar]
- 35.Batista TM, Marques ET Jr., Franco GR, Douradinha B. 2014. Draft Genome Sequence of the Probiotic Yeast Saccharomyces cerevisiae var. boulardii Strain ATCC MYA-796. Genome Announc 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castagliuolo I, Riegler MF, Valenick L, LaMont JT, Pothoulakis C. 1999. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect. Immun 67: 302–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buts JP, Dekeyser N, Stilmant C, Delem E, Smets F, Sokal E. 2006. Saccharomyces boulardii produces in rat small intestine a novel protein phosphatase that inhibits Escherichia coli endotoxin by dephosphorylation. Pediatr. Res 60: 24–9 [DOI] [PubMed] [Google Scholar]
- 38.Ducluzeau R, Bensaada M. 1982. [Comparative effect of a single or continuous administration of “Saccharomyces boulardii” on the establishment of various strains of “candida” in the digestive tract of gnotobiotic mice]. Ann. Microbiol 133: 491–501 [PubMed] [Google Scholar]
- 39.Zbinden R, Gonczi EE, Altwegg M. 1999. Inhibition of Saccharomyces boulardii (nom. inval.) on cell invasion of Salmonella typhimurium and Yersinia enterocolitica. Microb. Ecol. Health Dis 11: 158–62 [Google Scholar]
- 40.Qamar A, Aboudola S, Warny M, Michetti P, Pothoulakis C, et al. 2001. Saccharomyces boulardii stimulates intestinal immunoglobulin A immune response to Clostridium difficile toxin A in mice. Infect. Immun 69: 2762–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas S, Metzke D, Schmitz J, Dorffel Y, Baumgart DC. 2011. Anti-inflammatory effects of Saccharomyces boulardii mediated by myeloid dendritic cells from patients with Crohn’s disease and ulcerative colitis. Am. J. Physiol. Gastrointest. Liver Physiol 301: G1083–92 [DOI] [PubMed] [Google Scholar]
- 42.Guslandi M, Mezzi G, Sorghi M, Testoni PA. 2000. Saccharomyces boulardii in maintenance treatment of Crohn’s disease. Dig. Dis. Sci 45: 1462–4 [DOI] [PubMed] [Google Scholar]
- 43.Plein K, Hotz J. 1993. Therapeutic effects of Saccharomyces boulardii on mild residual symptoms in a stable phase of Crohn’s disease with special respect to chronic diarrhea--a pilot study. Z. Gastroenterol 31: 129–34 [PubMed] [Google Scholar]
- 44.Guslandi M, Giollo P, Testoni PA. 2003. A pilot trial of Saccharomyces boulardii in ulcerative colitis. Eur. J. Gastroenterol. Hepatol 15: 697–8 [DOI] [PubMed] [Google Scholar]
- 45.Dalmasso G, Cottrez F, Imbert V, Lagadec P, Peyron JF, et al. 2006. Saccharomyces boulardii inhibits inflammatory bowel disease by trapping T cells in mesenteric lymph nodes. Gastroenterology 131: 1812–25 [DOI] [PubMed] [Google Scholar]
- 46.Jawhara S, Poulain D. 2007. Saccharomyces boulardii decreases inflammation and intestinal colonization by Candida albicans in a mouse model of chemically-induced colitis. Med. Mycol 45: 691–700 [DOI] [PubMed] [Google Scholar]
- 47.Wu X, Vallance BA, Boyer L, Bergstrom KS, Walker J, et al. 2008. Saccharomyces boulardii ameliorates Citrobacter rodentium-induced colitis through actions on bacterial virulence factors. Am. J. Physiol. Gastrointest. Liver Physiol 294: G295–306 [DOI] [PubMed] [Google Scholar]
- 48.Takata K, Tomita T, Okuno T, Kinoshita M, Koda T, et al. 2015. Dietary Yeasts Reduce Inflammation in Central Nerve System via Microflora. Ann. Clin. Transl. Neurol 2: 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffmann C, Dollive S, Grunberg S, Chen J, Li H, et al. 2013. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One 8: e66019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Findley K, Oh J, Yang J, Conlan S, Deming C, et al. 2013. Topographic diversity of fungal and bacterial communities in human skin. Nature 498: 367–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rizzetto L, Ifrim DC, Moretti S, Tocci N, Cheng SC, et al. 2016. Fungal Chitin Induces Trained Immunity in Human Monocytes during Cross-talk of the Host with Saccharomyces cerevisiae. J. Biol. Chem 291: 7961–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quintin J, Saeed S, Martens JH, Giamarellos-Bourboulis EJ, Ifrim DC, et al. 2012. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12: 223–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagener J, Malireddi RK, Lenardon MD, Koberle M, Vautier S, et al. 2014. Fungal chitin dampens inflammation through IL-10 induction mediated by NOD2 and TLR9 activation. PLoS Pathog 10: e1004050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z, Li J, Zheng W, Zhao G, Zhang H, et al. 2016. Peripheral Lymphoid Volume Expansion and Maintenance Are Controlled by Gut Microbiota via RALDH(+) Dendritic Cells. Immunity 44: 330–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bauer H, Horowitz RE, Levenson SM, Popper H. 1963. The response of the lymphatic tissue to the microbial flora. Studies on germfree mice. Am. J. Pathol 42: 471–83 [PMC free article] [PubMed] [Google Scholar]
- 56.De Jesus M, Rodriguez AE, Yagita H, Ostroff GR, Mantis NJ. 2015. Sampling of Candida albicans and Candida tropicalis by Langerin-positive dendritic cells in mouse Peyer’s patches. Immunol. Lett 168: 64–72 [DOI] [PubMed] [Google Scholar]
- 57.Wheeler ML, Bar AS, Leal CA, Tang J, Brown J, et al. 2016. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe IN PRESS [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bacher P, Kniemeyer O, Schonbrunn A, Sawitzki B, Assenmacher M, et al. 2014. Antigen-specific expansion of human regulatory T cells as a major tolerance mechanism against mucosal fungi. Mucosal Immunol 7: 916–28 [DOI] [PubMed] [Google Scholar]
- 59.Bonifazi P, Zelante T, D’Angelo C, De Luca A, Moretti S, et al. 2009. Balancing inflammation and tolerance in vivo through dendritic cells by the commensal Candida albicans. Mucosal Immunol 2: 362–74 [DOI] [PubMed] [Google Scholar]
- 60.Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, et al. 2010. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat. Immunol 11: 846–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, et al. 2008. Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell-mediated effects on regulatory T cells. Blood 112: 1214–22 [DOI] [PubMed] [Google Scholar]
- 62.Pierce JV, Kumamoto CA. 2012. Variation in Candida albicans EFG1 expression enables host-dependent changes in colonizing fungal populations. MBio 3: e00117–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tyc KM, Herwald SE, Hogan JA, Pierce JV, Klipp E, Kumamoto CA. 2016. The game theory of Candida albicans colonization dynamics reveals host status-responsive gene expression. BMC Syst. Biol 10: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zelante T, Iannitti RG, De Luca A, Arroyo J, Blanco N, et al. 2012. Sensing of mammalian IL-17A regulates fungal adaptation and virulence. Nat. Commun 3: 683. [DOI] [PubMed] [Google Scholar]
- 65.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci. Transl. Med 4: 165rv13 [DOI] [PubMed] [Google Scholar]
- 66.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis 39: 309–17 [DOI] [PubMed] [Google Scholar]
- 67.Li Z, Jiang C, Dong D, Zhang L, Tian Y, et al. 2016. The Correlation Between Candida Colonization of Distinct Body Sites and Invasive Candidiasis in Emergency Intensive Care Units: Statistical and Molecular Biological Analysis. Mycopathologia [DOI] [PubMed] [Google Scholar]
- 68.Gouba N, Drancourt M. 2015. Digestive tract mycobiota: a source of infection. Med. Mal. Infect 45: 9–16 [DOI] [PubMed] [Google Scholar]
- 69.Miranda LN, van der Heijden IM, Costa SF, Sousa AP, Sienra RA, et al. 2009. Candida colonisation as a source for candidaemia. J. Hosp. Infect 72: 9–16 [DOI] [PubMed] [Google Scholar]
- 70.Nucci M, Anaissie E. 2001. Revisiting the source of candidemia: skin or gut? Clin. Infect. Dis 33: 1959–67 [DOI] [PubMed] [Google Scholar]
- 71.Kullberg BJ, Arendrup MC. 2015. Invasive Candidiasis. N. Engl. J. Med 373: 1445–56 [DOI] [PubMed] [Google Scholar]
- 72.Sobel JD. 2007. Vulvovaginal candidosis. Lancet 369: 1961–71 [DOI] [PubMed] [Google Scholar]
- 73.Tajima M, Sugita T, Nishikawa A, Tsuboi R. 2008. Molecular analysis of Malassezia microflora in seborrheic dermatitis patients: comparison with other diseases and healthy subjects. J. Invest. Dermatol 128: 345–51 [DOI] [PubMed] [Google Scholar]
- 74.Kaneko T, Murotani M, Ohkusu K, Sugita T, Makimura K. 2012. Genetic and biological features of catheter-associated Malassezia furfur from hospitalized adults. Med. Mycol 50: 74–80 [DOI] [PubMed] [Google Scholar]
- 75.Curvale-Fauchet N, Botterel F, Legrand P, Guillot J, Bretagne S. 2004. Frequency of intravascular catheter colonization by Malassezia spp. in adult patients. Mycoses 47: 491–4 [DOI] [PubMed] [Google Scholar]
- 76.Trick WE, Fridkin SK, Edwards JR, Hajjeh RA, Gaynes RP, National Nosocomial Infections Surveillance System H. 2002. Secular trend of hospital-acquired candidemia among intensive care unit patients in the United States during 1989–1999. Clin. Infect. Dis 35: 627–30 [DOI] [PubMed] [Google Scholar]
- 77.Koh AY, Kohler JR, Coggshall KT, Van Rooijen N, Pier GB. 2008. Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog 4: e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kozel TR. 1996. Activation of the complement system by pathogenic fungi. Clin. Microbiol. Rev 9: 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ram S, Lewis LA, Rice PA. 2010. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin. Microbiol. Rev 23: 740–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van Bruggen R, Drewniak A, Jansen M, van Houdt M, Roos D, et al. 2009. Complement receptor 3, not Dectin-1, is the major receptor on human neutrophils for beta-glucan-bearing particles. Mol. Immunol 47: 575–81 [DOI] [PubMed] [Google Scholar]
- 81.Soloviev DA, Fonzi WA, Sentandreu R, Pluskota E, Forsyth CB, et al. 2007. Identification of pH-regulated antigen 1 released from Candida albicans as the major ligand for leukocyte integrin alphaMbeta2. J. Immunol 178: 2038–46 [DOI] [PubMed] [Google Scholar]
- 82.Uzun O, Ascioglu S, Anaissie EJ, Rex JH. 2001. Risk factors and predictors of outcome in patients with cancer and breakthrough candidemia. Clin. Infect. Dis 32: 1713–7 [DOI] [PubMed] [Google Scholar]
- 83.Lehrer RI, Cline MJ. 1969. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: the role of myeloperoxidase in resistance to Candida infection. J. Clin. Invest 48: 1478–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Winkelstein JA, Marino MC, Johnston RB Jr., Boyle J, Curnutte J, et al. 2000. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine 79: 155–69 [DOI] [PubMed] [Google Scholar]
- 85.Swamydas M, Gao JL, Break TJ, Johnson MD, Jaeger M, et al. 2016. CXCR1-mediated neutrophil degranulation and fungal killing promote Candida clearance and host survival. Sci. Transl. Med 8: 322ra10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. 2012. Neutrophil function: from mechanisms to disease. Annu. Rev. Immunol 30: 459–89 [DOI] [PubMed] [Google Scholar]
- 87.Wozniok I, Hornbach A, Schmitt C, Frosch M, Einsele H, et al. 2008. Induction of ERK-kinase signalling triggers morphotype-specific killing of Candida albicans filaments by human neutrophils. Cell. Microbiol 10: 807–20 [DOI] [PubMed] [Google Scholar]
- 88.Ermert D, Niemiec MJ, Rohm M, Glenthoj A, Borregaard N, Urban CF. 2013. Candida albicans escapes from mouse neutrophils. J. Leukoc. Biol 94: 223–36 [DOI] [PubMed] [Google Scholar]
- 89.Lionakis MS, Lim JK, Lee CC, Murphy PM. 2011. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J. Innate Immun 3: 180–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lionakis MS, Fischer BG, Lim JK, Swamydas M, Wan W, et al. 2012. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog 8: e1002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vinh DC, Patel SY, Uzel G, Anderson VL, Freeman AF, et al. 2010. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood 115: 1519–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hunniger K, Lehnert T, Bieber K, Martin R, Figge MT, Kurzai O. 2014. A virtual infection model quantifies innate effector mechanisms and Candida albicans immune escape in human blood. PLoS Comput. Biol 10: e1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Qian Q, Jutila MA, Van Rooijen N, Cutler JE. 1994. Elimination of mouse splenic macrophages correlates with increased susceptibility to experimental disseminated candidiasis. J. Immunol 152: 5000–8 [PubMed] [Google Scholar]
- 94.Ngo LY, Kasahara S, Kumasaka DK, Knoblaugh SE, Jhingran A, Hohl TM. 2014. Inflammatory monocytes mediate early and organ-specific innate defense during systemic candidiasis. J. Infect. Dis 209: 109–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lionakis MS, Swamydas M, Fischer BG, Plantinga TS, Johnson MD, et al. 2013. CX3CR1-dependent renal macrophage survival promotes Candida control and host survival. J. Clin. Invest 123: 5035–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, et al. 2012. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog 8: e1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bar E, Whitney PG, Moor K, Reis e Sousa C, LeibundGut-Landmann S. 2014. IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity 40: 117–27 [DOI] [PubMed] [Google Scholar]
- 98.Whitney PG, Bar E, Osorio F, Rogers NC, Schraml BU, et al. 2014. Syk signaling in dendritic cells orchestrates innate resistance to systemic fungal infection. PLoS Pathog 10: e1004276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Voigt J, Hunniger K, Bouzani M, Jacobsen ID, Barz D, et al. 2014. Human natural killer cells acting as phagocytes against Candida albicans and mounting an inflammatory response that modulates neutrophil antifungal activity. J. Infect. Dis 209: 616–26 [DOI] [PubMed] [Google Scholar]
- 100.Lionakis MS. 2012. Genetic Susceptibility to Fungal Infections in Humans. Curr. Fungal Infect. Rep 6: 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lavigne LM, Schopf LR, Chung CL, Maylor R, Sypek JP. 1998. The role of recombinant murine IL-12 and IFN-gamma in the pathogenesis of a murine systemic Candida albicans infection. J. Immunol 160: 284–92 [PubMed] [Google Scholar]
- 102.Balish E, Wagner RD, Vazquez-Torres A, Pierson C, Warner T. 1998. Candidiasis in interferon-gamma knockout (IFN-gamma−/−) mice. J. Infect. Dis 178: 478–87 [DOI] [PubMed] [Google Scholar]
- 103.Netea MG, Vonk AG, van den Hoven M, Verschueren I, Joosten LA, et al. 2003. Differential role of IL-18 and IL-12 in the host defense against disseminated Candida albicans infection. Eur. J. Immunol 33: 3409–17 [DOI] [PubMed] [Google Scholar]
- 104.Kashem SW, Igyarto BZ, Gerami-Nejad M, Kumamoto Y, Mohammed J, et al. 2015. Candida albicans Morphology and Dendritic Cell Subsets Determine T Helper Cell Differentiation. Immunity 42: 356–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Johnson MD, Plantinga TS, van de Vosse E, Velez Edwards DR, Smith PB, et al. 2012. Cytokine gene polymorphisms and the outcome of invasive candidiasis: a prospective cohort study. Clin. Infect. Dis 54: 502–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grice EA, Segre JA. 2011. The skin microbiome. Nat. Rev. Microbiol 9: 244–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hannigan GD, Meisel JS, Tyldsley AS, Zheng Q, Hodkinson BP, et al. 2015. The human skin double-stranded DNA virome: topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome. MBio 6: e01578–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sanford JA, Gallo RL. 2013. Functions of the skin microbiota in health and disease. Semin. Immunol 25: 370–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, et al. 2012. Compartmentalized control of skin immunity by resident commensals. Science 337: 1115–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brasch J, Christophers E. 1993. Azelaic acid has antimycotic properties in vitro. Dermatology 186: 55–8 [DOI] [PubMed] [Google Scholar]
- 111.Mayser P 2015. Medium chain fatty acid ethyl esters - activation of antimicrobial effects by Malassezia enzymes. Mycoses 58: 215–9 [DOI] [PubMed] [Google Scholar]
- 112.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, et al. 2013. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504: 446–50 [DOI] [PubMed] [Google Scholar]
- 113.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, et al. 2013. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504: 451–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, et al. 2013. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341: 569–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stalhberger T, Simenel C, Clavaud C, Eijsink VG, Jourdain R, et al. 2014. Chemical organization of the cell wall polysaccharide core of Malassezia restricta. J. Biol. Chem 289: 12647–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ishikawa T, Itoh F, Yoshida S, Saijo S, Matsuzawa T, et al. 2013. Identification of distinct ligands for the C-type lectin receptors Mincle and Dectin-2 in the pathogenic fungus Malassezia. Cell Host Microbe 13: 477–88 [DOI] [PubMed] [Google Scholar]
- 117.Selander C, Engblom C, Nilsson G, Scheynius A, Andersson CL. 2009. TLR2/MyD88-dependent and -independent activation of mast cell IgE responses by the skin commensal yeast Malassezia sympodialis. J. Immunol 182: 4208–16 [DOI] [PubMed] [Google Scholar]
- 118.Gaitanis G, Magiatis P, Hantschke M, Bassukas ID, Velegraki A. 2012. The Malassezia genus in skin and systemic diseases. Clin. Microbiol. Rev 25: 106–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gaitanis G, Velegraki A, Mayser P, Bassukas ID. 2013. Skin diseases associated with Malassezia yeasts: facts and controversies. Clin. Dermatol 31: 455–63 [DOI] [PubMed] [Google Scholar]
- 120.Mexia N, Gaitanis G, Velegraki A, Soshilov A, Denison MS, Magiatis P. 2015. Pityriazepin and other potent AhR ligands isolated from Malassezia furfur yeast. Arch. Biochem. Biophys 571: 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Magiatis P, Pappas P, Gaitanis G, Mexia N, Melliou E, et al. 2013. Malassezia yeasts produce a collection of exceptionally potent activators of the Ah (dioxin) receptor detected in diseased human skin. J. Invest. Dermatol 133: 2023–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Barouti N, Mainetti C, Fontao L, Sorg O. 2015. L-Tryptophan as a Novel Potential Pharmacological Treatment for Wound Healing via Aryl Hydrocarbon Receptor Activation. Dermatology 230: 332–9 [DOI] [PubMed] [Google Scholar]
- 123.Bock KW, Kohle C. 2009. The mammalian aryl hydrocarbon (Ah) receptor: from mediator of dioxin toxicity toward physiological functions in skin and liver. Biol. Chem 390: 1225–35 [DOI] [PubMed] [Google Scholar]
- 124.Di Meglio P, Duarte JH, Ahlfors H, Owens ND, Li Y, et al. 2014. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 40: 989–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, et al. 2008. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453: 106–9 [DOI] [PubMed] [Google Scholar]
- 126.Vlachos C, Schulte BM, Magiatis P, Adema GJ, Gaitanis G. 2012. Malassezia-derived indoles activate the aryl hydrocarbon receptor and inhibit Toll-like receptor-induced maturation in monocyte-derived dendritic cells. Br. J. Dermatol 167: 496–505 [DOI] [PubMed] [Google Scholar]
- 127.Kadow S, Jux B, Zahner SP, Wingerath B, Chmill S, et al. 2011. Aryl hydrocarbon receptor is critical for homeostasis of invariant gammadelta T cells in the murine epidermis. J. Immunol 187: 3104–10 [DOI] [PubMed] [Google Scholar]
- 128.Zhang E, Tanaka T, Tajima M, Tsuboi R, Nishikawa A, Sugita T. 2011. Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiol. Immunol 55: 625–32 [DOI] [PubMed] [Google Scholar]
- 129.Marcinkiewicz M, Majewski S. 2016. The role of antimicrobial peptides in chronic inflammatory skin diseases. Postepy Dermatol. Alergol 33: 6–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rapala-Kozik M, Bochenska O, Zawrotniak M, Wolak N, Trebacz G, et al. 2015. Inactivation of the antifungal and immunomodulatory properties of human cathelicidin LL-37 by aspartic proteases produced by the pathogenic yeast Candida albicans. Infect. Immun 83: 2518–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lopez CM, Wallich R, Riesbeck K, Skerka C, Zipfel PF. 2014. Candida albicans uses the surface protein Gpm1 to attach to human endothelial cells and to keratinocytes via the adhesive protein vitronectin. PLoS One 9: e90796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ran Y, Yamazaki M, Tsuboi R, Ogawa H. 2014. Adherence and proliferation of keratinocytes cultured with Candida albicans. J. Dermatol 41: 554–6 [DOI] [PubMed] [Google Scholar]
- 133.Mohammed J, Beura LK, Bobr A, Astry B, Chicoine B, et al. 2016. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nat. Immunol 17: 414–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kaplan DH. 2010. In vivo function of Langerhans cells and dermal dendritic cells. Trends Immunol 31: 446–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Igyarto BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, et al. 2011. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 35: 260–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH. 2015. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 43: 515–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, et al. 2014. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 510: 157–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cassone A, Cauda R. 2012. Candida and candidiasis in HIV-infected patients: where commensalism, opportunistic behavior and frank pathogenicity lose their borders. AIDS 26: 1457–72 [DOI] [PubMed] [Google Scholar]
- 139.Mukherjee PK, Chandra J, Retuerto M, Sikaroodi M, Brown RE, et al. 2014. Oral mycobiome analysis of HIV-infected patients: identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog 10: e1003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, et al. 2016. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532: 64–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kirkpatrick CH. 2001. Chronic mucocutaneous candidiasis. Pediatr. Infect. Dis. J 20: 197–206 [DOI] [PubMed] [Google Scholar]
- 142.Puel A, Cypowyj S, Marodi L, Abel L, Picard C, Casanova JL. 2012. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr. Opin. Allergy Clin. Immunol 12: 616–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gaffen SL, Jain R, Garg AV, Cua DJ. 2014. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol 14: 585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Minegishi Y 2009. Hyper-IgE syndrome. Curr. Opin. Immunol 21: 487–92 [DOI] [PubMed] [Google Scholar]
- 145.Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, et al. 2009. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med 361: 1760–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, et al. 2009. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med 361: 1727–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.McGovern DP, Kugathasan S, Cho JH. 2015. Genetics of Inflammatory Bowel Diseases. Gastroenterology 149: 1163–76 e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Joossens S, Reinisch W, Vermeire S, Sendid B, Poulain D, et al. 2002. The value of serologic markers in indeterminate colitis: a prospective follow-up study. Gastroenterology 122: 1242–7 [DOI] [PubMed] [Google Scholar]
- 149.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, et al. 2012. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491: 119–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, et al. 2011. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet 43: 1066–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ott SJ, Kuhbacher T, Musfeldt M, Rosenstiel P, Hellmig S, et al. 2008. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand. J. Gastroenterol 43: 831–41 [DOI] [PubMed] [Google Scholar]
- 152.Becker KL, Ifrim DC, Quintin J, Netea MG, van de Veerdonk FL. 2015. Antifungal innate immunity: recognition and inflammatory networks. Semin. Immunopathol 37: 107–16 [DOI] [PubMed] [Google Scholar]
- 153.Cunha C, Di Ianni M, Bozza S, Giovannini G, Zagarella S, et al. 2010. Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 116: 5394–402 [DOI] [PubMed] [Google Scholar]
- 154.Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, et al. 2007. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol 8: 39–46 [DOI] [PubMed] [Google Scholar]
- 155.Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, et al. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol 8: 31–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kerscher B, Willment JA, Brown GD. 2013. The Dectin-2 family of C-type lectin-like receptors: an update. Int. Immunol 25: 271–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ifrim DC, Bain JM, Reid DM, Oosting M, Verschueren I, et al. 2014. Role of Dectin-2 for host defense against systemic infection with Candida glabrata. Infect. Immun 82: 1064–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhu LL, Zhao XQ, Jiang C, You Y, Chen XP, et al. 2013. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 39: 324–34 [DOI] [PubMed] [Google Scholar]
- 159.Wells CA, Salvage-Jones JA, Li X, Hitchens K, Butcher S, et al. 2008. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J. Immunol 180: 7404–13 [DOI] [PubMed] [Google Scholar]
- 160.Yamasaki S, Matsumoto M, Takeuchi O, Matsuzawa T, Ishikawa E, et al. 2009. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc. Natl. Acad. Sci. U. S. A 106: 1897–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sainz J, Lupianez CB, Segura-Catena J, Vazquez L, Rios R, et al. 2012. Dectin-1 and DC-SIGN polymorphisms associated with invasive pulmonary Aspergillosis infection. PLoS One 7: e32273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.de Jong MA, Geijtenbeek TB. 2010. Langerhans cells in innate defense against pathogens. Trends Immunol 31: 452–9 [DOI] [PubMed] [Google Scholar]
- 163.Dan JM, Kelly RM, Lee CK, Levitz SM. 2008. Role of the mannose receptor in a murine model of Cryptococcus neoformans infection. Infect. Immun 76: 2362–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Netea MG, Van Der Graaf CA, Vonk AG, Verschueren I, Van Der Meer JW, Kullberg BJ. 2002. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis 185: 1483–9 [DOI] [PubMed] [Google Scholar]
- 165.Plantinga TS, Johnson MD, Scott WK, van de Vosse E, Velez Edwards DR, et al. 2012. Toll-like receptor 1 polymorphisms increase susceptibility to candidemia. J. Infect. Dis 205: 934–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kesh S, Mensah NY, Peterlongo P, Jaffe D, Hsu K, et al. 2005. TLR1 and TLR6 polymorphisms are associated with susceptibility to invasive aspergillosis after allogeneic stem cell transplantation. Ann. N. Y. Acad. Sci 1062: 95–103 [DOI] [PubMed] [Google Scholar]
- 167.Zarember KA, Godowski PJ. 2002. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol 168: 554–61 [DOI] [PubMed] [Google Scholar]
- 168.Bochud PY, Chien JW, Marr KA, Leisenring WM, Upton A, et al. 2008. Toll-like receptor 4 polymorphisms and aspergillosis in stem-cell transplantation. N. Engl. J. Med 359: 1766–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Linden JR, De Paepe ME, Laforce-Nesbitt SS, Bliss JM. 2013. Galectin-3 plays an important role in protection against disseminated candidiasis. Med. Mycol 51: 641–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chen HY, Liu FT, Yang RY. 2005. Roles of galectin-3 in immune responses. Arch. Immunol. Ther. Exp 53: 497–504 [PubMed] [Google Scholar]
- 171.Means TK, Mylonakis E, Tampakakis E, Colvin RA, Seung E, et al. 2009. Evolutionarily conserved recognition and innate immunity to fungal pathogens by the scavenger receptors SCARF1 and CD36. J. Exp. Med 206: 637–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Soloviev DA, Jawhara S, Fonzi WA. 2011. Regulation of innate immune response to Candida albicans infections by alphaMbeta2-Pra1p interaction. Infect. Immun 79: 1546–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Donders GG, Babula O, Bellen G, Linhares IM, Witkin SS. 2008. Mannose-binding lectin gene polymorphism and resistance to therapy in women with recurrent vulvovaginal candidiasis. BJOG 115: 1225–31 [DOI] [PubMed] [Google Scholar]
- 174.Granell M, Urbano-Ispizua A, Suarez B, Rovira M, Fernandez-Aviles F, et al. 2006. Mannan-binding lectin pathway deficiencies and invasive fungal infections following allogeneic stem cell transplantation. Exp. Hematol 34: 1435–41 [DOI] [PubMed] [Google Scholar]
- 175.Cunha C, Aversa F, Lacerda JF, Busca A, Kurzai O, et al. 2014. Genetic PTX3 deficiency and aspergillosis in stem-cell transplantation. N. Engl. J. Med 370: 421–32 [DOI] [PubMed] [Google Scholar]
- 176.Garlanda C, Hirsch E, Bozza S, Salustri A, De Acetis M, et al. 2002. Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature 420: 182–6 [DOI] [PubMed] [Google Scholar]
- 177.Garlanda C, Bottazzi B, Bastone A, Mantovani A. 2005. Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu. Rev. Immunol 23: 337–66 [DOI] [PubMed] [Google Scholar]