

Abstract
Cyclo-oxygenase-2 (COX-2), a rate-limiting enzyme for prostanoid synthesis, is induced during inflammation and participates in inflammation-mediated cytotoxicity. Cerebral ischemia is followed by an inflammatory reaction that plays a role in the evolution of the tissue damage. We studied whether COX-2 is induced after cerebral ischemia and if so, whether such expression contributes to ischemic brain damage. The middle cerebral artery was occluded in rats, and the ischemic area was sampled for analysis 3–96 hr later. COX-2 mRNA was determined by the competitive reverse-transcription PCR. COX-2 mRNA was upregulated in the ischemic hemisphere, but not contralaterally, beginning 6 hr after ischemia. The upregulation reached a maximum at 12 hr, at which time a fivefold induction of the message occurred. Twenty-four hours after ischemia, the concentration of prostaglandin E2 was elevated in the injured brain by 292 ± 57% (n = 6). COX-2 immunoreactivity was observed in neurons at the medial edge of the ischemic area. Administration of the COX-2 inhibitor NS-398 attenuated the elevation in prostaglandin E2 in the postischemic brain and reduced the volume of the infarct by 29 ± 6% (p < 0.05). Thus, cerebral ischemia leads to upregulation of COX-2 message, protein, and reaction products in the injured hemisphere. The data implicate COX-2 in the mechanisms of delayed neuronal death at the infarct border and provide the rationale for neuroprotective strategies employing COX-2 inhibitors.
Keywords: stroke, prostanoids, prostaglandin H2synthase, gene expression, NS-398, reverse-transcription polymerase chain reaction, iNOS, inflammation
There is increasing evidence that the brain damage produced by cerebral ischemia develops over a period longer than previously believed. In the center of the ischemic territory, where the flow reduction is most severe, energy failure is followed by rapid cell death. However, in the surrounding region, neurons remain viable for a prolonged period of time, perhaps days (Dereski et al., 1993; Garcia et al., 1993; Marchal et al., 1996). Most research efforts to date have focused on the acute stages of cerebral ischemia (Choi, 1994; Chan, 1996), and less emphasis has been placed on the factors that contribute to the delayed progression of the injury occurring at the periphery of the infarct. The identification of such factors is important, because it might suggest new therapeutic strategies targeted at the late phase of the damage.
One of the processes that may play a role in the delayed progression of the damage is postischemic inflammation (Kochanek and Hallenbeck, 1992;Feuerstein et al., 1997). Cerebral ischemia is followed by infiltration of blood-borne neutrophils in the ischemic brain, a process initiated by local expression of cytokines, chemokines, and adhesion molecules (Pozzilli et al., 1985; Liu et al., 1994; Wang et al., 1994, 1995; Kim et al., 1995; for review, see Feuerstein et al., 1997). Although there is evidence that postischemic inflammation is deleterious to the ischemic brain, the mechanisms of its pathogenic effect have not been clearly defined (Kochanek and Hallenbeck, 1992). Expression of cyclo-oxygenase-2 (COX-2) recently has emerged as an important determinant of the cytotoxicity associated with inflammation (for review, see Seibert et al., 1995; Smith and DeWitt, 1995). COX, also known as prostaglandin H2 synthase, is a rate-limiting enzyme for prostanoid synthesis that is present in at least two isoforms: COX-1 and COX-2 (Smith and DeWitt, 1995). COX-1 is constitutively expressed in many cell types in which it produces prostanoids that subserve normal physiological functions (Smith and DeWitt, 1995). Although COX-2 normally is not present in most cells, its expression can be induced by endotoxins and cytokines (Smith and DeWitt, 1995). COX-2 is rapidly induced in inflamed tissues, and its reaction products are responsible for many of the cytotoxic effects of inflammation (Seibert et al., 1995).
In this study, we investigated whether cerebral ischemia is associated with upregulation of the COX-2 gene and if so, we sought to define whether COX-2 expression contributes to cerebral ischemic damage. We found that focal cerebral ischemia upregulates COX-2 in neurons at the periphery of the infarct and that a COX-2 inhibitor attenuates postischemic prostaglandin accumulation and reduces cerebral ischemic damage. The data provide strong evidence that COX-2 is implicated in the mechanisms of delayed neuronal death at the infarct border and suggest new neuroprotective strategies targeted at the progression of ischemic brain damage.
MATERIALS AND METHODS
Procedures for transient middle cerebral artery (MCA) occlusion. The MCA was transiently occluded in 86 Sprague Dawley rats (300–400 gm; Harlan, Indianapolis, IN) using an intravascular occlusion model (Zea Longa et al., 1989) that has been previously described in detail (Iadecola et al., 1996; Zhang et al., 1996). Under halothane anesthesia (induction, 5%; maintenance, 1%), a 4–0 nylon monofilament with a rounded tip was inserted centripetally into the external carotid artery and advanced into the internal carotid artery until it reached the circle of Willis. Throughout the procedure, body temperature was maintained at 37° ± 0.5°C by a thermostatically controlled lamp. Two hours after induction of ischemia, rats were reanesthetized, and the filament was withdrawn (Zhang et al., 1996). Animals were then returned to their cages and closely monitored until recovery from anesthesia. In sham-operated rats, the external carotid artery was surgically prepared for insertion of the filament, but the filament was not inserted (Zhang et al., 1996). Rats were killed at different time points after transient ischemia for mRNA determination, measurement of prostaglandin E2 (PGE2), and immunocytochemistry (see below).
Reverse-transcription PCR (RT-PCR). mRNA for COX-1, COX-2, and inducible nitric oxide synthase (iNOS) were detected by the RT-PCR (Kawasaki et al., 1988) as described previously (Iadecola et al., 1995;Ross and Iadecola, 1996). Animals were killed 3, 6, 12, 24, 48, and 96 hr after ischemia (n = 4 per time point) and their brains removed. Sham-operated rats served as controls (n = 4). A 4-mm-thick coronal brain slice was cut at the level of the optic chiasm, and the infarcted cortex was dissected using the corpus callosum as a ventral landmark. The corresponding region of the contralateral cortex was also sampled. Total RNA was extracted from the samples according to the method of Chomczynski and Sacchi (Chomczynski and Sacchi, 1987). RNA integrity was determined on denaturing formaldehyde gels. Aliquots of total RNA (0.25 μg) were used in the RT reaction mixed with 0.5 μg of oligo (dT) primer as directed (18 mer; New England Biolabs, Beverly, MA). First-strand cDNA synthesis was then performed using 0.25 μg of total RNA and M-MuLV reverse transcriptase (New England Biolabs) according to the manufacturer’s instructions. After heating at 95°C for 10 min, 5 μl from each RT reaction mixture was used for PCR amplification. Primers (0.2 μm each) for the sequence of interest and for porphobilinogen deaminase (PBD), a ubiquitously expressed sequence, were used in a final volume of 50 μl. The COX-2 primers were: forward, 5′-CCATGTCAAAACCGTGGTGAATG-3′; reverse: 5′-ATGGGAGTTGGGCAGTCATCAG-3′, which result in a PCR product of 374 bp. The COX-1 primers were: forward, 5′-TCTGATGCTCTTCTCCACGATCTG-3′; reverse, 5′-CAAAGTTCCTACCCCCACCAATC-3′, which result in a 431 bp PCR product. The iNOS primers were: forward, 5′-ACAACGTGGAGAAAACCCCAGGTG-3′; reverse, 5′-ACAGCTCCGGGCATCGAAGACC-3′, which result in a PCR product of 557 bp (Iadecola et al., 1996). The “hot start” method was used (Stratagene, La Jolla, CA) with the following cycle parameters: 94°C, 15 sec; 68°C, 30 sec; 73°C, 20 sec × 5 cycles, then 94°C, 15 sec; 64°C, 30 sec; 73°C, 20 sec × 35 cycles, and 73°C, 15 min. Reaction products were then separated on a 8% polyacrylamide gel, stained with ethidium-bromide, and photographed. Each set of PCR reactions included control samples run without RNA or samples in which the RT step was omitted to ensure that PCR products resulted from amplification from the COX-2 mRNA rather than from genomic DNA. The optical density of the bands was determined by an image analysis system (MCID, M4; Image Research). Measurements were normalized to the optical density of the PBD band used as an internal standard.
Competitive RT-PCR. Competitive RT-PCR was used to determine more accurately the magnitude of mRNA induction (Wang et al., 1989;Siebert and Larrick, 1992; Ross and Iadecola, 1996). A deletion construct was synthesized consisting of the same sequence amplified from the endogenous COX-2 message but missing an internal 81 nucleotide fragment. To generate the construct, another pair of COX-2 primers was prepared: forward: 5′-CCAGATGCTATCTTTGGGGAGAC-3′; reverse: 5′-ACTTGCGTTGATGGTGGCTG-3′, which result in a 249 bp PCR product. The PCR product was then digested with two restriction enzymes,HaeIII and AlwI (New England Biolabs). After inactivating the restriction endonucleases, the sample was religated at 15°C overnight with T4 DNA ligase (New England Biolabs). A 5 μl aliquot of the religated sample was then amplified using the COX-2 forward and reverse primers described above. The products were separated on an acrylamide gel, and a main product of 168 bp was excised from the gel for use in the competition assay. The RT reaction mixtures (5 μl each) of the animals killed 12 hr after ischemia (n = 4) and sham-operated (n = 4) were co-amplified with known amounts of deletion construct (1–100 fg). The PCR products were then separated on a gel and the gel stained with ethidium-bromide and photographed (see Fig. 2A). The optical density of the bands was determined by image analysis. For data analysis, the logarithm of the ratio of the density (COX-2/construct) was plotted as a function of the log of the concentration of the construct and fitted by linear regression analysis (see Fig.2B). The 0 value of the log of the ratio (COX-2/construct) (y-axis) represents the point at which the COX-2 PCR product and construct are present in equal amounts. Therefore, the amount of the construct corresponding to the 0 ratio (x-axis) corresponds to the amount of the COX-2 PCR product before PCR amplification (Diviacco et al., 1992; Galea and Feinstein, 1992) (see Fig. 2B,C).
Fig. 2.
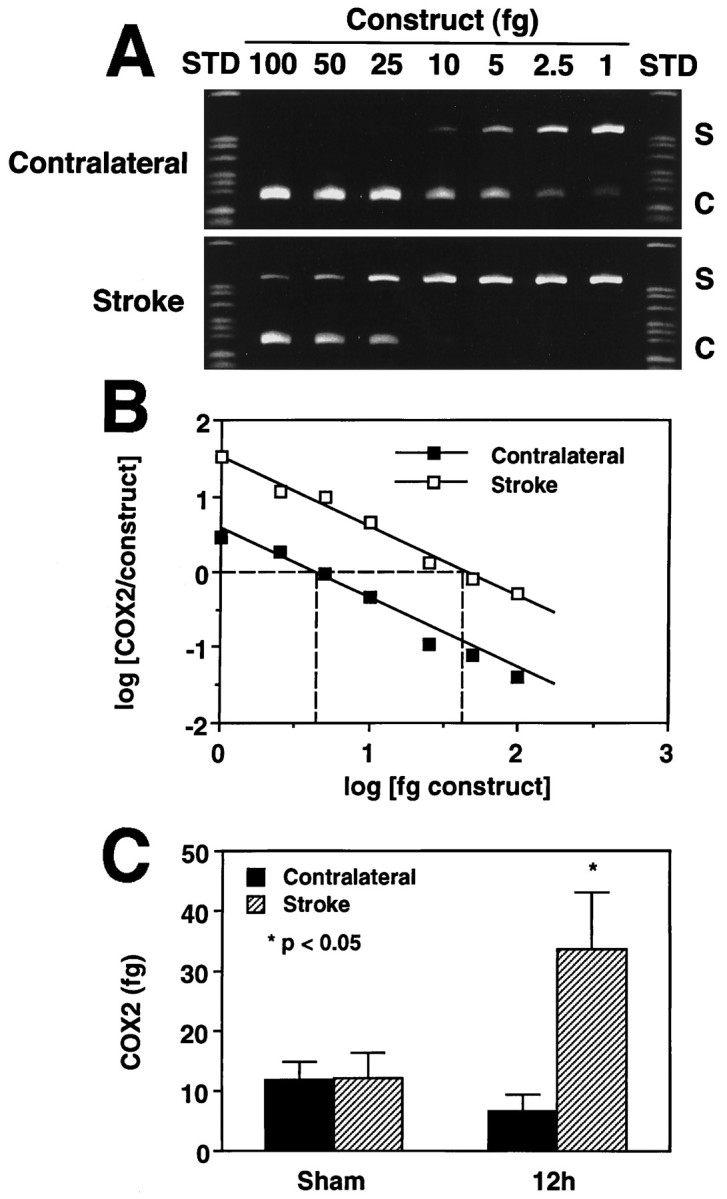
Competitive PCR to quantify the magnitude of COX-2 expression after cerebral ischemia. Samples were obtained from animals killed 12 hr after stroke. A, Competition between the COX-2 PCR product (S) and increasing amounts of a construct (C) produced by deletion of an internal portion of the COX-2 PCR product. Notice that a higher amount of construct is needed to compete out the COX-2 PCR product on the stroke side than on the contralateral side. B, Quantitative analysis of the gel presented in A. The log of the ratio of the density (COX-2/construct) was plotted as a function of the log of the concentration of the construct and fitted by linear regression analysis. The 0 value of the log of the ratio (COX-2/construct) (y-axis) represents the point at which the COX-2 PCR product and construct are present in equal amounts. Therefore, the amount of the construct corresponding to the 0 ratio (x-axis) corresponds to the amount of the COX-2 PCR product before PCR amplification. C, Group data on COX-2 expression in sham-operated rats and in rats 12 hr after transient MCA occlusion. In sham-operated rats (n = 4), COX-2 does not differ between sides. After ischemia (n = 4), there is a marked increase in the COX-2 PCR product.
COX-2 immunohistochemistry. Immunocytochemical procedures were identical to those described previously (Iadecola et al., 1995,1996). Six, 12, 24, 48, and 96 hours after ischemia, rats (n = 3 per time point) were anesthetized (pentobarbital, 100 mg/kg, i.p.) and perfused through the heart with 4% paraformaldehyde. Brains were removed, post-fixed, and embedded in paraffin. Coronal sections (7 μm thick) through the infarct were cut using a microtome and mounted on microscope slides. After removing paraffin, sections were quenched with hydrogen peroxide, washed, and incubated with horse serum (Vector Laboratories, Burlingame, CA) for 3 hr, before incubation overnight (4°C) with a polyclonal COX-2 antibody (Cayman Chemical, Ann Arbor, MI; dilution 1:200). After incubation with the secondary antibody (Vector) for 30 min, the immunocomplex was visualized using diaminobenzidine as a chromogen in a peroxidase reaction (ABC; Vectastain Elite Kit, Vector). To assist in the determination of the cellular localization of the label, some sections were counterstained with hematoxylin and eosin. Adjacent sections were stained for the astroglial marker GFAP as described previously (Iadecola et al., 1995). Slides were viewed and photographed using a Nikon Optiphot microscope.
PGE2 enzyme immunoassay. Tissue concentration of PGE2, one of the major COX reaction products, was determined using an enzyme immunoassay kit (Cayman Chemical) (Pradelles et al., 1985; Salvemini et al., 1995). Samples from the infarct and contralateral brain were collected 24 hr after ischemia, as described above for RT-PCR, and frozen in liquid nitrogen. The tissue was homogenized in 0.05 m Tris-HCl, pH 7.4 (4 ml/g) and extracted with 100% methanol (Powell, 1982). After centrifugation, the supernatant was diluted with acidified 0.1 m phosphate buffer, pH 4, (final methanol concentration, 15%) and applied to activated ODS-silica reverse-phase columns (Sep-Pak C18, Waters Associates, Milford, MA). The columns were rinsed with 5 ml of distilled water followed by 5 ml of hexane, and PGE2 was eluted twice with 2 ml of ethyl acetate containing 1% methanol. The ethyl acetate fraction was evaporated and resuspended in 1 ml of buffer. The recovery rate of this extraction procedure, determined using [3H]PGE2, was 74.7 ± 1.2% (n = 10). PGE2 concentration was determined spectrophotometrically after incubation with tracer and PGE2 monoclonal antibody in a microplate according to the manufacturer’s instructions.
Effect of NS-398 on ischemic damage and PGE2elevation. Under halothane anesthesia, the femoral artery was cannulated, and rats were placed on a stereotaxic frame. The arterial catheter was used for monitoring of arterial pressure and other parameters at different times after MCA occlusion (see below). The MCA was occluded for 2 hr, as described above, and treatments were begun 6 hr after induction of ischemia. In one group of rats (n= 6), the COX-2 inhibitor NS-398 (Futaki et al., 1993a; Masferrer et al., 1994) was administered. NS-398 inhibits COX-2 with an IC50 that is >1000 smaller than that of COX-1 (Reitz et al., 1994). NS-398 (Cayman Chemical; 20 mg/kg) was administered intraperitoneally at 1000 and 1800 hr for 3 consecutive days. A second group of rats (n = 7) was treated with vehicle (saline; 1 ml at 1000 and 1800 hr) for 3 d. Arterial pressure, rectal temperature, and plasma glucose were measured daily at 0900, 1300, 1700, and 2100 hr. Arterial hematocrit and blood gases were measured before injection and 24, 48, and 72 hr after ischemia (Table 1). Three days after MCA occlusion, brains were removed and frozen in cooled isopentane (−30°C). Coronal forebrain sections (30 μm thick) were serially cut in a cryostat, collected at 300 μm intervals, and stained with thionin for determination of infarct volume by an image analyzer (MCID, Imaging Research) (Iadecola et al., 1995). Infarct volume in cerebral cortex was corrected for swelling according to the method of Lin et al. (Lin et al., 1993), which is based on comparing the volumes of neocortex ipsilateral and contralateral to the stroke. The correction for swelling was needed to factor out the contribution of ischemic swelling to the total volume of the lesion (see Zhang and Iadecola, 1994b).
Table 1.
Arterial blood gases and hematocrit in rats treated with NS-398 after focal cerebral ischemia
| Time (hr) | Treatment | Arterial blood gases | Hematocrit (%) | ||
|---|---|---|---|---|---|
| pH | pCO2 (mmHg) | pO2 (mmHg) | |||
| 6 | Vehicle | 7.35 ± 0.02 | 41 ± 1 | 86 ± 4 | 49.3 ± 0.9 |
| NS-398 | 7.35 ± 0.03 | 40 ± 1 | 83 ± 5 | 49.2 ± 0.5 | |
| 24 | Vehicle | 7.39 ± 0.03 | 39 ± 2 | 99 ± 4 | 48.3 ± 0.6 |
| NS-398 | 7.39 ± 0.03 | 40 ± 1 | 104 ± 5 | 48.5 ± 0.4 | |
| 48 | Vehicle | 7.37 ± 0.07 | 39 ± 3 | 90 ± 8 | 48.5 ± 0.5 |
| NS-398 | 7.33 ± 0.04 | 39 ± 1 | 86 ± 3 | 48.5 ± 0.6 | |
| 72 | Vehicle | 7.34 ± 0.05 | 41 ± 2 | 88 ± 7 | 48.0 ± 0.9 |
| NS-398 | 7.31 ± 0.05 | 44 ± 2 | 90 ± 4 | 48.0 ± 0.7 | |
No statistically significant differences were found between rats treated with vehicle or NS-398 (p > 0.05,t test).
In separate rats, the effect of NS-398 on the elevation in the COX-2 product PGE2 was studied. In these experiments, PGE2 concentration was determined 24 hr after induction of ischemia, because at this time the concentration of this prostaglandin is elevated substantially in the ischemic brain (see Results). NS-398 was administered intraperitoneally (20 mg/kg) 6, 14, and 22 hr after induction of ischemia. Rats were killed for PGE2determination 2 hr after the last NS-398 administration.
Data analysis. Data are expressed as mean ± SE. Two group comparisons were evaluated by the paired or unpaired ttest, as appropriate. Multiple comparisons were analyzed by the ANOVA and Tukey’s test. Differences were considered statistically significant for p < 0.05.
RESULTS
Cerebral ischemia and COX-2 mRNA expression
In agreement with previous reports (Yamagata et al., 1993), low levels of COX-2 PCR product were observed in the brain of sham-operated rats (n = 4; Figs. 1A,2C). After transient MCA occlusion, a marked upregulation of COX-2 mRNA was observed in the postischemic brain, but not contralaterally (Fig. 1A). The upregulation began 6 hr after ischemia, reached a maximum at 12–24 hr, and subsided at 48 hr (Fig. 1A). In contrast, COX-1 remained unchanged in the postischemic period (Fig. 1B). To determine more accurately the magnitude of the mRNA upregulation, we used competitive PCR (Fig. 2). In sham-operated rats, low levels of COX-2 signal were detected on both sides of the brain. Twelve hours after ischemia, COX-2 was upregulated in the injured brain by fivefold (Fig. 2C). Therefore, cerebral ischemia results in upregulation of COX-2 message in the postischemic brain.
Fig. 1.
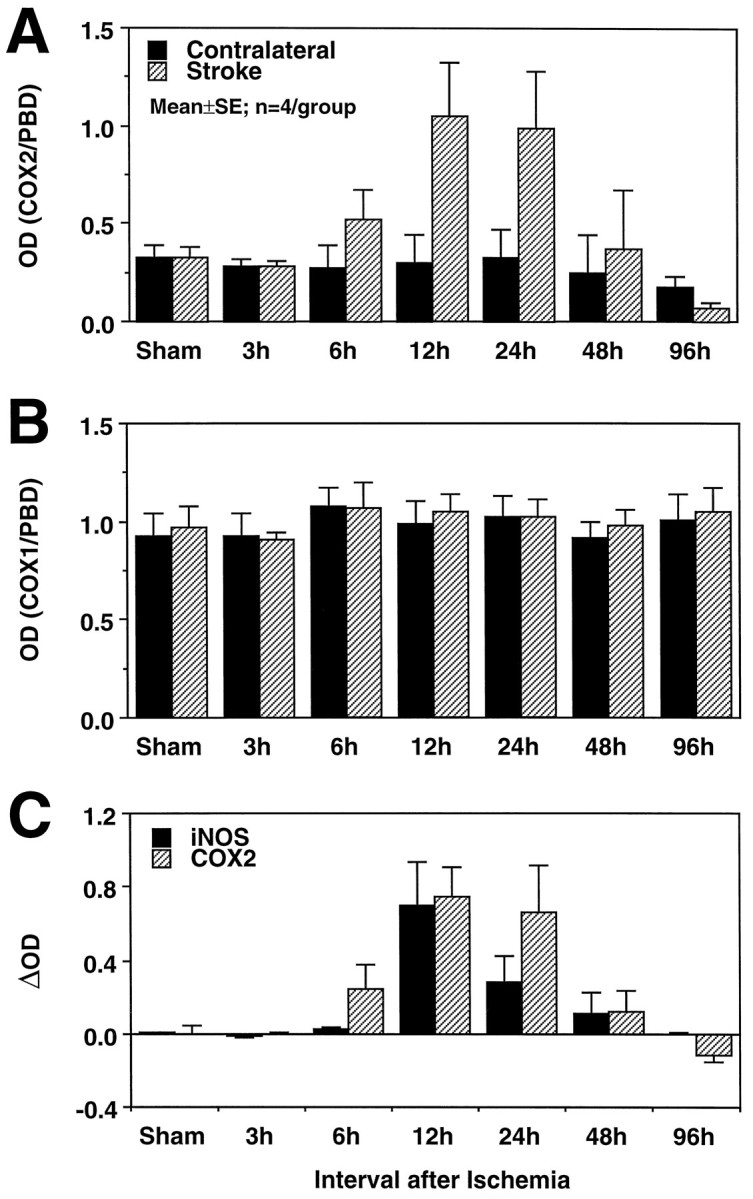
A, Effect of transient focal ischemia on COX-2 mRNA expression detected by RT-PCR. PCR products were run on a gel, and the optical density of the bands was measured by image analysis. The density of the COX-2 band was divided by the density of the band of a ubiquitous gene, PBD, used as a normalization factor. Each time point represents the average of four rats. The COX-2 signal increases at 6 hr, reaches a peak at 12–24 hr, and returns to baseline at 48–96 hr. No changes in COX-2 expression are seen in the brain contralateral to the stroke. B, In contrast to COX-2, COX-1 mRNA does not increase after cerebral ischemia on either side of the brain. The density of the COX-1 band was normalized by the PBD band as described in A. The fact that the mRNA for COX-1, an enzyme closely related to COX-2, was not increased attests to the selectivity of the COX-2 upregulation and to the selectivity of the RT-PCR technique used in the present study. C, Comparison of the time course of COX-2 and iNOS expression after transient cerebral ischemia. Data are presented as δ, obtained by subtracting the density of the COX-2 or iNOS bands in the contralateral nonischemic side from that of the stroke side. The time course of COX-2 and iNOS expression in the postischemic period is similar.
Cerebral ischemia also leads to iNOS expression in the injured brain (Iadecola et al., 1995, 1996). Because iNOS may be co-expressed with COX-2 (Salvemini et al., 1993; Vane et al., 1994), we compared the temporal profile of COX-2 and iNOS mRNA upregulation after ischemia. As illustrated in Figure 1C, the time courses of COX-2 and iNOS mRNA expression were similar. Therefore, iNOS and COX-2 are expressed over the same time period after cerebral ischemia.
Cerebral ischemia and COX-2 immunocytochemistry
We then used immunocytochemistry to determine whether upregulation of COX-2 mRNA resulted in increased synthesis of the COX-2 protein and to identify the cells in which COX-2 is upregulated. In the brain of sham- operated rats, COX-2-immunoreactive neurons were observed sparsely in cerebral cortex, hippocampus, piriform cortex, and amygdala (Yamagata et al., 1993; Breder et al., 1995). Cerebral ischemia produced a marked upregulation of COX-2 immunoreactivity, which was first observed 6 hr after MCA occlusion. The most marked upregulation occurred 12–24 hr after ischemia. At this time, numerous COX-2-immunoreactive cells were observed only on the stroke side (Fig.3). The majority of COX-2-immunoreactive cells were localized at the infarct border (Figs. 3A, 4). COX-2 cells had the morphological characteristics of neurons with a large round nucleus and a prominent nucleolus. GFAP-positive astrocytes were not observed in the region in which COX-2-positive cells were located. Some immunoreactive cells resided in normal brain near the medial edge of the infarct (Fig. 3A). In these neurons, the intracellular distribution of the immunoreactivity was predominantly perinuclear (Fig. 3B). Other COX-2-positive cells were located in the transitional region between normal and infarcted brain (Fig.3C). These cells had the morphological characteristics of ischemic neurons with a shrunken cytoplasm and condensed nucleus (Fig.3D). An increased number of COX-2-positive neurons were observed in the ipsilateral piriform cortex (Fig. 4). COX-2-immunoreactive cells were not increased in the contralateral cerebral cortex (Fig. 3A). COX-2 immunoreactivity returned to baseline 4 d after ischemia.
Fig. 3.
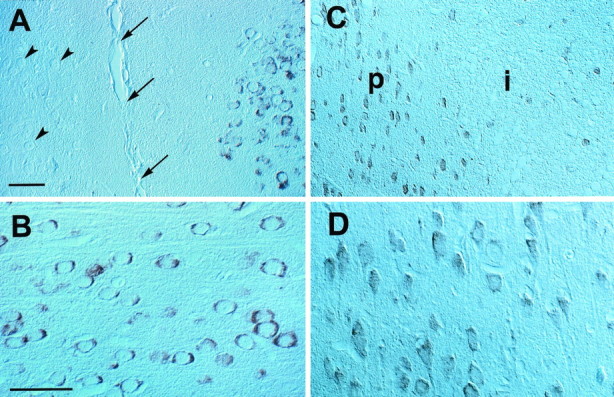
Effect of focal cerebral ischemia on COX-2 immunoreactivity in paraffin-embedded sections (7 μm thickness) of rats subjected to a 2 hr occlusion of the MCA. A, Some COX-2-immunoreactive cells are located in the intact cingulate cortex medial to ischemic lesion. This region corresponds to the border zone between the anterior cerebral artery and MCA. The arrowspoint to the midline (intrahemispheric fissure); thearrowheads point to unstained neurons in the contralateral side. B, High-power view of the cells depicted in A. These cells have the morphological characteristics of normal neurons, with a large round nucleus and a prominent nucleolus. The immunoreactivity is characteristically perinuclear (cf. Yamagata et al., 1993). C, Other COX-2-positive cells are located in the transitional region between normal and infarcted brain. These cells have an angular appearance, with a shrunken cytoplasm and nucleus. In alternate sections stained with hematoxylin and eosin, these cells correspond to neurons exhibiting distinct ischemic changes (“red neurons”). Therefore, COX-2 is expressed also in injured neurons at the periphery of the ischemic territory. The region in which these COX-2 neurons are located corresponds to the so-called ischemic penumbra. The positive cells observed within the infarct are most likely shrunken neurons.P, Anatomical location of the ischemic penumbra;i, infarct. D, High-power view of the COX-2-immunoreactive neurons depicted in C. Notice the cell shrinkage. Scale bars: A, C, 500 μm; B, D, 150 μm.
Fig. 4.

Spatial distribution of COX-2-immunoreactive cells throughout the brain after transient occlusion of the MCA. The diagram depicts data from four rats. The black area represents the region of infarction identified by counterstaining the sections processed for COX-2 immunocytochemistry with hematoxylin and eosin. The darker and lighter shades of gray indicate, respectively, higher and lower density and stain intensity of positive cells. The majority of COX-2-immunoreactive neurons are located medial to the infarct in a region corresponding to the border zone of the territories of the anterior cerebral artery and MCA. Some neurons are located in the normal cortex medial to the infarcted region (cingulate cortex). Other neurons are located at the transition between normal and infarcted tissue (see Fig. 3). An increased number of COX-2-immunoreactive cells is also observed in the ipsilateral piriform cortex. In this region, the cells closer to the rhinal fissure are near the inferolateral aspect of the cortical infarct. Rare lightly stained neurons are seen in the medial border of the striatal infarct. Therefore, the majority of COX-2 positive neurons are located at the medial border of the cortical infarct.
PGE2 concentration in the postischemic brain
To determine whether the upregulation of COX-2 protein corresponded to an increase in COX-2 enzymatic activity, the concentration of PGE2 was measured in the postischemic brain 24 hr after induction of ischemia. Cerebral ischemia increased PGE2 concentration in the injured brain by 292 ± 57% (p < 0.05; n = 6; Fig.5A). No increase was observed in the contralateral cortex or in the cortex of sham-operated rats (p> 0.05; n = 6; Fig. 5A). These data suggest that COX-2 enzymatic activity is increased in the postischemic brain.
Fig. 5.
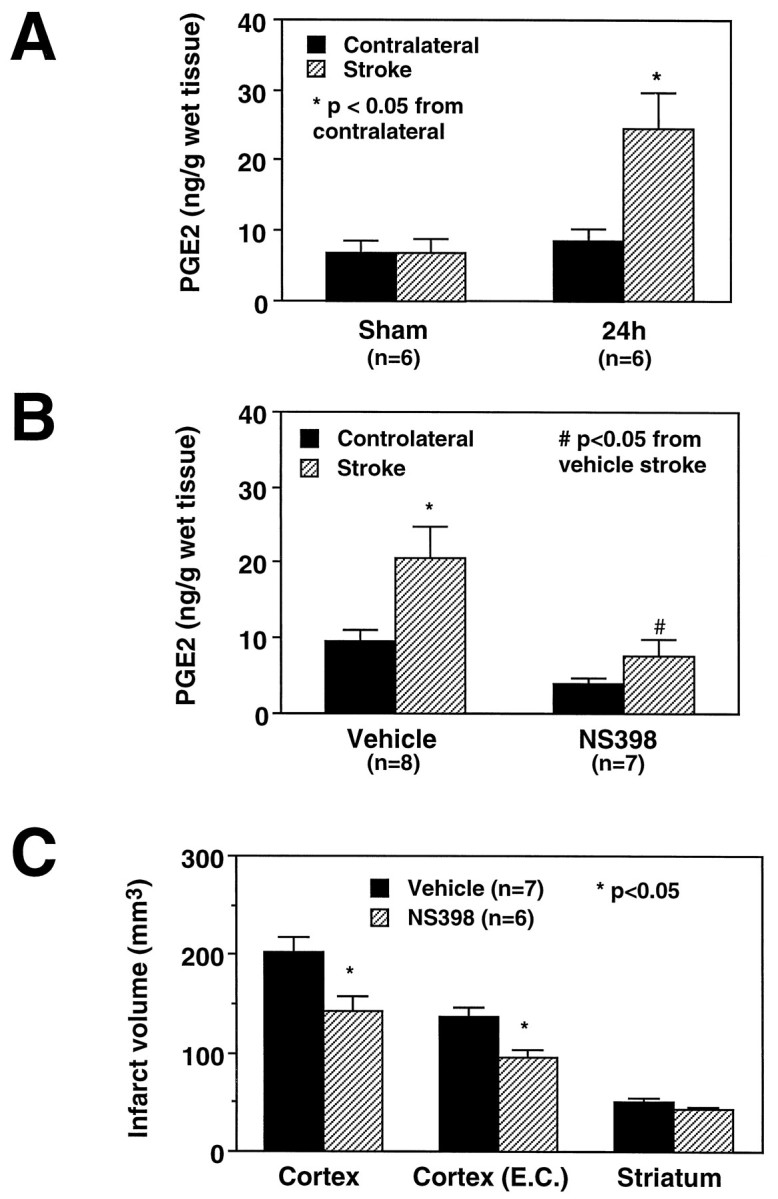
A, Effect of transient MCA occlusion on (PGE2) in the postischemic brain. In sham-operated rats, low levels of PGE2 are present in the brain. Cerebral ischemia increases PGE2 concentration 24 hr after stroke only on the ischemic side (p < 0.05, t test; n = 6).B, Effect of the COX-2 inhibitor NS-398 on postischemic increase in PGE2 in the injured brain. NS-398 (20 mg/kg, i.p.) was administered starting 6 hr after transient MCA occlusion. Rats were killed 24 hr after ischemia. At the time of death, the rats had received three doses. NS-398 attenuates the postischemic increases in PGE2 (p < 0.05 from vehicle; ANOVA and Tukey’s test). The residual increase in PGE2after NS-398 did not reach statistical significance (p > 0.05). NS-398 slightly reduced resting levels of PGE2 in the cerebral cortex contralateral to the stroke. However, such reduction did not reach statistical significance (p > 0.05). C, Effect of NS-398 on the volume of the infarct produced by transient MCA occlusion in the rat. Rats were treated for 3 d (20 mg/kg, i.p., twice per day) starting 6 hr after induction of ischemia. NS-398 reduced the volume of the infarct in the cerebral cortex but not in the striatum. The reduction in infarct volume persists after correction for ischemic swelling [Cortex (E.C.)], suggesting that the reduction in the lesion volume is not attributable to an effect of NS-398 on ischemic edema.
Effect of COX-2 inhibition on cerebral ischemic damage
To determine whether COX-2 expression contributes to cerebral ischemic damage, we used the relatively selective COX-2 inhibitor NS-398 (Futaki et al., 1993a; Masferrer et al., 1994). Treatment with NS-398 (20 mg/kg, i.p., 2 times per day for 3 d) did not affect arterial pressure, rectal temperature, plasma glucose, arterial blood gases, and hematocrit (Fig. 6; Table 1). However, in rats treated with NS-398, the size of the infarct was smaller than that of vehicle-treated controls (Figs. 5C, 7). The reduction averaged 29 ± 6% in the cerebral cortex (Fig. 5C;p < 0.05). The area spared from infarction was located at the periphery of the ischemic lesion and involved the border zone between the vascular territories of the anterior cerebral and MCAs (Fig. 7). Infarct size was not reduced in the striatum (Fig. 3C).
Fig. 6.

Arterial pressure, plasma glucose, and rectal temperature of the rats in which the effect of NS-398 on cerebral ischemic damage was studied (see Fig. 5C). NS-398 does not affect these parameters at any of the time points studied (p > 0.05; t test from vehicle).
Fig. 7.

Spatial distribution of the infarct produced by transient MCA occlusion in vehicle-treated rats and in rats treated with the COX-2 inhibitor NS-398. In NS-398-treated rats, the lesion is smaller at all rostro-caudal levels. The area “rescued” from infarction involves primarily the medial edge of the lesion and includes the region in which the COX-2-positive neurons are located (cf. Fig. 4)
To determine whether treatment with NS-398 was effective in reducing COX-2 activity in the postischemic brain, PGE2concentration was measured 24 hr after transient MCA occlusion in rats treated with NS-398 (n = 6) or vehicle (n = 7). NS-398 attenuated the postischemic increases in PGE2 (p < 0.05 from vehicle; ANOVA and Tukey’s test) (Fig. 5B). After NS-398 treatment, the PGE2 concentration in the injured brain was not statistically different from that in the contralateral (intact) side (p > 0.05). NS-398 slightly reduced resting levels of PGE2 in the cerebral cortex contralateral to the stroke (Fig. 5B). However, such reduction did not reach statistical significance (p > 0.05). These data suggest that NS-398 inhibits postischemic COX-2 activity and ameliorates cerebral ischemic damage.
DISCUSSION
The development of ischemic cell death is asynchronous in the different regions of the ischemic territory (Dereski et al., 1993). Whereas in the center of the lesion, severe ischemia leads to rapid pan-necrosis, in the surrounding regions, the tissue damage evolves slowly over many hours (Dereski et al., 1993; Garcia et al., 1993;Marchal et al., 1996). The factors involved in the secondary progression of the injury at the infarct border have not been fully elucidated (Kochanek and Hallenbeck, 1992; Feuerstein et al., 1997). After cerebral ischemia, there is infiltration of the affected brain by inflammatory cells, a process initiated by expression of inflammation-related genes in the postischemic brain (for review, seeFeuerstein et al., 1997). Although there is evidence that such inflammation contributes to the progression of cerebral ischemic damage, the mechanisms of the effect remain unclear (Kochanek and Hallenbeck, 1992; Feuerstein et al., 1997). The prostanoid-synthesizing enzyme COX-2 recently has emerged as an important factor in the cytotoxicity associated with inflammation (Seibert et al., 1995). Therefore, in this study, we investigated whether COX-2 is expressed in the postischemic brain and if so, whether its reaction products contribute to the secondary evolution of the damage. We found that focal cerebral ischemia is associated with marked upregulation of COX-2 mRNA in the affected hemisphere starting between 3 and 6 hr after cerebral ischemia. This finding is in agreement with a recent report in which COX-2 mRNA was detected by Northern analysis after transient focal ischemia (Collaco-Moraes et al., 1996). The upregulation is restricted to COX-2 and does not involve the closely related prostaglandin-synthesizing enzyme COX-1. Immunocytochemical experiments showed that COX-2 protein is also upregulated, the expression occurring in neurons located primarily at the infarct border. Some COX-2- positive neurons are devoid of pathological changes and are located in the intact brain. Other COX-2 neurons exhibit ischemic changes and reside in the transitional region between normal and infarcted brain. We did not observe cells the morphology of which is consistent with that of neutrophils, the inflammatory cell present at this time after ischemia. However, this evidence is far from conclusive, and the potential localization of COX-2 to other cell types will have to be explored further in future studies. The COX-2 message and protein upregulation is associated with increased tissue concentration of PGE2, one of the COX-2 reaction products. These observations indicate that COX-2 mRNA is translated in a functional enzyme. Thus, cerebral ischemia leads to upregulation of COX-2 mRNA, protein, and reaction products.
We then sought to determine whether the COX-2 upregulation, through reaction products of the COX-2 enzymatic pathway, contributes to the progression of cerebral ischemic damage occurring in the postischemic period. Although COX reaction products have long been implicated in the mechanisms of ischemic stroke (for review, see Hsu et al., 1989), previous studies focused on the effects of prostanoids in the acute stages of cerebral ischemia. In addition, previous studies have reported the effect of nonselective COX inhibitors, e.g., indomethacin and ibuprofen, on cerebral ischemic damage. The results of these investigations have been contradictory; some report protection and others show no effect or worsening (Harris et al., 1982; Johshita et al., 1989; Cole et al., 1993). These conflicting observations are likely to result from the fact that the effects of COX-2 inhibition were confounded by effects of COX-1 inhibition, an enzyme involved in normal cellular function. Furthermore, indomethacin, one of the agents studied most extensively, has profound effects on cerebral blood flow and vascular reactivity (for review, see Busija and Heistad, 1984). Because indomethacin was administered before or shortly after induction of ischemia (Harris et al., 1982; Johshita et al., 1989), it is likely that the attendant cerebrovascular effects of this agent influenced the outcome of cerebral ischemia.
To avoid confounding effects resulting from COX-1 inhibition, we used NS-398, a relatively selective inhibitor of COX-2 (Futaki et al., 1993b; Masferrer et al., 1994). In vitro, this agent inhibits COX-2 1000-fold more potently than COX-1 (Reitz et al., 1994).In vivo, NS-398, at a dose similar to that used in the present study, inhibits COX-2 but not COX-1 (Futaki et al., 1993a;Masferrer et al., 1994). We found that delayed treatment of rats with NS-398 reduces the size of the infarct produced by MCA occlusion. The brain area spared from infarction includes the region in which COX-2-positive neurons are located. These results are consistent with the hypothesis that COX-2 reaction products contribute to the delayed progression of the tissue damage that occurs in the postischemic brain. To confirm that NS-398 inhibited COX-2 activity in brain, we studied the effect of NS-398 on the elevation in PGE2 produced by cerebral ischemia. It was found that NS-398 markedly attenuates the postischemic elevation in PGE2. This observation indicates that NS-398 is able to enter the postischemic brain and to inhibit cerebral COX activity. The alteration in blood–brain barrier permeability that follows focal cerebral ischemia (Anwar et al., 1993) is likely to facilitate the penetration of NS-398 into the ischemic region. The protection exerted by NS-398 is not attributable to effects on body temperature, arterial pressure, blood gases, plasma glucose, or hematocrit, because these parameters were monitored and did not differ between treated and untreated groups. It is also unlikely that NS-398 reduced cerebral ischemic damage by improving postischemic blood flow, because at the time when the NS-398 treatment was instituted, i.e., 6 hr after ischemia, vascular-hemodynamic factors no longer influence tissue outcome (Overgard et al., 1994; Zhang and Iadecola, 1994a). Effects of NS-398 on platelet aggregation are also unlikely, because platelets contain COX-1 and not COX-2 (Klein et al., 1994). However, additional studies are needed to better define the cerebrovascular effects of NS-398 and to characterize the dose–response and temporal relationships of its protective effect in cerebral ischemia.
The mechanisms responsible for postischemic COX-2 induction remain to be defined. In the normal brain, COX-2 is expressed in selected neurons (Yamagata et al., 1993; Breder et al., 1995). COX-2 is induced in granule neurons during high-frequency hippocampal stimulation, suggesting that COX-2 expression may be regulated by normal synaptic activity (Yamagata et al., 1993). In experimental seizures, COX-2 is upregulated in neurons, an effect blocked by the NMDA receptor antagonist MK801 (Yamagata et al., 1993). The latter observation suggests that COX-2 expression may be induced by activation of glutamate receptors. Because glutamate is also released in cerebral ischemia, it is conceivable that activation of glutamate receptors participates in postischemic COX-2 induction. However, considering that glutamate is released in the penumbra only within the first 2 hr after MCA occlusion (Takagi et al., 1993), activation of glutamate receptors is unlikely to mediate the upregulation of COX-2 observed at 12 and 24 hr after ischemia. The time course of COX-2 expression closely follows the temporal profile of inflammatory genes, including genes encoding for cytokines, adhesion molecules, and iNOS (Feuerstein et al., 1997; present study). Cytokines are well known to induce COX-2 expressionin vitro (Jones et al., 1993). Therefore, cytokines could also contribute to postischemic COX-2 induction, particularly in neurons located in the ischemic territory where cytokine levels are increased (Liu et al., 1993; Wang et al., 1995). Another agent released in the ischemic brain that could also participate in COX-2 expression is the platelet-activating factor (Bazan et al., 1994). Therefore, it is likely that multiple factors are responsible for the upregulation in COX-2 observed in the postischemic period. The molecular mechanisms of COX-2 expression after ischemia are likely to involve interactions with regulatory elements in the 5′ flanking region of the COX-2 gene that include NFκb and NF-IL6/C/EBP binding sequences (Sirois and Richards, 1993).
Of interest is the observation that COX-2 and iNOS are induced over a similar time period after cerebral ischemia. Co-induction of iNOS and COX-2 has also been reported in other models of inflammation (Corbett et al., 1993; Salvemini et al., 1993; Vane et al., 1994). Because COX-2 is a heme-containing enzyme, its enzymatic activity is modulated by NO, a gas with high affinity for heme iron (Ignarro, 1991). In some models of inflammation, NO produced by iNOS has been shown to activate COX-2 and to increase its output of proinflammatory prostaglandins (Salvemini et al., 1993, 1995). We have demonstrated previously that iNOS induction and NO production contribute to focal cerebral ischemic damage (Iadecola et al., 1995, 1996). The finding that iNOS and COX-2 are co-induced after stroke raises the possibility that NO activates COX-2, thereby increasing the toxic output of the enzyme. Therefore, COX-2 activation could be another mechanism by which NO exerts its pathogenic effect on the ischemic brain.
Although the factors responsible for the cytotoxicity of COX-2 have not been clearly defined, it is likely that one of the mechanisms is related to production of reactive oxygen species (ROS). ROS are considered to be one of the major determinants of ischemic brain death (Chan, 1996). ROS are produced by the peroxidase step of the COX reaction in which prostaglandin G2 is converted to prostaglandin H2 (Chan and Fishman, 1980; Kontos et al., 1980; Armstead et al., 1988; Tsai et al., 1994). Cerebral ischemia results in an increase in the availability of arachidonic acid, the substrate for the COX enzymatic pathway (Chan et al., 1985). Our finding that COX-2 is markedly upregulated after cerebral ischemia suggests that the COX-2 pathway is an important route for arachidonic acid metabolism and free radical production in the postischemic brain. COX-2 enzymatic activity can also mediate tissue damage by producing proinflammatory prostanoids (Seibert et al., 1995). A third mechanism by which COX-2 could contribute to cell death is related to induction of apoptosis. In thymocytes, the COX-2 reaction product PGE2 induces apoptosis (Juzan et al., 1992). This finding raises the possibility that COX-2 contributes to postischemic apoptosis (Li et al., 1995). However, evidence that COX-2 overexpression in intestinal cells prevents apoptosis has also been presented (Tsujii and DuBois, 1995). Additional studies are needed to define the relative contribution of these pathogenic mechanisms to ischemic cell death related to COX-2 upregulation.
The finding that the COX-2 inhibitor NS-398 reduces cerebral ischemic damage when administered 6 hr after induction of ischemia has important implications for the treatment of stroke. Most patients with ischemic stroke reach the emergency room several hours after the onset of symptoms, at a time when most experimental therapeutic interventions are no longer effective (Marshall and Mohr, 1993). Therefore, in addition to therapeutic interventions targeted to the early stages of the damage, it would be highly desirable to develop strategies aimed at the delayed phase of the injury. In this context, COX-2 inhibitors would be valuable, because they could be used to target the delayed progression of the damage. However, additional studies are required to better characterize the effect of COX-2 inhibitors on cerebral ischemic damage and to define their potential use in human stroke.
In conclusion, we have demonstrated that focal cerebral ischemia induces expression of COX-2 mRNA, protein, and reaction products in the postischemic brain. The expression occurs in neurons at the periphery of the infarct. The relatively selective COX-2 inhibitor NS-398, administered 6 hr after induction of ischemia, reduces cerebral ischemic damage at the periphery of the infarct. The findings provide evidence that COX-2 expression is deleterious to the ischemic brain. In particular, COX-2 reaction products may contribute to recruit potentially salvageable regions into infarction at the border of the ischemic territory. Inhibition of COX-2 may be a valuable therapeutic strategy targeted specifically to the delayed progression of the infarct that occurs in the postischemic period.
Footnotes
This work was supported by National Institutes of Health Grants NS34179 and NS35806. S.N. is a Fellow of the American Heart Association (Minnesota). C.I. is an Established Investigator of the American Heart Association (National). We thank Dr. H. Brent Clark for his help with the immunocytochemical analysis and Ms. Karen MacEwan for editorial assistance.
Correspondence should be addressed to Dr. C. Iadecola, University of Minnesota Medical School, Department of Neurology, Box 295 UMHC, 420 Delaware Street SE, Minneapolis, MN 55455.
REFERENCES
- 1.Anwar M, Costa O, Shina AK, Weiss HR. Middle cerebral artery occlusion increases cerebral capillary permeability. Neurol Res. 1993;15:232–236. doi: 10.1080/01616412.1993.11740142. [DOI] [PubMed] [Google Scholar]
- 2.Armstead WM, Mirro R, Busija DW, Leffler CW. Postischemic generation of superoxide anion by newborn pig brain. Am J Physiol. 1988;255:H401–H403. doi: 10.1152/ajpheart.1988.255.2.H401. [DOI] [PubMed] [Google Scholar]
- 3.Bazan NG, Fletcher BS, Herschman HR, Mukherjee PK. Platelet-activating factor and retinoic acid synergistically activate the inducible prostaglandin synthase gene. Proc Natl Acad Sci USA. 1994;91:5252–5256. doi: 10.1073/pnas.91.12.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breder CD, Dewitt D, Kraig RP. Characterization of inducible cyclooxygenase in rat brain. J Comp Neurol. 1995;355:296–315. doi: 10.1002/cne.903550208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busija DW, Heistad DD. Factors involved in the physiological regulation of the cerebral circulation. Rev Physiol Biochem Pharmacol. 1984;101:161–211. doi: 10.1007/BFb0027696. [DOI] [PubMed] [Google Scholar]
- 6.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 7.Chan PH, Fishman RA. Transient formation of superoxide radicals in polyunsaturated fatty acid-induced brain swelling. J Neurochem. 1980;35:1004–1007. doi: 10.1111/j.1471-4159.1980.tb07100.x. [DOI] [PubMed] [Google Scholar]
- 8.Chan PH, Fishman RA, Longar S, Chen S, Yu A. Cellular and molecular effects of polyunsaturated fatty acids in brain ischemia and injury. Prog Brain Res. 1985;63:227–235. doi: 10.1016/S0079-6123(08)61986-X. [DOI] [PubMed] [Google Scholar]
- 9.Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- 10.Chomczynski P, Sacchi N. Single-step method for RNA isolation by acid guanidinium thiocyanate-phenol chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Cole DJ, Patel PM, Reynolds L, Drummond JC, Marcantonio S. Temporary focal cerebral ischemia in spontaneously hypertensive rats: the effect of ibuprofen on infarct volume. J Pharmacol Exp Ther. 1993;266:1713–1717. [PubMed] [Google Scholar]
- 12.Collaco-Moraes Y, Aspey B, Harrison M, de Belleroche J. Cyclo-oxygenase-2 messenger RNA induction in focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:1366–1372. doi: 10.1097/00004647-199611000-00035. [DOI] [PubMed] [Google Scholar]
- 13.Corbett JA, Kwon G, Turk J, McDaniel ML. IL-1 beta induces the coexpression of both nitric oxide synthase and cyclooxygenase by islets of Langerhans: activation of cyclooxygenase by nitric oxide. Biochemistry. 1993;32:13767–13770. doi: 10.1021/bi00213a002. [DOI] [PubMed] [Google Scholar]
- 14.Dereski MO, Chopp M, Knight RA, Rodolosi LC, Garcia JH. The heterogeneous temporal evolution of focal ischemic neuronal damage in the rat. Acta Neuropathol. 1993;85:327–333. doi: 10.1007/BF00227730. [DOI] [PubMed] [Google Scholar]
- 15.Diviacco S, Norio P, Sentilin L, Menzo S, Clementi M, Biamonti G, Riva S, Falaschi A, Giacca M. A novel procedure for quantitative polymerase chain reaction by coamplification of competitive templates. Gene. 1992;122:313–320. doi: 10.1016/0378-1119(92)90220-j. [DOI] [PubMed] [Google Scholar]
- 16. Feuerstein GZ, Wang X, Barone FC. Inflammatory mediators and brain injury: the role of cytokines and chemokines in stroke and CNS diseases. Cerebrovascular diseases Ginsberg MD, Bogousslavsky J. 1997. Blackwell; Cambridge, MA: Science. [Google Scholar]
- 17.Futaki N, Arai I, Hamasaka Y, Takahashi S, Higuchi S, Otomo S. Selective inhibition of NS-398 on prostanoid production in inflamed tissue in rat carrageenan-air-pouch inflammation. J Pharm Pharmacol. 1993a;45:753–755. doi: 10.1111/j.2042-7158.1993.tb07103.x. [DOI] [PubMed] [Google Scholar]
- 18.Futaki N, Yoshikawa K, Hamasaka Y, Arai I, Higuchi S, Iizuka H, Otomo S. NS-398, a novel non-steroidal anti-inflammatory drug with potent analgesic and antipyretic effects, which causes minimal stomach lesions. Gen Pharmacol. 1993b;24:105–110. doi: 10.1016/0306-3623(93)90018-s. [DOI] [PubMed] [Google Scholar]
- 19.Galea E, Feinstein DL. Rapid synthesis of DNA deletion constructs for mRNA quantitation: analysis of astrocyte mRNA. PCR Methods Appl. 1992;2:66–69. doi: 10.1101/gr.2.1.66. [DOI] [PubMed] [Google Scholar]
- 20.Garcia JH, Yoshida Y, Chen H, Li Y, Zhang ZG, Lian J, Chen S, Chopp M. Progression from ischemic injury to infarct following middle cerebral artery occlusion in the rat. Am J Pathol. 1993;142:623–635. [PMC free article] [PubMed] [Google Scholar]
- 21.Harris R, Bayhan M, Branston NM, Watson A, Symon L. Modulation of pathophysiology of primate focal cerebral ischemia by indomethacin. Stroke. 1982;13:17–24. doi: 10.1161/01.str.13.1.17. [DOI] [PubMed] [Google Scholar]
- 22.Hsu CY, Liu TH, Xu J, Hogan EL, Chao J, Sun G, Tai HH, Beckman JS, Freeman BA. Arachidonic acid and its metabolites in cerebral ischemia. Ann NY Acad Sci. 1989;559:282–295. doi: 10.1111/j.1749-6632.1989.tb22616.x. [DOI] [PubMed] [Google Scholar]
- 23.Iadecola C, Zhang F, Xu X, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following focal cerebral ischemia. J Cereb Blood Flow Metab. 1995;15:378–384. doi: 10.1038/jcbfm.1995.47. [DOI] [PubMed] [Google Scholar]
- 24.Iadecola C, Zhang F, Casey R, Clark HB, Ross ME. Inducible nitric oxide synthase gene expression in vascular cells following transient focal cerebral ischemia. Stroke. 1996;27:1373–1380. doi: 10.1161/01.str.27.8.1373. [DOI] [PubMed] [Google Scholar]
- 25.Ignarro LJ. Heme-dependent activation of guanylate cyclase by nitric oxide: a novel signal transduction mechanism. Blood Vessels. 1991;28:67–73. doi: 10.1159/000158845. [DOI] [PubMed] [Google Scholar]
- 26.Johshita H, Asano T, Hanamura T, Takakura K. Effect of indomethacin and a free radical scavenger on cerebral blood flow and edema after cerebral artery occlusion in cats. Stroke. 1989;20:788–794. doi: 10.1161/01.str.20.6.788. [DOI] [PubMed] [Google Scholar]
- 27.Jones DA, Carlton DP, McIntyre TM, Zimmerman GA, Prescott SM. Molecular cloning of human prostaglandin endoperoxide synthase type II and demonstration of expression in response to cytokines. J Biol Chem. 1993;268:9049–9054. [PubMed] [Google Scholar]
- 28.Juzan M, Hostein I, Gualde N. Role of thymus-eicosanoids in the immune response. Prostaglandins Leukot Essent Fatty Acids. 1992;46:247–255. doi: 10.1016/0952-3278(92)90030-m. [DOI] [PubMed] [Google Scholar]
- 29.Kawasaki ES, Clark SS, Coyne MY, Smith SD, Champlin R, Witte ON, McCormick FP. Diagnosis of chronic myeloid and acute lymphocytic leukemia by detection of leukemia-specific mRNA sequence amplified in vitro. Proc Natl Acad Sci USA. 1988;85:5698–5702. doi: 10.1073/pnas.85.15.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JS, Gautam SC, Chopp M, Zaloga C, Jones ML, Ward PA, Welch KM. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J Neuroimmunol. 1995;56:127–134. doi: 10.1016/0165-5728(94)00138-e. [DOI] [PubMed] [Google Scholar]
- 31.Klein T, Nusing RM, Pfeilschifter J, Ullrich V. Selective inhibition of cyclooxygenase 2. Biochem Pharmacol. 1994;48:1605–1610. doi: 10.1016/0006-2952(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 32.Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and monocyte/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23:1367–1379. doi: 10.1161/01.str.23.9.1367. [DOI] [PubMed] [Google Scholar]
- 33.Kontos HA, Wei EP, Povlishock JT, Dietrich WD, Magiera CJ, Ellis EF. Cerebral arteriolar damage by arachidonic acid and prostaglandin G2. Science. 1980;209:1242–1245. doi: 10.1126/science.7403881. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Sharov VG, Jiang N, Zaloga C, Sabbah HN, Chopp M. Ultrastructural and light microscopic evidence of apoptosis after middle cerebral artery occlusion in the rat. Am J Pathol. 1995;146:1045–1051. [PMC free article] [PubMed] [Google Scholar]
- 35.Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- 36.Liu T, McDonnell PC, Young PR, White RF, Siren AL, Hallenbeck JM, Barone FC, Feuerstein GZ. Interleukin-1 beta mRNA expression in ischemic rat cortex. Stroke. 1993;24:1746–1750. doi: 10.1161/01.str.24.11.1746. [DOI] [PubMed] [Google Scholar]
- 37.Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, Feuerstein GZ. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 38.Marchal G, Beaudouin V, Rioux P, de la Sayette V, Le Doze F, Viader F, Derlon J-M, Baron J-C. Prolonged persistence of substantial volumes of potentially viable brain tissue after stroke. Stroke. 1996;27:599–606. doi: 10.1161/01.str.27.4.599. [DOI] [PubMed] [Google Scholar]
- 39.Marshall RS, Mohr JP. Current management of ischaemic stroke. J Neurol Neurosurg Psychiatry. 1993;56:6–16. doi: 10.1136/jnnp.56.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Overgard K, Sereghy T, Pedersen H, Boysen G. Effect of delayed thrombolysis with rt-PA in a rat embolic stroke model. J Cereb Blood Flow Metab. 1994;14:472–477. doi: 10.1038/jcbfm.1994.58. [DOI] [PubMed] [Google Scholar]
- 42.Powell WS. Rapid extraction of arachidonic acid metabolites from biological samples using octadecylsilyl silica. In: Lands WEM, Smith WL, editors. Methods in enzymology. Academic; Orlando, FL: 1982. pp. 466–477. [DOI] [PubMed] [Google Scholar]
- 43.Pozzilli C, Lenzi GL, Argentino C, Carolei A, Rasura M, Signore A, Bozzao L, Pozzilli P. Imaging of leukocytic infiltration in human cerebral infarcts. Stroke. 1985;16:251–255. doi: 10.1161/01.str.16.2.251. [DOI] [PubMed] [Google Scholar]
- 44.Pradelles P, Grassi J, Maclouf J. Enzyme immunoassay of eicosanoids using acetylcholine esterase as a label: an alternative to radioimmunoassay. Anal Chem. 1985;57:1170–1173. doi: 10.1021/ac00284a003. [DOI] [PubMed] [Google Scholar]
- 45.Reitz DB, Li JJ, Norton MB, Reinhard EJ, Collins JT, Anderson GD, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Isakson PC. Selective cyclooxygenase inhibitors: novel 1,2-diarylcyclopentenes are potent and orally active COX-2 inhibitors. J Med Chem. 1994;37:3878–3881. doi: 10.1021/jm00049a005. [DOI] [PubMed] [Google Scholar]
- 46.Ross ME, Iadecola C. Nitric oxide synthase expression in cerebral ischemia: neurochemical, immunocytochemical and molecular approaches. In: Packer L, editor. Methods in enzymology. Academic; Orlando, FL: 1996. pp. 408–426. [DOI] [PubMed] [Google Scholar]
- 47.Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA. 1993;90:7240–7244. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salvemini D, Settle SL, Masferrer JL, Seibert K, Currie MG, Needleman P. Regulation of prostaglandin production by nitric oxide: an in vivo analysis. Br J Pharmacol. 1995;114:1171–1178. doi: 10.1111/j.1476-5381.1995.tb13330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seibert K, Masferrer J, Zhang Y, Gregory S, Olson G, Hauser S, Leahy K, Perkins W, Isakson P. Mediation of inflammation by cyclooxygenase-2. Agents Actions Suppl. 1995;46:41–50. doi: 10.1007/978-3-0348-7276-8_5. [DOI] [PubMed] [Google Scholar]
- 50.Siebert PD, Larrick JW. Competitive PCR. Nature. 1992;359:557–558. doi: 10.1038/359557a0. [DOI] [PubMed] [Google Scholar]
- 51.Sirois J, Richards JS. Transcriptional regulation of the rat prostaglandin endoperoxide synthase 2 gene in granulosa cells. Evidence for the role of a cis-acting C/EBP beta promoter element. J Biol Chem. 1993;268:21931–21938. [PubMed] [Google Scholar]
- 52.Smith WL, DeWitt DL. Biochemistry of prostaglandin endoperoxide H synthase-1 and synthase-2 and their differential susceptibility to nonsteroidal anti-inflammatory drugs. Semin Nephrol. 1995;15:179–194. [PubMed] [Google Scholar]
- 53.Takagi K, Ginsberg MD, Globus MY, Dietrich WD, Martinez E, Kraydieh S, Busto R. Changes in amino acid neurotransmitters and cerebral blood flow in the ischemic penumbral region following middle cerebral artery occlusion in the rat: correlation with histopathology. J Cereb Blood Flow Metab. 1993;13:575–585. doi: 10.1038/jcbfm.1993.75. [DOI] [PubMed] [Google Scholar]
- 54.Tsai A, Hsi LC, Kulmacz RJ, Palmer G, Smith WL. Characterization of the tyrosyl radicals in ovine prostaglandin H synthase-1 by isotope replacement and site-directed mutagenesis. J Biol Chem. 1994;269:5085–5091. [PubMed] [Google Scholar]
- 55.Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 56.Vane JR, Mitchell JA, Appleton I, Tomlinson A, Bishop BD, Croxtall J, Willoughby DA. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc Natl Acad Sci USA. 1994;91:2046–2050. doi: 10.1073/pnas.91.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang AM, Doyle MV, Mark DF. Quantitation of mRNA by the polymerase chain reaction. Proc Natl Acad Sci USA. 1989;86:9717–9721. doi: 10.1073/pnas.86.24.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, Siren AL, Liu Y, Yue TL, Barone FC, Feuerstein GZ. Upregulation of intercellular adhesion molecule 1 (ICAM-1) on brain microvascular endothelial cells in rat ischemic cortex. Mol Brain Res. 1994;26:61–68. doi: 10.1016/0169-328x(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 59.Wang X, Yue TL, Young PR, Barone FC, Feuerstein GZ. Expression of interleukin-6, c-fos, and zif268 mRNAs in rat ischemic cortex. J Cereb Blood Flow Metab. 1995;15:166–171. doi: 10.1038/jcbfm.1995.18. [DOI] [PubMed] [Google Scholar]
- 60.Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- 61.Zea Longa E, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rat. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 62.Zhang F, Iadecola C. Reduction of focal cerebral ischemic damage by delayed treatment with nitric oxide donors. J Cereb Blood Flow Metab. 1994a;14:574–580. doi: 10.1038/jcbfm.1994.71. [DOI] [PubMed] [Google Scholar]
- 63.Zhang F, Iadecola C. Infarct measurement methodology. J Cereb Blood Flow Metab. 1994b;14:697–698. doi: 10.1038/jcbfm.1994.88. [DOI] [PubMed] [Google Scholar]
- 64.Zhang F, Casey R, Ross ME, Iadecola C. Aminoguanidine ameliorates and l-arginine worsens brain damage from intraluminal middle cerebral artery occlusion. Stroke. 1996;27:317–323. doi: 10.1161/01.str.27.2.317. [DOI] [PubMed] [Google Scholar]