

Abstract
Increasing evidence suggests that oxidative damage to proteins and other macromolecules is a salient feature of the pathology of Alzheimer’s disease. Establishing the source of oxidants is key to understanding what role they play in the pathogenesis of Alzheimer’s disease, and one way to examine this issue is to determine which oxidants are involved in damage.
In this study, we examine whether peroxynitrite, a powerful oxidant produced from the reaction of superoxide with nitric oxide, is involved in Alzheimer’s disease. Peroxynitrite is a source of hydroxyl radical-like reactivity, and it directly oxidizes proteins and other macromolecules with resultant carbonyl formation from side-chain and peptide-bond cleavage. Although carbonyl formation is a major oxidative modification induced by peroxynitrite, nitration of tyrosine residues is an indicator of peroxynitrite involvement. In brain tissue from cases of Alzheimer’s disease, we found increased protein nitration in neurons, including but certainly not restricted to those containing neurofibrillary tangles (NFTs). Conversely, nitrotyrosine was undetectable in the cerebral cortex of age-matched control brains. This distribution is essentially identical to that of free carbonyls.
These findings provide strong evidence that peroxynitrite is involved in oxidative damage of Alzheimer’s disease. Moreover, the widespread occurrence of nitrotyrosine in neurons suggests that oxidative damage is not restricted to long-lived polymers such as NFTs, but instead reflects a generalized oxidative stress that is important in disease pathogenesis.
Keywords: Alzheimer’s disease, carbonyls, glycation, nitrotyrosine, oxidative stress, protein modification
During the past 2 years, a number of oxidative modifications have been found in association with the pathological lesions of Alzheimer’s disease. For example, advanced glycation end products (AGE), lipid peroxidation adducts, and free carbonyls are detected in both neurofibrillary tangles (NFTs) and senile plaques (Ledesma et al., 1994; Smith et al., 1994, 1996a; Vitek et al., 1994;Yan et al., 1994; Sayre et al., 1997). Such oxidative modifications of proteins may define many of the unique properties of the lesions, in particular their sparing solubility (Smith et al., 1996b) and association with degenerating neurons (Yan et al., 1994, 1995;Praprotnik et al., 1996). Nonetheless, despite this evidence, the source of the oxygen radicals in vivo is not completely understood. In this study, we examine this issue by determining whether peroxynitrite, a powerful oxidant produced as a result of the diffusion-limited reaction of superoxide with nitric oxide, is involved in oxidation of proteins, lipids, and nucleic acids in Alzheimer’s disease. Peroxynitrite is a source of hydroxyl radical-like reactivity that directly oxidizes proteins and other macromolecules, with resultant carbonyl formation from side-chain and peptide-bond cleavage. Peroxynitrite also causes the nitration of tyrosine residues, and this can be used as an index of peroxynitrite involvement. Here we show that nitrotyrosine immunoreactivity in Alzheimer’s disease is increased in the neuronal cytoplasm of the cerebral cortex within regions of neurodegeneration, whereas it is undetectable in the same brain regions of controls.
MATERIALS AND METHODS
Tissue. Hippocampal tissue, including the adjacent entorhinal and neocortex from 16 cases of Alzheimer’s disease (ages 60–91 years; average 79) and five control cases with no clinical or pathological history of neurological disease (ages 32–82 years; average 55), and cerebellar tissue from three Alzheimer’s disease (ages 69–84) and two control (ages 64 and 74) cases with similar postmortem interval were fixed in methacarn (chloroform/methanol/acetic acid, 60:30:10) at 4°C overnight. The apolipoprotein E genotype of the cases of Alzheimer’s disease was ε4/ε4 (n = 2); ε3/ε4 (n = 3); ε3/ε3 (n = 4); ε2/ε3 (n = 1); unknown (n = 5). After fixation, tissue was dehydrated through ascending ethanol and embedded in paraffin, and 6-μm-thick sections were placed on silane-coated slides (Sigma, St. Louis, MO).
Antibodies and immunocytochemistry. Affinity-purified rabbit antiserum 4708 and mouse monoclonal antibodies 7A2 and 1A6, raised to nitrated keyhole limpet hemocyanin, were used at a 1:100 dilution in 1% normal goat serum, 150 mm NaCl, 50 mmTris-HCl, pH 7.6 (Beckman et al., 1994). After incubation at 4°C for 16 hr, sections were immunostained with the peroxidase–antiperoxidase method, with 3,3′-diaminobenzidine as cosubstrate (Sternberger, 1986). Adjacent sections were immunostained with antiserum to ubiquitin (Manetto et al., 1988) to confirm the identity and location of pathological structures.
Controls consisted of (1) omission of the primary antibody; (2) adsorption of the antibody with 50 μm nitrated BSA or 15 μm nitrated glycine-tyrosine-alanine (Gly-Tyr-Ala) peptide at 4°C overnight before application to the section; and (3) chemical reduction of nitrotyrosine by treating sections with 15 mm sodium hydrosulfite in 50 mm carbonate buffer, pH 8.0, for 15 min at room temperature (Cuatrecasas et al., 1968) before immunostaining. These procedures were performed in parallel with the antisera to ubiquitin as controls against artifactual inactivation of either primary or secondary antibodies from use of sodium hydrosulfite-reduced sections and against nonspecific adsorption with nitrated BSA or nitrated Gly-Tyr-Ala. After they were immunostained, in some cases the sections were stained with Congo red and viewed under cross-polarized light to show NFTs and amyloid-β deposits of senile plaques.
RESULTS
In all 16 cases of Alzheimer’s disease, all three antibodies to nitrotyrosine recognized neuronal cytoplasm and nuclei in hippocampal sections (Fig. 1A,C), whereas, conversely, in the five control cases no specific structures were stained (Fig. 1B,D). In sections taken from cases of Alzheimer’s disease that were counterstained with Congo red, although the most intensely nitrotyrosine-positive neurons often contained NFTs, many neurons lacking NFTs were also intensely stained (Fig. 1C). Nuclei of glia were also stained in the cases of Alzheimer’s disease. No reaction was noted to extracellular NFTs, amyloid deposits, or dystrophic neurites of senile plaques (Fig.1C). This distribution is essentially identical to our findings of increased free carbonyls in Alzheimer’s disease that were also localized to neuronal cell bodies and nuclei of both neurons and glia but, as here, was restricted to regions of Alzheimer’s disease pathology (Smith et al., 1996a). Although differences were noted in the intensity of immunoreactivity between cases of Alzheimer’s disease, we found no correlations to apolipoprotein E genotype, age, or postmortem interval for those cases where this information was available. Statistically, for the 21 cases analyzed, neuronal nitrotyrosine immunoreactivity showed complete sensitivity for detecting Alzheimer’s disease and complete specificity in not detecting any of the control cases. Additional studies will be required to evaluate whether the specificity is unique to Alzheimer’s disease or whether neuronal nitrotyrosine immunoreactivity is also increased in other neurological diseases.
Fig. 1.
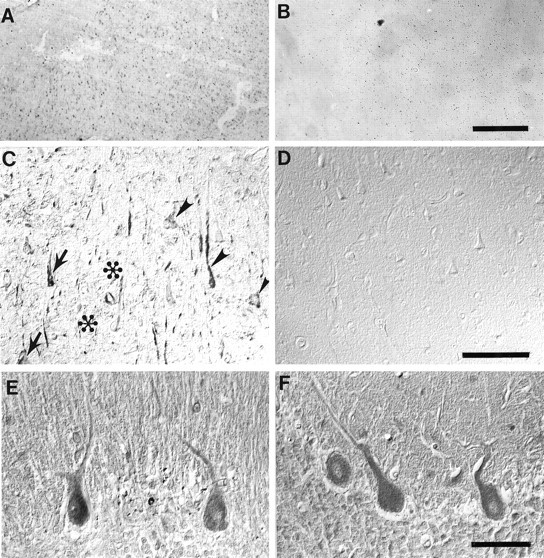
Nitrotyrosine immunoreactivity is prominent in the cytoplasm and nuclei of hippocampal neurons in cases of Alzheimer’s disease (A, C), whereas it is absent from control cases (B, D). Immunoreactivity was often but not always more intense in neurons containing NFTs (arrows) than in those lacking NFTs, which were also intensely stained (arrowheads). In contrast, amyloid-β deposits (✻) and surrounding dystrophic neurites of senile plaques as well as extracellular-NFTs (unmarked) were unstained. The location of NFTs and amyloid-β deposits was determined by Congo red counterstaining. In contrast, in the cerebellum, nitrotyrosine immunoreactivity was present at the same level in cases of Alzheimer’s disease (E) as it was in controls (F). Scale bars: A, B, 500 μm; C, D, 100 μm; E, F, 50 μm.
To understand whether increased nitrotyrosine represents a global alteration of the oxidative stress in Alzheimer’s disease, we studied the cerebellum. In Alzheimer’s disease, the cerebellum is spared from neuronal degeneration. Pathology in the cerebellum is restricted to diffuse amyloid-β deposits. Cerebellum from controls and Alzheimer’s disease (Fig. 1E,F) showed nitrotyrosine immunoreactivity in Purkinje cells, with no apparent difference between controls and the cases of Alzheimer’s disease. This finding supports the idea that peroxynitrite-dependent damage is restricted to regions of Alzheimer’s disease pathology, although the basal level of protein nitration is dependent on the physiology of the neurons involved.
Nitrotyrosine immunoreactivity is specific, because (1) all three antibodies recognized similar structures, with 7A2 showing the strongest immunoreactivity; (2) no immunostaining was noted with omission of the primary antibody; (3) preabsorption of the antibodies with nitrated BSA or nitrated Gly-Tyr-Ala blocked recognition (Fig.2); and (4) chemical reduction of nitrotyrosine with sodium hydrosulfite abolished immunoreactivity (Fig. 3). Importantly, there was no diminution of labeling with ubiquitin antisera after hydrosulfite reduction of the section, indicating that chemical reduction did not interfere with the peroxidase–antiperoxidase reaction or antibody recognition of other antigens (results not shown) (Kooy et al., 1995). Moreover, absorption of ubiquitin antisera with nitrated BSA or nitrated Gly-Tyr-Ala had no effect on consequent immunoreactivity (results not shown).
Fig. 2.
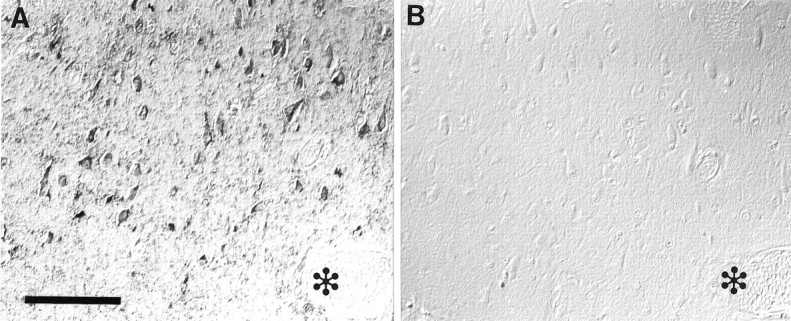
Nitrotyrosine immunoreactivity (A) is completely blocked by adsorption with nitrated Gly-Tyr-Ala (B). Adjacent serial section with a blood vessel is indicated by ✻ in each section. Scale bar, 100 μm.
Fig. 3.
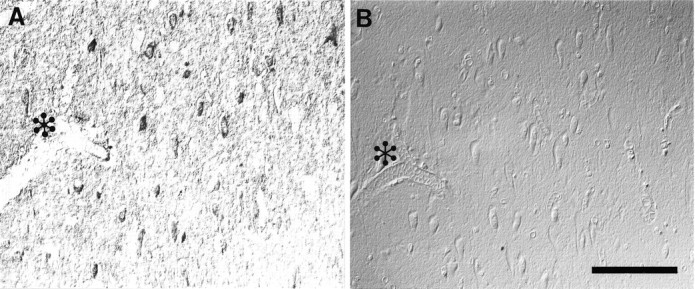
Nitrotyrosine immunoreactivity (A) is completely abolished by chemical reduction of nitrotyrosine with sodium hydrosulfite (B). Adjacent serial section with a blood vessel is indicated by ✻ in each section. Scale bar, 100 μm.
DISCUSSION
This study demonstrates that tyrosine nitration is increased in the neuronal cytoplasm as well as in the nuclei of both neurons and glia in regions of Alzheimer’s disease pathology. In stark contrast, nitrotyrosine immunoreactivity is undetectable in the cerebral cortex of controls. These findings implicate oxidants derived from nitric oxide, most likely peroxynitrite, in the pathogenesis of Alzheimer’s disease. Although one of the major oxidative modifications of proteins resulting from peroxynitrite is carbonyl formation from side-chain and peptide-bond cleavage, electrophilic nitration of tyrosine phenols is a signature of peroxynitrite involvement. Significantly, peroxynitrite formation is dependent on nitric oxide as well as superoxide, suggesting that nitric oxide synthase (NOS)-containing neurons or microglia may play a role in oxidative damage. NOS production increases after excitotoxicity, and it is suggested that neurodegeneration in Alzheimer’s disease is brought about by excitotoxicity resulting from overstimulation (Gibson, 1989). NOS-containing neurons are relatively spared in Alzheimer’s disease (Hyman et al., 1992), perhaps suggesting that NOS-containing neurons are better able to deal with oxidative stress than other neurons or, alternatively, that nitric oxide, from NOS-positive neurons, diffuses to other cells and reacts with superoxide to form peroxynitrite distal to the NOS-positive neurons. Activated microglia, present in most senile plaques in Alzheimer’s disease (Cras et al., 1991), can also produce nitric oxide (Goodwin et al., 1995; Nakashima et al., 1995; Paakkari and Lindsberg, 1995), and it is of note that the involvement of nitric oxide produced by microglia may provide an additional link to the lower incidence of Alzheimer’s disease with use of anti-inflammatory agents (Marx, 1996).
The strong association of peroxynitrite-related damage to regions of pathology indicates that the source for superoxide must lie in close proximity, because superoxide either reacts with tissue components (Hausladen and Fridovich, 1994) or is readily dismutated by superoxide dismutase, known to be associated with NFTs (Pappolla et al., 1992). The pathological lesions are a likely source, because glycated proteins (Yim et al., 1995), and particularly glycated τ in NFTs, produce superoxide (Yan et al., 1995). Therefore, this offers the possibility that NFT-containing neurons play a role in the oxidative damage of adjacent neurons. This hypothesis is consistent with the coordinate neuronal degeneration seen in sites of Alzheimer’s disease pathology and its dependence on NFT formation.
Although all of the Alzheimer’s disease cases examined show nitrotyrosine immunoreactivity, there was some case-to-case variation in intensity of staining, suggesting that nitrotyrosine may represent a specific pathological stage, that the epitope is often “masked,” or that the extent of involvement of nitrotyrosine is case-dependent. In this latter regard, we found no relationship between the extent of immunostaining and apolipoprotein E genotype. Significantly, our results differ from a recent report using a different antibody and formalin-fixed sections in which nitrotyrosine is described as being limited to NFTs (Good et al., 1996). By marked contrast, using methacarn-fixed material, which is not carbonyl cross-linked, we found evidence of widespread neuronal oxidative damage in Alzheimer’s disease. We were unable to obtain consistent nitrotyrosine immunostaining using formalin-fixed material, a peculiarity that may be restricted to brain, because nitrotyrosine has been localized in other tissues with these same antibodies after formalin fixation (Beckman et al., 1994). In preliminary immunoblotting studies, we were unable to establish whether there is a preferential nitration of specific neuronal proteins (our unpublished data). Nonetheless, by extending beyond the lesions, our findings suggest that oxidative damage in Alzheimer’s disease may result from a chronic abnormality of oxidative balance that affects neurons regardless of whether they themselves contain an NFT.
The distribution of nitrotyrosine presented here is essentially identical to the distribution of free carbonyls (Smith et al., 1996a), although additional studies will be necessary to establish the relative contribution of peroxynitrite or other oxidants to protein oxidation. The distinction between neurons, which show damage throughout the cytoplasm and nucleus, and glia, whose damage is limited to the nucleus, suggests cell type-dependent differences with respect to oxidative damage. An understanding of these distinctions may provide an insight into the neuronal specificity of cell degeneration in Alzheimer’s disease.
Footnotes
This work was supported by National Institutes of Health (AG09287 and NS22688), the American Health Assistance Foundation, the American Federation for Aging Research, and a Daland Fellowship from the American Philosophical Society.
Correspondence should be addressed to Dr. Mark A. Smith, Institute of Pathology, 2085 Adelbert Road, Case Western Reserve University, Cleveland, OH 44106.
REFERENCES
- 1.Beckman JS, Ye YZ, Anderson PG, Chen J, Accavitti MA, Tarpey MM, White CR. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- 2.Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci USA. 1991;88:7552–7556. doi: 10.1073/pnas.88.17.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cuatrecasas P, Fuchs S, Anfinsen CB. The tyrosyl residues at the active site of staphylococcal nuclease: modifications by tetranitromethane. J Biol Chem. 1968;243:4787–4798. [PubMed] [Google Scholar]
- 4.Gibson G. Causes of cell damage in hypoxia/ischemia, aging and Alzheimer’s disease. Neurobiol Aging. 1989;10:608–609. doi: 10.1016/0197-4580(89)90147-4. [DOI] [PubMed] [Google Scholar]
- 5.Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Evidence for neuronal oxidative damage in Alzheimer’s disease. Am J Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- 6.Goodwin JL, Uemura E, Cunnick JE. Microglial release of nitric oxide by the synergistic action of β-amyloid and IFN-γ. Brain Res. 1995;692:207–214. doi: 10.1016/0006-8993(95)00646-8. [DOI] [PubMed] [Google Scholar]
- 7.Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994;269:29405–29408. [PubMed] [Google Scholar]
- 8.Hyman BT, Marzloff K, Wenniger JJ, Dawson TM, Bredt DS, Snyder SH. Relative sparing of nitric oxide synthase-containing neurons in the hippocampal formation in Alzheimer’s disease. Ann Neurol. 1992;32:818–820. doi: 10.1002/ana.410320618. [DOI] [PubMed] [Google Scholar]
- 9.Kooy NW, Royall JA, Ye YZ, Kelley DR, Beckman JS. Evidence for in vivo peroxynitrite production in human acute lung injury. Am Rev Respir Crit Care Med. 1995;151:1250–1254. doi: 10.1164/ajrccm/151.4.1250. [DOI] [PubMed] [Google Scholar]
- 10.Ledesma MD, Bonay P, Colaco C, Avila J. Analysis of microtubule-associated protein tau glycation in paired helical filaments. J Biol Chem. 1994;269:21614–21619. [PubMed] [Google Scholar]
- 11.Manetto V, Perry G, Tabaton M, Mulvihill P, Fried VA, Smith HT, Gambetti P, Autilio-Gambetti L. Ubiquitin is associated with abnormal cytoplasmic filaments characteristic of neurodegenerative diseases. Proc Natl Acad Sci USA. 1988;85:4501–4505. doi: 10.1073/pnas.85.12.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marx J. Searching for drugs that combat Alzheimer’s. Science. 1996;273:50–53. [PubMed] [Google Scholar]
- 13.Nakashima MN, Yamashita K, Kataoka Y, Yamashita YS, Niwa M. Time course of nitric oxide synthase activity in neuronal, glial, and endothelial cells of rat striatum following focal cerebral ischemia. Cell Mol Neurobiol. 1995;15:341–349. doi: 10.1007/BF02089944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paakkari I, Lindsberg P. Nitric oxide in the central nervous system. Ann Med. 1995;27:369–377. doi: 10.3109/07853899509002590. [DOI] [PubMed] [Google Scholar]
- 15.Pappolla MA, Omar RA, Kim KS, Robakis NK. Immunohistochemical evidence of antioxidant stress in Alzheimer’s disease. Am J Pathol. 1992;140:621–628. [PMC free article] [PubMed] [Google Scholar]
- 16.Praprotnik D, Smith MA, Richey PL, Vinters HV, Perry G. Plasma membrane fragility in dystrophic neurites in senile plaques of Alzheimer’s disease: an index of oxidative stress. Acta Neuropathol. 1996;91:1–5. doi: 10.1007/s004010050385. [DOI] [PubMed] [Google Scholar]
- 17.Sayre LM, Zelasko DA, Richey PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 18.Smith MA, Taneda S, Harris PLR, Miyata S, Yan S-D, Stern D, Sayre LM, Monnier VM, Perry G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci USA. 1994;91:5710–5714. doi: 10.1073/pnas.91.12.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, Kowall N. Oxidative damage in Alzheimer’s. Nature. 1996a;382:120–121. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- 20.Smith MA, Siedlak SL, Richey PL, Nagaraj RH, Elhammer A, Perry G. Quantitative solubilization and analysis of insoluble paired helical filaments from Alzheimer disease. Brain Res. 1996b;717:99–108. doi: 10.1016/0006-8993(95)01473-x. [DOI] [PubMed] [Google Scholar]
- 21.Sternberger LA. Immunocytochemistry, 3rd Ed. Wiley; New York: 1986. [Google Scholar]
- 22.Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:4766–4770. doi: 10.1073/pnas.91.11.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan S-D, Chen X, Schmidt A-M, Brett J, Godman G, Zou Y-S, Scott CW, Caputo C, Frappier T, Smith MA, Perry G, Yen S-H, Stern D. Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci USA. 1994;91:7787–7791. doi: 10.1073/pnas.91.16.7787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, Zweier JL, Stern D. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid β-peptide. Nature Med. 1995;1:693–699. doi: 10.1038/nm0795-693. [DOI] [PubMed] [Google Scholar]
- 25.Yim HS, Kang SO, Hah YC, Chock PB, Yim MB. Free radicals generated during the glycation reaction of amino acids by methylglyoxal: a model study of protein-cross-linked free radicals. J Biol Chem. 1995;270:28228–28233. doi: 10.1074/jbc.270.47.28228. [DOI] [PubMed] [Google Scholar]