

Abstract
To elucidate the molecular mechanisms underlying neuronal death after transient forebrain ischemia, we cloned genes expressed after transient forebrain ischemia in the Mongolian gerbil by a differential display method. A gerbil homolog of rat zinc transporter, ZnT-1, which transports intracellular Zn2+ out of cells, was isolated. Its expression became detectable exclusively in pyramidal neurons of the CA1 region 12 hr after ischemia and reached a maximum from day 1 to day 2 as shown by in situ hybridization. By day 7, expression had disappeared entirely from the cells in the CA1 region, because the neurons had died. No other brain regions exhibited such a significant level of ZnT-1 mRNA expression during this period. Zn2+ was shown to accumulate in CA1 pyramidal neurons expressing ZnT-1 mRNA after the ischemia by using zinquin, a zinc-specific fluorescent dye. When primary hippocampal neurons were exposed to a high dose of Zn2+, ZnT-1 mRNA accumulated. These results suggest that the induction of ZnT-1 mRNA observed in CA1 neurons was caused by an increase in the intracellular Zn2+ concentration. It was reported recently that Zn2+ chelator blocked neuronal death after ischemia and that the influx of Zn2+ might be a key mechanism underlying neuronal death. The induction of ZnT-1 mRNA in CA1 pyramidal neurons fated to die after transient ischemia is of interest to the study of postischemic events and the molecular mechanisms underlying delayed neuronal death.
Keywords: brain ischemia, Zn, Zn transporter, ZnT-1, differential display, delayed neuronal death
Transient forebrain ischemia results in the degeneration of specific populations of neurons, such as pyramidal cells in the CA1 subfield of the hippocampus (Kirino and Sano, 1984). This neuronal degeneration occurs a few days after the ischemia and is known as delayed neuronal death (DND) (Kirino, 1982). Expressions of c-fos proto-oncogene (Jorgensen et al., 1989), heat–shock proteins (HSPs) (Nowark, 1985), and brain-derived neurotrophic factor (BDNF) (Lindvall et al., 1992) are known to be induced in the brain after ischemia. However, it is unclear how these genes are associated with the progress of neuronal damage, and their expression has been observed not only in ischemia-vulnerable neurons but also in ischemia-resistant cells. Therefore, the molecular events associated with DND still are understood poorly. In our previous study we searched for ischemia-regulated genes, using a differential display technique (Liang and Pardee, 1992), and identified a gerbil homolog of a serine protease inhibitor, SPI-3 (Tsuda et al., 1996). We also isolated another ischemia-regulated gene, the zinc transporter ZnT-1 that is described here.
The first mammalian transporter for zinc, ZnT-1, was cloned recently from a rat cDNA library. ZnT-1 protein was predicted to contain six membrane-spanning domains and revealed to be localized in the plasma membrane. A study in baby hamster kidney (BHK) cells showed that ZnT-1 protein transports Zn2+ out of cells (Palmiter and Findley, 1995). However, little is known about the physiological role, much less its role after brain ischemia. The zinc released from presynaptic boutons by kainate stimulation accumulates specifically in perikarya and proximal dendrites of the postsynaptic neurons, which consequently die as a result of seizures (Frederickson et al., 1989). Recently, it was postulated that the toxic influx of Zn2+ into neurons is a key mechanism underlying DND in rat brains subjected to transient forebrain ischemia (Koh et al., 1996). Thus differential expression of ZnT-1 in CA1 neurons after ischemia could reflect changes in zinc distribution. Here we examine the expression patterns and putative biological function of ZnT-1 after transient forebrain ischemia and discuss the relationship between gene expression and postischemic neuronal death.
MATERIALS AND METHODS
Animals. Adult male Mongolian gerbils weighing 60–80 gm were used in the present study. Animals were divided randomly into a sham-operated group and a 5 min ischemic group. They were anesthetized lightly by ether inhalation, and both of their common carotid arteries were exposed. Bilateral cerebral ischemia was generated by common carotid occlusion with miniature aneurysm clips. The rectal temperature was monitored, and body temperature was maintained at 37°C by warming blankets and a heating lamp during the ischemic period. At specified time points after clip removal, the animals were anesthetized again by ether inhalation and decapitated, and their hippocampi were removed promptly for further study.
Differential display. Total RNA was isolated from the hippocampi, and the differential display was performed as previously described (Liang and Pardee, 1992; Liang et al., 1992; Tsuda et al., 1996). Briefly, total RNA (3 μg) was converted to cDNA with Maloney murine leukemia virus (MMLV) reverse transcriptase (Life Technologies, Grand Island, NY). Subsequently, each pool of cDNA was amplified by PCR with 104 different arbitrary primers. After separation by 5% polyacrylamide gel electrophoresis, cDNA bands that appeared to be upregulated in the ischemic group were recovered and reamplified with the corresponding primer. Reamplified cDNA fragments were cloned into pGEM-T vector (Promega, Madison, WI). Plasmid DNA sequencing of the cloned fragments was performed with the Taq dye primer cycle sequencing kit (Perkin-Elmer, Norwalk, CT) and the 373A DNA sequencer (Applied Biosystems, Foster City, CA) according to the suppliers’ recommendations.
Northern blot analysis. Total RNA (10 μg) was fractionated by electrophoresis through 1.0% agarose/formaldehyde gels and transferred onto Immobilon-N membranes (Millipore, Bedford, MA). The membranes were prehybridized for 3 hr at 65°C in hybridization buffer containing 6× SSC (0.9 m NaCl and 0.09 msodium citrate, pH 7.0), 5× Denhardt’s solution (0.5% Ficoll, 0.5% polyvinylpyrrolidone, and 0.5% BSA), 0.5% SDS, and 100 μg/ml of heat-denatured salmon sperm DNA and then hybridized for 16 hr at 65°C with 32P-labeled cDNA probe, which was generated from the cloned cDNA by the random hexamer procedure (TAKARA SHUZO, Kyoto, Japan). After being washed first in 2× SSC/0.1% SDS at 65°C and then in 0.1× SSC/0.1% SDS, the membranes were dried and autoradiographed.
Construction of a cDNA library. Poly(A+) RNA was extracted from total RNA of 14-d-old gerbil brains with the mRNA purification kit (Pharmacia, Uppsala, Sweden). First- and second-strand cDNAs were synthesized with the TimeSaver cDNA synthesis kit (Pharmacia) and ligated to EcoRI-digested λZAPII DNA (Stratagene, La Jolla, CA). Then the ligated DNAs were packaged into phage particles by a MaxPlax packaging extract (Epicentre, Madison, WI). All of the steps were performed as recommended by the manufacturers. Using this cDNA library, we performed cDNA screening by the standard methods.
In situ hybridization. Animals were decapitated 12 and 24 hr and 2, 3, and 7 d after the 5 min ischemia treatment.In situ hybridization with digoxigenin-labeled probes was performed as described previously (Imaizumi et al., 1994). Digoxigenin-labeled cRNA probes (antisense and sense) were made byin vitro transcription with cDNAs subcloned into pGEM-T vector as templates in the presence of digoxigenin-labeledd-uridine triphosphate (Boehringer Mannheim, Mannheim, Germany) according to the manufacturer’s instructions. Thaw-mounted 15 μm sections were post-fixed for 20 min in 4% formaldehyde and then given two 5 min washes in 0.1 m phosphate buffer, pH 7.2. The post-fixed sections were treated with 0.001% proteinase K (Boehringer Mannheim) and subsequently for 10 min in a solution of 0.1m triethanolamine and 0.225% acetic acid anhydrous. After being washed in 0.1 m phosphate buffer, sections were dehydrated via a series of increasing concentrations of ethanol and air-dried. The sections were prehybridized for 1 hr at 50°C in the hybridization buffer (10% sodium dextran sulfate, 20 mmTris-HCl, pH 8.0, 0.3 m NaCl, 0.2% sarcosyl, 0.02% heat-denatured salmon sperm DNA, 1× Denhardt’s solution, and 50% formamide) and then hybridized overnight at 50°C in the hybridization solution with 100 ng/ml of cRNA probe. After being rinsed in 5× SSC at 60°C for 20 min, the sections were washed in 50% formamide/2× SSC at 60°C for 30 min. Next, they were subjected to RNase digestion for 20 min at 37°C (1 μg/ml RNase A in a buffer of 10 mmTris-HCl, 1 mm EDTA, and 0.5 m NaCl, pH 7.5) and then washed in 50% formaldehyde/2× SSC at 60°C for 30 min. For detection of hybridized cRNA probes, anti-digoxigenin antibody conjugated to alkaline phosphatase (Boehringer Mannheim) was reacted at 1:500 dilution, and color was developed by incubation with 4-nitro blue tetrazolium chloride (NBT) and X-phosphate solution.
Cell culture. Primary cultures of neuronal cells were prepared from the hippocampi of fetal rats at 17 d gestation. The dissected tissues were digested at 37°C for 10 min in Dulbecco’s PBS containing 180 U of papain (Sigma, St. Louis, MO), 0.02%dl-cysteine-HCl, 0.02% BSA, 0.5% glucose, and 0.1% DNase. Cells were plated in 10 cm dishes coated with poly-l-lysine and maintained at 37°C in DMEM containing the N2 supplement (Life Technologies) in an atmosphere of 5% CO2 in air.
To examine the induction of ZnT-1 mRNA, we incubated cells in the growth medium supplemented with 150 μmZn2+ for various times; then total RNA was prepared from the cells and used in Northern blot analysis.
Cells expressing rat ZnT-1. An expression plasmid was made from the rat ZnT-1 cDNA (provided by Dr. Palmiter, University of Washington). Recognition sites of XbaI were added to both ends of the ZnT-1 coding region by PCR, using primers of 5′-CACTCTAGAATGGGCTGCTGGGGCCGC-3′ and 5′-CACTCTAGATCACAAAGATGATTCGGG-3′. The product was ligated into theXbaI–cut DNA of vector pEF–BOS (Mizushima and Nagata, 1990) in which the cloned cDNA is expressed under the promoter for human polypeptide chain elongation factor 1α (pEFRZnT-1).
Neuro2A cells derived from mouse neuroblastoma (Klebe and Ruddle, 1969) were cultured in Hank’s–MEM with 10% FCS in an atmosphere of 5% CO2 in air. The cells were cotransfected with pMAMneo (Clontech, Palo Alto, CA) and pEFRZnT-1 DNAs with Lipofectamine (Life Technologies), and transfectants were selected in growth medium supplemented with Geneticin (G418 disulfate, Life Technologies, 400 μg/ml). Individual G418-resistant colonies were picked, grown, and screened for expression of ZnT-1 mRNA by Northern blotting.
To test zinc toxicity, we cultured cells in the growth medium for 2 d. Zn2+ stimulation was performed by changing to medium containing various concentrations of Zn2+. After 48 hr, cell numbers were counted in several representative culture fields, and the ratio of cells that survived, as judged by morphology and trypan blue staining, was calculated.
Visualization of intracellular Zn2+ of postischemic CA1 neurons. To stain intracellular Zn2+, we applied PBS containing 5 μmzinquin ester (Dojindo, Kumamoto, Japan), a zinc-specific fluorescence dye, to the sections at room temperature for 30 min (Zalewski et al., 1993; Palmiter et al., 1996). After being washed with PBS, the sections were examined under an inverted microscope with a UV-2A filter, and color images were obtained with a confocal laser scanning microscope ACAS ultima (Meridian Instruments, Okemos, MI).
RESULTS
Differential display and cDNA cloning
Via the differential display screening, four cDNA fragments were found to be amplified to a greater degree in the hippocampi of the ischemic gerbils as compared with the control. One of these clones, P28, was subjected to further analysis. By Northern blot analysis the DNA probe synthesized from the P28 cDNA fragment (200 bp) was shown to hybridize with a single mRNA species of ∼4 kb, and its expression increased a little in the ischemic hippocampi (Fig.1). The nucleotide sequence of the clone was AT-rich and showed no homology with known genes, suggestive of a 3′-untranslated region. To obtain cDNA containing the coding region of P28 gene, we screened a gerbil brain cDNA library, using a radiolabeled P28 fragment as a probe, and obtained a cDNA fragment of ∼1.1 kb. The amino acid sequence predicted from the cDNA clone showed 92% identity with rat ZnT-1 protein on a homology search with the current sequence data bases: European Molecular Biology Laboratory, DNA Data Bank of Japan, and GenBank (Fig. 2). Thus we concluded that P28 gene was a gerbil homolog of rat ZnT-1.
Fig. 1.

Northern blot demonstrating ZnT-1 mRNA expression 24 hr after 5 min ischemia. A, Each lane was loaded with 15 μg of total RNA purified from hippocampi of sham-operated control brains (C) and 5 min ischemic brains (I). Ethidium bromide-stained ribosomal RNAs (18S and 28S) indicate that equal amounts of total RNA were loaded in each lane. B, Membrane-transferred RNAs were hybridized with gerbil ZnT-1 (clone P28) probe. The arrow indicates the ZnT-1 signal.
Fig. 2.

Alignment of rat ZnT-1 and P28 amino acid sequences. The amino acid sequence of rat ZnT-1 is given along with that reported by Palmiter et al. (1996), whereas that of clone P28 was deduced from the cDNA sequence. Identical amino acids are indicated byasterisks, and conservative changes are shown asperiods.
Expression pattern of gerbil ZnT-1 after transient forebrain ischemia
In situ hybridization with cRNA probes and cresyl violet staining were performed to analyze the distribution of cells expressing ZnT-1 mRNA after the transient forebrain ischemia. Although no signals of ZnT-1 mRNA were detected in the CA1 pyramidal cells in sham-operated control brains (Fig.3A), positive signals were weakly detected in CA1 pyramidal neurons 12 hr after ischemia (data not shown). At 24 hr after ischemia the signal intensity became more vivid (Fig. 3C,I), and the same strength of signals was observed at day 2 (data not shown). By day 2 CA1 pyramidal neurons were morphologically intact, judging from the cresyl violet staining (Fig.3B,D), in accord with a previous report (Kirino, 1982). Three days after ischemia, the intensity of cresyl violet staining decreased at the medial side of the CA1 region, indicating neuronal death (Fig. 3F), and the signals of ZnT-1 mRNA disappeared (Fig. 3E). ZnT-1 mRNA expression was absent in the CA1 region 7 d after the ischemic insult (Fig. 3G), because almost all neurons in the subfield had died (Fig.3H). High magnification of the brain at 24 hr after ischemia showed that the ZnT-1 mRNA-positive cells were CA1 pyramidal neurons, but not glia (Fig. 3I,J). In other areas, such as the striatum and thalamus where DND does not occur in this model, ZnT-1 gene expression was undetectable by in situhybridization. Taken together, ZnT-1 mRNA was induced only in the CA1 neurons damaged by the 5 min ischemia.
Fig. 3.
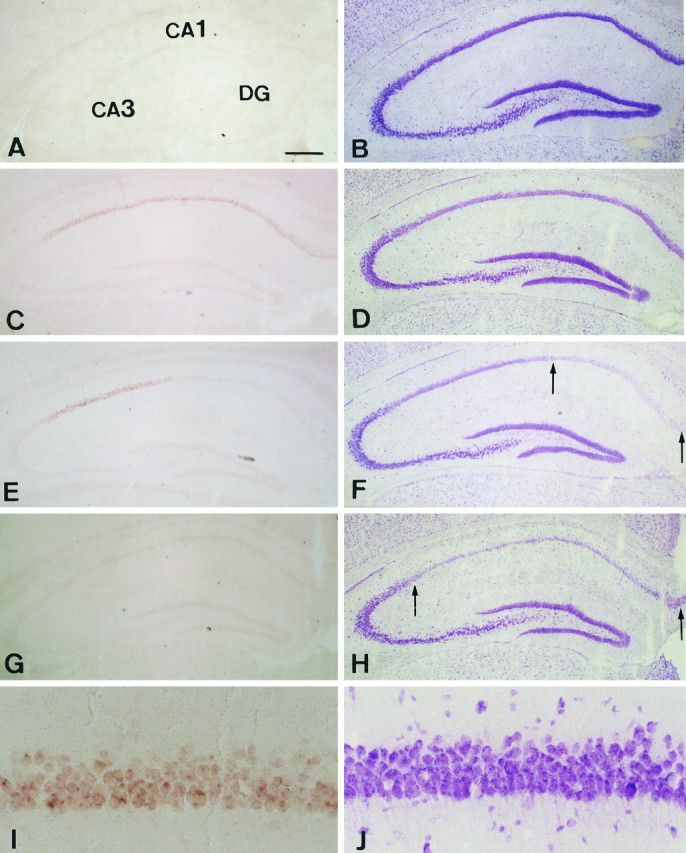
Expression of ZnT-1 mRNA (A, C, E, G, I) and cresyl violet staining (B, D, F, H, J). A, B, Sham-operated control brains.C, D, At 24 hr after ischemic insult. E, F, At 3 d after ischemic insult. G, H, At 7 d after ischemic insult. I, J, High magnification of results at 24 hr after ischemic insult. The signals of ZnT-1 are seen only in CA1 pyramidal neurons that undergo DND. The staining intensity of the neurons between the two arrows is reduced, indicating cell death. Scale bars: 300 μm for A–H; 20 μm for I, J.
Zinc accumulation in degenerating neurons
To visualize intracellular Zn2+, we incubated frozen sections of brain from sham-operated and ischemic gerbils with zinquin ester. In the brains from the sham-operated group, zinquin revealed intense fluorescence in the mossy fiber projections from the dentate granule cells and the CA3 region of the hippocampus (Fig.4A); however, the intensity was very low in the CA1 region (Fig. 4A,C). These findings support previous observations in rat brains (Koh et al., 1996). Three days after 5 min ischemia no changes were observed in CA3 and mossy fibers; however, fluorescence in CA1 was prominent (Fig.4B). This CA1 fluorescence was judged to be from pyramidal neurons, based on the high-magnification picture (Fig.4D). These results suggested that ZnT-1 mRNA was expressed in neurons in which Zn2+ accumulated after ischemia.
Fig. 4.

Zinquin staining of CA1 pyramidal neurons in the control sham-operated gerbil brain (A, C) and 24 hr after 5 min ischemic insult (B, D). The color images were obtained with ACAS ultima (A, B). High-magnification photographs of the CA1 pyramidal cell layer are shown (C, D). The control brain exhibited zinc fluorescence in CA3 and dentate gyrus, but not in CA1. In the 5 min ischemic brains the fluorescence appeared in pyramidal cells in CA1. Scale bars: 300 μm for A, B; 20 μm for C, D.
Induction of ZnT-1 mRNA in rat hippocampal primary culture
From the in vivo analysis described above, the induction of ZnT-1 mRNA appeared to be caused by Zn2+ accumulation. To examine responses of rat ZnT-1 mRNA expression, we added a high dose of Zn2+ to rat hippocampal primary cultures. Many of the neuronal cells were damaged after 24 hr exposure to Zn2+ at >150 μm (Fig. 5A–C). Less than 120 μm Zn2+ did not damage neurons (data not shown). The expression of ZnT-1 mRNA was examined in the primary hippocampal neurons exposed to 150 μmZn2+ by Northern blotting. The cells cultured in the growth medium expressed ZnT-1 mRNA at very low levels. After the addition of 150 μm Zn2+, ZnT-1 mRNA expression was induced within 1 hr, remained at high levels for 8 hr, and then decreased to pretreatment levels by 24 hr. The signal intensity of GAPDH mRNA as an internal control gradually decreased after Zn2+ exposure, paralleling neuronal degeneration (Fig. 5D).
Fig. 5.

Neurotoxicity of Zn2+ and the induction of ZnT-1 mRNA expression by exposure to Zn2+. A, Control hippocampal neurons cultured in growth medium. B, Hippocampal neurons cultured in growth medium containing 150 μmZn2+ for 12 hr. C, Hippocampal neurons cultured in 200 μm Zn2+ for 12 hr. Scale bar, 60 μm. D, Pattern of ratZnT-1 mRNA expression in primary cultures after exposure to 150 μm Zn2+. Rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA was used as a control.
Neuro2A cells overexpressing ZnT-1 became resistant to zinc
Neuro2A cells transfected with the rat ZnT-1 expression plasmid pEFRZnT-1 were isolated. We confirmed that these clones overexpressed ZnT-1 mRNA by Northern blotting (data not shown). All clones overexpressing ZnT-1 mRNA were more resistant to Zn2+ than parental Neuro2A. The parental cells were damaged after 48 hr exposure to 150 μmZn2+, and almost all cells died at 210 μm Zn2+. On the other hand, up to 210 μm Zn2+ had little effect on clone 309 (Fig. 6A–C). Thus, ZnT-1 gene overexpression significantly reduced zinc toxicity.
Fig. 6.

Effects of ZnT-1 overexpression on zinc toxicity.A, Parental Neuro2A after 48 hr exposure to 210 μm Zn2+. B, Clone 309 overexpressing ZnT-1 after 48 hr exposure to 210 μmZn2+. C, Parental Neuro2A (open squares) and clone 309 (filled squares) were cultured in growth medium containing various concentrations of Zn2+ for 48 hr. Cell survival is given as a percentage. Values are expressed as the mean of five experiments ±SE.
DISCUSSION
ZnT-1 cDNA recently was isolated from a rat kidney cDNA library as the first mammalian zinc transporter (Palmiter and Findley, 1995). It has been proposed that ZnT-1 protein transports Zn2+out of cells; however, its function in the brain is not known. In the present study we found that ZnT-1 mRNA was induced in the hippocampi after forebrain ischemia. ZnT-1 mRNA was expressed in all organs tested: brain, intestine, kidney, liver, lung, muscle, spleen, testis, and thymus, although at very low levels (data not shown).
Although Northern blot analyses that used total RNA from whole hippocampus tissues revealed only a small difference in the level of ZnT-1 mRNA expression between normal and ischemic brains, in situ hybridization signals of the expression were increased markedly in CA1 pyramidal neurons that are vulnerable to ischemic insult. Cresyl violet staining showed that all ZnT-1 mRNA-positive cells eventually died.
Recently, Koh et al. (1996) reported that Zn2+accumulated specifically in degenerating neurons in the CA1 region of rats after transient forebrain ischemia and that injection of a zinc chelator into the lateral ventricles blocked the neuronal death. In the present study we also observed Zn2+ accumulation in CA1 pyramidal cells vulnerable to ischemic insult. The concentration of Zn2+ is thought to be much higher in CA3 and dentate gyrus (DG), where mossy fibers contain higher amounts of Zn2+, than in the CA1 region. When rats were given kainic acid (KA) and underwent seizures, Zn2+accumulated in the neurons of the CA3 region, which subsequently died of Zn2+ toxicity (Frederickson et al., 1989). However, when gerbils were subjected to ischemic insult, no significant change of zinc fluorescence intensity in CA3 and DG was seen, and those neurons did not degenerate. These results suggest that KA and ischemia affect different neuronal populations; after KA treatment, CA3 are exposed to excess zinc, whereas after ischemia, the CA1 neurons are more at risk.
In primary cultures of hippocampal neurons the addition of a high dose of Zn2+ to the culture medium caused the neurons to die (Choi et al., 1988). Under these conditions expression of ZnT-1 mRNA was elevated greatly and remained at a high level until the cells degenerated because of zinc toxicity. It is known that the function of ZnT-1 protein is to transport Zn2+ out of cells (Palmiter and Findley, 1995). The transfectants overexpressing ZnT-1 mRNA were more resistant against extracellular Zn2+than parental cells (Fig. 6). We confirmed that the transfectants extrude Zn2+ more effectively (data not shown). Thus, it is likely that the induction of ZnT-1 mRNA after transient ischemia is a response to the increase in intracellular Zn2+ concentration in the postsynaptic neurons and may be an attempt by those cells to extrude the Zn2+.
To detect the expression of ZnT-1 protein, we generated a specific antibody against ZnT-1 and performed immunohistochemical studies (data not shown). However, no ZnT-1-immunoreactive neurons were observed in the CA1 region after transient ischemia. Protein synthesis is inhibited in CA1 neurons of postischemic gerbil brains (Thilmann et al., 1986). If ZnT-1 protein is not expressed in CA1 neurons, the Zn2+ that has accumulated cannot be extruded from the cells, resulting in an increase of intracellular Zn2+ concentration and, finally, neuronal death. Further analysis about ZnT-1 protein is necessary, however; it may protect CA1 neurons that are vulnerable to ischemic insult.
Footnotes
We thank Dr. R. D. Palmiter (University of Washington) for providing rat ZnT-1 cDNA.
Correspondence should be addressed to Dr. Manabu Tsuda, Department of Molecular Neurobiology (TANABE), Osaka University Medical School, 2-2 Yamadaoka, Suita, Osaka 565, Japan.
REFERENCES
- 1.Choi DW, Yokoyama M, Koh J. Zinc neurotoxicity in cortical cell culture. Neuroscience. 1988;24:67–69. doi: 10.1016/0306-4522(88)90312-0. [DOI] [PubMed] [Google Scholar]
- 2.Frederickson CJ, Hernandez MD, McGinty JF. Translocation of zinc may contribute to seizure-induced death of neurons. Brain Res. 1989;480:317–321. doi: 10.1016/0006-8993(89)90199-6. [DOI] [PubMed] [Google Scholar]
- 3.Imaizumi K, Tsuda M, Wanaka A, Tohyama M, Takagi T. Differential expression of sgk mRNA, a member of the Ser/Thr protein gene family, in rat brain after CNS injury. Mol Brain Res. 1994;26:189–196. doi: 10.1016/0169-328x(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 4.Jorgensen MB, Deckert J, Wright DC, Gehlert DR. Delayed c-fos proto-oncogene expression in the rat hippocampus induced by transient global ischemia: an in situ hybridization study. Brain Res. 1989;484:393–398. doi: 10.1016/0006-8993(89)90388-0. [DOI] [PubMed] [Google Scholar]
- 5.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 6.Kirino T, Sano K. Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol (Berl) 1984;62:201–208. doi: 10.1007/BF00691853. [DOI] [PubMed] [Google Scholar]
- 7.Klebe RJ, Ruddle FH. Neuroblastoma: cell culture analysis of a differentiating stem cell system. J Cell Biol. 1969;43:69A. [Google Scholar]
- 8.Koh J, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 9.Liang P, Pardee AB. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 10.Liang P, Averboukh L, Keyomarsi K, Sager R, Pardee AB. Differential display and cloning of messenger RNAs from human breast cancer versus mammary epithelial cells. Cancer Res. 1992;52:6966–6968. [PubMed] [Google Scholar]
- 11.Lindvall O, Ernfors P, Bengzon J, Kokaia Z, Smith ML, Siesjo BK, Persson H. Differential regulation of mRNAs for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin 3 in the adult rat brain following cerebral ischemia. Proc Natl Acad Sci USA. 1992;89:648–652. doi: 10.1073/pnas.89.2.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nowark TS. Synthesis of a stress protein following transient ischemia in the gerbil. J Neurochem. 1985;45:1635–1641. doi: 10.1111/j.1471-4159.1985.tb07236.x. [DOI] [PubMed] [Google Scholar]
- 14.Palmiter RD, Findley SD. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 1995;14:639–649. doi: 10.1002/j.1460-2075.1995.tb07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palmiter RD, Cole BC, Findley SD. ZnT-2, a mammalian protein that confers resistance to zinc by facilitating vesicular sequestration. EMBO J. 1996;15:1784–1791. [PMC free article] [PubMed] [Google Scholar]
- 16.Thilmann R, Xie Y, Kleiheus P, Plum F. Persistent inhibition of protein synthesis precedes delayed neuronal death in postischemic gerbil hippocampus. Acta Neuropathol (Berl) 1986;71:88–93. doi: 10.1007/BF00687967. [DOI] [PubMed] [Google Scholar]
- 17.Tsuda M, Kitagawa T, Imaizumi K, Wanaka A, Tohyama M, Takagi T. Induction of SPI-3 mRNA, encoding a serine protease inhibitor, in gerbil hippocampus after transient forebrain ischemia. Mol Brain Res. 1996;35:314–318. doi: 10.1016/0169-328x(95)00211-a. [DOI] [PubMed] [Google Scholar]
- 18.Zalewski PD, Forbes IJ, Betts WH. Correlation of apoptosis with change in intracellular labile Zn(II) using zinquin [(2-methyl-8-p-toluenesulphonoamido-6-quinolyloxy) acetic acid], a new specific fluorescent probe for Zn(II). Biochem J. 1993;296:403–408. doi: 10.1042/bj2960403. [DOI] [PMC free article] [PubMed] [Google Scholar]