

Abstract
Cytosolic calcium ([Ca2+]i) is an important mediator of neuronal signal transduction, participating in diverse biochemical reactions that elicit changes in synaptic efficacy, metabolic rate, and gene transcription. Excessive [Ca2+]i also has been implicated as a cause of acute neuronal injury, although measurement of [Ca2+]i in living neurons by fluorescent calcium indicators has not consistently demonstrated a correlation between [Ca2+]i and the likelihood of neuronal death after a variety of potentially lethal insults. Using fluorescence videomicroscopy and microinjected calcium indicators, we measured [Ca2+]i in cultured cortical neurons during intense activation with either NMDA (300 μm) or AMPA (450 μm). At these concentrations NMDA killed >80% of the cultured neurons by the next day, whereas neuronal death from AMPA was <20%. Using the conventional calcium indicator, fura-2/AM, we estimated [Ca2+]i elevations to be ∼300–400 nm during exposure to either glutamate agonist. In contrast, indicators with lower affinity for calcium, benzothiazole coumarin (BTC), and fura-2/dextran reported [Ca2+]i levels >5 μmduring lethal NMDA exposure, but [Ca2+]i levels were <1.5 μm during nonlethal activation of AMPA receptors or voltage-gated calcium channels. Fura-2 reported [Ca2+]i responses during brief exposure to glutamate, NMDA, AMPA, kainate, and elevated extracellular K+ between 0.5 and 1 μm. With the use of BTC, only NMDA and glutamate exposures resulted in micromolar [Ca2+]i levels. Neurotoxic glutamate receptor activation is associated with sustained, micromolar [Ca2+]i elevation. The widely used calcium indicator fura-2 selectively underestimates [Ca2+]i, depending on the route of entry, even at levels that appear to be within its range of detection.
Keywords: AMPA, calcium, excitotoxicity, fura-2, glutamate, kainate, NMDA, videomicroscopy
Neuronal injury in stroke or trauma may be mediated by excessive stimulation of glutamate receptors (Rothman and Olney, 1986; Choi, 1992). Studies using primary neuronal culture systems have shown that cultured neurons are selectively vulnerable to brief applications of glutamate (Rothman, 1985; Choi et al., 1987) or to selective agonists of the NMDA class of glutamate receptors (Rothman, 1985; Choi et al., 1988; Hartley and Choi, 1989). In contrast, prolonged activation of the AMPA or kainate receptors is required to produce neurotoxicity (Koh et al., 1990).
Schanne and colleagues (1979) originally described the dependence of extracellular calcium in hepatocyte toxicity, which provided a potential mechanism for neuronal toxicity with the subsequent discovery that glutamate receptor activation produced neuronal calcium influx (Connor et al., 1987, 1988; Murphy et al., 1987). Additional studies by other investigators led to the hypothesis that calcium entry was responsible for excitotoxic neuronal injury (Choi, 1987) (for review, see Dubinsky, 1993a). A correlation between intracellular calcium levels and glutamate toxicity was suggested by the dependence of the latter on extracellular calcium (Choi, 1987; Rothman et al., 1987). Studies with radioactive 45Ca2+ isotope demonstrated substantial calcium entry during toxic NMDA receptor activation but much lower 45Ca2+ influx during nonlethal AMPA treatment (Marcoux et al., 1988; Hartley et al., 1993; Eimerl and Schramm, 1994; Lu et al., 1996). The study by Hartley and coworkers (1993) suggested that a direct relationship existed between calcium accumulation and subsequent death in neurons exposed to NMDA. Although previous studies using fluorescent intracellular calcium indicators have noted that lethal excitotoxic insults may be followed by prolonged or delayed [Ca2+]ielevation (Ogura et al., 1988; de Erausquin et al., 1990; Randall and Thayer, 1992; Dubinsky, 1993b; Tymianski et al., 1993a; Limbrick et al., 1995), such indicators have failed to demonstrate a consistent difference between intracellular free calcium concentration ([Ca2+]i) in cellsduring lethal and nonlethal challenges (de Erausquin et al., 1990; Michaels and Rothman, 1990; Tymianski et al., 1993a).
One potential interpretation of these results is that there is not a direct relationship between the magnitude of acute [Ca2+]i elevation and the extent of subsequent neuronal death. Another possibility is that current methods of [Ca2+]i measurement fail to distinguish toxic levels of [Ca2+]i. In the present study we examine the hypothesis that conventional, high-affinity calcium indicators (e.g., fura-2) are unable to measure accurately the [Ca2+]i in the micromolar range and therefore fail to detect differences in [Ca2+]i between lethal and nonlethal excitotoxic exposures. We used fura-2/AM ester, fura-2/K+ salt, fura-2/dextran, or a benzothiazole coumarin-based indicator, BTC, to determine [Ca2+]i in neurons subjected to lethal and nonlethal challenges. The low affinity of BTC for calcium (KD ∼7–26 μm) allows detection of [Ca2+]i in the micromolar range (Iatridou et al., 1994; Zhao et al., 1996).
Some of the information in this paper has been published in abstract form (Hyrc et al., 1996, 1997).
MATERIALS AND METHODS
Neuronal cultures. We used dissociated neocortical neurons prepared from brains of embryonic Swiss–Webster mice at gestational days 15–16 (Rose et al., 1993). Gravid mice were anesthetized with halothane, and the uteri were removed under sterile conditions. After the embryo brains were removed, the neocortices were digested with trypsin (0.9%) and separated into a single-cell suspension by mechanical trituration through Pasteur pipettes. Cells were plated (0.5 hemisphere per dish) onto glass-bottom 35 mm dishes (Mattek, Ashland, MA) previously coated with a mixture of poly-d-lysine (0.5 mg/ml) and laminin (4 μg/ml). The culture medium consisted of Eagle’s Minimal Essential Medium containing 25 mm glucose, 2 mm glutamine, 5% fetal bovine serum, and 5% horse serum. After 8 d in vitro, the proliferation of non-neuronal cells was inhibited by the addition of 10 μm cytosine arabinoside. Cultures were fed twice weekly and were used 14–17 d after plating.
Excitotoxin exposure. Cells were exposed to elevated KCl (50 mm) or to glutamate receptor agonists (300 μmNMDA or glutamate; 450 μm AMPA or kainate) at room temperature in a HEPES- and bicarbonate-buffered salt solution (HBBSS) containing (in mm): 116 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgSO4, 1 NaH2PO4, 25 NaHCO3, 12 HEPES, and 5.5 d-glucose, pH 7.45 ± 0.2. Bicarbonate was included in the experimental solution to minimize glutamate receptor-mediated intracellular acidification (Canzoniero et al., 1996). The NMDA receptor antagonist MK-801 (10 μm) was included in all treatments except NMDA and glutamate to prevent secondary activation of NMDA receptors. For toxicity assessment, cultures subjected to experimental treatments were washed with HBBSS containing 10 μm MK-801 and returned to the incubator. Neuronal survival was determined 20–28 hr later by 0.4% trypan blue exclusion. The fraction of trypan blue-positive neurons in each culture dish was calculated in 5–10 random fields containing 20–50 neurons.
Calcium indicators. Cytosolic calcium determination was performed with fluorescent calcium indicators: fura-2/AM ester, fura-2/K+ salt, fura-2 coupled to dextran [fura-2/dextran; molecular weight (MW) 3000], and benzothiazole coumarin (BTC) (Iatridou et al., 1994; Konishi and Watanabe, 1995). All indicators were purchased from Molecular Probes (Eugene, OR).
Fura-2/AM (6 μm) was bath-loaded into neurons for 30 min in the presence of 0.12% of Pluronic 127, followed by another 30 min incubation at 37°C to allow for hydrolysis of the AM ester. All of the other indicators were loaded into the neurons by transient application of patch micropipettes in whole-cell recording configuration. We used 2–6 MΩ micropipettes fabricated from 1.2 mm outer diameter, thin-wall glass tubing (World Precision Instruments, Sarasota, FL), using a two-stage microelectrode puller (Sutter P-87, Sutter Instrument, Novato, CA). The tip of the micropipette was backfilled by capillary action with 500 μm calcium indicator dissolved in an intracellular solution. This solution contained (in mm): 115 K+ gluconate, 20 KCl, 10 HEPES, and 2 Mg-ATP, pH 7.4; the remainder of the micropipette was filled with intracellular solution alone.
We used a standard bridge amplifier (M707, World Precision Instruments) for monitoring resting membrane potential in the whole-cell configuration (Hamill et al., 1981). The neuron was filled with indicator for 60 sec by diffusion, after which the micropipette was withdrawn carefully. All neurons were incubated for 30 min at room temperature after pipette removal, and only those that maintained their preinjection morphology and retained dye were used for [Ca2+]i imaging (n = 289 neurons of 599 attempts, for an approximate success rate of 48%). In our hands, cultured cortical neurons tolerated indicator injection better with patch micropipettes than with high-impedance microelectrodes. In preliminary experiments the loading conditions were selected to provide comparable fluorescence intensity to that of bath-loaded fura-2/AM, using rapid diffusion (60 sec) and the smallest micropipette indicator concentration (500 μm). Previous studies indicate that the intracellular concentration of indicator is actually much lower than that of the pipette, because considerably longer periods generally are required for dye equilibration from patch pipettes (Pusch and Neher, 1988; Sala and Hernandez-Cruz, 1990).
Calcium imaging. Calcium measurements were performed with standard ratio imaging techniques (Grynkiewicz et al., 1985). Indicator-filled neurons were imaged on an inverted microscope (Nikon Diaphot, Nikon, Melville, NY), using a 40×, 1.3 numerical aperture (NA) fluorite oil immersion objective (Nikon) and an intensified charge-coupled device (ICCD) camera (Hamamatsu Photonics, Oak Brook, IL) operating at moderate gain (typical settings 0.5–3). A 75 W xenon arc lamp was used to provide fluorescence excitation. Ratio images were obtained by acquiring pairs of images at alternate excitation wavelengths (340/380 nm for fura-2 and 400/485 nm for BTC) and filtering the emission at 530 nm. Image acquisition and processing were controlled by a personal computer connected to the camera and filter wheel (MetaFluor, Universal Imaging Corporation, West Chester, PA). Image pairs were captured and digitized every 10 sec, and the images at individual wavelengths were averaged over four frames. We corrected for background fluorescence by imaging a field lacking an injected neuron and subtracting the paired images from the images of the indicator-loaded neuron.
We calculated actual [Ca2+]i in regions of interest in individual neurons with the formula:
where KD is the indicator’s dissociation constant for Ca2+; R is ratio of fluorescence intensity at two different wavelengths (340/380 nm for fura-2 and 400/485 nm for BTC); Rmax andRmin are the ratios at saturating Ca2+ and zero Ca2+, respectively; and B is the ratio of the fluorescence intensity of the second excitation wavelength at zero and saturating Ca2+ (Grynkiewicz et al., 1985).Rmin, Rmax, and B for all indicators were determined for our system by imaging large droplets (100 μl) evenly filling the microscopic field that contained 0 and 39.6 μm Ca2+ and 25 μm fura-2/K+ orfura-2/dextran (Calcium Calibration Buffer Kit I with magnesium, Molecular Probes). Rmax for BTC was determined with a 1 mm Ca2+ standard solution (Molecular Probes). The concentration of indicators in the calibration solution was selected to provide similar fluorescence intensity to that of dye-loaded neurons.
Spectrofluorimetric indicator calibration. We determined theKD values of the indicators used in this study with a spectrofluorimeter (Perkin-Elmer LS 50, Perkin-Elmer, Norwalk, CT). We recorded the spectra of the indicator solution (2–5 μm) in calibrating buffers containing (in mm): 100 KCl, 1 Mg2+, and 10 MOPS, with [Ca2+] ranging from 0 to 39 μm(Calcium Calibration Buffer Kit I with Mg2+, Molecular Probes). The spectra were excited over the range 320–400 nm for fura-2 and fura-2/dextran and 380–500 nm for BTC. Emission was detected at 525 nm, and excitation slits were set at 6–10 nm. All calibrations were done at room temperature.
The increasing calcium concentrations were obtained by cross-diluting 10 mm EGTA and 10 mm CaEGTA solutions, using the protocol provided by Molecular Probes. Free calcium concentrations were calculated from the equation: [Ca2+] =KD · [CaEGTA]/[EGTA]. Buffers containing more than the 40 μm [Ca2+] necessary to saturate BTC were prepared by adding aliquots of 10 mmCaCl2 stock solution to 10 mm CaEGTA solution. Subsequently, free calcium concentration was calculated with the method described by Tsien and Pozzan (1989). The pH of calibration buffers was adjusted to 7.2, and the dissociation constants of EGTA for Ca2+ and Mg2+ used in the calculations were assumed to be 150.5 nm and 19.7 mm, respectively (Tsien and Pozzan, 1989).
The indicator KDvalues were determined from the spectra by first calculating the ratios of indicator emission intensities over a range of [Ca2+] at appropriate excitation wavelengths (340/380 for fura-2 and fura-2/dextran and 400/485 nm for BTC). We used the basic equation relating ratio (R), KD, F, and [Ca2+]: [Ca2+] =KD B(R −Rmin)/(Rmax −R) and regressed log [Ca2+] against logB(R −Rmin)/(Rmax −R) to obtain each KD (Grynkiewicz et al., 1985).
We observed a KD value for fura-2 (0.24 μm; Fig. 1), which was close to the previously published value of 0.22 μm(Grynkiewicz et al., 1985). Our KD for BTC (15.2 μm; Fig. 1) lies between the values reported by Iatridou et al. (1994) (7 μm) and Zhao et al. (1996) (26 μm). Because the KD of fura-2/dextran varies between lots, there are no available data to compare with our estimate of 0.94 μm (Fig. 1) (Haugland, 1995). We used these KD values to convert ratio values to estimated [Ca2+]i in all subsequent experiments.
Fig. 1.

Ca2+ dissociation constants for fura-2, fura-2/dextran (MW 3000), and BTC. Spectrofluorimetric calibration curves were obtained for each indicator at 5.0 μm in buffered solutions over an appropriate range of calcium concentrations. The data shown are averages of three calibration experiments performed for each indicator (mean ± SD). To facilitate comparisons, the x-intercept and corresponding log[Ca2+] for each indicator are shown as dashed and dotted lines, respectively. Calculated KD values are as follows: fura-2 (•), 0.24 ± 0.04 μm; fura-2/dextran (○), 0.94 ± 0.25 μm; BTC (▴), 15.2 ± 0.12 μm.
Confocal microscopy. Optical sectioning through BTC-filled neurons under resting conditions was performed with a Noran Odyssey laser scanning confocal microscope (Noran Instruments, Middleton, WI) coupled to an inverted microscope (Nikon Diaphot), using a 60×, 1.2 NA water immersion objective lens (Nikon). BTC was illuminated with 488 nm excitation light. Frame-averaged (32 frames) images were collected at 0.5 μm axial intervals with a confocal slit aperture of 25 μm. Images were saved to computer hard disk, and computer reconstruction was performed with the Metamorph image analysis system (Universal Imaging). A differential interference contrast (DIC) image of the neuron was collected by a transmitted light diode detector with DIC optics and 488 nm laser illumination.
Statistics. Comparisons between two groups used an unpaired Student’s t test (SigmaPlot, Jandel Scientific, SPSS, Chicago, IL).
RESULTS
When cortical cultures at 14–18 d in vitro were exposed to 300 μm NMDA for 20 min, there was widespread neuronal death by the next day, whereas the application of 450 μm AMPA produced little neuronal loss (Fig.2, inset). Despite this difference in toxicity, the [Ca2+]ielevation, measured by conventional fura-2/AM ratiometric imaging, was similar in response to NMDA or AMPA, reaching 300–400 nm(Fig. 2, Table 1). These results are similar to previous reports from our own and other laboratories (Ogura et al., 1988; de Erausquin et al., 1990; Michaels and Rothman, 1990;Dubinsky and Rothman, 1991; Tymianski et al., 1993b; Lu et al., 1996).
Fig. 2.

[Ca2+]i levels measured using fura-2/AM do not differ during nontoxic AMPA and toxic NMDA exposures. Cultured cortical neurons were loaded by incubation with 6 μm fura-2/AM, and neuronal [Ca2+]i was determined by fluorescence ratio imaging as described (see Materials and Methods) in neurons exposed to 300 μm NMDA (with 10 μm glycine) or 450 μm AMPA (with 10 μm MK-801 to prevent NMDA receptor activation). Values represent mean (± SE) [Ca2+]i derived from 15–20 bath-loaded neurons in each of four to six dishes.Inset, Cultures were exposed for 20 min either to 300 μm NMDA (with 10 μm glycine) or to 450 μm AMPA (with 10 μm MK-801 to prevent NMDA receptor activation). After treatment, cells were washed with HBBSS with 10 μm MK-801 and returned to the incubator. Cell viability was assessed after 20–28 hr by trypan blue exclusion. Values represent mean (± SD) percentage of nonviable neurons counted in 5–10 random fields in 10–15 dishes.
Table 1.
Maximal [Ca2+]i (in μm) observed during 20 min drug treatment
| 450 μm AMPA | 300 μmNMDA | pa | |
|---|---|---|---|
| Fura-2/AM | 0.3 ± 0.2b (4)c | 0.4 ± 0.1 (3) | p > 0.26 |
| Fura-2/K+ | 0.8 ± 0.3 (7) | 1.2 ± 0.5 (11) | p = 0.065 |
| BTC | 2.8 ± 1.4 (6) | 11.4 ± 3.9 (6) | p < 0.0005 |
| Fura-2/dextran | 2.5 ± 0.2 (4) | 6.8 ± 0.5 (4) | p < 0.005 |
Student’s t test;bmean ± SD; cnumber of dishes.
One interpretation of these findings is that NMDA and AMPA elevate [Ca2+]i to similar, submicromolar levels. Because the [Ca2+]i values of 300–400 nm are well below the theoretical limit of fura-2/AM detection, ∼1–2 μm, we would not anticipate a problem of indicator saturation. However, previous reports on calcium permeability of NMDA receptors suggest that it is unlikely that calcium entry is similar after NMDA and AMPA receptor activation (Mayer and Westbrook, 1987; Goldberg and Choi, 1993; Hartley et al., 1993; Eimerl and Schramm, 1994). Rather, we examined the possibility that the method of ratio imaging that used fura-2/AM might be under-reporting [Ca2+]i. There are several factors that might complicate the measurement of [Ca2+]i with fura-2/AM. These include the high affinity of fura-2 for calcium, incomplete hydrolysis of the acetoxymethyl ester, compartmentalization of fura-2/AM into organelles, and the contribution of fluorescence signal from underlying glial cells (Tsien, 1988; Roe et al., 1990; Kao, 1994). Unesterified molecules exhibit the spectral properties of unbound (i.e., calcium-free) fura-2 and result in underestimation of [Ca2+]i. A significant indicator sequestration into calcium-insensitive compartments or the collection of fura-2 fluorescence from underlying glia also would affect the quantitation of [Ca2+]i, because in these cases fluorescence is collected from dye molecules that are not located in the intraneuronal cytosolic compartment. Dye quenching with Mn2+ ions (2–5 mmMn2+ with 50 μm ionomycin) or cell membrane lysis with 10 μm digitonin reduced fura-2 fluorescence to ∼10–20% of that of resting levels (data not shown), suggesting that a small proportion of the fura-2 was present in unesterified or compartmentalized forms, respectively. Signals from the glial monolayer may play a larger role in underestimating NMDA-induced [Ca2+]i elevation.
To avoid these problems, we repeated the NMDA and AMPA exposures after individual neurons had been loaded by transient patch electrode application with the potassium salt of fura-2 (fura-2/K+). Neuronal recovery after fura-2/K+ delivery via the patch pipette application was robust, as indicated by a healthy appearance under differential interference contrast imaging, uniform labeling of cytoplasm and processes, and typical low calcium at baseline. The [Ca2+]i response in individual neurons loaded with fura-2/K+ and exposed to NMDA and AMPA was different from the [Ca2+]iresponse of neurons bath-loaded with fura-2/AM. First, the peak [Ca2+]i observed for both NMDA and AMPA was higher, ranging from 0.4 to 1 μm (Fig.3). Second, NMDA produced a prolonged [Ca2+]i elevation that remained at 1 μm, whereas AMPA elicited a transient [Ca2+]i elevation, approaching 1 μm, followed by a rapid return to nm values. The peak [Ca2+]i levels were not statistically different, although the integrated calcium load was significantly greater for NMDA (Table 2). Although this measure of [Ca2+]icorrelated better with neuronal loss when fura-2/K+was injected directly into the neurons, the absolute difference in [Ca2+]i levels triggered by NMDA and AMPA was not very large (Table 2).
Fig. 3.

[Ca2+]i measured by microinjected fura-2. Single neurons were filled with 500 μm fura-2 by transient patch micropipette application and exposed to AMPA or NMDA, as in Figure 2. Values represent mean ± SE [Ca2+]i from four to six neurons.
Table 2.
Total calcium load (in μm • min) calculated as the integral of calcium during 20 min drug treatment
| 450 μmAMPA | 300 μm NMDA | pa | |
|---|---|---|---|
| Fura-2/AM | 2.6 ± 1.6b (4)c | 5.0 ± 1.5 (3) | p > 0.12 |
| Fura-2/K+ | 9.1 ± 2.7 (7) | 18.5 ± 8.8 (11) | p = 0.015 |
| BTC | 11.3 ± 4.1 (6) | 180 ± 48 (6) | p < 0.0001 |
| Fura-2/dextran | 23.5 ± 4.2 (4) | 108 ± 11.5 (4) | p < 0.0001 |
Student’s t test;bmean ± SD; cnumber of dishes.
Although differences in calcium buffering (Sala and Hernandez-Cruz, 1990; Connor, 1993) potentially could contribute to differences in [Ca2+]i measurements with fura-2/AM and injected fura-2, this does not seem likely to contribute to the present results. Despite the apparent difference in loading conditions (30 min incubation with 6 μm fura-2/AM vs 60 sec diffusion with 500 μm fura-2/K+), the fluorescence intensities of injected and bath-loaded cells were similar, indicating that the intracellular dye concentrations and, consequently, distortions because of calcium buffering were comparable. Comparison of the fluorescence intensity of fura-2 injected cells and fura-2 in 20 μm microslides suggested that the actual dye concentration in injected cells was on the order of 25 μm, less than the amount predicted to cause significant buffering effect (Sala and Hernandez-Cruz, 1990).
We were concerned that the high affinity of fura-2 for calcium (KD ∼0.22 μm) might preclude an accurate estimate of [Ca2+]i exceeding 1 μm. Therefore, we repeated the NMDA and AMPA exposures in individual neurons loaded with BTC, which has aKD for calcium of ∼15 μm (see Materials and Methods). In contrast to either form of fura-2, BTC revealed a large difference in the [Ca2+]i response to NMDA and AMPA treatment (Fig. 4). After NMDA exposure, BTC reported a rapid increase in [Ca2+]i to 5–10 μm, whereas AMPA exposure produced a transient elevation not exceeding 3 μm, which decreased to <0.5 μm within 5 min (Fig. 4). Toxic NMDA receptor activation provoked peak [Ca2+]i at least 2.5-fold greater than that of AMPA receptor activation (Table 1). This difference was magnified when [Ca2+]i was integrated for the 20 min exposure period (Table 2). The integrated response to AMPA application was similar with fura-2/K+ and BTC measurement, 9.1 versus 11.3 μm/min, respectively (not significant; Table 2). We used confocal microscopy to ensure that the BTC results did not reflect localized distribution of the novel indicator. Confocal optical sections through BTC-filled neurons revealed a relatively homogeneous distribution of BTC fluorescence, with minimal indicator compartmentalization (Fig.5).
Fig. 4.

[Ca2+]i measured by microinjected BTC. Single neurons were filled with the low-affinity calcium indicator BTC (500 μm;KD = 15 μm) and exposed to AMPA or NMDA, as in Figure 2. Values represent mean ± SE [Ca2+]i from four to six neurons. BTC reveals large differences between NMDA- and AMPA-induced [Ca2+]i elevation.
Fig. 5.

Cellular distribution of BTC in an individual dye-filled cortical neuron. Optical sections were taken at 0.5 μm intervals, using a laser scanning confocal microscope.A, A through-focus confocal image of BTC (green) overlayed on the Nomarski image (scale bar, 15 μm). B, The BTC fluorescence data (xz cross section) are shown in the upper right, with the indicated xy planes shownbelow (scale for these images is 12 μm). BTC appears uniformly distributed throughout the neuronal cytoplasm.
We also have compared [Ca2+]iresponses to NMDA and AMPA with two other low-affinity indicators, mag-fura-2 and Calcium Green-5N. The changes in mag-fura-2 ratio values or Calcium Green-5N intensity values were much greater after exposure to NMDA than AMPA (data not shown), consistent with the BTC result. We did not use these indicators extensively, because quantification of [Ca2+]i can be problematic with both. Intracellular ionized magnesium contaminates the mag-fura-2 signal, and calibration of single excitation indicators like Calcium Green-5N is difficult (Brocard et al., 1993; Rajdev and Reynolds, 1993; Stout et al., 1996). BTC ratio values are not affected significantly by alterations in [Mg2+] in the 0–10 mmrange (Iatridou et al., 1994; Hyrc et al., 1997).
To determine whether the differential reporting of [Ca2+]i by fura-2 and BTC was the result of differences in intracellular distribution, we filled neurons with fura-2 coupled to a dextran (3000 MW; fura-2/dextran), which restricts the distribution of the indicator to the cytoplasm (Kao, 1994). Fura-2/dextran had an intermediate affinity for calcium,KD 0.9 μm (see Fig. 1). In response to NMDA or AMPA exposure, the difference in ratio values reported by fura-2/dextran was substantially larger than that of fura-2/K+ (Fig. 6), and the [Ca2+]i levels after AMPA exposure were similar to the levels reported by BTC. The peak [Ca2+]i reported by fura-2/dextran was significantly higher after NMDA exposure, reaching levels of 7 μm, as compared with 2.5 μm with AMPA treatment (Table 1); this difference was substantial at all time points (Fig. 6). Like BTC, fura-2/dextran reported a highly significant difference in integrated [Ca2+]iduring NMDA and AMPA exposure (Table 2).
Fig. 6.

[Ca2+]i levels measured with fura-2/dextran. Neurons were filled with fura-2 conjugated to 3000 MW dextran (500 μm;KD = 0.94 μm) and exposed to NMDA or AMPA as in Figure 2. Values represent mean ± SE [Ca2+]i from four to six neurons.
We considered the possibility that fura-2 and BTC might provide distinctly different results only under conditions of lethal NMDA receptor activation. Therefore, we examined [Ca2+]i during brief, nontoxic exposures to different excitatory amino acids or elevated extracellular potassium. Two minute exposures to 50 mm KCl, 450 μm kainate, 450 μm AMPA, 300 μm glutamate, or 300 μm NMDA produced little neuronal death after 1 d (data not shown). With fura-2/K+, estimated [Ca2+]i reached ∼0.5–1.0 μm in the second minute of exposure in all conditions (Fig. 7A) and did not differ significantly among conditions (Fig. 7C). With BTC as the calcium indicator, KCl, AMPA, or kainate transiently elevated [Ca2+]i to 2 μm, with a return to nanomolar levels within the 2 min (Fig. 7B). Glutamate and NMDA provoked substantially higher, sustained [Ca2+]i elevations to 8 μm (Fig. 7B,C).
Fig. 7.
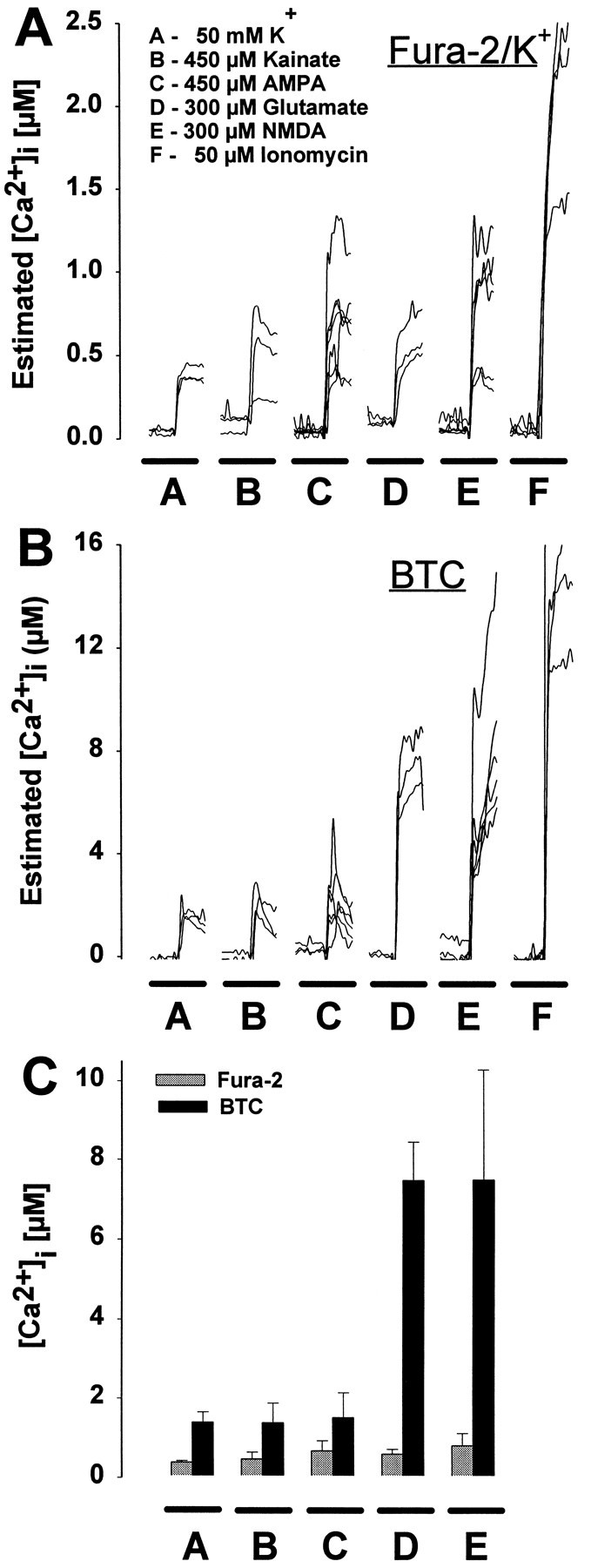
Fura-2 and BTC report markedly different [Ca2+]i levels even during sublethal exposure conditions. Neurons were exposed for 2 min to 50 mm KCl, 450 μm kainate or AMPA, 300 μm glutamate or NMDA, or 50 μm ionomycin after microinjection with either fura-2 (A) or BTC (B). Each trace represents estimated [Ca2+]i from a single neuron. C, Mean ± SD [Ca2+]i elevation during the second minute of drug application in A or B. With the use of fura-2, measured [Ca2+]i increased to a similar level for each treatment except ionomycin (A, C). In contrast, measurements using BTC revealed a significant difference in [Ca2+]i among cells treated with KCl, kainate or AMPA, and cells exposed to NMDA or glutamate (B, C). In fura-2-filled neurons, ionomycin application elevated [Ca2+]i to a significantly greater extent than other drug treatments (A), suggesting that fura-2 was not saturated in these experiments.
It is well recognized that fluorescent ion indicators provide little information under saturating conditions in which virtually all indicator molecules are already bound (in the case of fura-2, [Ca2+]i above 1–2 μm). However, that situation did not seem to apply in these experiments. When we applied the calcium ionophore ionomycin (50 μm) to fura-2/K+-injected neurons, [Ca2+]i rose to 2.5 μm, a concentration much higher than that produced by NMDA or glutamate (Fig. 7A). These results suggest that fura-2 wasnot saturated fully during NMDA- or glutamate-induced calcium influx.
DISCUSSION
The role of [Ca2+]i in excitotoxic neuronal death has been a topic of considerable debate, primarily because of conflicting evidence obtained by different methods of [Ca2+]i measurement. A lack of correlation between the magnitude of early [Ca2+]i elevation and subsequent excitotoxic death was noted by us originally and confirmed by others (Michaels and Rothman, 1990; Dubinsky and Rothman, 1991; Tymianski et al., 1993a,b; Lu et al., 1996). We replicated our earlier observations in this paper: maximum [Ca2+]i(measured with fura-2/AM) was no higher during an NMDA exposure that was ultimately lethal than during a nonlethal exposure to AMPA. These results stand in sharp contrast to calcium flux experiments that report substantially greater 45Ca2+ uptake during NMDA receptor activation (Marcoux et al., 1988; Hartley et al., 1993; Eimerl and Schramm, 1994; Lu et al., 1996). However, glutamate receptor activation can lead to marked elevation of [Ca2+]i (MacDermott et al., 1986;Rajdev and Reynolds, 1993), and biophysical investigations indicate much higher calcium permeability of NMDA receptors (MacDermott et al., 1986; Mayer and Westbrook, 1987; McBain and Mayer, 1994).
In view of this apparent discrepancy, we tested the hypothesis that the choice of indicator is critically important in obtaining an accurate estimate of [Ca2+]i. When we switched from fura-2 to lower-affinity indicators, the correlation between [Ca2+]i rise and neuronal death markedly improved. We found sustained micromolar levels of [Ca2+]i during toxic NMDA receptor activation, whereas nonlethal perturbations (e.g., K+ depolarization, AMPA, or kainate receptor activation), resulted in transient [Ca2+]i elevations that rapidly returned to nanomolar levels.
There are several reasons why our older [Ca2+]i determinations with fura-2 failed to anticipate correctly the outcome of excitotoxic paradigms. The usual method of indicator loading by bath application of the acetoxymethyl ester (fura-2/AM) contributed to underestimation of the differences between AMPA- and NMDA-induced [Ca2+]i elevation; this is apparent in the different results obtained by using microinjected free indicator (fura-2/K+) under identical exposure conditions (compare Figs. 2 and 3). By allowing active indicator to diffuse directly into neurons through patch pipettes, we eliminated several potential problems associated with the use of the acetoxymethyl ester form of fura-2 (Tsien, 1988; Roe et al., 1990; Kao, 1994), including incomplete deesterification, indicator compartmentalization, and contributing stray light from the underlying glial layer. Each of these problems results in underestimation of peak [Ca2+]i because of fluorescence signals from fura-2 molecules that are insensitive to calcium (unesterified fura-2/AM) or that are not exposed to neuron-specific changes in calcium (fura-2 in astrocytes or in noncytosolic intracellular compartments).
Our results suggest that the most important limitations of fura-2 in previous experiments stem from its high calcium affinity (KD ∼0.22 μm). At micromolar calcium concentrations, almost all of fura-2 is bound, and therefore it cannot distinguish accurately the differences in [Ca2+]i (Tsien, 1988). Our observations using two low-affinity indicators, BTC and fura-2/dextran, suggest that AMPA-induced [Ca2+]ielevation is measured accurately by fura-2 but that NMDA-induced [Ca2+]i elevation (7–12 μm, Table 1) is considerably higher than this range. We believe these results reflect a real difference in cytosolic calcium levels rather than an artifact caused by the indicator or imaging properties. For example, because in vitro calibration methods cannot reproduce the exact intracellular milieu, it is possible that inaccurate determination of indicator KDartificially might elevate the estimated [Ca2+]i. However, these considerations are not likely to account for the marked discrepancy between NMDA- and AMPA-induced [Ca2+]i elevation, which should be affected to the same extent by any errors in calibration. Independent of the calculated [Ca2+]i, the ratiochanges with BTC and fura-2/dextran were substantially larger during NMDA than AMPA exposure.
We specifically considered effects of magnesium and pH, because these are known to alter the properties of many calcium indicators. The calcium affinity and excitation ratios of BTC are not affected substantially by Mg2+ in the millimolar range (Iatridou et al., 1994; Hyrc et al., 1997). We avoided using two other low-affinity ratiometric Ca2+ indicators, mag-fura-2 and mag-fura-5, because of their higher sensitivity to physiological levels of magnesium (Brocard et al., 1993; Stout et al., 1996). Although many calcium indicators are also sensitive to pH and glutamate receptor activation lowers intracellular pH (Hartley and Dubinsky, 1993), alterations in pHi did not account for the large [Ca2+]i elevation observed with BTC during NMDA, but not AMPA, application. In preliminary experiments with the fluorescent pH indicator BCECF, NMDA and AMPA exposures reduced neuronal pHi to a similar extent, pH ∼6.7–6.8. Further, spectrofluorimetric measurements with BTC indicate that acidic pH levels would lead to moderate underestimation of [Ca2+]i and that actual calcium concentrations may be 30–40% higher than levels estimated by assuming pH 7.2 (Hyrc et al., 1997).
Microinjected BTC has several desirable properties for future studies requiring a low-affinity calcium indicator: it has a high selectivity for calcium over magnesium and other divalent cations, it provides ratiometric calcium determination, and it allows visible wavelength excitation, avoiding the requirement for special UV-transparent optics. Fura-2/dextran also demonstrates promising features as a microinjectable indicator with cytosolic distribution and intermediate calcium affinity. In preliminary experiments, bath application of BTC by its acetoxymethyl ester (BTC/AM) provided results qualitatively similar to those obtained with the use of injected BTC. The quantitative analysis of these results is, however, subject to the same restrictions that apply to fura-2/AM and is likely to yield underestimated [Ca2+]i (Hyrc et al., 1997).
Although affinity differences among indicators partially explain the inability of earlier research with fura-2 and indo-1 to correlate reliably the [Ca2+]i and outcome in neurotoxicity experiments, there are other factors that complicate this analysis. Our initial observations with fura-2 suggested that calcium concentrations after NMDA receptor activation did not reach micromolar levels expected to saturate this indicator (e.g., Fig. 7A). Indeed, ionomycin application doubled the [Ca2+]i levels reported by fura-2/K+ (Fig. 7A). Therefore, we were surprised to find much higher [Ca2+]ireported with BTC and fura-2/dextran during NMDA exposure, concentrations that should be more than sufficient to saturate fura-2.
There are at least two possible reasons for this apparent paradox. First, discrepant results might be caused by differences inindicator distribution, such that fura-2 is sequestered in calcium-insensitive compartments or BTC in high calcium areas. However, digitonin (10 μm) treatment led to the loss of 85–95% of fluorescence of microinjected fura-2 and BTC, suggesting that both indicators were confined primarily in the cytoplasm. Confocal imaging of the distribution of BTC (Fig. 5) and of fura-2 (S. D. Handran and R. S. Hotchkiss, unpublished results) confirmed that neither indicator was compartmentalized substantially. Furthermore, similar results were obtained with fura-2/dextran, which is predicted to have a uniform cytosolic distribution. Second, the results may be explained by a spatially heterogeneous calcium distribution during NMDA receptor activation. Activation of calcium-permeable membrane channels elevates [Ca2+]i in microdomains, with [Ca2+]i reaching 50–100 μm (Smith and Augustine, 1988; Augustine and Neher, 1992;Llinás et al., 1992a,b; Neher and Augustine, 1992; Petrozzino et al., 1995). All high-affinity indicator molecules located within such microdomains will be saturated rapidly, leaving a portion of the indicator in other areas unbound. The fura-2 response to ionomycin, which forms a calcium/proton shuttle (Nicholls and Ferguson, 1992), could result in a higher reported [Ca2+]i if ionomycin globally elevates [Ca2+]i. Thus, fura-2 fails to measure [Ca2+]i accurately when high calcium microdomains are present because a large portion of the indicator pool is saturated. Although this may occur with any calcium indicator, a low-affinity indicator such as BTC is less likely to be saturated under the same conditions (Petrozzino et al., 1995; Ito et al., 1997). Under our imaging conditions we were unable to visualize such microdomains; a histogram of the distribution of pixel ratio values within the cell body during peak NMDA receptor activation did not support the presence of two populations of [Ca2+]i (data not shown). Therefore, any calcium heterogeneity occurred below the spatial and temporal resolution of our imaging apparatus.
These results have immediate consequences for a large number of experiments that use fura-2 and other high-affinity calcium indicators. Many such experiments may be accurate qualitatively but are unlikely to provide a correct determination of [Ca2+]i beyond the normal physiological range, ∼50–500 nm, if substantial calcium microdomains are present. Importantly, our results indicate that widely used high-affinity indicators, such as fura-2, mayselectively underestimate [Ca2+]i, depending on the route of calcium entry. This can occur under lethal or nonlethal conditions and at calcium levels that appear to be well within the range of accurate detection.
One clear message from the present experiments is that [Ca2+]i can rise to concentrations reaching 5–10 μm during excitotoxic exposures. Our results further establish that [Ca2+]imeasurement with low-affinity indicators distinguishes the vulnerability of neurons in excitotoxicity paradigms. This result has implications for investigators attempting to unravel the intracellular pathways that play an early role in triggering or propagating the signals that ultimately are responsible for neuronal death. We now may appreciate the possibility that catabolic enzymes and modulators with relatively low affinity for calcium are activated in these cascades.
Footnotes
This work was supported by National Institutes of Health Grants R01 19988 (S.M.R.), P20 NS32568 (S.M.R.), R29 NS32140 (M.P.G.), and P01 NS32626 (M.P.G.) and was done during the tenure of a Grant-in-Aid Award (to M.P.G.) from the American Heart Association and William Randolph Hearst Foundation. Dr. Hyrc is on leave from the Institute of Molecular Biology, Jagiellonian University, Krakow, Poland. We thank Sandy P. Althomsons for expert assistance with cell cultures, Joanna Bownik for performing spectrofluorimetric experiments, and Richard S. Hotchkiss for assistance with confocal microscopy.
K.H. and S.D.H. contributed equally to this work.
Correspondence should be addressed to Dr. Mark P. Goldberg, Department of Neurology, Box 8111, 660 South Euclid Avenue, St. Louis, MO 63110.
REFERENCES
- 1.Augustine GJ, Neher E. Neuronal Ca2+ signaling takes the local route. Curr Opin Neurobiol. 1992;2:302–307. doi: 10.1016/0959-4388(92)90119-6. [DOI] [PubMed] [Google Scholar]
- 2.Brocard JB, Rajdev S, Reynolds IJ. Glutamate-induced increases in intracellular free Mg2+ in cultured cortical neurons. Neuron. 1993;11:751–757. doi: 10.1016/0896-6273(93)90084-5. [DOI] [PubMed] [Google Scholar]
- 3.Canzoniero LMT, Sensi SL, Choi DW. Recovery from NMDA-induced intracellular acidification is delayed and dependent on extracellular bicarbonate. Am J Physiol. 1996;39:C593–C599. doi: 10.1152/ajpcell.1996.270.2.C593. [DOI] [PubMed] [Google Scholar]
- 4.Choi DW. Ionic dependence of glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 6.Choi DW, Maulucci-Gedde MA, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi DW, Koh J, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connor JA. Intracellular calcium mobilization by inositol 1,4,5-trisphosphate: intracellular movements and compartmentalization. Cell Calcium. 1993;14:185–200. doi: 10.1016/0143-4160(93)90066-f. [DOI] [PubMed] [Google Scholar]
- 9.Connor JA, Tseng HY, Hockberger PE. Depolarization- and transmitter-induced changes in intracellular Ca2+ of rat cerebellar granule cells in explant cultures. J Neurosci. 1987;7:1384–1400. doi: 10.1523/JNEUROSCI.07-05-01384.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connor JA, Wadman WJ, Hockberger PE, Wong RK. Sustained dendritic gradients of Ca2+ induced by excitatory amino acids in CA1 hippocampal neurons. Science. 1988;240:649–653. doi: 10.1126/science.2452481. [DOI] [PubMed] [Google Scholar]
- 11.de Erausquin GA, Manev H, Guidotti A, Costa E, Brooker G. Gangliosides normalize distorted single-cell intracellular free Ca2+ dynamics after toxic doses of glutamate in cerebellar granule cells. Proc Natl Acad Sci USA. 1990;87:8017–8021. doi: 10.1073/pnas.87.20.8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubinsky JM. Examination of the role of calcium in neuronal death. Ann NY Acad Sci. 1993a;679:34–42. doi: 10.1111/j.1749-6632.1993.tb18287.x. [DOI] [PubMed] [Google Scholar]
- 13.Dubinsky JM. Intracellular calcium levels during the period of delayed excitotoxicity. J Neurosci. 1993b;13:623–631. doi: 10.1523/JNEUROSCI.13-02-00623.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubinsky JM, Rothman SM. Intracellular calcium concentrations during “chemical hypoxia” and excitotoxic neuronal injury. J Neurosci. 1991;11:2545–2551. doi: 10.1523/JNEUROSCI.11-08-02545.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eimerl S, Schramm M. The quantity of calcium that appears to induce neuronal death. J Neurochem. 1994;62:1223–1226. doi: 10.1046/j.1471-4159.1994.62031223.x. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 18.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 19.Hartley DM, Choi DW. Delayed rescue of N-methyl-d-aspartate receptor-mediated neuronal injury in cortical culture. J Pharmacol Exp Ther. 1989;250:752–758. [PubMed] [Google Scholar]
- 20.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hartley Z, Dubinsky JM. Changes in intracellular pH associated with glutamate excitotoxicity. J Neurosci. 1993;13:4690–4699. doi: 10.1523/JNEUROSCI.13-11-04690.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haugland RP. Molecular Probes handbook of fluorescent probes and research chemicals. Molecular Probes; Portland, OR: 1995. [Google Scholar]
- 23.Hyrc KH, Handran SD, Rothman SM, Goldberg MP. Low-affinity calcium indicators distinguish lethal calcium entry during glutamate receptor activation in cultured neurons. Soc Neurosci Abstr. 1996;22:798. [Google Scholar]
- 24.Hyrc KH, Bownik JM, Handran SD, Rothman SM, Goldberg MP (1997) Properties of BTC, a novel low-affinity calcium indicator. Soc Neurosci Abstr, in press.
- 25.Iatridou H, Foukaraki E, Kuhn MA, Marcus EM, Haugland RP, Katerinopoulos HE. The development of a new family of intracellular calcium probes. Cell Calcium. 1994;15:190–198. doi: 10.1016/0143-4160(94)90058-2. [DOI] [PubMed] [Google Scholar]
- 26.Ito K, Miyashita Y, Kasai H. Micromolar and submicromolar Ca2+ spikes regulating distinct cellular functions in pancreatic acinar cells. EMBO J. 1997;16:242–251. doi: 10.1093/emboj/16.2.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kao JP. Practical aspects of measuring [Ca2+] with fluorescent indicators. Methods Cell Biol. 1994;40:155–181. doi: 10.1016/s0091-679x(08)61114-0. [DOI] [PubMed] [Google Scholar]
- 28.Koh J, Goldberg MP, Hartley DM, Choi DW. Non-NMDA receptor-mediated neurotoxicity in cortical culture. J Neurosci. 1990;10:693–705. doi: 10.1523/JNEUROSCI.10-02-00693.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konishi M, Watanabe M. Resting cytoplasmic free Ca2+ concentration in frog skeletal muscle measured with fura-2 conjugated to high molecular weight dextran. J Gen Physiol. 1995;106:1123–1150. doi: 10.1085/jgp.106.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Limbrick DD, Jr, Churn SB, Sombati S, DeLorenzo RJ. Inability to restore resting intracellular calcium levels as an early indicator of delayed neuronal cell death. Brain Res. 1995;690:145–156. doi: 10.1016/0006-8993(95)00552-2. [DOI] [PubMed] [Google Scholar]
- 31.Llinás R, Sugimori M, Silver RB. Microdomains of high calcium concentration in presynaptic terminal. Science. 1992a;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- 32.Llinás R, Sugimori M, Silver RB. Presynaptic calcium concentration microdomains and transmitter release. J Physiol (Paris) 1992b;86:135–138. doi: 10.1016/s0928-4257(05)80018-x. [DOI] [PubMed] [Google Scholar]
- 33.Lu YM, Yin HZ, Chiang J, Weiss JH. Ca2+-permeable AMPA/kainate and NMDA channels: high rate of Ca2+ influx underlies potent induction of injury. J Neurosci. 1996;16:5457–5465. doi: 10.1523/JNEUROSCI.16-17-05457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 35.Marcoux FW, Goodrich JE, Probert AWJ, Dominick MA. Ketamine prevents glutamate-induced calcium influx and ischemic nerve cell injury. In: Domino EF, Kamenka JM, editors. Sigma and phencylidine-like compounds as molecular probes in biology. NPP Books; Ann Arbor, MI: 1988. pp. 735–746. [Google Scholar]
- 36.Mayer ML, Westbrook GL. Permeation and block of N-methyl-d-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol (Lond) 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McBain CJ, Mayer ML. N-methyl-d-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- 38.Michaels RL, Rothman SM. Glutamate neurotoxicity in vitro: antagonist pharmacology and intracellular calcium concentrations. J Neurosci. 1990;10:283–292. doi: 10.1523/JNEUROSCI.10-01-00283.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy SN, Thayer SA, Miller RJ. The effects of excitatory amino acids on intracellular calcium in single mouse striatal neurons in vitro. J Neurosci. 1987;7:4145–4158. doi: 10.1523/JNEUROSCI.07-12-04145.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol (Lond) 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicholls DG, Ferguson SJ. Bioenergetics 2, pp 23–37. Academic; San Diego: 1992. Ion transport across energy-conserving membranes. [Google Scholar]
- 42.Ogura A, Miyamoto M, Kudo Y. Neuronal death in vitro: parallelism between survivability of hippocampal neurones and sustained elevation of cytosolic Ca2+ after exposure to glutamate receptor agonist. Exp Brain Res. 1988;73:447–458. doi: 10.1007/BF00406601. [DOI] [PubMed] [Google Scholar]
- 43.Petrozzino JJ, Pozzo Miller LD, Connor JA. Micromolar Ca2+ transients in dendritic spines of hippocampal pyramidal neurons in brain slice. Neuron. 1995;14:1223–1231. doi: 10.1016/0896-6273(95)90269-4. [DOI] [PubMed] [Google Scholar]
- 44.Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflügers Arch. 1988;411:204–211. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- 45.Rajdev S, Reynolds IJ. Calcium green-5N, a novel fluorescent probe for monitoring high intracellular free Ca2+ concentrations associated with glutamate excitotoxicity in cultured rat brain neurons. Neurosci Lett. 1993;162:149–152. doi: 10.1016/0304-3940(93)90582-6. [DOI] [PubMed] [Google Scholar]
- 46.Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Regehr WG, Atluri PP. Calcium transients in cerebellar granule cell presynaptic terminals. Biophys J. 1995;68:2156–2170. doi: 10.1016/S0006-3495(95)80398-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roe MW, Lemasters JJ, Herman B. Assessment of fura-2 for measurements of cytosolic free calcium. Cell Calcium. 1990;11:63–73. doi: 10.1016/0143-4160(90)90060-8. [DOI] [PubMed] [Google Scholar]
- 49.Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine cortical cell culture. In: Tyson CA, Frazier JM, editors. In vitro biological methods. Methods in toxicology. Academic; San Diego: 1993. pp. 46–60. [Google Scholar]
- 50.Rothman SM. The neurotoxicity of excitatory amino acids is produced by passive chloride influx. J Neurosci. 1985;5:1483–1489. doi: 10.1523/JNEUROSCI.05-06-01483.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- 52.Rothman SM, Thurston JH, Hauhart RE. Delayed neurotoxicity of excitatory amino acids in vitro. Neuroscience. 1987;22:471–480. doi: 10.1016/0306-4522(87)90347-2. [DOI] [PubMed] [Google Scholar]
- 53.Sala F, Hernandez-Cruz A. Calcium diffusion modeling in a spherical neuron. Relevance of buffering properties. Biophys J. 1990;57:313–324. doi: 10.1016/S0006-3495(90)82533-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schanne FAX, Kane AB, Young EE, Farber JL. Calcium dependence of toxic cell death: a final common pathway. Science. 1979;206:700–702. doi: 10.1126/science.386513. [DOI] [PubMed] [Google Scholar]
- 55.Smith SJ, Augustine GJ. Calcium ions, active zones, and synaptic transmitter release. Trends Neurosci. 1988;11:458–464. doi: 10.1016/0166-2236(88)90199-3. [DOI] [PubMed] [Google Scholar]
- 56.Stout AK, Li-Smerin Y, Johnson JW, Reynolds IJ. Mechanisms of glutamate-stimulated Mg2+ influx and subsequent Mg2+ efflux in rat forebrain neurones in culture. J Physiol (Lond) 1996;492:641–657. doi: 10.1113/jphysiol.1996.sp021334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsien R, Pozzan T. Measurement of cytosolic free Ca2+ with Quin2. Methods Enzymol. 1989;172:230–262. doi: 10.1016/s0076-6879(89)72017-6. [DOI] [PubMed] [Google Scholar]
- 58.Tsien RY. Fluorescence measurement and photochemical manipulation of cytosolic free calcium. Trends Neurosci. 1988;11:419–424. doi: 10.1016/0166-2236(88)90192-0. [DOI] [PubMed] [Google Scholar]
- 59.Tymianski M, Charlton MP, Carlen PL, Tator CH. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 1993a;607:319–323. doi: 10.1016/0006-8993(93)91523-u. [DOI] [PubMed] [Google Scholar]
- 60.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993b;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao M, Hollingworth S, Baylor SM. Properties of tri- and tetracarboxylate Ca2+ indicators in frog skeletal muscle fibers. Biophys J. 1996;70:896–916. doi: 10.1016/S0006-3495(96)79633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]