

Abstract
Subanesthetic doses of ketamine, a noncompetitive NMDA receptor antagonist, impair prefrontal cortex (PFC) function in the rat and produce symptoms in humans similar to those observed in schizophrenia and dissociative states, including impaired performance of frontal lobe-sensitive tests. Several lines of evidence suggest that ketamine may impair PFC function in part by interacting with dopamine neurotransmission in this region. This study sought to determine the mechanism by which ketamine may disrupt dopaminergic neurotransmission in, and cognitive functions associated with, the PFC. A thorough dose-response study using microdialysis in conscious rats indicated that low doses of ketamine (10, 20, and 30 mg/kg) increase glutamate outflow in the PFC, suggesting that at these doses ketamine may increase glutamatergic neurotransmission in the PFC at non-NMDA glutamate receptors. An anesthetic dose of ketamine (200 mg/kg) decreased, and an intermediate dose of 50 mg/kg did not affect, glutamate levels. Ketamine, at 30 mg/kg, also increased the release of dopamine in the PFC. This increase was blocked by intra-PFC application of the AMPA/kainate receptor antagonist, 6-cyano-7-nitroquinoxaline-2,3-dione CNQX. Furthermore, ketamine-induced activation of dopamine release and impairment of spatial delayed alternation in the rodent, a PFC-sensitive cognitive task, was ameliorated by systemic pretreatment with AMPA/kainate receptor antagonist LY293558. These findings suggest that ketamine may disrupt dopaminergic neurotransmission in the PFC as well as cognitive functions associated with this region, in part, by increasing the release of glutamate, thereby stimulating postsynaptic non-NMDA glutamate receptors.
Keywords: microdialysis, phencyclidine, schizophrenia, working memory, antipsychotic drugs, AMPA receptors
Recent clinical trials with ketamine, a noncompetitive antagonist of the NMDA receptor (Thomson et al., 1985), have demonstrated that subanesthetic doses of this drug exacerbate preexisting symptoms of schizophrenia (Lahti et al., 1995a,b) and produce behaviors in healthy individuals that bear a resemblance to a broad range of symptoms associated with schizophrenia, including impaired performance in psychological tests sensitive to prefrontal cortex (PFC) function (Ghoneim et al., 1985; Oye et al., 1992; Krystal et al., 1994; Malhorta et al., 1996). In agreement with clinical studies, basic findings indicate that subanesthetic doses of ketamine and other noncompetitive NMDA receptor antagonists produce deficits in acquisition and performance of cognitive tasks, including those that are sensitive to the functional integrity of the PFC (Danysz et al., 1988; Hauber and Schmidt, 1989; Heale and Harley, 1990; Parada-Turska and Turski, 1990; Wesierska et al., 1990; Hauber, 1993; Verma and Moghaddam, 1996). These actions of ketamine are similar to those observed decades ago with phencyclidine (Luby et al., 1959; Davies and Beech, 1960; Bakker and Amini, 1961), which led to the “phencyclidine model of schizophrenia” and, more recently, to several glutamate- and NMDA-related hypotheses of schizophrenia (Kim et al., 1980; Deakin et al., 1989; Wachtel and Turski, 1990; Javitt and Zukin, 1991; Ulas and Cotman, 1993; Olney and Farber, 1995; Akbarian et al., 1996; Coyle, 1996).
Although the behavioral deficits that are produced by ketamine (and PCP) suggest a role for the NMDA receptor in the etiology of schizophrenia, several lines of evidence have pointed to a dysfunctional dopamine system (Matthysse, 1973; Angrist et al., 1974;Snyder et al., 1974; Seeman et al., 1976). Interestingly, dopamine neurotransmission contributes to the behavioral deficits produced by low doses of ketamine because dopamine D2 receptor antagonists ameliorate ketamine-induced impairment of some PFC-dependent functions in humans and in the rodent (Krystal et al., 1995; Verma and Moghaddam, 1996). Furthermore, low doses of NMDA receptor antagonists, including ketamine and PCP, preferentially increase dopamine release in the PFC (Rao et al., 1989; Wedzony, et al., 1993; Hondo et al., 1994; Verma and Moghaddam, 1996).
At high doses, ketamine and PCP are known to interfere with the function of dopamine uptake sites (Smith et al., 1981); however, the mechanism for their dopamine release-activating properties at low doses remains unclear. Recent biochemical studies have indicated that NMDA receptor antagonists may increase the release of endogenous excitatory amino acids, glutamate, and aspartate (Bustos et al., 1992; Liu and Moghaddam, 1995). This increase may then activate glutamatergic neurotransmission at non-NMDA receptors, including AMPA and kainate receptors. Recent studies from our laboratory have indicated that stimulation of AMPA or kainate receptors potently increases the release of dopamine in the PFC (Jedema and Moghaddam, 1996).
The present study was performed to test the hypothesis that ketamine increases dopamine neurotransmission in the PFC and disrupts cognitive functions associated with this region by activation of glutamatergic neurotransmission at non-NMDA receptors. First, a thorough dose–response study on the effect of ketamine on the outflow of Glu in the PFC was performed using intracerebral microdialysis in the awake rat. Second, the effect of a subanesthetic dose of ketamine on the release of dopamine in the PFC and whether this effect is produced by activation of non-NMDA receptors in the PFC were examined. Finally, the effect of systemic pretreatment with a non-NMDA receptor antagonist on ketamine-induced impairment of a PFC-related behavioral paradigm and activation of dopamine release in this region was examined. To evaluate the previous reports on the regional selectivity of ketamine, some of the microdialysis experiments mentioned above were also performed in the striatum.
MATERIALS AND METHODS
Animal preparation. Animal use procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Yale University Animal Care and Use Committee. Microdialysis probes were implanted while rats were under halothane anesthesia. Animals were placed in a stereotaxic frame with blunt ear bars, and a small incision (3–5 mm) was made in the skin over the skull. The wound margin was infiltrated with lidocaine, and holes were drilled for skull screws and concentric microdialysis probes aimed (bilaterally) at the prefrontal cortex (anteroposterior, 3.0; lateral, 0.8; ventral, 5.5) or the striatum (anteroposterior, 0.0; lateral, 3.0; ventral, 6.5) according the atlas of Paxinos and Watson (1982). Probes and screws were secured in place with dental cement. Additional local anesthetic was then injected (under the skin) around the wound area. After surgery, animals were placed in the same container that they had been in before surgery and allowed to recover for 24 hr before the microdialysis measurements, which were performed in the same container.
Microdialysis procedure. Concentric microdialysis probes with an exposed tip length of 3.0 mm were used. A flow rate of 2 μl/min was used during sample collection (probes were perfused overnight at a rate of 0.5 μl/min. The perfusion solution was a 2 mm phosphate buffer, pH 7.4, containing the following (in mm): 145 NaCl, 2.7 KCl, 1.2 CaCl2, 1.0 MgCl2, and 0.1 ascorbic acid. Probes were calibrated in a solution containing 10−6m aspartate, glutamate, glutamine, serine, and homoserine or 10−7m DA. The probes exhibited a recovery of 14–18% (for all compounds). All probes were perfused at 2 μl/min for 2–3 hr before local drug perfusion.
Chromatographic analysis of amino acids and dopamine.For analysis of dopamine, samples were injected onto an HPLC with electrochemical detection. This system employed a narrow-bore column [2.1 mm inner diameter (i.d.), 3 μm C-18 particles, 10 cm long] and a Bioanalytical Systems LC-4C potentiostat (West Lafayette, IN). The Eapp was +0.55 V versus Ag/AgCl reference electrode. The mobile phase consisted of: 9 gm/l NaH2PO4, 575 mg/l octylsulfonic acid, 8% acetonitrile (v/v), 250 mg/l EDTA, and 250 μl/l triethylamine, pH 5.1. A detection limit of 5 fmol was routinely achieved.
The amount of glutamate and homoserine (internal standard) in the perfusate was determined by precolumn derivatization witho-pthalaldehyde and mercaptoethanol, followed by HPLC with fluorescence detection. This system used an Autosampler (Spectra System AS 3000, Spectra-Physics, Fremont, CA), a gradient pump (Spectra-Physics, model SP 8800), and a fluorescence detector (Spectra-Physics, model SP 8410). The excitation and emission wave lengths were set at 300 and 400 nm, respectively. A 3 μm, C-18 (10 cm long, 4.2 mm i.d.) analytical column was used. The mobile phase was a 0.1 m phosphate buffer containing 0.01m EDTA, pH 6.4, and acetonitrile (with a gradient profile of 13–22% acetonitrile over 12 min).
Data analysis. For statistical analysis, because of the variability in resting basal levels, the glutamate data were expressed as percentage of the mean of three basal values obtained immediately before the treatment. Different groups (e.g., ketamine- or saline-treated groups) were compared using Kruskal-Wallis ANOVA. Significant differences (p < 0.05) from saline-treated groups are depicted with asterisks on figures. The dopamine data (absolute values in fmol/μl) were analyzed using repeated measures ANOVA. In figures, levels are expressed as percentage of the mean (±SEM) of the three basal values obtained immediately before the treatment.
Histology. After the termination of each experiment, animals were anesthetized with chloral hydrate and perfused intracardially with saline, followed by 10% buffered formalin. Brains were removed and stored in formalin. Serial sections of the fixed brains were cut at 100 μm intervals and stained with cresyl violet for probe placement verification.
Materials. All reagents for the HPLC mobile phase and the perfusion fluid were analytical grade and were obtained from Eastman Kodak (Rochester, NY) or J. T. Baker Chemical Company (Phillipsburg, NJ). LY293558 was a gift from Eli Lilly. Ketamine and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) were purchased from Research Biochemicals (Natick, MA). A 10 mm CNQX stock solution was prepared by dissolving CNQX in 0.1 m NaOH. Before use, the stock solution was diluted to 50 μm with the normal perfusion solution.
Spatial delayed alternation. Spatial delayed alternation performance was tested using a T-maze as described previously (Verma and Moghaddam, 1996). The T-maze was constructed from Plexiglas and painted a medium gray. The walls were 30 cm high, and the alleys were 15 cm wide. The length of the main alley was 50 cm, and the length of the side alleys was 40 cm. The side alleys were closed off from the main alley by movable doors. At the end of each side alley was a 2 cm high barrier that concealed the food reward (one morsel of Froot Loops cereal, General Mills, Kankakee, IL) from view. A third movable door was mounted in the main alley. A holding cage was placed adjacent to the T-maze for use during experiments with intertrial intervals. A video camera was situated ∼1 m above the T-maze to videotape the test session. The T-maze was cleaned between different animals, but not between different trials. Animals were handled for 3 d and then allowed to explore the maze with all doors raised for 10 min for 2 d. After these 2 d, animals were food-restricted, each animal receiving ∼15 gm of food per day, and remained that way throughout the experiment. After 2 d of food restriction, the adaptation process continued for two more days by placing the animal in each side arm with food behind the barrier, for 5 min. The animal was then placed in the other arm, which also contained food behind the barrier, for a 5 min stay. Next, the actual spatial alternation testing started. Each rat received 10 trials per day. During the first trial of each day, a Froot Loop morsel was presented in both goal arms. During the next 10 trials, the arm opposite to the one the animal had entered in the previous trial was baited, except when the animal had gone to the empty arm on the last trial. In that case, the food pellet was left in place (hence, the baited side was changed only after the animal had alternated). Once the animal entered either arm, the door was closed. Ten seconds after entry to the arm, the animal was removed and returned to the holding cage for a 10 sec intertrial interval. This training continued until a criterion of 80% correct choices (of 10 trials) on two consecutive days was achieved. Animals took 10–14 d to reach the criterion. Animals that did not reach the criterion by 20 d were rejected from the study. Approximately 5% of animals belonged to this latter group.
RESULTS
Basal extracellular levels of glutamate or dopamine were measured in 91 rats. The average extracellular dopamine values were 0.29 ± 0.05 fmol/μl (n = 41) in the PFC and 0.98 ± 0.1 fmol/μl (n = 7) in the striatum. Average extracellular levels for glutamate were 0.8 ± 0.25 pmol/μl (n = 38) in the PFC and 0.64 ± 0.3 pmol/μl (n = 5) in the striatum. These values are not corrected for in vitro recovery.
Effect of ketamine on extracellular glutamate levels in the PFC
Figure 1 demonstrates the effect of subanesthetic doses of ketamine on the extracellular levels of glutamate in the PFC. All doses tested increased these levels significantly (p < 0.05). The effect of 30 mg/kg ketamine remained significant (compared with the saline-treated group) until 100 min after the injection, whereas the effect of 10 and 20 mg/kg ketamine subsided 60 and 80 min, respectively, after the injection. The stress of saline injection produced an immediate increase in glutamate efflux in some animals, similar to that observed before (Moghaddam and Bolinao, 1994).
Fig. 1.

Effect of intraperitoneal injections of subanesthetic doses of ketamine on the extracellular levels of glutamate in the prefrontal cortex. All doses tested increased these levels significantly compared with the saline injected groups.Asterisks (*) denote significant differences from saline-treated groups as determined by Kruskal–Wallis ANOVA (n = 6 for 30 mg/kg; n = 7 for 20 mg/kg; n = 7 for 10 mg/kg groups).
Injection of 50 mg/kg ketamine, which produced a short-lasting sedation, did not increase the extracellular levels of glutamate significantly (Fig. 2). Furthermore, the anesthetic dose of 200 mg/kg significantly decreased these levels. These levels returned to baseline after the rebound from anesthesia, about 1 hr after the injection. It should be noted that in some animals higher than baseline glutamate levels were observed after the cessation of anesthesia; however, because of the variability of this response, there was no significant increase.
Fig. 2.

Effect of intraperitoneal injections of anesthetic doses of ketamine on the extracellular levels of glutamate in the prefrontal cortex. Asterisks (*) denote significant differences as compared with saline-treated group (n = 6 for 50 mg/kg;n = 5 for 200 mg/kg).
Effect of ketamine on cortical release of dopamine
In separate groups of animals, injection of a subanesthetic dose (30 mg/kg, i.p.) of ketamine significantly increased the extracellular levels of dopamine in this region (p < 0.001; see Fig. 3). To determine whether the ketamine-induced increase in the PFC is in part because of an increase in endogenous glutamate and subsequent stimulation of non-NMDA receptors within the PFC, the experiment above was repeated while the non-NMDA antagonist CNQX (50 μm) was locally perfused through the microdialysis probe. Injection of ketamine in the CNQX-treated group produced only a trend toward an increase (p = 0.07). Furthermore, the response of this group was significantly (p < 0.05) less than those animals that received ketamine in the absence of CNQX, suggesting that intra-PFC application of CNQX reduced the stimulatory effect of ketamine on dopamine release.
Fig. 3.

Effect of intraperitoneal injections of 30 mg/kg ketamine on the extracellular levels of dopamine in the prefrontal cortex in the presence or absence of local infusion of CNQX. Ketamine increased dopamine release significantly compared with saline-injected groups as determined by two-way repeated- measures ANOVA (p < 0.001, n = 9 for ketamine-treated group; n = 5 for saline-treated group). Infusion of CNQX (50 μm) started 1 hr before ketamine injection (n = 6). After this treatment, injection of ketamine only produced a trend toward an increase (p = 0.07). Furthermore, the effect of ketamine in CNQX-treated animals was significantly lower than CNQX-untreated animals (p < 0.05).
Effect of ketamine in the striatum
To assess the regional specificity of subanesthetic doses of ketamine, the effect of 30 mg/kg ketamine on the outflow of glutamate and dopamine in the striatum was examined (Fig. 4). The effect of ketamine on extracellular levels of Glu was highly variable, and although a trend toward an increase was evident, it did not reach significance. A significant (p < 0.01), but small, increase of ∼25% in dopamine release was observed in the striatum.
Fig. 4.

Effect of intraperitoneal injections of 30 mg/kg ketamine on the extracellular levels of dopamine (n = 7) and glutamate (n = 5) in the striatum. This treatment caused a trend toward an increase in extracellular levels of glutamate; however, because of high variability, there was no statistically significant effect. The striatal extracellular levels of dopamine did increase after the injection of 30 mg/kg ketamine (p < 0.01); however, this increase was significantly smaller than that observed in the prefrontal cortex (demonstrated on Fig. 3, p < 0.005).
Effect of systemic pretreatment with the AMPA/kainate receptor antagonist, L Y293558, on ketamine-induced performance decrement of spatial delayed alternation and dopamine release
As demonstrated on Figure 5, treatment with 30 mg/kg ketamine produces a deficit in spatial delayed alternation performance, whereas systemic treatment with 0.1 and 1.0 mg/kg LY293558, an AMPA/kainate receptor antagonist, did not affect the performance of this task. However, pretreatment with LY293558 significantly attenuated the deficit produced by ketamine as compared with saline pretreatment. Accordingly, pretreatment with LY293558 produced a significantly (p < 0.05) lower increase of dopamine release in response to ketamine as compared with saline pretreatment (Fig. 6).
Fig. 5.
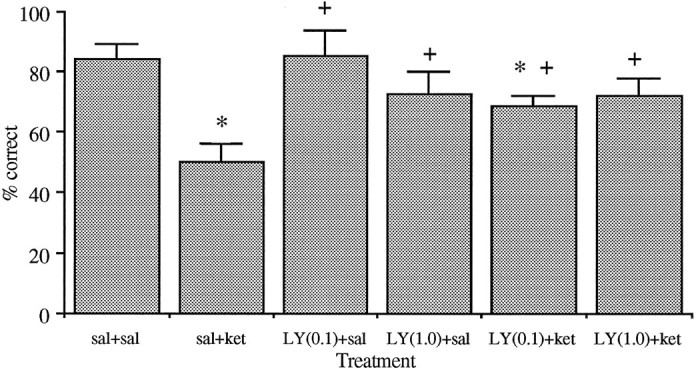
Effect of ketamine (30 mg/kg) and LY293558 (0.1 and 1.0 mg/kg) on spatial delayed alternation performance in rats. Each animal received 10 trials per day, and the mean ± SEM percentage of correct responses per session is presented. The trials were performed with a delay of 10 sec. The values inparentheses represent the doses in mg/kg. Saline(veh) and LY293558 (LY) were injected 15–20 min before ketamine (ket) in groups designated as veh/ket and LY/ket, respectively. Pretreatment with LY293558 produced a significant reversal of ketamine-induced decrease in percentage of correct choice in saline-treated rats (*p < 0.01, as compared with the veh group, + p < 0.01 as compared with the veh/ket group).
Fig. 6.

Effect of intraperitoneal pretreatment with saline or LY293558 (1.0 mg/kg) on ketamine (30 mg/kg)-induced increase in dopamine release in the prefrontal cortex. Treatment groups paralleled the behavioral studies depicted on Figure 5. Injections were 20 min apart. Significant increases in dopamine release were observed in both LY293558+ketamine and saline+ketamine group (n = 7,p < 0.01; n = 8, p < 0.01, respectively). However, the increase after LY293558+ketamine was significantly smaller than that observed after saline+ketamine (p < 0.05, as determined by two-way repeated-measures ANOVA).
DISCUSSION
Effect of ketamine on glutamate efflux
The present study demonstrates that ketamine has a biphasic influence on the outflow of Glu in the prefrontal cortex: low subanesthetic doses increase these levels, whereas an anesthetic dose decreases these levels. Several mechanisms may account for the stimulatory effect of low doses of ketamine on glutamate outflow. First, several lines of evidence suggest that NMDA receptors are located presynaptically and may be regulating the release of glutamate and other endogenous excitatory amino acids (Miwa et al., 1992;Smirnova et al., 1993; Huntley et al., 1994; Liu and Moghaddam, 1995;Wang and Thukral, 1996). Second, ketamine, as well as other NMDA antagonists, may produce disinhibition of GABAergic or other inhibitory inputs to the glutamate-containing neurons, thereby enhancing the firing rate of these glutamatergic neurons. Such disinhibition may occur locally at the PFC and/or at regions with ascending glutamatergic projections to the PFC.
The stimulatory effects of low doses of ketamine were dose-dependent to the extent that the higher doses produced longer durations of response. However, the magnitude of response did not vary among doses at early time points. This may be expected in that during basal conditions the excitatory effects mediated by glutamate at the NMDA receptor may be partially blocked by the hyperpolarizing influence of tonically active GABA neurons that impinge on PFC afferents and PFC pyramidal cells (Houser et al., 1984; Smiley and Goldman-Rakic, 1993). Thus, low doses of NMDA antagonists may be sufficient to reach maximum blockade of these receptors. In that case, higher doses of ketamine will have similar magnitudes, but longer duration of effect in light of the short half-life of ketamine.
The inhibitory effect of an anesthetic dose of ketamine on extracellular levels of glutamate agrees with previous reports indicating that anesthesia decreases the extracellular levels of glutamate (Moghaddam and Bolinao, 1994). This decrease may be caused by inhibition of other neuronal systems postsynaptic to NMDA receptor-mediated neurotransmission. Nevertheless, the present finding suggests that a decrease in the outflow of endogenous excitatory amino acids may be common occurrence during anesthesia.
Ketamine (30 mg/kg) did not significantly increase Glu efflux in the striatum. Although in some animals an increase in these levels was evident, the high variability of response prevented this effect from reaching statistical significance. We have frequently observed a higher degree of variability in extracellular levels of Glu in the striatum as compared with cortical regions (Moghaddam, 1993).
Mechanism by which ketamine increases dopamine release in the prefrontal cortex
The stimulatory effect of ketamine on dopamine release in the PFC was reduced by local application of the AMPA/kainate receptor antagonist CNQX at a concentration (50 μm) that by itself does not affect basal release of dopamine in the PFC (Jedema and Moghaddam, 1994). This finding suggests that ketamine may increase dopamine release in the PFC by producing an initial increase in the release of endogenous excitatory amino acid, such as glutamate, which in turn increases the release of dopamine by stimulating non-NMDA excitatory amino acid receptors. Indeed, our previous studies have indicated that stimulation of AMPA or kainate receptors, but not the NMDA receptors, readily increases dopamine release in the PFC (Jedema and Moghaddam, 1996). Furthermore, similar reversal by CNQX was observed previously when assessing the effect of stress, a treatment that increases the outflow of glutamate (Moghaddam, 1993) and dopamine (Abercrombie et al., 1989) in the PFC. Specifically, it was found that pretreatment with CNQX significantly attenuated the stress-induced increase of dopamine in this region (Jedema and Moghaddam, 1994), suggesting that physiologically released glutamate (in response to stress) activated dopaminergic neurotransmission in the PFC via AMPA/kainate receptors.
Subanesthetic doses of ketamine increased dopamine release in the PFC in a manner that was larger in magnitude and longer in duration than in the striatum. Numerous other studies have indicated similar findings with regard to regional specificity of PCP in activating dopamine neurotransmission (Rao et al., 1989; Hondo et al., 1994; Hertel et al., 1996; Nishijima et al., 1996). One reason for the regional specificity of ketamine and PCP may be that, as suggested by the nonsignificant effect of ketamine in the striatum, the NMDA receptor antagonists are more effective in releasing glutamate in the PFC relative to the striatum. Another may be differences in the manner in which glutamate, released by these antagonists, may stimulate the release of dopamine in the PFC versus the striatum. Previous studies investigating this interaction indicate that stimulation of kainate or AMPA receptors is much more potent in increasing dopamine release in the PFC as compared with the striatum (Westerink et al., 1992; Jedema and Moghaddam, 1996).
Systemic AMPA/kainate receptor antagonists attenuate the influence of ketamine on dopamine release and spatial delayed alternation performance
Behavioral tests that impose a delay between a cue and execution of a motor response are thought to assess “working memory,” a cognitive function that is dependent on the functional integrity of the prefrontal cortex (Goldman-Rakic, 1987). In the rat, T-maze-delayed alternation (Simon et al., 1980; Brito et al., 1982; Stam et al., 1989;De Brabander et al., 1991) as well as operant delayed-matching-to-position (Dunnett, 1985; Broersen et al., 1994), are routinely used to assess this function. Previous studies from our laboratory have reported that subanesthetic doses of ketamine impair the performance of rats in a T-maze-delayed alternation paradigm (Verma and Moghaddam, 1996). Furthermore, it was demonstrated that this impairment is reversed by pretreatment with raclopride and haloperidol, which have in common their antagonistic properties at dopamine D2 receptors, suggesting that activation of dopamine neurotransmission at the D2 receptor contributes to the behavioral impairment of ketamine. This observation is in line with the work of Arnsten and coworkers that activation of dopamine neurotransmission in the PFC impairs performance of delayed alternation tasks (Arnsten et al., 1994; Murphy et al., 1996).
The neurochemical studies presented here that demonstrate that activation of dopamine neurotransmission by ketamine may be secondary to activation of AMPA/kainate receptors would predict that the behavioral decrement produced by ketamine should be ameliorated by AMPA/kainate antagonists. This hypothesis was tested by examining the effect of pretreatment with an AMPA/kainate receptor antagonist on the ketamine-induced T-maze-delayed alternation performance decrement. LY293558 (Schoepp et al., 1995) was chosen because, unlike CNQX, it readily crosses the blood brain barrier. The doses of LY293558 used in the present study (0.1 and 1.0 mg/kg) were much lower than the doses (≥10 mg/kg) that produce motor retardation and sedation (Schoepp et al., 1995). It was found that LY293558 by itself did not affect delayed alternation performance, in agreement with previous studies demonstrating that other AMPA/kainate receptor antagonists do not affect learning and memory in the rodent (Parada et al., 1992). However, LY293558 ameliorated the performance decrement elicited by ketamine, suggesting that activation of AMPA/kainate receptors may contribute to the NMDA receptor antagonist-induced performance decrement. In agreement with these behavioral findings, parallel microdialysis studies demonstrated that pretreatment with LY293558 reduces the ketamine-induced increase of dopamine release in the PFC. It is important to note that pretreatment with LY293558 only partially reversed the behavioral and dopaminergic effects of ketamine. This may be because of the relatively small doses of LY293558 used in the present study. Furthermore, other mechanisms that are independent of AMPA/kainate receptor activation are likely to contribute to the behavioral effects of ketamine. Nonetheless, the findings above suggest that enhancement of glutamatergic neurotransmission at AMPA/kainate receptors contributes, at least in part, to cognitive impairments and cortical dopaminergic activation elicited by ketamine.
CONCLUSIONS
The behavioral syndrome produced by ketamine (and PCP) in humans and laboratory animals has been primarily attributed to postsynapticreduction of glutamatergic neurotransmission at the NMDA receptor. The present findings demonstrate that ketamine and PCP may exert at least part of their effect in the PFC by activationof glutamatergic neurotransmission at AMPA/kainate receptors. This is supported by findings that subanesthetic doses of ketamine increase Glu efflux and that AMPA/kainate receptor antagonists attenuate ketamine-induced PFC dopamine release and cognitive impairment. It is noteworthy that antagonists of AMPA/kainate receptors reverse NMDA receptor antagonist-induced neurodegeneration (Olney and Farber, 1995;Sharp et al., 1995), hyperlocomotion (Hauber and Andersen 1993; Willins et al., 1993; Bubser et al., 1995), and stereotypy (our unpublished observations). An important implication from these findings, therefore, may be that hyperactivation of non-NMDA receptors, as opposed to a “glutamatergic deficiency,” may account for some of the cognitive deficits and schizophrenia-like symptoms of NMDA receptor antagonists. Hence, low doses of non-NMDA glutamate antagonists may be effective for treatment of cognitive disorders in which NMDA receptor hypofunction is suspect. Importantly, AMPA/kainate receptor antagonists also reduce the psychostimulant-induced activation of dopaminergic neurotransmission in limbic regions (Pap and Bradberry, 1995) and thus may be effective antipsychotics not only by reversing the NMDA receptor-induced deficiencies, but also by reducing the putative limbic hyperdopaminergic state in schizophrenia.
Footnotes
This work was supported in part by PHS Award MH48404 (B.M.), the Scottish Rite Foundation (B.M.), the Veterans Administration Merit Award (J.K.), and Centers for Schizophrenia and PTSD. We thank Drs. John Krystal and Steve Bunney for helpful discussions and Malu Bolinao for technical assistance.
Correspondence should be addressed to Dr. Bita Moghaddam, Department of Psychiatry, Yale University School of Medicine, VA Medical Center 116A12, West Haven, CT 06516.
REFERENCES
- 1.Abercrombie ED, Keefe KA, DiFrischia DS, Zigmond MJ. Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J Neurochem. 1989;52:1655–1658. doi: 10.1111/j.1471-4159.1989.tb09224.x. [DOI] [PubMed] [Google Scholar]
- 2.Akbarian S, Sucher NJ, Bradley D, Tafazzoli A, Trinh D, Hetrick WP, Potkin SG, Sandman CA, Bunney WE, Jones EG., Jr Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J Neurosci. 1996;16:19–30. doi: 10.1523/JNEUROSCI.16-01-00019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angrist B, Sathananthan G, Wilk S, Gershon S. Amphetamine psychosis: behavioral and biochemical aspects. J Psychiatr Res. 1974;11:13–23. doi: 10.1016/0022-3956(74)90064-8. [DOI] [PubMed] [Google Scholar]
- 4.Arnsten AF, Cai JX, Murphy BL, Goldman-Rakic PS. Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology. 1994;116:143–151. doi: 10.1007/BF02245056. [DOI] [PubMed] [Google Scholar]
- 5.Bakker CB, Amini FB. Observations on the psychotomimetic effects of sernyl. Comp Psychiatr. 1961;2:269–280. doi: 10.1016/s0010-440x(61)80033-3. [DOI] [PubMed] [Google Scholar]
- 6.Brito G, Thomas G, Davis B, Gingold S. Prelimbic cortex, mediodorsal thalamus, septum, and delayed alternation in rats. Exp Brain Res. 1982;46:52–58. doi: 10.1007/BF00238097. [DOI] [PubMed] [Google Scholar]
- 7.Broersen LM, Heinsbroek RPW, De Bruin JPC, Van Hest A, Olivier B. Effects of local application of dopaminergic drugs into the dorsal part of the medial prefrontal cortex of rats in a delayed matching to position task: comparison with local cholinergic blockade. Brain Res. 1994;645:113–122. doi: 10.1016/0006-8993(94)91644-6. [DOI] [PubMed] [Google Scholar]
- 8.Bubser M, Tzschentke T, Hauber W. Behavioral and neurochemical interactions of the AMPA antagonist GYKI 52466 and the non-competitive NMDA antagonist dizocilpine in rats. J Neural Transm Gen Sect. 1995;101:115–126. doi: 10.1007/BF01271550. [DOI] [PubMed] [Google Scholar]
- 9.Bustos G, Abarca J, Forrey MI, Gysling K, Bradberry CW, Roth RH. Regulation excitatory amino acid release by N-methyl-d-asparate receptors in rat striatum: in vivo microdialysis studies. Brain Res. 1992;585:105–115. doi: 10.1016/0006-8993(92)91195-k. [DOI] [PubMed] [Google Scholar]
- 10.Coyle JT. The glutamatergic dysfunction hypothesis for schizophrenia. Harvard Rev Psychiatr. 1996;3:241–253. doi: 10.3109/10673229609017192. [DOI] [PubMed] [Google Scholar]
- 11.Danysz W, Wroblewski JT, Costa E. Learning impairment in rats by n-methyl-d-aspartate receptor antagonists. Neuropharmacology. 1988;27:653–656. doi: 10.1016/0028-3908(88)90189-x. [DOI] [PubMed] [Google Scholar]
- 12.Davies BM, Beech DR. The effect of the 1-arylcyclohexylamine (sernyl) on twelve normal volunteers. J Ment Sci. 1960;106:912–924. doi: 10.1192/bjp.106.444.912. [DOI] [PubMed] [Google Scholar]
- 13.Deakin JFW, Slater P, Simpson MDC, Gilchrist AC, Skan WJ, Royston MC, Reynolds GP, Cross AJ. Frontal cortical and left temporal glutamatergic dysfunction in schizophrenia. J Neurochem. 1989;52:1781–1786. doi: 10.1111/j.1471-4159.1989.tb07257.x. [DOI] [PubMed] [Google Scholar]
- 14.De Brabander JM, De Bruin JPC, Van Eden CG. Comparison of the effects of neonatal and adult medial prefrontal cortex lesions on food hoarding and spatial delayed alternation. Behav Brain Res. 1991;42:67–75. doi: 10.1016/s0166-4328(05)80041-5. [DOI] [PubMed] [Google Scholar]
- 15.Dunnett SB. Comparative effects of cholinergic drugs and lesions of nucleus basalis or fimbria-formix on delayed matching in rats. Psychopharmacology. 1985;87:357–363. doi: 10.1007/BF00432721. [DOI] [PubMed] [Google Scholar]
- 16.Ghoneim MM, Hinrichs JV, Mewaldt SP, Petersen RC. Ketamine: behavioral effects of subanesthetic doses. J Clin Psychopharmacol. 1985;5:70–77. [PubMed] [Google Scholar]
- 17.Goldman-Rakic PS. Curcuitry of primate prefrontal cortex and regulation of behavior by representational memory. In: Plum F, editor. Handbook of physiology, Sec I, Higher functions of the brain. Oxford; New York: 1987. pp. 373–417. [Google Scholar]
- 18.Hauber W. Clozapine improves dizocilpine-induced delayed alternation impairments in rats. J Neural Transm Gen Sec. 1993;94:223–233. doi: 10.1007/BF01277027. [DOI] [PubMed] [Google Scholar]
- 19.Hauber W, Andersen R. The non-NMA glutamate receptor antagonist GYKI 52466 counteracts locomotor stimulation and anticataleptic activity induced by the NMDA antagonist dizocilpine. Naunyn Schmiedebergs Arch Pharmacol. 1993;348:486–490. doi: 10.1007/BF00173207. [DOI] [PubMed] [Google Scholar]
- 20.Hauber W, Schmidt WJ. Effects of intrastriatal blockade of glutamatergic transmission on the acquisition of T-maze and radial maze tasks. J Neural Transm Gen Sec. 1989;78:29–41. doi: 10.1007/BF01247111. [DOI] [PubMed] [Google Scholar]
- 21.Heale V, Harley C. MK-801 and AP5 impair acquisition, but not retention of the Morris milk maze. Pharmacol Biochem Behav. 1990;36:145–149. doi: 10.1016/0091-3057(90)90140-d. [DOI] [PubMed] [Google Scholar]
- 22.Hertel P, Mathé JM, Nomikos GG, Iurlo M, Mathé AA, Svensson TH. Effects of d-Amphetamine and phencyclidine on behavior and extracellular concentrations of neurotensin and dopamine in the ventral striatum and the medial prefrontal cortex of the rat. Behav Brain Res. 1996;72:103–114. doi: 10.1016/0166-4328(96)00138-6. [DOI] [PubMed] [Google Scholar]
- 23.Hondo H, Yonezawa Y, Nakahara T, Nakamura KHM, Mirano M, Uchimura H, Tashiro N. Effects of phencyclidine on dopamine release in the rat prefrontal cortex: an in vivo microdialysis study. Brain Res. 1994;633:337–342. doi: 10.1016/0006-8993(94)91558-x. [DOI] [PubMed] [Google Scholar]
- 24.Houser CR, Vaughn JE, Hendry SHC, Jones EG, Peters A. GABA neurons in the cerebral cortes. In: Peters A, Jones EG, editors. Cerebral cortex: functional properties of cortical cells, Vol 2. Plenum; New York: 1984. pp. 63–90. [Google Scholar]
- 25.Huntley GW, Vickers JC, Morrison JH. Cellular and synaptic localization of NMDA and non-NMDA receptor subunits in neocortex: organization features related to cortical circuitry, function and disease. Trends Neurosci. 1994;17:536–542. doi: 10.1016/0166-2236(94)90158-9. [DOI] [PubMed] [Google Scholar]
- 26.Javitt D, Zukin S. Recent advances in phencyclidine model of schizophrenia. Amer J Psychiatr. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 27.Jedema HP, Moghaddam B. Glutamatergic control of dopamine release during stress in rat prefrontal cortex. J Neurochem. 1994;63:785–788. doi: 10.1046/j.1471-4159.1994.63020785.x. [DOI] [PubMed] [Google Scholar]
- 28.Jedema HP, Moghaddam B. Characterization of excitatory amino acid modulation of dopamine release in the prefrontal cortex of conscious rats. J Neurochem. 1996;66:1448–1453. doi: 10.1046/j.1471-4159.1996.66041448.x. [DOI] [PubMed] [Google Scholar]
- 29.Kim JS, Kornhuber HH, Schmid-Burgk W, Holzmuller B. Low cerebrospinal fluid glutamate in schizophrenia patients and a new hypothesis on schizophrenia. Neurosci Lett. 1980;20:379–382. doi: 10.1016/0304-3940(80)90178-0. [DOI] [PubMed] [Google Scholar]
- 30.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Arch Gen Psychiatr. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 31.Krystal J, Karper L, Bennett A, Abi-Saab D, Souza C, Abi-Dargham A, Charney D. Modulating ketamine-induced thought disorder with lorazepam and haloperidol in humans. Schizo Res. 1995;15:156a. [Google Scholar]
- 32.Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995b;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- 33.Lahti AC, Holcomb HH, Medoff DR, Tamminga CA (1995a) Ketamine activates psychosis and alters limbic blood flow in schizophrenia. NeuroReport 6(6):869–872. [DOI] [PubMed]
- 34.Lalonde R, Joyal CC. Effects of ketamine and l-glutamic acid diethyl ester on concept learning in rats. Pharmacol Biochem Behav. 1991;39:829–833. doi: 10.1016/0091-3057(91)90039-5. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, Moghaddam B. Regulation of glutamate efflux by excitatory amino acid receptors: evidence for tonic inhibitory and phasic excitatory regulation. J Pharmacol Exp Ther. 1995;274:1209–1215. [PubMed] [Google Scholar]
- 36.Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. Study of a new schizophrenomimetic drug-sernyl. Amer Med Assoc Arch Neurol Psychiatr. 1959;81:363–369. doi: 10.1001/archneurpsyc.1959.02340150095011. [DOI] [PubMed] [Google Scholar]
- 37.Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, Breier A. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology. 1996;14:301–307. doi: 10.1016/0893-133X(95)00137-3. [DOI] [PubMed] [Google Scholar]
- 38.Matthysse S. Antipsychotic drug actions: a clue to the neuropharmacology of schizophrenia? Fed Proc. 1973;32:200–205. [PubMed] [Google Scholar]
- 39.Miwa A, Robinson HPC, Kawai N. Presynaptic glutamate receptors depress inhibitory postsynaptic transmission in lobster neuromuscular sypase. J Neurophysiol. 1992;70:1159–1167. doi: 10.1152/jn.1993.70.3.1159. [DOI] [PubMed] [Google Scholar]
- 40.Moghaddam B. Stress preferentially activates extraneuronal levels of excitatory amino acids in the prefrontal cortex: a comparison to hippocampus and basal ganglia. J Neurochem. 1993;60:1650–1657. doi: 10.1111/j.1471-4159.1993.tb13387.x. [DOI] [PubMed] [Google Scholar]
- 41.Moghaddam B, Bolinao ML. Glutamatergic antagonists attenuate ability of dopamine uptake blockers to increase extracellular levels of dopamine: implications for tonic influence of glutamate on dopamine release. Synapse. 1994;18:337–342. doi: 10.1002/syn.890180409. [DOI] [PubMed] [Google Scholar]
- 42.Murphy BL, Roth RH, Arnsten AFT. Effect of FG 7142 on prefrontal cortex dopamine and spatial working memory in rat and monkey. Proc Natl Acad Sci USA. 1996;92:1325–1329. doi: 10.1073/pnas.93.3.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishijima K, Kashiwa A, Hashimoto A, Iwama H, Umino A, Nishikawa T. Differential effects of phencyclidine and methamphetamine on dopamine metabolism in rat frontal cortex and striatum as revealed by in vivo dialysis. Synapse. 1996;22:304–312. doi: 10.1002/(SICI)1098-2396(199604)22:4<304::AID-SYN2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 44.Olney JW, Labruyere J, Pric MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244:1360–1362. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- 45.Oye N, Paulsen O, Maurset A. Effects of ketamine on sensory preception: evidence for a role of N-methyl-d-asparate receptors. J Pharmacol Exp Ther. 1992;260:1209–1213. [PubMed] [Google Scholar]
- 46.Pap A, Bradberry CW. Excitatory amino acid antagonists attenuate the effects of cocaine on extracellular dopamine in the nucleus accumbens. J Pharmacol Exp Ther. 1995;274:127–133. [PubMed] [Google Scholar]
- 47.Parada J, Czuczwar SJ, Turski WA. NBQX does not affect learning and memory tasks in mice: a comparison with d-CPPene and ifenprodil. Brain Res Cogn Brain Res. 1992;1:67–71. doi: 10.1016/0926-6410(92)90006-d. [DOI] [PubMed] [Google Scholar]
- 48.Parada-Turska J, Turski W. Excitatory amino acid antagonists and memory: effect of drugs acting at N-methyl-d-aspartate receptors in learning and memory tasks. Neuropharmacology. 1990;29:1111–1116. doi: 10.1016/0028-3908(90)90034-o. [DOI] [PubMed] [Google Scholar]
- 49.Paxinos G, Watson C. The brain in stereotaxic coordinates. Academic; New York: 1982. [Google Scholar]
- 50.Pulvirentia L, Swerdlow NR, Koob GF. Microinjection of a glutamate antagonist into the nucleus accumbens reduces psychostimulant locomotion in rats. Neurosci Lett. 1989;103:213–218. doi: 10.1016/0304-3940(89)90578-8. [DOI] [PubMed] [Google Scholar]
- 51.Rao T, Kim H, Lehman J, Martin L, Wood P. Differential effects of phencyclidine (PCP) and ketamine on mesocortical and mesostriatal dopamine release in vivo. Life Sci. 1989;45:1065–1072. doi: 10.1016/0024-3205(89)90163-x. [DOI] [PubMed] [Google Scholar]
- 52.Schoepp DD, Lodge D, Bleakman D, Leander JD, Tizzano JP, Wright RA, Palmer AJ, Salhoff CR, Ornstein PL. In vitro and in vivo antagonism of AMPA receptor activation by (3S,4aR,6R,8aR)-6-[2-(I(2)H-tetrazole-5-yl)etheyl]decahydroisoquinoline-3-carboxylic acid. Neuropharmacology. 1995;34:1059–1168. doi: 10.1016/0028-3908(95)00099-r. [DOI] [PubMed] [Google Scholar]
- 53.Seeman P, Lee T, Chau-Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261:717–719. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]
- 54.Sharp JW, Petersen DL, Langford MT. DNQX inhibits phencyclidine (PCP) and ketamine induction of the hsp70 heat shock gene in the rat cingulate and retrosplenial cortex. Brain Res. 1995;687:114–124. doi: 10.1016/0006-8993(95)00477-8. [DOI] [PubMed] [Google Scholar]
- 55.Simon H, Scatton B, LeMoal M. Dopaminergic A10 neurons are involved in cognitive functions. Nature. 1980;286:150–151. doi: 10.1038/286150a0. [DOI] [PubMed] [Google Scholar]
- 56.Smiley J, Goldman-Rakic P. Heterogeneous targets of dopamine synapses in monkey prefrontal cortex demonstrated by serial section electron microscopy: a laminar analysis using the silver-enhanced diaminobenzidine sulfide (SEDS) immunolabeling technique. Cereb Cortex. 1993;3:223–238. doi: 10.1093/cercor/3.3.223. [DOI] [PubMed] [Google Scholar]
- 57.Smirnova T, Stinnakre J, Mallet J. Characterization of a presynaptic glutamate receptor. Science. 1993;262:430–433. doi: 10.1126/science.8105537. [DOI] [PubMed] [Google Scholar]
- 58.Smith DJ, Azzaro AJ, Zaldivar SB, Palmer S, Lee HS. Properties of the optical isomers and metabolites of ketamine on the high affinity transport and catabolism of monoamines. Neuropharmacology. 1981;20:391–396. doi: 10.1016/0028-3908(81)90015-0. [DOI] [PubMed] [Google Scholar]
- 59.Snyder SH, Banerjee SP, Yamamura HI. Drugs, neurotransmitters and schizophrenia. Science. 1974;184:1243–1253. doi: 10.1126/science.184.4143.1243. [DOI] [PubMed] [Google Scholar]
- 60.Stam C, deBruin J, van Haelst A, van der Gugten J, Kalsbeck A. Influence of the mesocortical dopaminergic system on activity, food hoarding, social-agonistic behavior, and spatial delayed alternation in male rats. Behav Neurosci. 1989;103:24–35. doi: 10.1037//0735-7044.103.1.24. [DOI] [PubMed] [Google Scholar]
- 61.Thomson AM, West DC, Lodge D. An N-methyl-d-asparte receptor-mediated synapse in rat cerebral cortex: a site of action of ketamien? Nature. 1985;313:479–481. doi: 10.1038/313479a0. [DOI] [PubMed] [Google Scholar]
- 62.Ulas J, Cotman CW. Excitatory amino acid receptors in schizophrenia. Schizophr Bull. 1993;19:105–117. doi: 10.1093/schbul/19.1.105. [DOI] [PubMed] [Google Scholar]
- 63.Verma A, Moghaddam B. The role of excitatory amino acids in prefrontal cortex function as assessed by spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci. 1996;16:373–379. doi: 10.1523/JNEUROSCI.16-01-00373.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wachtel H, Turski L. Glutamate: a new target in schizophrenia? Trends Pharmacol. 1990;11:219–220. doi: 10.1016/0165-6147(90)90243-2. [DOI] [PubMed] [Google Scholar]
- 65.Wang JK, Thukral V. Presynaptic NMDA receptors display physiological characteristics of homomeric complexes of NRI subunits that contain the exon 5 insert in the N-terminal domain. J Neurochem. 1996;66:865–868. doi: 10.1046/j.1471-4159.1996.66020865.x. [DOI] [PubMed] [Google Scholar]
- 66.Wedzony K, Klimek V, Golembiowska W. MK-801 elevates the extracellular concentration of dopamine in the rat prefrontal cortex and increases the density of striatal dopamine D1 receptors. Brain Res. 1993;622:325–329. doi: 10.1016/0006-8993(93)90839-f. [DOI] [PubMed] [Google Scholar]
- 67.Wesierska M, Macias-Gonzalez R, Bures A. Differential effect of ketamine on the reference and working memory versions of the morris water maze task. Behav Neurosci. 1990;104:74–83. doi: 10.1037//0735-7044.104.1.74. [DOI] [PubMed] [Google Scholar]
- 68.Westerink BHC, Santiago M, De Vries JB. The release of dopamine from nerve terminals and dendrites of nigrostriatal neurons induced by excitatory amino acids in the conscious rat. Naunyn Schmiedebergs Arch Pharmacol. 1992;345:523–529. doi: 10.1007/BF00168943. [DOI] [PubMed] [Google Scholar]
- 69.Willins DL, Narayanan S, Wallace LJ, Uretsky NJ. The role of dopamine and AMPA/kainate receptors in the nucleus accumbens in the hypermotility response to MK801. Pharmacol Biochem Behav. 1993;46:881–887. doi: 10.1016/0091-3057(93)90217-h. [DOI] [PubMed] [Google Scholar]