

Abstract
This study aimed to examine changes of presynaptic voltage-sensitive calcium channel (VSCC) subtypes during synapse formation and regeneration in relation to transmitter release at the neuromuscular junction (NMJ). Synaptic potentials were recorded from developing rat NMJs and from regenerating mouse and frog NMJs. As in normal adult NMJs, evoked transmitter release was reduced by an N-type VSCC blocker in the frog and by a P/Q-type VSCC blocker in the mammal at immature NMJs; however, various L-type VSCC blockers, both dihydropyridine and nondihydropyridine antagonists,increased evoked but not spontaneous release in a dose-dependent manner at newly formed NMJs. This presynaptic potentiation disappeared as NMJs matured. A rapid intracellular Ca2+ buffer, bis(O-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid-AM, prevented the potentiation effect of nifedipine, but a slow Ca2+ buffer, EGTA-AM, did not. Thus, the potentiation effect of L-type blockers requires Ca2+ transients. Pretreatment with Ca2+-activated K+ channel blockers, iberiotoxin or charybdotoxin, did not prevent potentiation by nifedipine at regenerating frog NMJs. Thus, Ca2+-activated K+ channels were not likely involved in this potentiation. In contrast, no additional potentiation by nifedipine was seen in muscles pretreated with pertussis toxin (PTX), a G-protein blocker, which by itself enhances evoked transmitter release at regenerating frog NMJs. These results suggest the existence of multiple subtypes of VSCCs at newly formed motor nerve terminals. In addition to the normal N- or P/Q-type VSCCs that mediate transmitter release, L-type VSCCs may play a novel modulatory role in evoked transmitter release by activating a mechanism linked to PTX-sensitive G-proteins during synapse maturation.
Keywords: voltage-sensitive calcium channels, dihydropyridine, neuromuscular junctions, PTX-sensitive G-proteins, synapse formation, transmitter release
The entry of Ca2+ through voltage-sensitive calcium channels (VSCCs) triggers the release of neurotransmitters from nerve terminals (Katz, 1969; Augustine et al., 1987). Several subtypes of VSCCs, such as T, L, N, P and Q, have been identified on the basis of electrophysiological and pharmacological characteristics (Tsien et al., 1988; Hille, 1992; Zhang et al., 1993). Accordingly, it is important to identify which subtypes of VSCCs are crucial for presynaptic Ca2+ entry and transmitter release (Olivera et al., 1994; Dunlap et al., 1995).
Because of its relative simplicity and accessibility, the neuromuscular junction (NMJ) has been used to study mechanisms of synaptic transmission and formation. It has been shown that the motor nerve terminal uses N-type (Kerr and Yoshikami, 1984) and P/Q-type VSCCs (Uchitel et al., 1992; Protti and Uchitel, 1993; Bowersox et al., 1995;Sugiura et al., 1995) to mediate transmitter release at adult frog and mammalian NMJs, respectively; however, whether NMJs use the same and/or other subtypes of VSCCs for evoked transmitter release during synapse formation has not been examined. It is known that ion channels in excitable membranes often undergo changes during development (Spitzer, 1994). One of the best-studied ion channels in terms of developmental change is the postsynaptic acetylcholine receptor, which shows different characteristics during development and regeneration of the NMJ (Brehm, 1989; Steinbach, 1989). Investigation of these developmental changes at the NMJ has led to a better understanding of the general mechanisms of synapse formation.
Studies of developmental changes in the populations of VSCCs in neuronal soma have yielded interesting findings, i.e., developmental alterations from T-type to L- or N-type VSCCs have been found (McCobb et al., 1989; O’Dowd et al., 1988). Such alterations may be correlated with the appearance of neurites in cultured rat hippocampal neurons (Yaari et al., 1987). In contrast, very little is known about the developmental expression of VSCC subtypes at thenerve terminal. Gray et al. (1992) showed that, in the chick ciliary ganglion, acetylcholine secretion changes during the course of development. At stage 40, release is dihydropyridine (DHP, a class of L-type VSCC blockers)-sensitive and partially ω-conotoxin GVIA (an N-type VSCC blocker)-sensitive. Posthatch release is insensitive to DHP but sensitive to ω-conotoxin GVIA; however, whether this kind of developmental change in VSCCs occurs at the NMJ is not known.
The present study aimed to examine changes of VSCC subtypes in relation to transmitter release during the development and regeneration of NMJs. We found that L-type VSCC blockers consistently increasedevoked but not spontaneous transmitter release at newly developed or regenerated, but not at mature, NMJs. This presynaptic potentiation involves Ca2+ transients and may activate a regulatory mechanism linked to pertussis toxin (PTX)-sensitive G-proteins during synapse formation. The results suggest that L-type VSCCs modulate evoked transmitter release and may play a role in synapse maturation.
Some of these results have been published previously in abstract form (Sugiura and Ko, 1995).
MATERIALS AND METHODS
Animals and preparations. Isolated phrenic nerve–diaphragm muscle preparations from embryonic day 17 (E17) to postnatal 1-month-old Sprague Dawley rats were used. The rats were anesthetized with ether and decapitated. The nerves and muscles were dissected, pinned on a Sylgard-coated dish, and bathed in normal mammalian Ringer’s solution (NMR) consisting of 135 mmNaCl, 5 mm KCl, 15 mm NaHCO3, 1 mm Na2HPO4, 1 mmMgSO4, 2.5 mm calcium gluconate, and 11 mm glucose, pH 7.2. Regenerating NMJs were studied using the sternomastoid muscle of adult male Swiss Webster mice (20–30 gm body weight) and the cutaneous pectoris muscle of the frog Rana pipiens (6–7 cm body length). The mice were anesthetized with pentobarbital, and a midline incision was made in the neck. The left sternomastoid muscle was exposed by lateral reflection of the salivary glands. With use of fine forceps, the nerve to the sternomastoid muscle was crushed near its entry into the muscle, and then the wound was closed with sutures. Seven days later, the muscles were dissected and bathed in NMR. The intact contralateral muscle was used as the control. A similar nerve crush operation was carried out on the cutaneous pectoris muscle of frogs. The frogs were anesthetized with 0.1% tricaine methanesulfonate (Sigma, St. Louis), and the nerve was crushed at the site of nerve entry. Two or 6 weeks after nerve crush, the muscles were dissected in normal frog Ringer’s solution (NFR) containing 120 mm NaCl, 2 mm KCl, 1 mm NaHCO3, 1.8 mmCaCl2, and 5 mm HEPES, pH 7.2, and examined. All of the bath solutions were bubbled continuously with a mixture of 95% O2/5% CO2.
Electrophysiology. Conventional methods for intracellular recording from skeletal muscles were used. Glass microelectrodes were filled with 3 m KCl (30–50 MΩ resistance). The isolated phrenic nerve–diaphragm muscle preparations from E17 to postnatal 1-month-old rats were bathed in 10 mm Ca2+Ringer’s solution (according to Redfern, 1970), with the remaining ions the same as in NMR. Raising the Ca2+ concentration to at least twice its normal level increased the stability of the recordings and the responsiveness of motor axons in developing muscles (Redfern, 1970; Dennis et al., 1981). To prevent muscle contractions,d-tubocurare (1–5 μm) was added to the bath solution. Transmitter release was evoked by suprathreshold stimulation of the motor nerve via a suction electrode. Evoked endplate potentials (EPPs) were recorded continuously in the same muscle fiber with stimulation at 0.1 Hz before and during drug application. Stock solutions of each drug (see Drug preparations) were added to the bath solution and allowed to achieve specific drug concentrations by diffusion. The resting membrane potential was monitored continuously, and an experiment was rejected when more than a 10 mV change of membrane potential was observed. The change of EPP amplitude was expressed as the ratio of the average EPP amplitude when a drug showed its full effect divided by the average EPP amplitude before drug application at the same NMJ.
Reinnervating adult mouse and frog muscles after nerve crush were studied by procedures similar to those described above. The mouse sternomastoid muscles were bathed in mammalian Ringer’s solution containing 1.2 mm Ca2+/6.0 mmMg2+. The frog cutaneous pectoris muscles were bathed in 0.7 mm Ca2+/4.0 mm Mg2+frog Ringer’s solution. EPPs and spontaneous miniature EPPs (mEPPs) were recorded in low Ca2+/high Mg2+ saline. EPPs were recorded either with continuous stimulation at 0.03 Hz (once every 30 sec) or stimulation at 0.5 Hz stimulation for 48 sec every 15 min. This low frequency of stimulation was chosen to avoid synaptic depression at regenerating NMJs. Changes in EPP amplitude were calculated using the same procedure described above. All recordings were performed at room temperature (22–24°C). Data were collected and analyzed using PClamp programs (Version 6.2, Axon Instruments, Foster City, CA).
Drug preparations. Synthetic ω-conotoxin MVIIC (SNX-230) and ω-conotoxin GVIA (SNX-124) were generous gifts from Dr. G. Miljanich (Neurex). Isradipine (PN200-110) was kindly provided by Research Biochemicals International (Natick, MA). The DHPs, nifedipine, nimodipine (Sigma), and isradipine, were dissolved in ethanol to make a 20 mm stock solution, and various concentrations of drugs were cumulatively applied to the bath solution. The maximal concentration of ethanol was 0.05%, which had no effect on EPPs in embryonic muscles (see Results). Verapamil (Sigma) was dissolved in water. Membrane-permeable Ca2+ chelators, EGTA-AM, and bis (O-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid-AM (BAPTA-AM) (Molecular Probes, Eugene, OR) were prepared in dimethylsulfoxide (DMSO) to make a 25 mm stock solution. Both EGTA-AM and BAPTA-AM were used at a final concentration of 25 μm. Charybdotoxin (Alomone Labs) was reconstituted with 1 mg/ml bovine serum albumin (BSA), 0.1 m NaCl, 10 mm Tris buffer, pH 7.5, to make a 10 μm stock solution. Iberiotoxin (Bachem, Torrance, CA) was reconstituted with 5 mm HEPES buffer, pH 6.2, to make 20 μmstock solution . Both toxins were used at a final concentration of 5, 10, 20, and 40 nm.
Pretreatment with PTX. PTX (Sigma) was reconstituted with 5 mg/ml BSA in water. This reconstituted PTX (100 μg/ml) stock solution also contained 0.1 m phosphate, 0.16 m NaCl, and 6 mm lactose, pH 7.4. The control solution for the PTX preincubation experiment was the same as the PTX stock solution but lacked the toxin. Experiments were performed 2 weeks after the nerves to both the right and left sides of frog cutaneous pectoris muscles were crushed. One side of the muscle preparation was incubated with 2 μg/ml PTX for 22–24 hr at 4°C in NFR. As a control, the other side from the same animal was incubated in the same solution without PTX under the identical conditions. EPPs and mEPPs were recorded either continuously from the same NMJs or from randomly sampled NMJs. To quantify the effect of nifedipine, 100 EPPs and 50 mEPPs were recorded from randomly sampled NMJs before and after application of nifedipine.
Statistical analyses. Each set of data represents mean ± SEM of an indicated number (n) of experiments or NMJs. Statistical significant difference was evaluated by Student Newman–Keuls test for mean values of multiple groups and by Kolmogorov–Smirnov test for cumulative probability curves (Sokal and Rohlf, 1969).
RESULTS
ω-Conotoxin MVIIC blocks evoked transmitter release at developing rat NMJs
To determine whether developing rat NMJs are similar to mature mammalian NMJs in their use of P/Q-type VSCCs to mediate evoked transmitter release, the effect of ω-conotoxin MVIIC, a P/Q-type VSCC blocker (Hillyard et al., 1992) was examined. As shown in Figure1, synthetic ω-conotoxin MVIIC (SNX-230) inhibited evoked transmitter release at newborn rat NMJs (postnatal day 0). EPPs were recorded from the same muscle fiber in the presence of curare, before (a) and after treatment with 1 μm(b) and 3 μm (c, d) ω-conotoxin MVIIC (Fig. 1). Application of 1 μm ω-conotoxin MVIIC reduced the EPP amplitude to 80%, and 3 μm nearly completely blocked the transmitter release. This result is similar to that in adult mice (Bowersox et al., 1995; Sugiura et al., 1995) and suggests that developing rat NMJs also primarily use P/Q-type VSCC to mediate transmitter release.
Fig. 1.

ω-Conotoxin MVIIC blocks evoked transmitter release at developing rat NMJs. Superimposed EPPs recorded from the same NMJ of a postnatal day 0 (P0) rat. Bath application of P/Q-type VSCC blocker, synthetic ω-conotoxin MVIIC (SNX-230), reduced EPP amplitude to 80% at 1 μm (b) and nearly caused blockade of transmitter release at 3 μm(c, d). Each trace represents an average of 20 EPPs recorded 30 min (b), 45 min (c), and 75 min (d) after application of ω-conotoxin MVIIC from a curare-blocked phrenic nerve–diaphragm muscle preparation. Similar results were obtained in three experiments.
L-type VSCC blockers increase EPP amplitude at developing rat NMJs
To test whether DHP-sensitive L-type VSCCs are involved in synaptic transmission at developing NMJs, we examined the effect of nifedipine, a DHP-antagonist for L-type VSCCs (Miller, 1987), on evoked transmitter release at embryonic rat NMJs. If DHP-sensitive VSCCs directly mediate evoked transmitter release, one would expect a reduction in the amplitude of EPPs by nifedipine. We found, however, that nifedipine enhanced transmitter release at developing NMJs (Fig. 2A). Application of 1, 5, and 10 μm nifedipine increased EPP amplitude at an E17 rat NMJ in a dose-dependent manner. The long duration of the EPP seen in Figure 2A is a typical feature of embryonic NMJs (Diamond and Miledi, 1962; Bennett and Pettigrew, 1974; Dennis et al., 1981) attributable to the small amount of acetylcholine esterase, the large input resistance of smaller muscle fibers, and the different kinetics of embryonic acetylcholine receptors (Vicini and Schuetze, 1985).
Fig. 2.

Various L-type VSCC blockersincrease evoked transmitter release at developing rat NMJs. A, Superimposed EPPs recorded from the same NMJ of an E17 rat. Nifedipine, a DHP-antagonist,increased EPP amplitude to 1.6-fold at 1 μm (b), 2.3-fold at 5 μm(c), and 8.3-fold at 10 μm(d) compared with the control (a) at this NMJ. Each trace represents an average of 20 EPPs recorded 30 (b, c) and 15 min (d) after bath application of each concentration of nifedipine. B, The effect of nifedipine on EPP amplitude at E17–E19 rat NMJs was dose-dependent. The percentage of EPP amplitude was calculated as the ratio of after/before nifedipine application at the same NMJ. The control, 0.05% ethanol, the highest concentration of solvent for nifedipine, had no effect on EPP amplitude. Each error bar represents mean ± SEM of the indicated number of experiments.C, Isradipine (PN200-110), another DHP antagonist, alsoincreased EPP amplitude at developing rat NMJ. This NMJ showed a 1.7-fold (b) and a 2.8-fold (c) enhancement of EPP amplitude 40 min after application of 0.5 μm and 16 min after 1 μm isradipine, respectively. Each trace represents an average of 24 EPPs recorded from the same NMJ in a P0 rat. D, Verapamil, a non-DHP L-type channel blocker, alsoincreased EPP amplitude at developing rat NMJs. This NMJ showed a 1.7-fold (b) and a 3.3-fold (c) enhancement of EPP amplitude 30 min after application of 1 and 3 μm verapamil, respectively. The shortened latency observed after application of 3 μm verapamil (c) may be related to the multiple actions of this drug, such as the alteration of membrane action potentials (Kass and Tsien, 1975). Each trace represents an average of 24 EPPs recorded from the same NMJ in a P2 rat.
The dose-dependence was quantified further by testing different concentrations of nifedipine in embryonic (E17–E19) NMJs (Fig.2B). In each experiment, EPPs were recorded continuously from the same NMJ before and after nifedipine application. The effect of nifedipine was normalized using the ratio of the average EPP amplitude after/before nifedipine application. At 1 μm, nifedipine approximately doubled the size of EPPs. At 10 μm, it significantly increased EPP amplitude to ∼400% (p < 0.01). There was no correlation between EPP size before nifedipine application and the percentage increase after nifedipine application at individual NMJs (data not shown). The control experiments using the vehicle alone (0.05% ethanol) showed no effect on EPPs. Thus, the potentiation of EPPs observed at embryonic NMJs is not an artifact of ethanol.
To confirm that the enhancement of EPPs by nifedipine is attributable to the specific blockade of DHP-sensitive L-type VSCCs, we compared nifedipine with other L-type VSCC blockers using early postnatal rats. Nifedipine (10 μm) increased EPP amplitude to 265 ± 57.9% (n = 13) at P0–P3 rat NMJs. Nimodipine (10 μm), also a DHP-antagonist (McCarthy and Tan-Piengco, 1992), increased EPP amplitude to 195 ± 34.2% (n= 6) at developing rat (P1–P3) NMJs (data not shown). Isradipine (PN200-110), another DHP-antagonist that has a higher affinity to L-type channels than nifedipine (Yaney et al., 1991), showed a more potent effect (Fig. 2C). On average, isradipine enhanced EPP amplitude to 170 ± 35.9% (n = 5) at 0.5 μm and to 284 ± 80.0% (n = 5) at 1 μm at developing rat (P0–P2) NMJs.
Verapamil, a phenylalkylamine, is a different type of organic L-type channel blocker with a binding site distinct from that of DHPs (Yaney et al., 1991; Knaus et al., 1992). Figure 2D shows an example of the enhancement of EPP amplitude by verapamil at a P2 NMJ. Application of verapamil results in an increase of EPPs to 166 ± 19.1% (n = 6) at 1 μm and to 203 ± 31.4% (n = 6) at 3 μm at developing rat (P0–P2) NMJs. Thus, the enhancement of EPP amplitude is most likely attributable to the blockade of L-type VSCCs and is not an anomalous effect of nifedipine.
The effect of nifedipine is age dependent
Nifedipine induced enhancement of evoked transmitter release at NMJs in embryonic and newborn, but not in adult, rats. Figure3A shows a time course of the effect of nifedipine on EPP amplitude in an E17 rat in which EPPs were recorded continuously from the same NMJ. Typically, ∼10-20 min after an application of 5 μl of 20 mm nifedipine into 10 ml of bath solution (final concentration of nifedipine 10 μm), EPP amplitude began to increase, reached a plateau within 30–40 min, and persisted for at least 2 hr. In contrast, nifedipine (10 μm) had no effect on EPPs in a 1-month-old rat (Fig.3B). Thus, the enhancement of evoked transmitter release by nifedipine seems to be a phenomenon unique to developing NMJs.
Fig. 3.
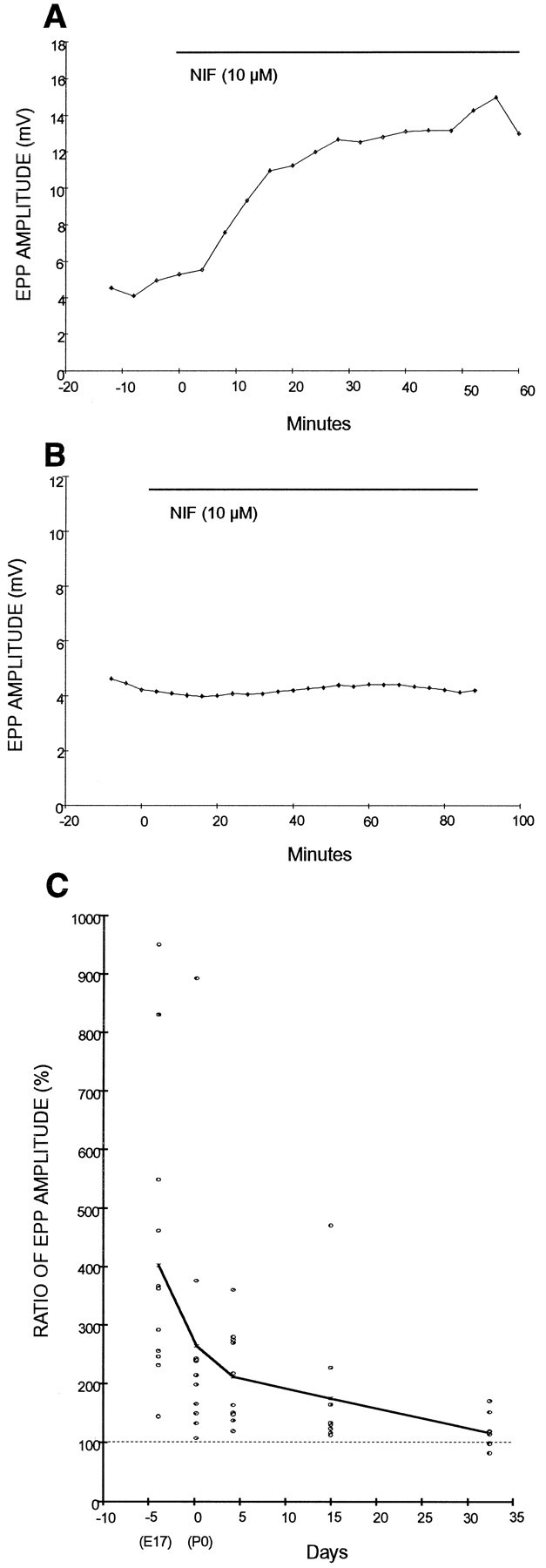
The potentiation effect of nifedipine is age-dependent. A, EPP amplitude before and during application of 10 μm nifedipine (indicated with thebar) in an E18 rat NMJ. Nifedipineincreased evoked transmitter release at immature NMJs. Each point represents an average of 24 EPPs. Similar results were obtained in 12 experiments. B, EPP amplitude before and during application of nifedipine (10 μm) in a 1-month-old rat NMJ. Nifedipine showed no effect at mature NMJs. Similar results were obtained in nine experiments. C, The effect of 10 μmnifedipine on EPP amplitude at developing rat NMJs ranging from E18 to P30: E18 (E18 ± 0.2 d; n = 12), P0 (0.3 ± 0.15 d; n = 12), P4 (4.3 ± 0.22 d; n = 10), 2-week-old (15 ± 0.44 d; n = 10), and 1-month-old (32 ± 2.3 d; n = 9) rats. Each pointrepresents individual data calculated as the ratio of after/before 10 μm nifedipine at the same NMJ. Mean values of each age are shown as X and joined with the solid line. The potentiation effect of nifedipine decreased as development progressed.
To explore the relationship between developmental age and the potentiation effect of nifedipine, we recorded EPP amplitude before and after application of 10 μm nifedipine in rats ranging in age from E18 to 1 month old. As summarized in Figure 3C, the potentiation effect of nifedipine was age-dependent. The largest potentiation by nifedipine was found in embryonic muscle and decreased as development proceeded. The potency of the effect of nifedipine diminished in the first 2 weeks after birth, after which the effect weakened gradually. The results obtained from E18 were significantly different from those from P4 (P < 0.05), P15 (P < 0.05), and 1-month-old (P < 0.01) rats.
Nifedipine increases EPP amplitude at regenerating adult mouse and frog NMJs
It has been shown that many aspects of synapse formation, such as polyneuronal innervation and the characteristics of transmitter release, also occur in the regeneration of adult synapses after injury (Miledi, 1960; McArdle, 1975; DeCino, 1981). Thus, we tested whether nifedipine increased evoked transmitter release at regenerating adult NMJs. Figure 4 shows a similar potentiation effect of nifedipine observed at regenerating mouse (A) and frog (B) NMJs bathed in low Ca2+/high Mg2+ saline. Nifedipine (10 μm) increased EPP amplitude 2.2-fold in regenerating mouse sternomastoid muscle 7 d after nerve crush (Fig.4A-1), but did not affect EPPs in the contralateral control muscle of the same mouse (Fig. 4A-2). The slow time course of the EPP (Fig. 4A-1), typically observed at early stages of reinnervating NMJs, was probably attributable to a delay of restoration in acetylcholine esterase activity (Miledi, 1960; Bennett et al., 1973, 1974). Because mEPPs could not be recorded from developing rat muscle because of the use of curare to prevent spontaneous muscle contraction (Diamond and Miledi, 1962), the effect of nifedipine on spontaneous mEPPs was examined at regenerating NMJs. There was no significant effect on mEPP amplitude and frequency (0.69 mV and 0.126 Hz before and 0.79 mV and 0.171 Hz after nifedipine in this NMJ), suggesting that the potentiation effect of nifedipine was presynaptic. Similar to developing NMJs, the P/Q-type VSCC blocker, synthetic ω-conotoxin MVIIC (SNX-230), also blocked evoked transmitter release at regenerating NMJs in mouse sternomastoid muscle 7 d after nerve crush (data not shown).
Fig. 4.

Nifedipine increases EPP amplitude at regenerating mouse and frog NMJs. Superimposed EPPs recorded from the same NMJ in low Ca2+/high Mg2+ saline. Each trace represents an average of 24 EPPs before and after nifedipine treatment. A, Nifedipine (10 μm) increased EPP amplitude to 1.7-fold 7 d after nerve crush in the mouse sternomastoid (A-1); however, 10 μm nifedipine did not affect EPPs in the contralateral control muscle of the same mouse (A-2). Traces b in bothA-1 and A-2 were recorded 15 min after bath application of nifedipine. B, Nifedipine (10 μm) increased EPP amplitude to 2.8-fold 14 d after nerve crush in frog cutaneous pectoris muscles (B-1), but 10 μm nifedipine did not affect EPPs in the contralateral control muscle of the same frog (B-2). Traces b in bothB-1 and B-2 were recorded 50 min after bath application of nifedipine. C, The effect of nifedipine on EPPs was dependent on the stage of reinnervation. Nifedipine (10 μm) increased EPP amplitude significantly (p < 0.01) to 2.7-fold of control at 2 weeks after nerve- crushed NMJs, but to only 1.6-fold at 6 weeks after nerve crush. The latter was not significantly different from intact NMJs. Each error bar represents mean ± SEM of indicated number of experiments. D, Nifedipine had no effects on mEPPs at regenerating frog NMJs. Comparison of amplitude distribution of mEPPs before (•) and after (X) 10 μmnifedipine treatment. The cumulative probability is defined as the percentage of total events with amplitudes smaller than a given amplitude. The data were obtained from 10 identified NMJs 2 weeks after nerve crush in frog cutaneous pectoris muscles.
The enhancement of EPP amplitude by nifedipine was not unique to mammalian NMJs. It was also demonstrated at regenerating frog NMJs. Nifedipine (10 μm) increased EPP amplitude 2.8-fold 2 weeks after nerve crush in frog cutaneous pectoris muscle (Fig.4B-1). At this reinnervated NMJ, two separate EPPs, which indicate polyneuronal innervation, typical of regenerating (McArdle, 1975) as well as developing NMJs (Redfern, 1970; Letinsky, 1974), were observed. Both were enhanced by nifedipine. The potentiation effect by nifedipine was irreversible at regenerating NMJs; enhanced EPPs still persisted 2 hr after washout of nifedipine (data not shown). In contrast, 10 μm nifedipine did not affect EPPs in the intact contralateral muscle (Fig.4B-2). Similar to normal adult frog NMJs, the N-type channel blocker synthetic ω-conotoxin GVIA (SNX-124) blocked evoked transmitter release at regenerating NMJs (data not shown).
We examined whether the effect of nifedipine on EPPs also depends on the maturation of regenerating NMJs as it did in developing muscles. Regenerating NMJs in frog cutaneous pectoris muscles were examined 2 and 6 weeks after nerve crush. As summarized in Figure 4C, 2 weeks after nerve crush the EPP amplitude was significantly enhanced by 10 μm nifedipine on average to 274 ± 36.7% (p < 0.01; n = 17); however, 6 weeks after nerve crush, muscles showed only a 148 ± 14.3% (n = 16) enhancement, which was not statistically significant when compared with intact controls. Thus, nifedipine was more potent at potentiating EPPs at an earlier stage of regeneration.
Similar to the result we obtained from mouse NMJs, 10 μmnifedipine showed no significant effect on the amplitude and frequency of mEPPs. At the 10 identified NMJs that showed an increase in EPP amplitude with application of nifedipine, the average mEPP amplitude was 0.525 ± 0.066 mV before and 0.480 ± 0.053 mV after nifedipine. The average mEPP frequency was 0.193 ± 0.037 Hz before and 0.210 ± 0.032 Hz after nifedipine (n = 10). As shown in Figure 4D, no significant difference (Kolmogorov–Smirnov test) was observed in the cumulative probability for the amplitude distribution of mEPPs (Van der Kloot, 1991) before and after nifedipine treatments. The absence of effects on the mEPP suggests that the potentiation of EPP amplitude by nifedipine is a presynaptic effect.
BAPTA prevents the effect of nifedipine, but EGTA does not
If nifedipine acts specifically on L-type VSCCs, its potentiation of transmitter release should be attenuated by an intracellular Ca2+ buffer, because the presynaptic Ca2+transient through L-type VSCCs would be reduced. To test this hypothesis, we applied nifedipine after loading regenerating frog NMJs with BAPTA-AM, a membrane-permeant buffer with fast Ca2+-binding kinetics (Adler et al., 1991).
Figure 5A shows an example of the effect of BAPTA-AM on the potentiation effect of nifedipine at regenerating frog NMJs 2 weeks after nerve crush. Application of 25 μmBAPTA-AM decreased EPP amplitude to an average of 35.0 ± 4.8% (n = 9), whereas the vehicle 0.1% DMSO had no effect on EPPs (data not shown). The result is similar to that reported at normal adult frog NMJs (Robitaille et al., 1993) and is consistent with the known effects of Ca2+ buffers on transmitter release (Adler et al., 1991). After BAPTA-AM had its full effect on EPPs, application of 10 μm nifedipine did not enhance the EPP amplitude (n = 7). The failure of nifedipine to increase transmitter release in the presence of intracellular Ca2+ buffer suggests that the effect of nifedipine is related to the influx of Ca2+ through L-type VSCCs at the regenerating motor nerve terminal.
Fig. 5.

BAPTA prevents the potentiation effect of nifedipine, but EGTA does not. The changes of EPP amplitude before and during application of drugs (indicated with the bars) at regenerating frog NMJs 2 weeks after nerve crush. A, BAPTA-AM reduced EPP amplitude and prevented any potentiation by subsequent application of nifedipine. B, EGTA-AM reduced EPP amplitude but did not prevent potentiation of EPPs by subsequent application of nifedipine. Similar results were obtained in seven experiments treated with BAPTA-AM and in eight treated with EGTA-AM. Each point represents an average of 24 EPPs.
We also used EGTA-AM, a Ca2+ buffer with slow Ca2+ binding kinetics and with a Ca2+ affinity similar to that of BAPTA-AM (Adler et al., 1991), to confirm the involvement of a Ca2+ transient. As shown in Figure5B, loading of 25 μm EGTA-AM decreased evoked transmitter release at regenerating frog NMJs 2 weeks after nerve crush. An average of eight experiments showed a decrease to 56.8 ± 5.9% in EPP amplitude. The result is similar to the effect of EGTA-AM reported at normal adult frog NMJs (Robitaille et al., 1993). In contrast to the result of BAPTA-AM pretreatment, 10 μmnifedipine enhanced EPP amplitude to an average of 172.8 ± 19.8% (n = 8), even after EGTA-AM had it full effect on release (compare Fig. 5, A and B). Thus, BAPTA-AM but not EGTA-AM can nullify the potentiation effect by nifedipine at immature NMJs. These observations suggest that BAPTA-AM binds Ca2+ fast enough to intercept Ca2+ before it can activate an unknown modulatory mechanism. The results suggest that L-type VSCCs are closer to this unknown modulatory site than to the exocytotic site during synapse formation.
Are L-type channels coupling with Ca2+-activated K+ channels (gKCa) at immature NMJs?
To explore possible mechanisms by which L-type VSCCs modulate evoked transmitter release at immature NMJs, we examined the involvement of gKCa. It has been shown that gKCa are colocalized with VSCCs at the active zone and modulate transmitter release, probably by shortening the duration of the presynaptic action potential at normal frog NMJs (Robitaille and Charl-ton, 1992; Robitaille et al., 1993). Ca2+ entry through L-type VSCCs at immature motor nerve terminal that activated nearby gKCa (thereby shortening action potential duration and reducing transmitter release) could explain the potentiation of EPP amplitude by L-type VSCC blockers observed in the present study.
To test the hypothesis that L-type VSCCs modulate transmitter release by activating gKCa, we examined the effect of nifedipine in the presence of a gKCa blocker, charybdotoxin (Miller et al., 1985) or iberiotoxin (Galvez et al., 1990), at frog NMJs 2 weeks after nerve crush. If gKCa are colocalized with the N-type VSCCs that directly mediate transmitter release at regenerating frog NMJs, the enhancement of the EPP amplitude by a gKCa blocker would be expected, as seen in normal adult frog (Robitaille and Charlton, 1992). If Ca2+ entry through L-type VSCCs also activates nearby gKCa, nifedipine treatment should not show additional enhancement in EPP size after the potentiation by a gKCa blocker.
Similar to results in previous work (Robitaille et al., 1993), 20 nm iberiotoxin, which gave a saturated effect on transmitter release (data not shown), increased EPP amplitude to 279 ± 43% at normal frog NMJs (n = 9). In contrast, only two of eight regenerating NMJs showed potentiation by iberiotoxin. As shown in an example in Figure6A, EPP amplitude increased 2.6-fold in the presence of 20 nm iberiotoxin at a regenerating NMJ. Further enhancement of EPP amplitude (5.4-fold) was observed when 10 μm nifedipine was added to this iberiotoxin-pretreated NMJ. Similar results were obtained at regenerating NMJs pretreated with charybdotoxin (n = 4). The additive effect by gKCa and nifedipine suggests that the potentiation by nifedipine does not involve gKCa at immature motor nerve terminals.
Fig. 6.
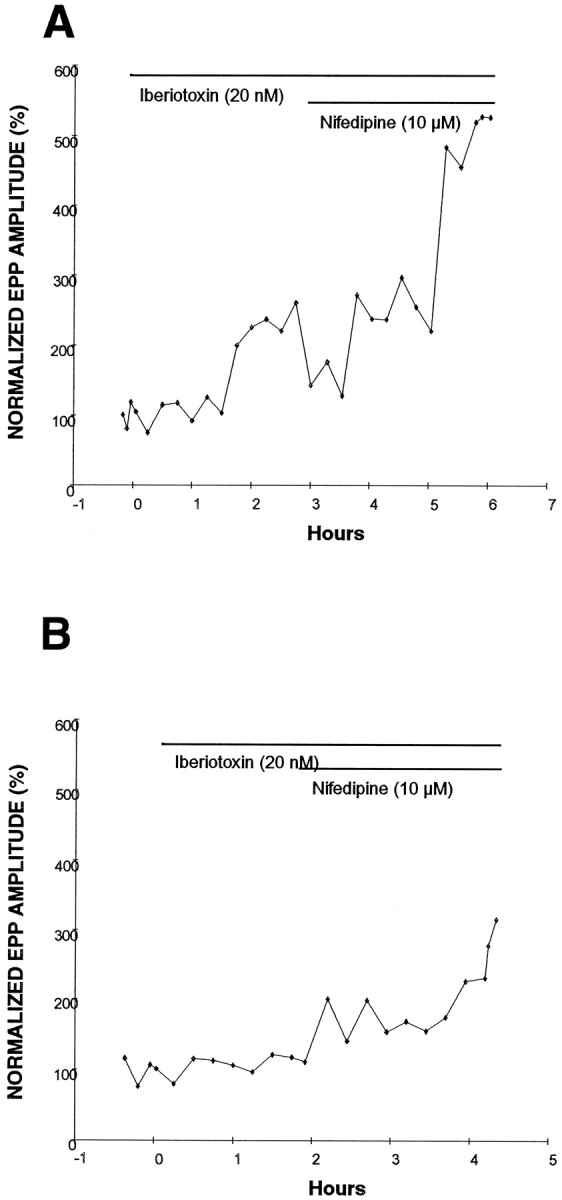
Ca2+-activated K+ channel (gKCa) blocker pretreatment does not prevent the potentiation effect of nifedipine. A, Iberiotoxin (20 nm) increased EPP amplitude (before, 0.6 mV; after, 1.5 mV) at a regenerating frog NMJ 2 weeks after nerve crush. After beriotoxin, nifedipine further increased EPP amplitude (3.1 mV) at this NMJ.B, Iberiotoxin did not increase EPP amplitude (1.0 mV) at this regenerating (2 weeks after nerve crush) frog NMJ. In the presence of iberiotoxin, this NMJ showed an enhancement of EPP amplitude to threefold after application of 10 μmnifedipine. Each point represents an average of 24 EPPs. For comparison, EPP sizes were normalized.
The lack of involvement of gKCa was supported further by an experiment in which iberiotoxin treatment did not show any potentiation at regenerating NMJs (Fig. 6B). Even at these NMJs, nifedipine enhanced EPP amplitude to 275 ± 25.4% (n = 3). The lack of effects by iberiotoxin may indicate that gKCa have not yet appeared at these regenerating NMJs. The potentiation of EPP amplitude by nifedipine, regardless of the presence or absence of gKCa, suggests that gKCa is probably not involved in this modulation of transmitter release at immature NMJs.
L-type VSCCs may involve an inhibitory mechanism linked to a G-protein at immature NMJs
An alternative mechanism for the modulation of transmitter release by L-type VSCCs may involve GTP-binding (G-) proteins. It has been shown that activating trimeric G-proteins by neuromodulators inhibits VSCCs that directly mediate transmitter release in various types of synapses (Tsien et al., 1988; Dolphin, 1995). If G-protein-mediated inhibition of VSCCs also operated at immature NMJs, it could explain the potentiation of EPP amplitude by L-type VSCC blockers. It has been shown that L-type VSCCs are involved in the release of neuropeptides in the hypothalamic neurosecretory cells (Cazalis et al., 1987; Lemos and Norwycky, 1989) and in dorsal root ganglion neurons (Perney et al., 1986; Rane et al., 1987; Holz et al., 1988). Blocking L-type VSCCs may prevent the release of a neuropeptide acting as a neuromodulator that normally activates a G-protein (Dolphin, 1996). The blockade of L-type VSCCs may thus reduce the G-protein-mediated inhibition of VSCCs and thereby lead to an increase in transmitter release.
To test whether L-type VSCCs involve a G-protein-mediated inhibition of transmitter release at immature NMJs, we studied the effect of PTX, which inhibits certain types of G-proteins (presumably Giand Go) (Reisine and Law, 1992), on the enhanced transmitter release by nifedipine. Overnight incubation of PTX was used for the internalization of PTX and inactivation of G-proteins (Reisine and Law, 1992). As shown in Figure 7, PTX pretreatment (2 μg/ml, 22 hr) prevents the enhancement of EPPs by nifedipine at frog NMJs 2 weeks after nerve crush. There was no significant difference in average EPP amplitude recorded before and after nifedipine treatment in randomly sampled NMJs (n = 26) incubated overnight with PTX in three reinnervated muscles. In contrast, nifedipine increased EPP amplitude 158% on average at regenerating frog NMJs (n = 25) in three control muscles incubated overnight without PTX. Thus, the absence of potentiation by nifedipine in PTX-preincubated regenerating NMJs is not simply attributable to a nonspecific deterioration of synaptic transmission caused by overnight incubation. Rather, the result suggests that inactivation of PTX-sensitive G-proteins prevents the potentiation of EPPs by nifedipine.
Fig. 7.

Pertussis toxin (PTX), a G-protein inhibitor, prevents the potentiation effect of nifedipine at regenerating frog NMJs 2 weeks after nerve crush. EPPs were recorded from randomly sampled NMJs in muscles preincubated with PTX or without PTX (Control). Control muscles showed a 1.6-fold enhancement of average EPP amplitude after exposure to nifedipine (p < 0.05). In contrast, nifedipine did not increase average EPP amplitude in PTX preincubated muscles. Each error bar represents mean ± SEM of the indicated number of NMJs from three independent experiments.
It was also noted that PTX pretreatment itself increased EPP amplitude, as indicated by comparing the EPP amplitude in reinnervated muscles treated with and without PTX (stippled bars in Fig. 7). The average EPP amplitude, 4.05 + 0.43 mV (n = 26) in PTX-pretreated muscles, was significantly larger (p < 0.01) than that in control (2.01 + 0.26 mV, n = 25); however, PTX treatment did not affect the amplitude (0.432 ± 0.028 mV, control; 0.478 ± 0.030 mV, PTX treated) and frequency (0.081 ± 0.016 Hz, control; 0.074 ± 0.011 Hz, PTX treated) of spontaneous mEPPs. Thus, the increase in the EPP amplitude by PTX is a presynaptic rather than a postsynaptic effect at regenerating frog NMJs.
DISCUSSION
The present study has demonstrated that there are at least two types of VSCCs involved in the evoked release of transmitter at newly formed NMJs. Similar to mature NMJs, one type of VSCC (N-type for frogs and P/Q-type for mammals) directly mediates evoked transmitter release at immature NMJs; however, an additional type of VSCC, L-type, also regulates transmitter release at developing and regenerating NMJs. The observation of potentiation, instead of inhibition, of EPP amplitude after blocking L-type VSCCs suggests that L-type VSCCs do not directly mediate evoked transmitter release at immature NMJs. Rather, L-type VSCCs may be linked to a modulatory mechanism for evoked transmitter release during synaptogenesis. As shown in a model (Fig.8) based on the present data, we hypothesize that Ca2+ entry through L-type VSCCs may trigger the release of a neuromodulator. This may in turn bind to a receptor that activates a PTX-sensitive G-protein that inhibits the release of acetylcholine at newly formed NMJs. The blockade of L-type VSCCs with nifedipine would prevent this G-protein-mediated inhibition and thus result in potentiation of EPPs during synapse maturation. We speculate that L-type VSCCs may play a role in self-limiting the amount of evoked transmitter release during the development and regeneration of NMJs.
Fig. 8.

A proposed model to explain the possible mechanism for the modulatory role of L-type VSCCs at newly formed motor nerve terminals. A transient of Ca2+ through L-type VSCCs may be responsible for the release of an unknown neuropeptide or neuromodulator, which activates a presynaptic receptor coupled to a PTX-sensitive G-protein. The activation of the G-protein inhibits N- or P/Q-type VSCCs and results in a reduction of transmitter release at immature NMJs. Blocking L-type VSCCs may prevent the release of the neuromodulator and result in the removal of an inhibition of transmitter release.
Specificity of L-type VSCC blockers
Cautious interpretation is warranted in identification of the subtypes of VSCCs at NMJs based solely on pharmacological characterization. For example, the specificity of nifedipine at concentrations over 10 μm as an L-type VSCC antagonist is questionable (Hagiwara and Byerly, 1981). We have found that nifedipine, even at 1 μm, enhances EPP amplitude but has no effect on the mEPP amplitude at immature NMJs. Thus, the potentiation of EPPs by nifedipine is a presynaptic rather than postsynaptic effect. In addition, although nimodipine showed similar potentiation, isradipine was approximately tenfold more potent than nifedipine, consistent with the higher binding affinities of isradipine for L-type VSCCs (Yaney et al., 1991). Furthermore, verapamil, a non-DHP antagonist, also showed an enhanced effect on EPPs. These results suggest that the potentiation is not an anomalous effect by nifedipine, but is attributable to a specific antagonistic effect of blocking L-type VSCCs.
The potentiation induced by L-type VSCC blocker requires Ca2+ transients
The specific effect of nifedipine was supported by the experiments using Ca2+ buffers. We have shown that the fast buffer BAPTA-AM, but not the slow buffer EGTA-AM, prevents the potentiation of EPPs by nifedipine. The result suggests that the nifedipine effect requires a transient rise in intracellular Ca2+. Furthermore, the target of the Ca2+ transient may be located close to the L-type VSCCs. The entry of Ca2+ would reach a nearby target before Ca2+ can be buffered by EGTA-AM. Because L-type VSCCs do not directly mediate transmitter release, they may be located some distance from the active zone where N- or P/Q-type VSCCs are clustered. This is consistent with the idea that Ca2+ signaling may be controlled by multiple VSCC subtypes, which are distributed into distinct subcellular regions and generate localized changes in ion concentrations for different roles in synaptic functions (Miller, 1987).
L-type VSCC and gKCa at immature NMJs
To examine the mechanism of synaptic modulation by L-type VSCCs, we first tested the involvement of gKCa at the regenerating frog NMJ. Fossier et al. (1993) have indicated that Ca2+ flowing through L-type VSCCs modulates the duration of the presynaptic action potential by controlling the gKCa current in Aplysia buccal ganglion. In addition, gKCa are colocalized with N-type VSCCs that mediate transmitter release at the active zone of the normal frog NMJ (Robitaille and Charlton, 1992; Robitaille et al., 1993). The present study showing the potentiation of EPP sizes by gKCa also suggests that gKCa are colocalized with N-type VSCCs at some regenerating frog NMJs.
If gKCa are also colocalized with L-type VSCCs, blocking gKCa should prevent the potentiation of EPPs by subsequent treatment with nifedipine; however, the present study showed additive potentiation by nifedipine after the enhancement of EPPs by gKCa blocker treatment at regenerating frog NMJs. Furthermore, nifedipine increased EPP amplitudes even at some regenerating NMJs that did not show potentiation by gKCa blockers. These results suggest that the potentiation by nifedipine does not require gKCa, and thus it is likely not the only mechanism to explain the modulation of transmitter release by L-type VSCCs at immature NMJs.
L-type VSCCs and G-protein-mediated inhibition of transmitter release
To explain the potentiation of EPP amplitude by nifedipine, we also tested an alternative mechanism involving G-protein-mediated inhibition of transmitter release. Gray et al. (1990) reported that DHP-sensitive VSCCs mediate secretion of somatostatin, which inhibits acetylcholine release via a PTX-sensitive G-protein pathway at the nerve terminal of avian choroid neurons. Our results are consistent with this idea and a proposed model (Fig. 8) that Ca2+entry through L-type VSCCs may trigger the release of a neuromodulator that in turn results in a G-protein-mediated inhibition of transmitter release. The enhancement of EPPs by PTX suggests that a G-protein-mediated inhibition of acetylcholine release also operates in the regenerating frog motor nerve terminal. In addition, we have found no potentiation of transmitter release by nifedipine in muscles pretreated with PTX. This lack of additive potentiation by nifedipine supports the idea that L-type VSCCs are serially linked to G-protein-mediated inhibition (Fig. 8); however, we cannot exclude other interpretations on the lack of additive potentiation. For example, L-type VSCC modulation may involve a cascade of intracellular events that run parallel to G-protein-mediated inhibition. These two processes may converge downstream on a common step that influences evoked transmitter release. If this common step can be fully activated by either nifedipine or PTX alone, an additive effect would not be expected.
It is not known how L-type VSCCs might be coupled with PTX-sensitive G-proteins at immature NMJs. Entry of Ca2+ through L-type VSCCs triggers the release of neuropeptides in various neural tissues. A neuropeptide, calcitonin gene-related peptide (CGRP), located in the large dense-core vesicles of the motor nerve terminal (Matteoli et al., 1988), potentiates the postsynaptic response at developing NMJs in culture (Lu et al., 1993). It is unknown, however, whether CGRP has any effect on evoked transmitter release at immature NMJs.
Purines are known to act as neural signaling substances in various systems and have receptors that are often coupled to G-proteins (Zimmermann, 1994). Thus, purines may be involved in the L-type VSCC modulation. In the hippocampus, nifedipine and nimodipine enhance transmission at the CA1 pyramidal neuron synapse, probably through the suppression of the release of adenosine (O’Regan et al., 1991). Exogenous ATP and adenosine inhibit acetylcholine release at mature NMJs (Ginsborg and Hirst, 1972; Ribeiro and Walker, 1975). In contrast, ATP potentiates spontaneous transmitter release at developing NMJs in culture (Fu and Poo, 1991), and this potentiation can be prevented by nifedipine (Fu and Huang, 1994). In the normal frog muscle, L-type VSCCs are found at perisynaptic Schwann cells, which possess ATP and adenosine receptors (Robitaille, 1995; Robitaille et al., 1996); however, whether the release of ATP is related to L-type VSCCs at developing and regenerating NMJs in vivo remains to be investigated.
Developmental changes in VSCC subtypes
In contrast to the switching of VSCC subtypes mediating transmitter release that is seen in terminals of the chick choroid neuron during development (Gray et al., 1992), we found that the immature NMJs use an additional subtype, L-type, to modulate evoked transmitter release. The observed potentiating effect of nifedipine decreases as developing and regenerating NMJs mature, with a time course (Fig. 3C) reminiscent of synapse elimination (Redfern, 1970; Bennett and Pettigrew, 1974; Betz et al., 1979; Dennis et al., 1981). It remains to be investigated whether these two phenomena are related.
Our results are consistent with previous studies that found no effects on evoked transmitter release by nifedipine at normal adult NMJs (Atchison, 1989; Pancrazio et al., 1989); however, these studies also showed that Bay K 8644, a DHP agonist, increased evoked transmitter release at normal adult NMJs. L-type VSCCs may exist at normal adult NMJs but play no role in transmitter release under physiological conditions (Miller, 1987).
A possible role of L-type VSCCs in immature nerve terminals may be related to the paucity of transmitter available. During synaptogenesis, the immature synapse is more vulnerable to depression after repetitive stimulation, probably because the amount of transmitter is smaller than that found in mature synapses (Dennis et al., 1981). The proposed involvement of L-type VSCCs in G-protein-mediated modulation of transmitter release may provide a self-limiting mechanism to conserve the amount of transmitter release at immature NMJs. Without this negative feedback modulation, the amount of transmitter might be depleted quickly from the immature motor nerve terminal, and the developing synapses might not function reliably. Whether the expression of L-type VSCCs plays a role in the stability and maturation of synaptic connections remains to be investigated.
Footnotes
This work was supported by National Institutes of Health Grant NS 30051. We thank Drs. S. H. Astrow, L. Byerly, G. Miljanich, and M.-M. Poo for critical comments on the manuscript, C. Li and R. Tolentino for technical assistance, and Dr. Y. Hayashi for help at the early stages of this project. Conopeptides were kindly supplied by Dr. G. Miljanich of Neurex, and isradipine (PN200-110) was provided by Research Biochemicals Internationals as part of the Chemical Synthesis Program of the National Institute of Mental Health, Contract N01MH30003.
Correspondence should be addressed to Dr. Chien-Ping Ko at the above address.
REFERENCES
- 1.Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atchison WD. Dihydropyridine-sensitive and -insensitive components of acetylcholine release from rat motor nerve terminals. J Pharmacol Exp Ther. 1989;251:672–678. [PubMed] [Google Scholar]
- 3.Augustine GJ, Charlton MP, Smith SJ. Calcium action in synaptic transmitter release. Annu Rev Neurosci. 1987;10:633–693. doi: 10.1146/annurev.ne.10.030187.003221. [DOI] [PubMed] [Google Scholar]
- 4.Bennett MR, Pettigrew AG. The formation of synapses in striated muscle during development. J Physiol (Lond) 1974;241:515–545. doi: 10.1113/jphysiol.1974.sp010670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett MR, McLachlan EM, Taylor RS. The formation of synapses in reinnervated mammalian striated muscle. J Physiol (Lond) 1973;233:481–500. doi: 10.1113/jphysiol.1973.sp010319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett MR, Florin T, Woog R. The formation of synapses in regenerating mammalian striated muscle. J Physiol (Lond) 1974;238:79–92. doi: 10.1113/jphysiol.1974.sp010511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Betz WJ, Caldwell JH, Ribchester RR. The size of motor units during postnatal development of rat lumbrical muscle. J Physiol (Lond) 1979;297:463–478. doi: 10.1113/jphysiol.1979.sp013051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowersox SS, Miljanich GP, Sugiura Y, Li C, Nadasdi L, Hoffman BB, Ramachandran J, Ko CP. Differential blockade of voltage-sensitive calcium channels at the mouse neuromuscular junction by novel omega-conopeptides and omega-agatoxin-IVA. J Pharmacol Exp Ther. 1995;273:248–256. [PubMed] [Google Scholar]
- 9.Brehm P. Resolving the structural basis for developmental changes in muscle ACh receptor function: it takes nerve. Trends Neurosci. 1989;12:174–176. doi: 10.1016/0166-2236(89)90064-7. [DOI] [PubMed] [Google Scholar]
- 10.Cazalis M, Dayanithi G, Nordmann JJ. Hormone-release from isolated nerve endings of the rat neurohypophysis. J Physiol (Lond) 1987;390:55–70. doi: 10.1113/jphysiol.1987.sp016686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeCino P. Transmitter release properties along regenerated nerve processes at the frog neuromuscular junction. J Neurosci. 1981;1:308–317. doi: 10.1523/JNEUROSCI.01-03-00308.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dennis MJ, Ziskind-Conhaim L, Harris AJ. Development of neuromuscular junctions in rat embryos. Dev Biol. 1981;81:266–279. doi: 10.1016/0012-1606(81)90290-6. [DOI] [PubMed] [Google Scholar]
- 13.Diamond J, Miledi R. A study of foetal and new-born rat muscle fibres. J Physiol (Lond) 1962;162:393–408. doi: 10.1113/jphysiol.1962.sp006941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dolphin AC. Voltage-dependent calcium channels and their modulation by neurotransmitters and G proteins. Exp Physiol. 1995;80:1–36. doi: 10.1113/expphysiol.1995.sp003825. [DOI] [PubMed] [Google Scholar]
- 15.Dolphin AC. Facilitation of Ca2+ current in excitable cells. Trends Neurosci. 1996;19:35–43. doi: 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- 16.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 17.Fossier P, Baux G, Tauc L. Role of different types of Ca2+ channels and a reticulum-like Ca2+ pump in neurotransmitter release. J Physiol (Paris) 1993;87:3–14. doi: 10.1016/0928-4257(93)90019-p. [DOI] [PubMed] [Google Scholar]
- 18.Fu W-M, Huang F-L. L-type Ca channel is involved in the regulation of spontaneous transmitter release at developing neuromuscular synapses. Neuroscience. 1994;58:131–140. doi: 10.1016/0306-4522(94)90160-0. [DOI] [PubMed] [Google Scholar]
- 19.Fu W-M, Poo M-M. ATP potentiates spontaneous transmitter release at developing neuromuscular synapses. Neuron. 1991;6:837–843. doi: 10.1016/0896-6273(91)90179-4. [DOI] [PubMed] [Google Scholar]
- 20.Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom on the scorpion Buthus tamulus. J Biol Chem. 1990;265:11083–11090. [PubMed] [Google Scholar]
- 21.Ginsborg BL, Hirst GDS. The effect of adenosine on the release of the transmitter from the phrenic nerve of the rat. J Physiol (Lond) 1972;224:629–645. doi: 10.1113/jphysiol.1972.sp009916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gray DB, Zelezny D, Manthay N, Pilar G. Endogenous modulation of ACh release by somatostatin and the differential roles of Ca2+ channels. J Neurosci. 1990;10:2687–2698. doi: 10.1523/JNEUROSCI.10-08-02687.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gray DB, Brusés JL, Pilar GR. Developmental switch in the pharmacology of Ca2+ channels coupled to acetylcholine release. Neuron. 1992;8:715–724. doi: 10.1016/0896-6273(92)90092-r. [DOI] [PubMed] [Google Scholar]
- 24.Hagiwara S, Byerly L. Calcium channel. Annu Rev Neurosci. 1981;4:69–125. doi: 10.1146/annurev.ne.04.030181.000441. [DOI] [PubMed] [Google Scholar]
- 25.Hille B. Ionic channels of excitable membranes. Sinauer; Sunderland, MA: 1992. [Google Scholar]
- 26.Hillyard DR, Monje VD, Mintz IM, Bean BP, Nadasdi L, Ramachandran J, Miljanich G, Azimi ZA, McIntosh JM, Cruz LJ, Imperial JS, Olivera BM. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron. 1992;9:69–77. doi: 10.1016/0896-6273(92)90221-x. [DOI] [PubMed] [Google Scholar]
- 27.Holz GG, Dunlap K, Kream RM. Characterization of the electrically evoked release of substance P from dorsal root ganglion neurons: methods and dihydropyridine sensitivity. J Neurosci. 1988;8:463–471. doi: 10.1523/JNEUROSCI.08-02-00463.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kass RS, Tsien RW. Multiple effects of calcium antagonists on plateau currents in cardiac Purkinje fibers. J Gen Physiol. 1975;66:169–192. doi: 10.1085/jgp.66.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katz B. The release of neural transmitter substances. Liverpool UP; Liverpool, England: 1969. [Google Scholar]
- 30.Kerr LM, Yoshikami D. A venom peptide with a novel presynaptic blocking action. Nature. 1984;308:282–284. doi: 10.1038/308282a0. [DOI] [PubMed] [Google Scholar]
- 31.Knaus H-G, Moshammer T, Kang HC, Haugland RP, Glossmann H. A unique fluorescent phenylalkylamine probe for L-type Ca2+ channels. J Biol Chem. 1992;267:2179–2189. [PubMed] [Google Scholar]
- 32.Letinsky MS. The development of nerve-muscle junctions in Rana catesbeiana tadpoles. Dev Biol. 1974;40:129–153. doi: 10.1016/0012-1606(74)90114-6. [DOI] [PubMed] [Google Scholar]
- 33.Lemos JR, Nowycky MC. Two types of calcium channels coexist in peptide-releasing vertebrate nerve terminals. Neuron. 1989;2:1419–1426. doi: 10.1016/0896-6273(89)90187-6. [DOI] [PubMed] [Google Scholar]
- 34.Lu B, Fu W-M, Greengard P, Poo M-M. Calcitonin gene-related peptide potentiates synaptic responses at developing neuromuscular junction. Nature. 1993;363:76–79. doi: 10.1038/363076a0. [DOI] [PubMed] [Google Scholar]
- 35.Matteoli M, Haimann C, Torri-Tarelli F, Polak JM, Ceccarelli B, Camilli PD. Differential effect of α-latrotoxin on exocytosis from small synaptic vesicles and from large dense-core vesicles containing calcitonin gene-related peptide at the frog neuromuscular junction. Proc Natl Acad Sci USA. 1988;85:7366–7370. doi: 10.1073/pnas.85.19.7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McArdle JJ. Complex end-plate potentials at the regenerating neuromuscular junction of the rat. Exp Neurol. 1975;49:629–638. doi: 10.1016/0014-4886(75)90048-5. [DOI] [PubMed] [Google Scholar]
- 37.McCarthy RT, TanPiengco PE. Multiple types of high-threshold calcium channels in rabbit sensory neurons: high-affinity block of neuronal L-type by nimodipine. J Neurosci. 1992;12:2225–2234. doi: 10.1523/JNEUROSCI.12-06-02225.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCobb DP, Best PM, Beam KG. Development alters the expression of calcium currents in chick limb motoneurons. Neuron. 1989;2:1633–1643. doi: 10.1016/0896-6273(89)90052-4. [DOI] [PubMed] [Google Scholar]
- 39.Miller C, Moczydlowski E, Latorre R, Phillips M. Charybdotoxin, a protein inhibitor of single Ca2+-activated K+ channels from mammalian skeletal muscle. Nature. 1985;313:316–318. doi: 10.1038/313316a0. [DOI] [PubMed] [Google Scholar]
- 40.Miller RJ. Multiple calcium channels and neuronal function. Science. 1987;235:46–52. doi: 10.1126/science.2432656. [DOI] [PubMed] [Google Scholar]
- 41.Miledi R. Properties of regenerating neuromuscular synapses in the frog. J Physiol (Lond) 1960;154:190–205. doi: 10.1113/jphysiol.1960.sp006573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Dowd DK, Ribera AB, Spitzer NC. Development of voltage-dependent calcium, sodium, and potassium currents in Xenopus spinal neurons. J Neurosci. 1988;8:792–805. doi: 10.1523/JNEUROSCI.08-03-00792.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olivera BM, Miljanich G, Ramachandran J, Adams ME. Calcium channel diversity and neurotransmitter release: the ω-conotoxins and ω-agatoxins. Annu Rev Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- 44.O’Regan MH, Kocsis JD, Waxman SG. Nimodipine and nifedipine enhance transmission at the Schaffer collateral CA1 pyramidal neuron synapse. Exp Brain Res. 1991;84:224–228. doi: 10.1007/BF00231778. [DOI] [PubMed] [Google Scholar]
- 45.Pancrazio JJ, Viglione MP, Kim YI. Effects of Bay K 8644 on spontaneous and evoked transmitter release at the mouse neuromuscular junction. Neuroscience. 1989;30:215–221. doi: 10.1016/0306-4522(89)90366-7. [DOI] [PubMed] [Google Scholar]
- 46.Perney TM, Hirning SE, Leeman SE, Miller RJ. Dihydropyridine effects on release of substance P. Proc Natl Acad Sci USA. 1986;83:6656–6660. doi: 10.1073/pnas.83.17.6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Protti DA, Uchitel OD. Transmitter release and presynaptic Ca currents blocked by the spider toxin omega-Aga-IVA. NeuroReport. 1993;5:333–336. doi: 10.1097/00001756-199312000-00039. [DOI] [PubMed] [Google Scholar]
- 48.Rane SG, Holz GG, Dunlap K (1987) Dihydropyridine inhibition of neuronal calcium current and substance P release. Pflügers Arch. 409:361–366. [DOI] [PMC free article] [PubMed]
- 49.Redfern PA. Neuromuscular transmission in new-born rats. J Physiol (Lond) 1970;209:701–709. doi: 10.1113/jphysiol.1970.sp009187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reisine T, Law SF. Pertussis toxin in analysis of receptor mechanisms. In: Conn PM, editor. Neurotoxins, method in neurosciences, Vol 8. Academic; San Diego: 1992. pp. 358–367. [Google Scholar]
- 51.Ribeiro JA, Walker J. The effect of adenosine triphosphate and adenosine diphosphate on transmission at the rat and frog neuromuscular junctions. Br J Pharmacol. 1975;54:213–218. doi: 10.1111/j.1476-5381.1975.tb06931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robitaille R. Purinergic receptors and their activation by endogenous purines at perisynaptic glial cells of the frog neuromuscular junction. J Neurosci. 1995;15:7121–7131. doi: 10.1523/JNEUROSCI.15-11-07121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J Neurosci. 1992;12:297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- 55.Robitaille R, Bourque M-J, Vandaele S. Localization of L-type Ca channels at presynaptic glial cells of the frog neuromuscular junction. J Neurosci. 1996;16:148–158. doi: 10.1523/JNEUROSCI.16-01-00148.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sokal RR, Rohlf FJ. Biometry. W. H. Freeman; San Francisco: 1962. [Google Scholar]
- 57. Spitzer NC. Development of voltage-dependent and ligand-gated channels in excitable membranes. Progress in brain research Van Pelt J, Corner MA, Uylings HBM, de Silva FHL. 1994. 169 179 Elsevier; Amsterdam: Science BV. [DOI] [PubMed] [Google Scholar]
- 58.Steinbach JH. Structural and functional diversity in vertebrate skeletal muscle nicotinic acetylcholine receptors. Annu Rev Biol. 1989;51:353–365. doi: 10.1146/annurev.ph.51.030189.002033. [DOI] [PubMed] [Google Scholar]
- 59.Sugiura Y, Ko CP. L-type calcium channels modulate evoked transmitter release at newly formed neuromuscular junctions. Soc Neurosci Abstr. 1995;21:1571. doi: 10.1523/JNEUROSCI.17-03-01101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sugiura Y, Woppmann A, Miljanich GP, Ko CP. A novel omega-conopeptide for the presynaptic localization of calcium channels at the mammalian neuromuscular junction. J Neurocytol. 1995;24:15–27. doi: 10.1007/BF01370157. [DOI] [PubMed] [Google Scholar]
- 61.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 62.Uchitel OD, Protti DA, Sanchez V, Cherksey BD, Sugimori M, Llinas R. P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc Natl Acad Sci USA. 1992;89:3330–3333. doi: 10.1073/pnas.89.8.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van der Kloot W. The regulation of quantal size. Prog Neurobiol. 1991;36:93–130. doi: 10.1016/0301-0082(91)90019-w. [DOI] [PubMed] [Google Scholar]
- 64.Vicini S, Schuetze SM. Gating properties of acetylcholine receptors at developing rat endplates. J Neurosci. 1985;5:2212–2224. doi: 10.1523/JNEUROSCI.05-08-02212.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yaari Y, Hamon B, Lux HD. Development of two types of calcium channels in cultured mammalian hippocampal neurons. Science. 1987;235:680–682. doi: 10.1126/science.2433765. [DOI] [PubMed] [Google Scholar]
- 66.Yaney GC, Stafford GA, Henstenberg JD, Sharp GWG, Weiland GA. Binding of the dihydropyridine calcium channel blocker (+)-[3H]isopropyl-4-(2,1,3-benzoxadiazol-4-yl)-1,4-dihydro-5-methoxycarbonyl-2,6-dimethyl-3-pyridinecarboxylate (PN200-110) to RINm5F membranes and cells: characterization and functional significance. J Pharmacol Exp Ther. 1991;258:652–662. [PubMed] [Google Scholar]
- 67.Zhang J-F, Randall AD, Ellinor PT, Horne WA, Sather WA, Tanabe T, Schwarz TL, Tsien RW. Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology. 1993;32:1075–1088. doi: 10.1016/0028-3908(93)90003-l. [DOI] [PubMed] [Google Scholar]
- 68.Zimmermann H. Signalling via ATP in the nervous system. Trends Neurosci. 1994;17:420–426. doi: 10.1016/0166-2236(94)90016-7. [DOI] [PubMed] [Google Scholar]