

Abstract
The effects of a variety of antiproliferative agents on voltage-dependent K+ channel function in cortical oligodendrocyte progenitor (O-2A) cells were studied. Previously, we had shown that glutamate receptor activation reversibly inhibited O-2A cell proliferation stimulated by mitogenic factors and prevented lineage progression by attenuating outward K+ currents in O-2A cells. We now show that the antiproliferative actions of glutamate receptor activation are Ca2+-independent and arise from an increase in intracellular Na+ and subsequent block of outward K+ currents. In support of this mechanism, agents that acted to depolarize O-2A cells or increase intracellular sodium similarly had an antiproliferative effect, attributable at least in part to a reduction in voltage-gated K+ currents. Also, these effects were reversible and Ca2+-independent. Chronic treatment with glutamate agonists was without any long-term effect on K+ current function. Cells cultured in elevated K+, however, demonstrated an upregulation of inward rectifier K+ currents, concomitant with an hyperpolarization of the resting membrane potential. This culture condition therefore promoted a current phenotype typical of pro-oligodendroblasts. Finally, cells chronically treated with the mitotic inhibitor retinoic acid displayed a selective downregulation of outward K+ currents. In conclusion, signals that affect O-2A cell proliferation do so by regulating K+ channel function. These data indicate that the regulation of K+currents in cells of the oligodendrocyte lineage plays an important role in determining their proliferative potential and demonstrate that O-2A cell K+ current phenotype can be modified by long-term depolarization of the cell membrane.
Keywords: potassium channels, O-2A progenitors, cell proliferation, glial development, depolarization, lineage progression
In the mammalian CNS, both types of macroglial cells, astrocytes and oligodendrocytes, express virtually all the membrane channels that are found in neurons (for review, see Newman and Reichenbach, 1996; Ransom and Orkand, 1996; Sontheimer et al., 1996;Steinhauser and Gallo, 1996; Theodosis and MacVicar, 1996; Verkhratsky and Kettenmann, 1996). The physiological role of neurotransmitter receptors and voltage-dependent channels in glia, however, is primarily unknown, as well as the regulation of their expression during development. Delayed rectifier, transient, and inward rectifier K+ currents have been distinguished in both astrocytes and oligodendrocytes (Bevan and Raff, 1985; Barres et al., 1989; Sontheimer et al., 1989; Borges et al., 1994; Chvatal et al., 1995; Gallo et al., 1996). The functional expression of these currents changes during oligodendrocyte development between the highly proliferative progenitor (O-2A) and the differentiated oligodendrocyte stages (Barres et al., 1989; Sontheimer et al., 1989; Gallo et al., 1996). This makes cells of the oligodendrocyte lineage an ideal model to analyze the physiological role played by K+ channels during development and to study plastic changes in glial K+ current phenotype.
O-2A cell proliferation is modulated by a variety of factors (McMorris and Dubois-Dalcq, 1988; Raff et al., 1988; Bogler et al., 1990; Hunter and Bottenstein, 1990; McKinnon et al., 1990; Barres et al., 1993; Gard and Pfeiffer, 1993; Hardy and Reynolds, 1993; Canoll et al., 1996) including retinoic acid (RA), which has been identified as an antimitotic agent that also inhibits O-2A differentiation (Barres et al., 1993; Laeng et al., 1994; Noll and Miller, 1994). Similar to RA treatment, we found that activation of glutamate receptors (GluRs) in O-2A cells inhibits proliferation stimulated by mitogenic factors and prevents lineage progression (Gallo et al., 1996). We have also shown that the opening of GluR channels causes a blockage of K+currents in O-2A cells and that selective K+ channel blockers mimic the effects of GluR agonists on O-2A development (Gallo et al., 1996). Taken together, these findings indicate that modulation of voltage-gated K+ channels can modify the proliferative state of O-2A cells.
Because of the emerging role that K+ channels play in glial development, we sought to determine the following: (1) the mechanism of blockade of these channels on GluR activation; (2) which functional subtypes of K+ channels are expressed in oligodendrocyte lineage cells during their proliferative and postmitotic stages; (3) how these distinct subtypes of K+ channels are affected by acute treatment with antimitotic agents acting through different mechanisms; and (4) whether chronic treatment with antiproliferative agents induce changes in the functional expression of K+channels in O-2A cells. Our data indicate that membrane depolarization by itself and the subsequent reduction of delayed rectifier and transient K+ currents are sufficient to inhibit O-2A proliferation and lineage progression. Finally, we show that culturing O-2A cells in depolarizing concentrations of K+ not only blocks their proliferation but also promotes expression of a K+ channel phenotype that is typical of preoligodendroblasts, i.e., causes a strong upregulation of inward rectifier K+ channels.
MATERIALS AND METHODS
Materials. All cell culture media were obtained from Life Technologies (Gaithersburg, MD). Fetal bovine serum (FBS) was obtained from HyClone (Logan, UT). Platelet-derived growth factor (PDGF) and basic fibroblast growth factor (bFGF) were obtained from Upstate Biotechnology (Lake Placid, NY). Kainic acid, glutamate, TTX, veratridine, and all-trans RA were obtained from Sigma (St. Louis, MO), and AMPA and DNQX were obtained from Tocris Cookson (Bristol, UK). A23187 was obtained from Calbiochem (San Diego, CA). [Methyl-3H]thymidine was obtained from Amersham Life Science (Arlington Heights, IL). All secondary fluorochrome-conjugated antibodies used for immunocytochemistry were obtained from Cappel-Organon Teknika (Durham, NC).
Cell cultures. Purified cortical O-2A progenitor cultures were prepared by modifications of previously described methods (Armstrong et al., 1990; McKinnon et al., 1990). Briefly, E20 Sprague Dawley rats were killed following the National Institutes of Health Animal Welfare guidelines, and cortices were removed, mechanically dissociated, suspended in DMEM containing 10% FBS, and plated in plastic T75 flasks. After 12 d in culture, O-2A progenitor cells growing on top of a confluent monolayer of astrocytes were detached by overnight shaking (McCarthy and de Vellis, 1980). Contaminating microglial cells were further eliminated by plating this fraction on plastic culture dishes for 1 hr. The O-2A progenitor cells, which do not attach well to plastic, were collected by gently washing the dishes, replated (3 × 104 cells/cm2) onto poly-d-ornithine-coated plates (0.1 mg/ml) and cultured in DME-N1 biotin-containing medium. After 2 hr, either PDGF (human AB, heterodimer form; 10 ng/ml) or bFGF (human; 10 ng/ml), or PDGF + bFGF (10 ng/ml each) was added to the culture medium. O-2A cells were cultured for 1–3 d and treated every 24 hr with PDGF and/or bFGF. Differentiation into O4+, postmitotic pro-oligodendroblasts (Gallo and Armstrong, 1995) was promoted by growing the O-2A progenitors for 2–3 d in DME-N1 medium containing 0.5% FBS. The culture media containing high KCl were modified DMEM containing 25 mm KCl and 89 mm NaCl (25 mmK+ medium), or 45 mm KCl and 69 mmNaCl (45 mm K+ medium) (Life Technologies). The 25 mm K+/no Ca2+ medium contained 91.3 mm NaCl, whereas the 45 mmK+/no Ca2+ medium contained 71.3 mmNaCl (Life Technologies).
Cultures enriched with different cell types were characterized immunocytochemically by using cell type-specific antibodies (see below). Cell cultures used for immunostaining were grown on glass coverslips precoated with poly-d-ornithine. In cortical cultures enriched in O-2A progenitors, >95% of the cells were labeled by the monoclonal LB1, anti-GD3 antibody (Levi et al., 1986; Curtis et al., 1988) after 24–48 hr in vitro with PDGF or PDGF + bFGF. In pro-oligodendroblast-enriched cultures, >85% of the cells were O4+ (Sommer and Schachner, 1981) after 48 hr of culture in the absence of mitogens.
Cell proliferation assays. Cell proliferation was assayed as described previously (Gallo et al., 1996). Purified cortical O-2A cells were plated in DME-N1 biotin-containing medium with 0.5% FBS in 24 multiwell plates at a density of 2 × 104cells/cm2. After 2 hr, PDGF and/or bFGF and test compounds were added to the cultures along with [methyl-3H]thymidine (0.5 μCi/ml; 85 Ci/mmol). After 22 hr, cells were lysed and [3H]thymidine incorporation was measured by precipitation with 10% trichloroacetic acid and scintillation counting.
Immunocytochemistry and counting of cell cultures. The primary antibodies used were LB1 (Levi et al., 1986; Curtis et al., 1988) and O4 (Sommer and Schachner, 1981). Indirect immunofluorescence experiments were performed as described previously (Gallo and Armstrong, 1995; Gallo et al., 1996). Live cells were incubated for 30 min with primary antibodies diluted in DMEM, followed by fluorescein-conjugated GAM IgG (for LB1) or IgM (for O4) for 20 min. Cells were then fixed in 4% paraformaldehyde and 0.2% glutaraldehyde, pH 7.4 in PBS, for 15 min and mounted in Vectashield (Vector Laboratories, Burlingame, CA). Controls for antibody specificity were performed by sequentially omitting each of the primary antibodies in the immunostaining protocols. The percentage of O4+ cells was determined on three independent sets of cultures as described previously (Gallo et al., 1996). After 2 d in culture, only a small percentage (<2%) of the total cells were stained with the monoclonal antibody O1 (Sommer and Schachner, 1981) (see also Gallo and Armstrong, 1995).
Electrophysiology. For electrophysiological experiments, cells were cultured either with 10 ng/ml PDGF (proliferating O-2A progenitor cells) or with PDGF for 2 d and then in N1 + O.5% FBS for 3 d (pro-oligodendroblasts). Cells were perfused with media of the following composition (in mm): NaCl 160, KCl 2.5, CaCl2 1.5, MgSO4 1.5, and glucose 10; HEPES 10; TTX 0.5–1 μm. In experiments in which Ca2+was omitted from the recording solution, 1 mmCo2+ or 200 μm Cd2+ was substituted. In the experiments using 45 mm[K+]o, K+ was substituted by an equimolar amount for [Na+]o. In experiments involving NMDG substitution, 160 mm NMDG was substituted for Na+. The solution was buffered to pH 7.3 using HCl to give a final [Cl−]o of 160 mm.
Tight-seal (>5 GΩ) whole-cell recordings (Hamill et al., 1981;Edwards et al., 1989) were made from GD3+ O-2A progenitors or O4+ pro-oligodendroblasts. Careful attention was paid to select only cells with a strict bipolar morphology for O-2A electrophysiological analysis, to ensure that a homogeneous population of cells was studied. Patch electrodes were fabricated from thin-walled borosilicate glass (WPI, Gaithersburg, MD, TW150F-6) and had resistances of 2–6 MΩ when filled (in mm) with K-gluconate 130; NaCl 10; Na2ATP 2; NaGTP 0.3; HEPES 10; ethylene glycol-bis-β-aminoethylether, EGTA 0.6; buffered to pH 7.4 and ∼275 mOsm. Glutathione (5 mm) was included and Mg2+ excluded from the intracellular solution to prevent a loss of N-type inactivation resulting from cysteine oxidation (Ruppersberg et al., 1991). Cell sealing and breakthrough into whole-cell mode were performed in current-clamp mode, permitting an accurate determination of cell resting membrane potential. Unless stated otherwise, cells were then voltage-clamped between −70 and −40 mV and test pulses delivered to −60 to +70 mV (0.1 Hz). Linear leak current and capacitive artifacts were digitally subtracted off-line using Clampfit (Axon Instruments, Burlingame CA) before analysis. Records were filtered at 2 kHz and digitized at 5–10 kHz. The series resistances were calculated from the capacitive current peak (filtered at 10–20 kHz) in a 10 mV voltage step and were in the range of 5–15 MΩ (mean 11.4 ± 1.3 MΩ; n = 47). Series resistances were compensated to at least 85%. Cell capacitance was measured directly from the amplifier after compensation of the series resistance and capacitance in response to a 5–10 mV voltage step. Current density was calculated by dividing current amplitude by the cell membrane capacitance. Plots of the voltage dependence of current activation were constructed by dividing the peak current by the driving force (Vtest − Vr), where Vtest was the step depolarizing potential and Vr was the calculated reversal potential (Ek = −95 mV). The activation profiles were fitted with a Boltzmann equation of the form:
where g/gmax is the conductance normalized to its maximum value, V is the membrane potential, V1/2 is the membrane potential at which the current amplitude is half maximum, and k is a constant. For the construction of activation curves of the sustained current component (e.g., see Fig. 1) the sum of two independent Boltzmann equations was used. All drug solutions were added directly to the bath via the perfusion system in known concentrations. All data are expressed as the mean ± SEM.
Fig. 1.

Both transient and sustained voltage-dependent outward K+ current phenotypes are observed in O-2A progenitor cells. A, Outward currents were activated by test potentials up to +70 mV (10 mV increments, 0.1 Hz,Vhold = −70 mV). A prepulse to either −110 or −40 mV (100 msec duration, see insets) permitted the isolation of both the sustained and transient current components. Currents evoked from −40 mV possessed only a sustained current phenotype (left panel). When a prepulse to −110 mV was included, an additional transient current component was observed in the total current activated (middle trace). Digital subtraction of the sustained current component (left panel) from the total outward current (middle panel) permitted the isolation of the inactivating transient current component (right panel). The sustained current demonstrated modest inactivation (10%) during a 500 msec test pulse. B, The sum of two Boltzmann distributions was required to adequately describe the voltage-dependence of sustained current activation. The mean half-activations of the two current components were −23.7 ± 0.5 mV (k = 5) and 12.9 ± 1.8 mV (k = 13) (n = 25). The two currents represented 37 and 63% of the total current, respectively. This suggests that the total sustained current in O-2A cells reflects the temporal overlap of two current components. C, Transient currents were observed in only 63% of cells. In contrast to the sustained current, a single Boltzmann distribution was sufficient to describe the voltage-dependence of activation. Transient currents had a mean half-activation of −10.4 ± 0.9 mV (n = 24).
RESULTS
Voltage-dependent potassium currents in O-2A progenitor cells
Sustained current component
Previous studies have characterized the variety of voltage-dependent currents present in A2B5+ O-2A progenitor cells (Sontheimer et al., 1989; Barres, 1990). However, because these cells possess multiple proliferative stages, all with distinct repertoires of voltage-gated currents (Sontheimer et al., 1989; Barres, 1990), we considered it appropriate to first perform a detailed analysis of the outward and inward voltage-dependent potassium currents present in GD3+, O-2A progenitors cultured with PDGF.
At a holding potential of −40 mV, a voltage close to the resting membrane potential (Table 1), voltage-gated, outward currents were activated by incremental test potentials (10 mV increments, 0.1 Hz) up to +70 mV (Fig. 1). Currents activated from this holding potential were of a sustained phenotype (Fig.1A,B; Table 1) and possessed properties similar, but not identical, to those described previously for A2B5+ cells (Sontheimer et al., 1989). The current time to peak was relatively rapid; at a test potential of +70 mV, the mean time to peak = 13.7 ± 2.0 msec (n = 10) and showed no voltage-dependence in its rate of activation over all potentials tested. The sustained current phenotype, observed in >90% of cells, demonstrated modest inactivation during a maintained depolarizing test pulse (500 msec). At a time point of 490 msec, the mean current inactivation was 34.1 ± 3.4% (n = 10, Vtest = +70 mV). The magnitude of current inactivation, however, varied from cell to cell (range, 0–50%; compare currents in Figs. 1, 3, and 4).
Table 1.
A comparison of 0-2A and pro-oligodendroblast K+ current properties
| O-2A (n = 47) | O4+ (n = 11) | |
|---|---|---|
| RMP (mV) | −49.6 ± 3.2 | −80.0 ± 0.9 |
| Membrane capacitance (pF) | 10.6 ± 0.6 | 35.0 ± 2.6 |
| Sustained current1-a(pA/pF) | 150.7 ± 12.6 | 8.9 ± 2.9 |
| Transient current1-a (pA/pF) | 148.2 ± 18.3c | ND |
| Kir (pA/pF)b | −3.9 ± 1.1d | −8.8 ± 2.2 |
Currents amplitude measured at +70 mV.bCurrent amplitude measured at −120 mV.cn = 28.dn = 16. ND, Not determined.
A comparison between O-2A progenitor cell (GD3+) properties and pro-oligodendroblasts (O4+) cultured for 24–48 hr. On attainment of the O4+ phenotype, a negative shift in the cell membrane potential was observed concomitant with an increase in the cell capacitance. A complete loss of the transient current and a marked downregulation of the sustained current components were observed compared to O-2A progenitor cells. In addition, an upregulation of the Kir current was observed, consistent with the negative shift in the membrane potential.
Fig. 3.

The block of outward K+ currents by GluR activation in O-2A cells is [Na+]i-dependent. A, Addition of kainate (KA, 200 μm) reversibly attenuated the sustained current by 48.5 ± 5.3% (n = 11). Currents were activated by a test pulse to +70 mV (with or without a prepulse to −110 or −40 mV; see Fig. 1). In addition, the isolated transient current was also blocked by GluR activation (41.2 ± 9%, n = 6,E). B, The AMPA-preferring receptor antagonist DNQX (20 μm) blocked the kainate-induced attenuation of K+ currents (B,E), confirming a requirement for AMPA receptor activation in K+ current attenuation. C, When Na+ in the extracellular medium was replaced by NMDG, kainate application no longer reduced O-2A K+ currents (94.2 ± 5.3% of control), confirming that an increase in [Na+]i alone was responsible for the K+ current block. D, A direct increase in [Na+]i by application of veratridine (50 μm) also attenuated both the transient and sustained current (53.7 ± 3.1% and 22.2 ± 7.1%, n = 10), respectively. E, Summary histogram of the effects shown in A–D for both the isolated sustained and transient current components.
Fig. 4.

An elevation of extracellular K+reduces outward and augments inward K+ currents in O-2A cells. Elevation of [K+]o from 2.5 to 45 mm reduced outward currents (A) and augmented the inward rectifying current (B). The magnitudes of both the sustained current reduction and the Kir augmentation were as predicted from a simple change in the K+ driving force. Aii, The current amplitude was reduced by 52 ± 5% (n = 6), a value close to the calculated value (56%). Bii, Likewise, the observed augmentation of the Cs+-sensitive Kir was 440 ± 60% (n = 5), a value close to the predicted value of 469%.
The voltage-dependence of activation of O-2A cell outward currents has not been analyzed previously. Figure 1B illustrates the mean conductance-voltage relationship of the sustained current obtained from 25 representative O-2A cells. The sum of two Boltzmann distributions was required to adequately describe the voltage-dependence of activation (see Materials and Methods); the mean half-activation potentials of the two currents were −23.7 ± 0.5 mV (k = 5) and +12.9 ± 1.8 mV (k = 13) (n = 25). Each current component represented 37 and 63% of the total current respectively. We considered it possible that one of these components may represent a Ca2+-dependent outward K+ current, similar to that shown in O4+ cells (Sontheimer et al., 1989). When experiments were performed in a nominally Ca2+-free solution (see Materials and Methods), however, the voltage-dependence of activation and the relative proportion of each current remained relatively unchanged (data not shown). These data suggest that the total sustained current in these cells reflects the temporal overlap of two Ca2+-independent sustained current components.
Transient current properties
When depolarizing test pulses were preceded by a prepulse to −110 mV (100 msec), a rapidly activating current component was observed in 63% of the cells (Fig. 1A; Table 1). Digital subtraction of the current family obtained with a prepulse to −40 mV from that obtained with a prepulse to −110 mV permitted the isolation of the transient “A-type” current component (Fig.1A, right panel). Figure 1 shows that transient currents were activated at potentials positive to −70 mV and had a half-activation potential of −10.4 ± 0.9 mV (n = 24). In many O-2A cells (37%) we were unable to detect an appreciable transient current component. We considered it possible that this may result from a loss of N-type inactivation of the channels underlying the transient current resulting from cysteine oxidation (Ruppersberg et al., 1991) during whole-cell recording. This was unlikely, however, because the reducing agent glutathione was included and Mg 2+ excluded from the pipette solution in all recordings.
Inward rectifying K+ currents (Kir)
GD3+ O-2A cells displayed Kir with properties similar to those described previously by Barres et al. (1990) (Fig.2; Table 1). In the present experiments, Kir were activated by either one of two protocols. Cells were voltage-clamped at −40 mV, and a voltage step was delivered to −120 mV (200 msec) to fully activate the Kir. A voltage ramp protocol was then used to cross the voltage range −120 mV to +50 mV (60 mV/sec; Fig.2A). This protocol was then repeated in the presence of extracellular Cs+ (5 mm) to selectively block the Kir (Barres et al., 1990). Kir were isolated by digitally subtracting the current in the presence of Cs+ from that obtained in control (Fig. 2B). The reversal potential of the Cs+-sensitive current was −96 mV, close to the calculated reversal potential for K+(EKcalc = −100 mV). At potentials positive to −90 mV, the Cs+-sensitive current became outward, but with additional depolarization, strong rectification was observed and no outward current was observed at the most positive potentials. Alternatively, Kir were activated by holding the cell at −70 mV and voltage steps to negative test potentials (5 mV increments, 0.1 Hz, Fig. 2B) were delivered, to a final test potential of −120 mV. Application of Cs+ removed all time-dependent currents and permitted the isolation of Kir. The inward rectifying current was also sensitive to extracellular Ba2+ (5 mm, n = 6; data not shown). The KirErev was shifted in a predictable manner when [K+]o was elevated to 45 mm(Erev = EKcalc = −26 mV; Fig. 2D), confirming that the the isolated current was indeed Kir and not the hyperpolarizing currentIh (Halliwell and Adams, 1982; Maccaferri et al., 1993).
Fig. 2.

Kir currents in O-2A progenitor cells are revealed at negative test potentials. Kir were activated by either one of two protocols. A, A ramp protocol (inset) delivered from −120 mV to +50 mV (60 mV/sec) evoked both inward and outward currents. Addition of Cs+ (5 mm) to the extracellular solution selectively blocked the Kir. B, Kir were isolated by digitally subtracting the current obtained in the presence of Cs+ from that obtained in control. The reversal potential of Kir was −96 mV, close to the calculated reversal potential for K+ (EKcalc = −100 mV). At potentials positive to −90 mV, the Cs+-sensitive current became outward until strong rectification was observed and no current was observed at the most positive potentials. C, Alternatively, Kir were activated by voltage-clamping cells at −70 mV and delivering test potentials to negative voltage-steps (5 mV increments, 0.1 Hz). Application of Cs+ removed all time-dependent currents (middle panel) and permitted the isolation of Kir by digital subtraction (bottom panel). D, The KirErev were shifted in a predictable manner when [K+]o was elevated from 2.5 to 45 mm (Erev =EKcalc = −26 mV, D). This confirms that the major permeant ion is K+, confirming that the isolated current was Kir and not the hyperpolarizing-activated current Ih.
K+ current properties in pro-oligodendroblasts
We reported previously that on attainment of the O4+pro-oligodendroblast phenotype, a downregulation of all outward K+ currents was observed (Gallo et al., 1996). A comparison of K+ current phenotypes and their corresponding current densities in both O-2A and pro-oligodendroblast cell types is shown in Table 1. In addition to our previous data, we now report an upregulation of the Kir current density concomitant with a negative shift in the cell resting membrane potential in O4+ cells (Table 1).
The observations that K+ currents in the O-2A lineage are developmentally downregulated and that the antiproliferative actions of glutamate occur through a reversible blockade of voltage-dependent K+ channels (Gallo et al., 1996) suggest that regulation of K+ channel activity might be intimately linked to the proliferative potential of the O-2A cells, similar to that observed in other neural (Chiu and Wilson, 1989; Pappas et al., 1994) and non-neural (DeCoursey et al., 1984) cell types.
[Na+]i-dependent block of K+currents in O-2A cells
In our previous study, we demonstrated that AMPA receptor activation attenuated O-2A cell K+ currents and suggested that the antiproliferative action of GluR activation may result from a mechanism involving a blockade or a downregulation of K+channels. A study by Kettenmann and co-workers (Borges and Kettenmann, 1995) also demonstrated that the GluR-induced K+ current reduction resulted from an increase in [Na+]i, presumably entering the cell as a consequence of GluR activation.
In the present experiments, we first confirmed our initial finding that GluR activation blocks K+ currents in O-2A cells and then extended this observation to analyze the role of depolarization and [Na+]i in the current attenuation. Figure3 demonstrates that at a test potential of +70 mV and in the presence of extracellular Na+ ions, 200 μm kainate attenuated the sustained current by 48.5 ± 5.3% (n = 11). In addition, we now show that the isolated transient current was also blocked by GluR activation (mean current block, 41.2 ± 9%, n = 6) (Fig.3E). The block of the K+ currents by kainate was weakly voltage-dependent; the block of the sustained current at +70 mV (48%) was similar to that observed at 0 mV (mean current block, 46.0 ± 6.0%, n = 6), but was greater than that seen at −20 mV (39.4 ± 9.8, n = 6). Inclusion of the non-NMDA receptor antagonist DNQX (20 μm) in the extracellular solution blocked the kainate-induced attenuation of K+ currents (Fig.3B,E), confirming a requirement for AMPA receptor activation in K+ current attenuation. Previously, we demonstrated that the effects of kainate on O-2A K+ currents were Ca2+-independent (Gallo et al., 1996). To confirm that an increase in [Na+]i alone was responsible for the K+ current block, we substituted extracellular Na+ with the membrane impermeant ion NMDG (160 mm). In NMDG-containing solution, kainate had no effect on O-2A K+ currents (94.2 ± 5.3% of control currents,n = 5). It is possible that NMDG may block current through AMPA receptor channels by physical occlusion of the channel pore; however, in the present experiments, a small inward current still could be observed in response to kainate in the presence of NMDG (22.1 ± 3.7 n = 14 compared with control 112.7 ± 20 pA n = 12), which was presumably attributable to Ca2+ entry. These data are in agreement with the findings of Borges and Kettenmann (1995) and confirm that Na+ entry after AMPA receptor activation underlies the attenuation of both the transient and sustained K+current.
Voltage-dependent Na+ currents have been described previously in cultured O-2A cells (Sontheimer et al., 1989; Barres et al., 1990). We therefore determined whether a direct increase in [Na+]i by the Na+ channel opener veratridine also blocked K+ currents in O-2A cells. Application of veratridine (50 μm) reversibly attenuated both the transient and sustained current (Fig.3D,E). The mean block of sustained and transient currents was 53.7 ± 3.1% and 22.2 ± 7.1% (n = 10), respectively. In addition, the action of veratridine was associated with a small inward current (17 ± 8pA,n = 9) consistent with an increase in [Na+]i. Finally, the Na+/K+ exchange inhibitor ouabain (1 mm), which causes a rise in the intracellular Na+ concentration, inhibited the sustained outward current by 46 ± 12% (n = 4).
We next investigated the impact of directly depolarizing O-2A cells with high [K+]o (45 mm) on K+ currents in O-2A cells. As expected, elevation of [K+]o from 2.5 to 45 mm decreased outward currents and augmented the inward rectifying current (Fig.4). Both the magnitude of the reduction of the sustained current and the augmentation of the Kir were as predicted from a simple change in the K+ driving force. The calculated reversal potential (EKcalc) for K+ currents in 45 mm [K+]o was −26 mV and predicted an attenuation of outward currents by 56%, assuming no change in the voltage-dependence of activation. Figure4A demonstrates that the current amplitude was reduced by 52 ± 5% (n = 6), a value close to the calculated value. Likewise, the observed augmentation of the Kir was 440 ± 60% (n = 5) (Fig. 4B), a value close to the predicted value of 469%.
The inhibition of O-2A cell proliferation by depolarization is Ca2+-independent
The data illustrated above indicate that activation of GluR receptors, an elevation in [Na+]i, and membrane depolarization with elevated [K+]oall reduce K+ currents in proliferating O-2A cells. Activation of GluRs in the same cells also inhibits their proliferation (Gallo et al., 1996). Because the opening of GluR channels causes a large influx of Na+ (and Ca2+) ions through the cell membrane and subsequent depolarization, we first sought to determine whether a direct increase in [Na+]ior membrane depolarization would be sufficient to inhibit O-2A cell proliferation. We performed cell proliferation assays in O-2A cells cultured for 24 hr in the presence of mitogenic factors (i.e., PDGF, bFGF, or PDGF + bFGF) in combination with veratridine or high concentrations (25–45 mm) of extracellular K+ions. Both agents mimicked the effects of GluR agonists on O-2A cell proliferation under all the culture conditions tested (Fig.5). Similarly to kainate or AMPA, high extracellular K+ (Fig. 5A) and veratridine (Fig.5B) strongly and dose-dependently decreased [3H]thymidine incorporation in O-2A cells. These results indicate that a direct increase in [Na+]iand/or depolarization of the cell membrane are sufficient to inhibit O-2A cell proliferation.
Fig. 5.

Membrane depolarization and increased [Na+]i inhibit O-2A cell proliferation—[3H]thymidine incorporation assays.A, High extracellular K+ inhibits O-2A cell proliferation. B, Veratridine inhibits O-2A cell proliferation. Cells were plated in 24-well plates at a density of 30,000 cells/well and cultured in DME-N1 + 0.5% FBS with PDGF and/or bFGF (both 10 ng/ml). Veratridine was added at a concentration of 10 or 50 μm, whereas the high K+ media contained 25 or 45 mm KCl, respectively. [3H]thymidine (0.5 μCi/ml) was added to the cultures 2 hr after plating the cells. After 22 hr, [3H]thymidine incorporation was measured by trichloroacetic acid precipitation and scintillation counting. Averages of three experiments in triplicate ± SEM are shown.A, *p < 0.001, **p < 0.05 compared with their respective controls (Student’s t test). B–D, Proliferation of cells treated with veratridine (50 and 10 μm) was significantly different from controls (p < 0.05 and p < 0.001, respectively). The antiproliferative effects of high K+ and veratridine are reversible. Time course after removal of high K+- (C) or veratridine-containing (D) medium. Progenitor cells were cultured in PDGF (10 ng/ml) in the absence (control condition) or presence of 45 mmK+- or veratridine-containing (20 μm) medium. After 22 hr, all cells were shifted to fresh culture medium without high K+ or veratridine, but containing PDGF (10 ng/ml) and [3H]thymidine (0.5 μCi/ml). At 22 hr, before the shift to low -K+ or veratridine-free medium, high K+ and veratridine inhibited [3H]thymidine incorporation by 70 and 47%, respectively. Cells were harvested after 6, 12, and 24 hr after shift to low-K+ or veratridine-free medium, and [3H]thymidine incorporation was measured by trichloroacetic acid precipitation and scintillation counting. Averages ± SEM (n = 3) are shown.
O-2A cells cultured with PDGF and 45 mm K+ for 24 hr, and then [3H]thymidine-pulsed in a low-K+ medium containing PDGF, reentered S-phase with a temporal pattern similar to cells that were never exposed to depolarizing concentrations of K+ (Fig. 5C). A similar pattern was also observed for cells cultured with PDGF and veratridine for 24 hr, and then [3H]thymidine-pulsed in a veratridine-free medium containing PDGF (Fig. 5D). It can be concluded, therefore, that the inhibitory effects of depolarization on O-2A cell proliferation were reversible.
The Na+/K+ exchange inhibitor ouabain, which causes a raise in the intracellular Na+ concentration, dose-dependently inhibited proliferation of O-2A cells cultured with PDGF or bFGF. Table 2 shows that ouabain (10–1000 μm) caused a 20–80% dose-dependent inhibition of both PDGF- and bFGF-stimulated O-2A cell proliferation.
Table 2.
Ouabain inhibits O-2A cell proliferation
| [3H]thymidine incorporation (% over N1, no growth factors) | ||
|---|---|---|
| PDGF | bFGF | |
| Control | 324 ± 30 | 310 ± 15 |
| Ouabain 10 | 260 ± 262-160 | 283 ± 8 |
| Ouabain 100 | 59 ± 8* | 75 ± 4* |
| Ouabain 1000 | 38 ± 6* | 50 ± 4* |
O-2A progenitor cells were purified and cultured as described previously at a density of 3 × 104cells/cm2 in DME-N1 medium, containing 0.5% FBS (Gallo and Armstrong, 1995). After 2 hr, PDGF or bFGF (both 10 ng/ml), ouabain (10–1000 μm), and [3H]thymidine were added to the culture medium. After 22 hr, [3H]thymidine was measured by trichloroacetic acid precipitation and scintillation counting. Averages of 3 experiments in triplicate ± SEM are shown.
p < 0.01;
F2-160: p < 0.005, compared with their respective controls (Student’s t test).
The antiproliferative effects of GluR agonists, agents that increase [Na+]i and elevated [K+]o, were independent of extracellular Ca2+. Figure 6A shows that in a nominally Ca2+-free culture medium, O-2A cell proliferation stimulated by mitogenic factors was still strongly inhibited by kainate, veratridine, and high [K+]o. In the nominally Ca2+-free medium, basal and growth factor-stimulated O-2A cell proliferation was lower than in the presence of Ca2+. [3H]thymidine incorporation in O-2A cells cultured in Ca2+-free N1 was 35.7 ± 1.7% of N1 + Ca2+ (n = 11). PDGF- and bFGF-stimulated O-2A cell proliferation in the absence of Ca2+ was 40.7 ± 4.3 and 41 ± 2.1% (n = 12 each), respectively, of that measured in a Ca2+-containing medium. However, growth factor-stimulated O-2A cell proliferation over control (N1) was unaltered by the absence of extracellular Ca2+ (Fig. 6A). These findings indicate that extracellular Ca2+ plays an important role in O-2A cell proliferation, but is not involved in the inhibitory effects of GluR agonists, agents that increase [Na+]i or elevate [K+]o. Consistent with this conclusion, the ionophore A23187, which strongly stimulates transmembrane Ca2+ influx in O-2A cells (Raff et al., 1988), did not affect cell proliferation under any of the culture conditions analyzed (Fig. 6B). In particular, 30 nm A23187 caused an appreciable [Ca2+]i elevation in the majority of the cells tested (P. Simpson and J. Russell, personal communication), but failed to modify [3H]thymidine incorporation in O-2A progenitors (Fig.6B). Finally, the voltage-dependent Ca2+channel blocker nifedipine did not reverse the inhibitory effects of kainate, veratridine, or high K+ on O-2A cell proliferation stimulated by PDGF or bFGF (data not shown).
Fig. 6.

The inhibitory effects of GluR agonists, agents that increase [Na+]i and membrane depolarization on O-2A cell proliferation, are independent on extracellular Ca2+. A, Absence of extracellular Ca2+ does not prevent kainate-, veratridine-, and high K+-induced inhibition of O-2A cell proliferation. Cells were plated in 24-well plates at a density of 30,000 cells/well and cultured in DME-N1 + 0.5% FBS with PDGF and/or bFGF (both 10 ng/ml), in the presence or absence of extracellular Ca2+(see Materials and Methods for media composition). Veratridine was added at a concentration of 25 μm, kainate was 100 μm, whereas the high K+ media contained 45 mm KCl. [3H]thymidine (0.5 μCi/ml) was added to the cultures 2 hr after plating the cells. After 22 hr, [3H]thymidine incorporation was measured by trichloroacetic acid precipitation and scintillation counting. Averages of two experiments in triplicate ± SEM are shown. All treatments (kainate, veratridine, and high K+) were significantly different from their respective controls, in both the presence and absence of Ca2+ (p < 0.001, Student’s t test). B, Treatment with the Ca2+ ionophore A23187 does not modify O-2A cell proliferation. Cells were plated in a DME-N1 medium containing Ca2+ with PDGF and/or bFGF (both 10 ng/ml), in the presence or absence of A23187 (1–30 nm) or AMPA (100 μm). [3H]thymidine (0.5 μCi/ml) was added to the cultures 2 hr after plating the cells. After 22 hr, [3H]thymidine incorporation was measured by trichloroacetic acid precipitation and scintillation counting. Averages of three experiments in triplicate ± SEM are shown.
Membrane depolarization prevents O-2A lineage progression
Our previous studies demonstrated that activation of GluRs in O-2A cells inhibited their lineage progression to the pro-oligodendroblast stage, identified by the monoclonal antibody O4 (Gallo et al., 1996). We therefore analyzed whether membrane depolarization also reproduced the effects of GluR activation on O-2A development. Under culture conditions that permitted O-2A lineage progression (N1, PDGF, or bFGF for 2 d), depolarizing concentrations (45 mm) of K+ significantly decreased the percentage of O4+ pro-oligodendroblasts (Fig. 7). In cells treated with PDGF + bFGF, a condition that prevents O-2A progenitor differentiation (Bogler et al., 1990; McKinnon et al., 1990; Gallo and Armstrong, 1995), membrane depolarization did not modify the small percentage of O4+ cells present in the cultures after 2 d. Under all the culture conditions tested, the small percentage (<2%) of O1+ cells was not affected by 45 mmKCl (data not shown).
Fig. 7.

Depolarization with high extracellular K+ prevents O-2A lineage progression, as detected by staining with O4 antibody. O-2A progenitor cells were purified and cultured on coverslips in DME-N1 medium with 0.5% FBS with PDGF (10 ng/ml), or bFGF (10 ng/ml), or PDGF + bFGF. K45indicates that cells were cultured in the same medium with the addition of 45 mm K+ (see Materials and Methods for media composition). After 48 hr, cells were immunostained with O4 antibody and counted. Averages ± SEM obtained from three experiments (n = 30) are shown. The total number of cells counted for each culture condition ranged from 1020 to 2046; *p < 0.001; **p < 0.005 (Student’s t test).
Chronic GluR activation does not alter K+current properties
Previous studies have indicated that cells of the oligodendrocyte lineage display changes in their K+ current phenotype that are related to their proliferative potential and their developmental stage (Sontheimer et al., 1989, Gallo et al., 1996). We therefore considered the possibility that long-term treatment of O-2A cells with antimitotic agents could induce plastic changes in the K+current phenotype similar to those observed during the transition from a proliferative to a nonproliferative state (Sontheimer et al., 1989;Barres et al., 1990). Our O-2A cell proliferation assays followed a time course of 24–48 hr. We therefore tested whether chronic exposure to any of the anti-proliferative agents might cause long-term changes in K+ channel function. In the next series of experiments, we monitored the effects of chronic exposure (24–48 hr) of O-2A cells to either GluR agonists or 45 mm[K+]o. After exposure to each antiproliferative agent, recordings were made from O-2A cells using standard recording conditions and solutions (i.e., 2.5 mmK+) to determine any changes in the K+ current phenotype.
Treatment of cultures with either 100 μm kainate or 100 μm AMPA for 24–48 hr had little effect on O-2A cell resting membrane parameters (Table 3). The mean current densities of both the sustained and Kir currents in O-2A cells were unaltered (Table 3). Likewise, the voltage-dependence of activation of the sustained current components were unaltered (data not shown). In kainate-treated cultures, the mean half-activation potentials of the sustained current components were −26.3 ± 1.3 and 16.7 ± 5.3 mV (n = 7). The contribution of each current to the total sustained current remained unchanged at 39 and 61%, respectively, (data not shown). These data suggest that long-term culture in either kainate or AMPA was without long-lasting effect on the O-2A cell K+ current phenotype. Interestingly, the transient current density was significantly reduced in cultures chronically treated with kainate. The reasons for the selective attenuation of the transient current are unclear at this time and were not studied further. These results support our previous data (Gallo et al., 1996), which demonstrated that on removal of kainate, O-2A cells continue to proliferate similarly to control cultures.
Table 3.
Effects of chronic treatment with antiproliferative agents on K+ current expression
| Control cells (n = 23) | Kainate-treated cells (n = 10) | 45 mm [K+]o-treated cells (n = 21) | RA-treated cells (n = 13) | |
|---|---|---|---|---|
| RMP (mV) | −42.0 ± 5.5 | −35.0 ± 3.4 | −74.2 ± 1.9 | −34.9 ± 4.6 |
| Membrane capacitance (pF) | 10.0 ± 0.7 | 9.1 ± 0.9 | 11.7 ± 0.7 | 9.2 ± 1.2 |
| Sustained current3-a (pA/pF) | 168.0 ± 14.7 | 168.2 ± 48.5 | 227.5 ± 22.9 | 118.9 ± 17.7 |
| Transient current3-a(pA/pF) | 131.6 ± 19.1 | 32.9 ± 8.5 | 169.0 ± 24.2 | 51.3 ± 16.2 |
| Kir (pA/pF)b | −3.1 ± 1.0c | −2.6 ± 0.6 | 41.3 ± 16.5d | −3.5 ± 1.2e |
Currents amplitude measured at +70 mV.bCurrent amplitude measured at −120 mV.cn = 6.dn = 6.en = 8.
Culturing O-2A cells in 45 mm[K+]o modifies the K+ current phenotype
Cells cultured in 45 mm[K+]o displayed physiological properties distinct from those of control O-2A cells. When cells cultured in 45 mm [K+]o were recorded in standard extracellular recording conditions (i.e., 2.5 mm[K+]o, see Materials and Methods), the resting membrane potential was significantly more negative than that seen in control control cultured cells, whereas no significant change in cell capacitance was observed (Table 3). This shift in the membrane potential likely reflected the upregulation of the Kir current density observed (Fig. 8B; Table 3), because Kir play a dominant role in setting resting membrane potential (Duffy et al., 1995). Normalization of the Kir currents obtained in control cultured cells versus the 45 mm[K+]o cultured cells revealed no change in the current–voltage relationship of the Kir in cells cultured in 45 mm [K+]o (Fig.8Bii). Likewise, both the sustained and transient outward current densities were upregulated after culture in 45 mm [K+]o (Fig.8A; Table 3). Despite a larger current density, the voltage-dependence of the sustained current components was unaffected. The mean half-activations of the two current components were −29.4 ± 0.2 (k = 6) and 16.3 ± 1.7 (k = 13) (n = 9, data not shown). Likewise, the contributions of both current components remained unchanged at 43 and 57%, respectively.
Fig. 8.

Chronic exposure of O-2A cells to 45 mm [K+]o upregulates the Kir current. Cells cultured in 45 mm[K+]o for 48 hr possessed properties distinct from those observed in control cultured O-2A cells. After removal of the elevated K+ culturing medium, cells were perfused with a control solution (i.e., 2.5 mm K+) for electrophysiological recordings. The resting membrane potential of cells cultured in 45 mm [K+]o was significantly more negative than that seen in control (Table 3).A, An increase in the sustained current density was observed after culture in 45 mm[K+]o for 48 hr (see also Table 1).Ai, Representative current traces obtained from two different cells cultured under control and 45 mm[K+]o conditions demonstrate the upregulation of the sustained current component after chronic exposure to 45 mm [K+]o. Aii, Summary histograms of data obtained at a test pulse of +70 mV reveals an upregulation of both the current amplitude and the current density when normalized for any changes in membrane capacitance.Bi, Similarly, an upregulation of the Kir current density was also observed after chronic exposure to 45 mm[K+]o culturing conditions (Bi, 1, 3,4) (see also Table 3). Bi,2, Normalization of the Kir currents obtained in control (n = 8) versus the 45 mm[K+]o (n = 6) revealed no change in the current–voltage relationship of the Kir after chronic 45 mm [K+]o culture.Aii, Bii, Summary histograms of sustained and Kir current data obtained from both control (2.5 mm[K+]o) and 45 mm[K+]o culturing conditions reveal an upregulation of both the total current and the current density in 45 mm [K+]o culturing conditions. Data shown in the histogram was obtained from a current step to −120 mV.
RA reversibly inhibits O-2A cell proliferation and downregulates K+ currents
Finally, we analyzed the effects of a different anti-proliferative agent, RA, on K+ channel function. RA is known to regulate oligodendrocyte precursor development (Barres et al., 1993; Laeng et al., 1994; Noll and Miller, 1994). In agreement with previous studies on brain and spinal cord O-2A cells cultured with bFGF (Laeng et al., 1994; Noll and Miller, 1994), micromolar concentrations of RA inhibited cortical O-2A proliferation triggered by PDGF, bFGF, or PDGF + bFGF, as measured by [3H]thymidine incorporation (Fig.9A). IC50 values for RA were 0.36 ± 0.1, 3.4 ± 0.6, and 1.7 ± 0.5 μmin PDGF, bFGF, and PDGF+bFGF, respectively (n = 9). The effects of RA on O-2A cell proliferation were reversible. O-2A cells cultured with PDGF and RA for 24 hr, and then [3H]thymidine-pulsed in an RA-free medium containing PDGF, reentered S-phase with a temporal pattern similar to that of untreated cells (Fig. 9B).
Fig. 9.
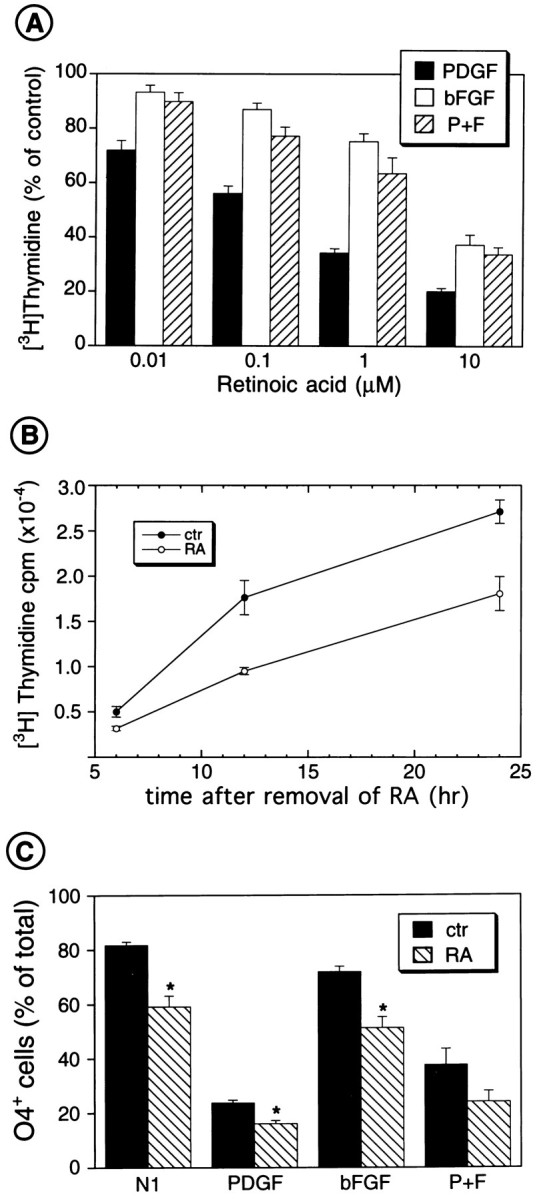
RA reversibly inhibits O-2A cell proliferation and prevents lineage progression. A, RA inhibits O-2A cell proliferation. Cells were plated in 24-well plates. After 2 hr, PDGF and/or bFGF (both 10 ng/ml), as well as RA (0.1–3 μm), were added to the cultures, together with [3H]thymidine (0.5 μCi/ml). After 22 hr, [3H]thymidine incorporation was measured by trichloroacetic acid precipitation and scintillation counting. Averages ± SEM obtained from three to six experiments run in triplicate are shown. All concentrations of RA significantly (p < 0.05) inhibited O-2A cell proliferation, except for 0.01 μm in bFGF and P + F (Student’st test). B, The antiproliferative effects of RA are reversible. Progenitor cells were cultured in PDGF (10 ng/ml) in the absence or presence of RA (1 μm). After 22 hr, all cells were shifted to fresh culture medium, without RA, containing PDGF (10 ng/ml) and [3H]thymidine (0.5 μCi/ml). Cells were harvested after 6, 12, and 24 hr, and [3H]thymidine incorporation was determined by trichloroacetic acid precipitation and scintillation counting. Averages ± SEM (n = 3) are shown. At 22 hr, before the shift to RA-free medium, RA inhibited [3H]thymidine incorporation by 47%.C, Treatment with RA prevents O-2A lineage progression, as detected by staining with O4 antibody. O-2A progenitor cells were purified and cultured on coverslips in DME-N1 medium with 0.5% FBS with PDGF (10 ng/ml), or bFGF (10 ng/ml), or PDGF + bFGF. RA (1 μm) was added to the culture medium 2 hr after plating. After 48 hr, cells were immunostained with O4 antibody and counted. Averages ± SEM obtained from three experiments (n = 20) are shown. The total number of cells counted for each culture condition ranged from 683 to 2176; *p < 0.001 (Student’s ttest).
Consistent with previous reports (Barres et al., 1994; Laeng et al., 1994; Noll and Miller, 1994), we found that RA treatment promoted a more bipolar morphology in O-2A cells (data not shown). Additionally, under culture conditions that promoted lineage progression (N1, PDGF, or bFGF), treatment with 1 μm RA significantly decreased the percentage of O-2A cells that developed into O4+pro-oligodendroblasts (Fig. 9C). In cells treated with PDGF + bFGF, the percentage of O4+ cells was not significantly affected by RA (Fig. 9C). In conclusion, RA, agents that increase [Na+]i, and membrane depolarization similarly modulate O-2A cell proliferation and lineage progression.
Like the other antiproliferative agents tested, RA also altered O-2A K+ currents. In cells cultured for periods of 24–48 hr in 1 μm RA-containing media, both a downregulation of the transient current component and the sustained current density were observed in O-2A cells recorded in the absence of RA (Fig.10). The mean current density reductions were 61 and 30%, respectively, (Table 3) and were not associated with any changes in the resting membrane potential or cell capacitance (Table 3). Similar to the other antiproliferative agents, RA had no effect on the voltage-dependent properties of activation of either the sustained or the transient current components. In cells treated with RA, two sustained current components still could be resolved, possessing half-activations of −22.4 ± 0.7 (k = 6;n = 13) and 16.1 ± 1.6 mV (k = 7), values similar to those seen in control. The relative contribution of each current component remained unchanged from control untreated cells. Likewise, despite a 60% attenuation of current density, the transient current component possessed voltage-dependent activation (−6 ± 1.3 mV) properties similar to those seen in untreated cells. In contrast, RA treatment was without effect on the Kir current density (Table 3).
Fig. 10.

Long-term treatment with RA downregulates both the transient and sustained outward K+ currents in O-2A cells. A, B, Cells cultured in RA-containing (1 μm) media for 48 hr displayed a downregulation of both the isolated sustained current (Ai) and transient current density (Aii) (see also Table 3). Representative traces from two different cells in both control and RA culture conditions are depicted inAi and Bi to illustrate the downregulation of both the transient and sustained currents in RA culturing conditions. Aii, Bii, Summary histograms of the effects of chronic exposure to RA on both the isolated sustained and transient current components. Both the transient and the sustained current densities were significantly reduced after chronic exposure to RA. Aiii, Biii, RA had no effect on the voltage-dependent properties of activation of either the sustained or the transient current components.Aiii, Two sustained current components could be resolved, possessing half-activations of −22.4 ± 0.7 (k = 6; n = 13) and 16.1 ± 1.6 mV (k = 7); values similar to those seen in control. The relative contribution of each current component was unchanged from control untreated cells. Biii, Despite a 60% attenuation of current density, the transient current component possessed voltage-dependent activation properties (−6 ± 1.3 mV,n = 13) identical to that seen in control treated cells.
DISCUSSION
The present report extends our previous findings in O-2A cells that GluR activation causes an indirect blockage of voltage-dependent K+ channels and through this mechanism, a reduction in cell proliferation (Gallo et al., 1996). Here we show that (1) the effects of GluR agonists on K+ channel activity are not direct but require receptor activation, as determined by their sensitivity to the non-NMDA receptor antagonist DNQX, and (2) GluR-mediated blockage of K+ channels in O-2A cells requires a transmembrane influx of Na+ ions, as demonstrated by experiments in which extracellular Na+ was replaced by NMDG.
Similar to GluR activation, agents that directly increase [Na+]i or depolarize the cell membrane caused marked antiproliferative effects on cultured O-2A progenitor cells. Our experiments indicate that all of these agents act through a similar mechanism to inhibit O-2A cell proliferation, i.e., through the reduction of voltage-dependent outward K+ currents. First, elevation of [Na+]i with the alkaloid veratridine or depolarization with high concentrations of [K+]o ions caused a reduction of K+ currents in O-2A cells, similar to that observed on GluR activation. Second, both veratridine and high [K+]o markedly and reversibly inhibited O-2A cell proliferation in the same concentration range that inhibited K+ currents in O-2A cells. Third, the antiproliferative effects of kainate, veratridine, and high [K+]o were independent of extracellular Ca2+ ions. Finally, another agent, ouabain, that acts to increase [Na+]i suppressed both the sustained and transient K+ currents and inhibited O-2A cell proliferation. These findings not only confirm that modulation of voltage-gated K+ channels can modify the proliferative state of O-2A cells, but indicate further that glial development can be controlled by signals that cause changes in the cell membrane potential and/or increase [Na+]i.
RA has been shown to regulate oligodendrocyte development in different areas of the nervous system. Our analysis in cortical O-2A cells confirms previous findings that both cell proliferation and lineage progression are partially inhibited by RA (Barres et al., 1994; Laeng et al., 1994; Noll and Miller, 1994). The effects of RA on cell proliferation appear to be attributable, at least in part, to its action on voltage-dependent K+ channels, because O-2A cells cultured in RA displayed a significant downregulation of both sustained and transient K+ currents. Although it is likely that RA inhibits K+ channel function through a mechanism distinct from that of veratridine and high [K+]o(Mangelsdorf et al., 1995), its action on voltage-dependent K+ currents is consistent with its effects on O-2A cell proliferation.
The present results provide some insight into the role of voltage-gated outward currents in an otherwise electrically inexcitable (i.e., absence of action potentials) cell type. The observation that the activation threshold for outward K+ currents (−40 mV) lies close to the cell resting membrane potential (−45 mV) suggests that O-2A cells are endowed with a mechanism to tightly regulate depolarizing perturbations in membrane potential. In the normal proliferative state, therefore, only small depolarizations around the resting membrane potential would be tolerated by the O-2A cell before activation of outward K+ currents. Depolarizing stimuli that exceed this narrow voltage range would activate outward K+ currents, which would tend to hyperpolarize the O-2A cell and prevent further depolarization. This tight regulation of the membrane resting membrane potential may act to ensure that O-2A cells remain in an active proliferative state. Voltage-dependent sodium channels have an activation threshold close to −35 mV (Barres et al., 1990), a voltage more positive to outward K+ current activation. Any agent that strongly depolarized the O-2A cell membrane to activate voltage-dependent Na+ channels and increase [Na+]i would consequently inhibit or downregulate K+ currents, compromising the ability of the cell to adequately regulate its membrane potential in response to depolarizing stimuli. Therefore, any reduction in outward K+ currents would permit a larger membrane depolarization to be experienced by the cell for a given stimulus. This larger depolarization may be sufficient to cause promotion of the antiproliferative state; however, we cannot rule out that mechanisms secondary to changes in membrane potential are also involved. These may include the modulation of expression of genes involved in cell cycle progression.
Previous studies have demonstrated that membrane depolarization and an increase in [Na+]i can stimulate a mitogenic response in cultured central neurons (Cone and Cone, 1976; Cone, 1980). Our findings agree with the analysis in distinct glial cell types. Proliferation of astrocytes (Pappas et al., 1994) and Schwann cells (Chiu and Wilson, 1989) is also inhibited by membrane depolarization or by K+ channel blockers. In cells of the oligodendrocyte lineage, suppression of K+ channel activity has important functional consequences, as demonstrated by the findings of Shrager and Novakovic (1995) that long-term treatment with the K+channel blocker TEA prevented myelination in spinal cord microexplants. We are currently studying the intracellular mechanism that links blockage of K+ channels and inhibition of O-2A proliferation, as well as the phase of the cell cycle affected by activation of GluRs or membrane depolarization. One likely mechanism was demonstrated in a series of elegant experiments by Pappas et al. (1994), who showed that inhibition of astrocyte proliferation by K+ channel blockers was attributable to (and can be mimicked by) alkaline shifts in intracellular pH (pHi). Consistent with this mechanism, depolarization by an elevation of [K+]o has been demonstrated to result in alkalinization of forebrain astrocytes (Boyarsky et al., 1993). Changes in pHi are known to affect progression through S-phase of the mitotic cycle (Hutchinson and Glover, 1995), and it is also likely that in O-2A cells, blockage of voltage-dependent K+channels reduces H+ leak through these channels, thereby causing an acidic shift in pHi, which is also known to have an antiproliferative effect (Pappas et al., 1994).
An important finding of the present study is that chronic exposure of O-2A cells to elevated [K+]ocauses a long-term change in K+ channel current expression, such that the resulting current phenotype is similar to that observed previously in pro-oligodendroblast (Barres et al., 1989; Sontheimer et al., 1989; Gallo et al., 1996). Importantly, this shift to a “pro-oligodendroblast-like” phenotype occurs independently of a developmental transition to a more differentiated stage of the lineage, as determined by the antigenic phenotype of the cells. O-2A progenitor cells cultured in 45 mm K+-containing medium displayed a marked upregulation of inward rectifier K+currents, but these culture conditions resulted in a decrease in the percentage of cells expressing the O4+ phenotype. Inward rectifying K+ channels in oligodendrocytes and Schwann cells have been proposed to play a major role in spatial buffering of [K+]o (Orkand et al., 1966; Chiu, 1991) by providing a rapid and efficient removal of ions by taking up K+. In agreement with this hypothesis, Wilson and Chiu (1990) demonstrated that inwardly rectifying K+ channels are concentrated in Schwann cell membranes in the vicinity of the nodes of Ranvier, an area associated with the extrusion of K+into the extracellular environment after action potential activity. In contrast, Kir were absent from the perinuclear region of the cell, i.e., an area not associated with the node. High levels of expression of Kir channels in cells of the oligodendrocyte lineage have been largely associated with the acquisition of the pro-oligodendroblast phenotype; however, our data suggest that the presence of Kir channels on cells of the oligodendrocyte lineage does not depend on the proliferative state of the cell per se, but perhaps is attributable simply to the concentrations of K+ in the surrounding medium. In our experiments, only cells exposed to elevated [K+]o demonstrated an upregulation of Kir channels. This observation predicts that a high level of expression of inwardly rectifying K+ channels would be induced and maintained only in membrane domains exposed to elevated [K+]o. This suggests that K+channels in O-2A cells possess a certain degree of plasticity in response to environmental cues, i.e., elevated K+ ions.
Similarly, RA partially reproduced a shift to a pro-oligodendroblast phenotype by selectively downregulating outward K+currents, but did not promote lineage progression to the O4+ stage. These results not only show that plastic changes in voltage-dependent K+ currents can be triggered by distinct extracellular signals in O-2A cells, but indicate additionally that the voltage-dependent channel phenotype of oligodendrocyte lineage cells does not correlate with their differentiation stage.
Depolarizing [K+]o not only affects glial cell proliferation (Canady et al., 1990; Pappas et al., 1994; present study), but also increases protein synthesis and decreases GFAP expression in astrocytes (Canady et al., 1990). Taken together, these data on distinct macroglial cell types indicate that elevated [K+]o accompanying neuronal activity may cause short- and long-term effects on glial cell development through changes in K+ channel activity and expression. Finally, high [K+]o ions may also impact oligodendrocyte development and function in pathological states, such as epileptic seizures, anoxia, and spreading depression, because significant increases in [K+]o have been reported under all these pathological conditions (Moody et al., 1974;Sugaya et al., 1975; Somjen, 1979; Hertz, 1986).
Footnotes
This work was supported by a National Institute of Child Health and Human Development (NICHD) Pre-IRTA Fellowship (P.K.) and partially supported by a fellowship from the National Research Council of Italy (C.A.G.). We thank Sotirios Keros for providing the Origin 4.0 macros used for the data analysis, Alex Eisen for providing the data concerning the effects of ouabain on outward K+ currents, Dr. Mark Mayer for critically reading this manuscript, and Drs. James Russell and Peter Simpson for communicating their data concerning the effects of A23187 on O-2A intracellular calcium levels.
Correspondence should be addressed to Dr. Chris J. McBain, Laboratory of Cellular and Molecular Neurophysiology, Room 5A72, Building 49, NICHD, National Institutes of Health, 49 Convent Drive, Bethesda, MD 20892-4495.
REFERENCES
- 1.Armstrong RC, Harvath L, Dubois-Dalcq M. Type 1 astrocytes and oligodendrocytes-type 2 astrocytes glial progenitors migrate toward distinct molecules. J Neurosci Res. 1990;27:400–407. doi: 10.1002/jnr.490270319. [DOI] [PubMed] [Google Scholar]
- 2.Barres BA, Chun LLY, Corey DP. Glial and neuronal forms of the voltage-dependent sodium channel: characteristics and cell-type distribution. Neuron. 1989;2:1375–1388. doi: 10.1016/0896-6273(89)90076-7. [DOI] [PubMed] [Google Scholar]
- 3.Barres BA, Koroshetz WJ, Swartz KJ, Chun LLY, Corey DP. Ion channel expression by white matter glia: the O-2A glial progenitor cell. Neuron. 1990;4:507–524. doi: 10.1016/0896-6273(90)90109-s. [DOI] [PubMed] [Google Scholar]
- 4.Barres BA, Schmid R, Sendtner M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118:283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- 5.Barres BA, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development. 1994;120:1097–1108. doi: 10.1242/dev.120.5.1097. [DOI] [PubMed] [Google Scholar]
- 6.Bevan S, Raff M. Voltage-dependent potassium currents in cultured astrocytes. Nature. 1985;315:229–232. doi: 10.1038/315229a0. [DOI] [PubMed] [Google Scholar]
- 7.Bogler O, Wren D, Barnett SC, Land H, Noble M. Cooperation between two growth factors promotes extended self-renewal and inhibits differentiation of oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells. Proc Natl Acad Sci USA. 1990;87:6368–6372. doi: 10.1073/pnas.87.16.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borges K, Kettenmann H. Blockade of K+ channels induced by AMPA/kainate receptor activation in mouse oligodendrocyte precursor cells is mediated by Na+ entry. J Neurosci Res. 1995;42:579–593. doi: 10.1002/jnr.490420416. [DOI] [PubMed] [Google Scholar]
- 9.Borges K, Ohlemeyer C, Trotter J, Kettenmann H. AMPA/kainate receptor activation in murine oligodendrocyte precursors leads to activation of a cation conductance, calcium influx and blockade of delayed rectifying K+ channels. Neuroscience. 1994;63:135–149. doi: 10.1016/0306-4522(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 10.Boyarsky G, Ransom B, Schlue WR, Davis MB, Boron WF. Intracellular pH regulation in single cultured astrocytes from rat forebrain. Glia. 1993;8:241–248. doi: 10.1002/glia.440080404. [DOI] [PubMed] [Google Scholar]
- 11.Canady KS, Ali-Osman F, Rubel EW. Extracellular potassium influences DNA and protein syntheses and glial fibrillary acidic protein expression in cultured glial cells. Glia. 1990;3:368–374. doi: 10.1002/glia.440030508. [DOI] [PubMed] [Google Scholar]
- 12.Canoll PD, Musacchio JM, Hardy R, Reynolds R, Marchionni MA, Salzer JL. GGF/neuregulin is a neuronal signal that promotes the proliferation and survival and inhibits the differentiation of oligodendrocyte progenitors. Neuron. 1996;17:229–243. doi: 10.1016/s0896-6273(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 13.Chiu SY. Functions and distribution of voltage-gated sodium and potassium channels in mammalian Schwann cells. Glia. 1991;4:541–558. doi: 10.1002/glia.440040602. [DOI] [PubMed] [Google Scholar]
- 14.Chiu SY, Wilson GF. The role of potassium channels in Schwann cell proliferation in Wallerian degeneration of explant rabbit sciatic nerves. J Physiol (Lond) 1989;408:199–222. doi: 10.1113/jphysiol.1989.sp017455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chvatal A, Pastor A, Mauch M, Sykova E, Kettenmann H. Distinct populations of identified glial cells in the developing rat spinal cord slice: ion channel properties and cell morphology. Eur J Neurosci. 1995;7:129–142. doi: 10.1111/j.1460-9568.1995.tb01027.x. [DOI] [PubMed] [Google Scholar]
- 16.Cone CD. Ionically mediated induction of mitogenesis in CNS neurons. Ann NY Acad Sci. 1980;339:115–131. doi: 10.1111/j.1749-6632.1980.tb15973.x. [DOI] [PubMed] [Google Scholar]
- 17.Cone CD, Cone CM. Induction of mitosis in mature neurons in central nervous system by sustained depolarization. Science. 1976;192:155–158. doi: 10.1126/science.56781. [DOI] [PubMed] [Google Scholar]
- 18.Curtis R, Cohen J, Fok-Seang J, Hanley MR, Gregson NA, Reynolds R, Wilkin GP. Development of macroglial cells in rat cerebellum. I. Use of antibodies to follow early in vivo development and migration of oligodendrocytes. J Neurocytol. 1988;17:43–54. doi: 10.1007/BF01735376. [DOI] [PubMed] [Google Scholar]
- 19.DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T-lymphocytes: a role in mitogenesis? Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- 20.Duffy S, Fraser DD, MacVicar BA. Potassium channels. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 185–201. [Google Scholar]
- 21.Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recording from synaptically connected neurones of the mammalian central nervous system. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 22.Gallo V, Armstrong R. Developmental and growth factor-induced regulation of nestin in oligodendrocyte lineage cells. J Neurosci. 1995;15:394–406. doi: 10.1523/JNEUROSCI.15-01-00394.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallo V, Zhou JM, McBain CJ, Wright P, Knutson PL, Armstrong R. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J Neurosci. 1996;16:2659–2670. doi: 10.1523/JNEUROSCI.16-08-02659.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gard AL, Pfeiffer SE. Glial cell mitogens bFGF and PDGF differentially regulate development of O4+GalC− oligodendrocyte progenitors. Dev Biol. 1993;159:618–630. doi: 10.1006/dbio.1993.1269. [DOI] [PubMed] [Google Scholar]
- 25.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recordings from cells and cell-free patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 26.Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Br Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- 27.Hardy R, Reynolds R. Rat cerebral cortical neurons in primary culture release a mitogen specific for early (GD3+/O4−) oligodendroglial progenitors. J Neurosci Res. 1993;34:589–600. doi: 10.1002/jnr.490340510. [DOI] [PubMed] [Google Scholar]
- 28.Hertz L. Potassium transport in astrocytes and neurons in primary cultures. Ann NY Acad Sci. 1986;481:318–333. doi: 10.1111/j.1749-6632.1986.tb27161.x. [DOI] [PubMed] [Google Scholar]
- 29.Hunter SF, Bottenstein JE. Growth factor responses of enriched bipotential glial progenitors. Brain Res. 1990;54:235–248. doi: 10.1016/0165-3806(90)90146-p. [DOI] [PubMed] [Google Scholar]
- 30.Hutchinson C, Glover DM. Cell cycle control. Oxford UP; New York: 1995. [Google Scholar]
- 31.Laeng P, Decimo D, Pettmann B, Janet T, Labourdette G. Retinoic acid regulates the development of oligodendrocyte precursor cells in vitro. J Neurosci Res. 1994;39:613–633. doi: 10.1002/jnr.490390602. [DOI] [PubMed] [Google Scholar]
- 32.Levi G, Gallo V, Ciotti MT. Bipotential precursors of putative fibrous astrocytes and oligodendrocytes in rat cerebellar cultures express distinct surface features and “neuron-like” GABA transport. Proc Natl Acad Sci USA. 1986;83:1504–1508. doi: 10.1073/pnas.83.5.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maccaferri G, Mangoni M, Lazzari A, DiFrancesco D. Properties of the hyperpolarization-activated current in rat hippocampal CA1 pyramidal cells. J Neurophysiol. 1993;69:2129–2136. doi: 10.1152/jn.1993.69.6.2129. [DOI] [PubMed] [Google Scholar]
- 34.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron. 1990;5:603–614. doi: 10.1016/0896-6273(90)90215-2. [DOI] [PubMed] [Google Scholar]
- 37.McMorris FA, Dubois-Dalcq M. Insulin-like growth factor I promotes cell proliferation and oligodendroglial commitment in rat glial progenitor cells developing in vitro. J Neurosci Res. 1988;21:199–209. doi: 10.1002/jnr.490210212. [DOI] [PubMed] [Google Scholar]
- 38.Moody WJ, Jr, Futamachi KJ, Prince DA. Extracellular potassium activity during epileptogenesis. Exp Neurol. 1974;42:248–263. doi: 10.1016/0014-4886(74)90023-5. [DOI] [PubMed] [Google Scholar]
- 39.Newman EA, Reichenbach A. The Muller cell: a functional element of the retina. Trends Neurosci. 1996;19:307–312. doi: 10.1016/0166-2236(96)10040-0. [DOI] [PubMed] [Google Scholar]
- 40.Noll E, Miller RH. Regulation of oligodendrocyte differentiation: a role for retinoic acid in the spinal cord. Development. 1994;120:649–660. doi: 10.1242/dev.120.3.649. [DOI] [PubMed] [Google Scholar]
- 41.Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- 42.Pappas CA, Ulrich N, Sontheimer H. Reduction of glial proliferation by K+ channel blockers is mediated by changes in pHi. NeuroReport. 1994;6:193–196. doi: 10.1097/00001756-199412300-00049. [DOI] [PubMed] [Google Scholar]
- 43.Raff MC, Lillien LE, Richardson WD, Burne FJ, Noble MD. Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature. 1988;333:562–565. doi: 10.1038/333562a0. [DOI] [PubMed] [Google Scholar]
- 44.Ransom BR, Orkand RK. Glial-neuronal interactions in non-synaptic areas of the brain: studies in the optic nerve. Trends Neurosci. 1996;19:352–358. doi: 10.1016/0166-2236(96)10045-x. [DOI] [PubMed] [Google Scholar]
- 45.Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of the fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- 46.Shrager P, Novakovic SD. Control of myelination, axonal growth, and synapse formation in spinal cord explants by ion channels and electrical activity. Dev Brain Res. 1995;88:68–78. doi: 10.1016/0165-3806(95)00081-n. [DOI] [PubMed] [Google Scholar]
- 47.Somjen GG. Extracellular potassium in the mammalian central nervous system. Annu Rev Physiol. 1979;41:159–177. doi: 10.1146/annurev.ph.41.030179.001111. [DOI] [PubMed] [Google Scholar]
- 48.Sommer I, Schachner M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces:an immunocytological study in the central nervous system. Dev Biol. 1981;83:311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- 49.Sontheimer H, Trotter J, Schachner M, Kettenmann H. Channel expression correlates with differentiation stage during the development of oligodendrocytes from their precursor cells in culture. Neuron. 1989;2:1135–1145. doi: 10.1016/0896-6273(89)90180-3. [DOI] [PubMed] [Google Scholar]
- 50.Sontheimer H, Black JA, Waxman SG. Voltage-gated Na+ channels in glia: properties and possible functions. Trends Neurosci. 1996;19:325–331. doi: 10.1016/0166-2236(96)10039-4. [DOI] [PubMed] [Google Scholar]
- 51.Steinhauser C, Gallo V. News on glutamate receptors in glial cells. Trends Neurosci. 1996;19:339–345. doi: 10.1016/0166-2236(96)10043-6. [DOI] [PubMed] [Google Scholar]
- 52.Sugaya E, Takato M, Noda Y. Neuronal and glial activity during spreading depression in the cerebral cortex of cat. J Neurophysiol. 1975;38:822–841. doi: 10.1152/jn.1975.38.4.822. [DOI] [PubMed] [Google Scholar]
- 53.Theodosis DT, MacVicar B. Neurone-glia interactions in the hypothalamus and pituitary. Trends Neurosci. 1996;19:363–367. doi: 10.1016/0166-2236(96)10055-2. [DOI] [PubMed] [Google Scholar]
- 54.Verkhratsky A, Kettenmann H. Calcium signalling in glial cells. Trends Neurosci. 1996;19:346–352. doi: 10.1016/0166-2236(96)10048-5. [DOI] [PubMed] [Google Scholar]
- 55.Wilson GF, Chiu SY. Ion channels in axon and Schwann cell membrane at paranodes of mammalian myelinated fibers studied with patch clamp. J Neurosci. 1990;10:3263–3274. doi: 10.1523/JNEUROSCI.10-10-03263.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]