

Abstract
Transgenic (NFHLacZ) mice expressing a fusion protein composed of a truncated high-molecular-weight mouse neurofilament (NF) protein (NFH) fused to β-galactosidase (LacZ) develop inclusions in neurons throughout the CNS. These inclusions persist from birth to advanced age and contain massive filamentous aggregates including all three endogenous NF proteins and the NFHLacZ fusion protein. Further, the levels of endogenous NF proteins are selectively reduced in NFHLacZ mice. Because these inclusions resemble NF-rich Lewy bodies (LBs) in Parkinson’s disease and LB dementia, we asked whether these lesions compromised the viability of affected neurons during aging. We studied hippocampal CA1 neurons, nearly all of which harbored inclusions (type I) devoid of cellular organelles, and cerebellar Purkinje cells, nearly all of which accumulated inclusions (type II) containing numerous entrapped organelles. Purkinje cells with type II inclusions began to degenerate in the NFHLacZ mice at ∼1 year of age, and most were eliminated by 18 months of age. In contrast, there was no significant loss of type I inclusion-bearing CA1 neurons with age. These data suggest that the sequestration of cellular organelles in type II inclusions may isolate and impair the function of these organelles, thereby rendering Purkinje cells selectively vulnerable to degeneration with age as in neurodegenerative diseases of the elderly characterized by accumulation of LBs.
Keywords: neurofilament, neurodegeneration, necrosis, phosphorylation, apoptosis, Lewy body, Purkinje cells, Parkinson’s disease
Neuronal death occurs under physiological and pathological conditions. As many as 50% of nascent neurons undergo programmed cell death (apoptosis) in the developing CNS (Vaux, 1993; Lo et al., 1995). Neuron death also is caused by excitotoxins (Beal, 1992), oxidative stress (Cleeter et al., 1992; Jenner et al., 1992;Olanow, 1993), mitochondrial dysfunction (Schapira and Cooper, 1992), ectopic oncogene expression (Feddersen et al., 1992), chromosomal abnormalities (Johnson, 1991; Holtzman, 1992; Gurney et al., 1994; Hong and Driscoll, 1994; Huang and Chalfie, 1994), and disruption of the neuronal cytoskeleton (Goldman and Yen, 1987; Pendlebury et al., 1987). Massive neuronal death is a prominent feature of human neurodegenerative diseases including Parkinson’s disease (PD), diffuse Lewy body (LB) disease (DLBD), and amyotrophic lateral sclerosis (ALS). A hallmark of these diseases is a disruption of the neurofilament (NF) network and the accumulation of NF-rich inclusions in selectively vulnerable neurons (Goldman and Yen, 1986; Ferrer et al., 1994; Itoh et al., 1992; Pollanen et al., 1993; Trojanowski and Lee, 1994).
NFs are the most abundant neuron-specific intermediate filaments (IFs) (Nixon, 1993; Tu and Lee, 1996) and are obligate heteropolymersin vivo of three subunits, i.e., low- (NFL), middle- (NFM), and high- (NFH) molecular-weight NF proteins (Ching and Liem, 1993; Lee et al., 1993). It appears that NFL forms the 10 nm filamentous shaft into which NFM and NFH are incorporated (Tokutake, 1990; Nixon, 1993). NF subunits have a tripartite structure including a head domain, a central rod domain, and a tail domain. The tail domains of NFM and NFH contain repeated KSP motifs that are extensively phosphorylated (Lee et al., 1988) by incompletely defined kinases (Nixon, 1993; Shetty et al., 1993; Lew and Wang, 1995). The most heavily phosphorylated isoforms of NFH and NFM are confined primarily to axons (Sternberger and Sternberger, 1983; Carden et al., 1987) and regulate axonal caliber (de Waegh et al., 1992; Cole et al., 1994; Tu et al., 1995).
Phosphorylated NF-rich inclusions accumulate in neurons in age-related neurodegenerative diseases (Goldstein et al., 1987; Schmidt et al., 1987; Nakazato et al., 1990; Gold and Austin, 1991; Hill et al., 1991;Tu et al., 1996), but their role in the pathogenesis of these diseases remains ill-defined (Calne et al., 1992; Trojanowski and Lee, 1994). Transgenic mice overexpressing NFL or NFH develop NF-rich inclusions in spinal cord motoneurons and an ALS-like disease (Côté et al., 1993; Xu et al., 1993; Lee et al., 1994). Because transgenic mice expressing a fusion protein consisting of NFH and β-galactosidase (LacZ) sequences also develop intraneuronal NF inclusions throughout the CNS (Eyer and Peterson, 1994), we investigated the biological significance of these NF inclusions in brain. Significantly, cerebellar Purkinje neurons, which accumulate NF-rich inclusions with numerous entrapped cellular organelles, selectively degenerated in aged NFHLacZ mice, whereas hippocampal CA1 neurons containing NF-rich inclusions devoid of organelles did not. Thus, the sequestration of organelles within NF inclusions may functionally isolate organelles, thereby compromising the viability of affected neurons.
MATERIALS AND METHODS
Generation of transgenic mice. The generation and initial characterization of the NFHLacZ construct and two lines of the NFHLacZ transgenic mice have been described (Eyer and Peterson, 1994). Briefly, the NFHLacZ fusion construct was generated by blunt-end ligation of the 3′ end of the mouse NFH gene digested withEcoRV to the 5′ end of LacZ, which was digested withXmnI. This ligation resulted in an in-frame ligation between these two genes, such that the fusion transcript encodes a protein of 1801 amino acids. The first 782 amino acids are from NFH and include the N-terminal domain, the central rod domain (which is essential for assembly of NFs), and the C-terminal domain, including up to the 45th KSP repeat. The remaining 1019 amino acids correspond to the completeEscherichia coli β-galactosidase monomer. The number and age of the transgenic and control mice used here are listed in Table1.
Table 1.
Summary of the NFHLacZ transgenic mice
| Line | Age (number pairs) | Phenotype | Loss of Purkinje cells |
|---|---|---|---|
| 44A | 1 week (2) | − | ND |
| 6 weeks (2) | − | ND | |
| 2 months (4) | + | ND | |
| 4 months (4) | + | ND | |
| 6 months (10) | + | − | |
| 12 months (3) | + | + | |
| 18 monrhs (3) | ++ | ++ | |
| 44B | 1 week (2) | − | ND |
| 6 weeks (2) | − | ND | |
| 2 months (2) | − | ND | |
| 4 months (2) | ± | ND | |
| 6 months (3) | + | ND | |
| 12 months (2) | + | ND | |
| 18 months (2) | ++ | ++ | |
Phenotype +, tremor and muscular wasting; Phenotype ++, tremor, muscular wasting, and ataxia; ND, not determined.
X-gal staining. Transgenic mice (1 week to 6 months old) were killed with carbon dioxide and transcardially perfused with PBS, and the brains were quickly removed and snap-frozen in O.C.T. compound (Miles, Elkhart, IN) at −20°C. Frozen sections were cut at a thickness of 8 μm with a cryostat and air-dried for 30 min. Samples were rinsed briefly in PBS and stained at 37°C with 1 mg/ml X-gal in 5 mm potassium ferrocyanide, 5 mm ferricyanide, and 1 mm MgCl2 in PBS until a bright blue color appeared (Pleasure et al., 1992).
Immunohistochemistry. Age-matched control and transgenic mice were killed with carbon dioxide and perfusion-fixed with isotonic 70% alcohol. The brains were dissected out, immersion-fixed in the same fixative for 16 hr, and embedded in paraffin as described (Trojanowski et al., 1989). Sections (6-μm-thick) were cut and incubated with primary antibodies for ∼16 hr at 4°C (see Table2 for a list of the antibodies used here and a summary of their properties). The peroxidase/anti-peroxidase method was used for the mouse mAbs, whereas the avidin–biotin complex method was used for the polyclonal antibodies, as described (Tu et al., 1995,1996).
Table 2.
List of antibodies for immunohistochemistry
| Antibody | Dilution | Epitope | Inclusion staining |
|---|---|---|---|
| Rabbit anti-β-Gal | 200× | NA | ++ |
| Rabbit Anti-NFL | 2000× | NFL, Pind/tail | ++ |
| RM03 | Neat | NFM, Pind/tail outside MPR | ++ |
| RM0189 | 100× | NFM, Pind/core | ++ |
| RM0255 | 200× | NFM, Pind/tail, last 20 amino acids | ++ |
| RM032 | 50× | NFM, P+++/tail within MPR | ++ |
| RM024 | 10× | NFH, P+++/tail within MPR | + |
| RM0217 | 10× | NFH, P++/tail within MPR | + |
| TA51 | 2000× | NFH, P+/tail within MPR | ++ |
| RMd09 | 1000× | NFH, P−/tail within MPR | ++ |
| Actin | 100× | NA | − |
| α-Tubulin | 10,000× | NA | − |
| β-Tubulin | 10,000× | NA | − |
| GFAP | 100× | NA | − |
| Ubiquitin | 20,000× | NA | Variable |
| MAP2 | Neat | NA | − |
| MAP5 | Neat | NA | − |
| Anti-synaptophysin | 50× | NA | − |
P+++, Heavily phosphorylated epitope in NFM or NFH; P++, moderately phosphorylated epitope in NFM or NFH; P+, lightly phosphorylated NFM or NFH epitope. P−, non- or poorly phosphorylated epitope in NFM or NFH; Pind, phosphorylation-independent epitope in NFM; MPR, multiphosphorylation repeat in NFM or NFH; NA, not applicable.
Terminal deoxynucleotidyl transferase-mediated bio-dUTP nick end labeling (TUNEL) staining. TUNEL staining was performed as described (Gavrieli et al., 1992) with minor modifications. Briefly, two pairs of 12- and 18-month-old transgenic mice were perfusion-fixed with 4% paraformaldehyde and then processed as described above for immunohistochemistry. The positive controls were brain sections of an 8-d-old rat. After treatment with 0.01% trypsin in PBS for 15 min at room temperature (RT), tissue sections were preincubated for 15 min at RT in TdT buffer (30 mm Tris, pH 7.2, 140 mmsodium cacodylate, 1 mm cobalt chloride) and then were incubated in a moist chamber for 1 hr at 37°C with 60 μl of the reaction mixture (0.25 U of TdT/μl of TdT buffer and 20 μm biotinylated 16-deoxyuridine triphosphate). The reaction was terminated by rinsing the sections in 2 × SSC buffer (300 mm NaCl, 30 mm sodium citrate) for 15 min at RT. The sections were washed three times in 0.1 m Tris buffer, pH 7.6, for 2 min each, blocked in 10% FBS in Tris buffer for 30 min, and incubated for 1 hr at RT in streptavidin peroxidase (BioGenex) diluted 1:100 in Tris buffer. The sections were developed with diaminobenzidine.
Electron microscopy. Age-matched control and transgenic mice were killed with carbon dioxide and perfusion-fixed with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4. Brains were dissected out, immersion-fixed for 16 hr in the same fixative, and processed as described (Hill et al., 1991; Tu et al., 1995). Briefly, the tissue was trimmed into 1 mm3cubes and post-fixed in osmium tetroxide for 1 hr. En bloc staining with uranyl acetate was performed for 20 min. After serial dehydration, tissue was embedded in epon, and ultrathin sections were examined under electron microscope (Hitachi).
Quantitative Western blot analysis. Tissue from different regions of the nervous system of age-matched control and transgenic mice was dissected and processed as described (Bramblett et al., 1994;Tu et al., 1995). Briefly, the samples were weighed, homogenized, and sonicated immediately in BUST buffer (50 mm Tris-HCl, pH 7.4, 8 m urea, 2% mercaptoethanol, and 0.5% SDS) with a ratio of 10 mg wet tissue/100 μl of BUST. Tissue was centrifuged at 40 × 103 rpm at 25°C for 30 min in a TL-100 Ultracentrifuge (Beckman). Supernatants were used for additional experiments. Coomassie protein assay (Pierce, Rockford, IL) was performed according to the manufacturer’s instruction, and the amounts of total proteins were determined using Ultraspec 4050 (LKB-Wallac, Gaithersburg, MD) at 595 nm wavelength by comparing with a standard curve of BSA with known concentrations. For quantitation, three identical lanes (40 μg/lane) of total protein from each sample of both control and transgenic mice were loaded into one gel. After transfer to nitrocellulose membranes, primary antibodies (see Table 2) were incubated with the membranes overnight. Then, one blot was incubated for 1 hr with 10 μCi125I-conjugated goat anti-mouse IgG for the mouse mAbs or 125I-conjugated A-protein for the rabbit polyclonal antiserum. The dried blots were exposed to PhosphorImager plates for various time, and then the immunobands were visualized and quantified with ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Neuronal counting. Three pairs of 12- and 18-month-old control and transgenic mice were perfusion-fixed and processed as described above for immunohistochemistry. Matched sections (6-μm-thick) of cerebellum and hippocampus were stained with Cresyl violet for 2 min, differentiated in 95% alcohol and used for neuronal counting as described (Arnold et al., 1991; Li et al., 1994). For the cerebellum, images magnified 100-fold were used, whereas for the hippocampus, the images were magnified 200-fold. Images captured from slides with a CCD-IRIS (Sony) camera were imported into ScionImage 1.54 (National Institutes of Health). The width of the computer monitor was arbitrarily defined as a unit length. Purkinje neurons (100 U lengths) in cerebellum and pyramidal neurons in the hippocampal CA1 region were analyzed for each mouse. Counting was performed with a manual counting macro. More than 8500 Purkinje cells and CA1 pyramidal neurons per mouse were counted (i.e., ∼2500 Purkinje cells and 6500 CA1 neurons).
Trypsin digestion of NFHLacZ inclusions. Trypsin digestion was performed as described by Yamada and Hazama (1993) with minor modifications. Briefly, 6-μm-thick paraffin sections of brains from age-matched control mice and transgenic mice were incubated for 30 min at 37°C with 0, 0.2, 0.5, and 1 mg/ml type I trypsin (Sigma, St. Louis, MO) in 50 mm Tris-HCl, pH 7.6, and then processed for immunohistochemical staining with different anti-NF antibodies. Images of stained sections were magnified 400-fold and captured into ScionImage 1.54. The staining intensity of NF-positive inclusions in large pyramidal neurons in the neocortex of transgenic mice and that of corpus callosum in control mice was converted to the gray scale and their values were measured by densitometry. All of the measurements were corrected using the average density of areas without tissue as the background. The ratio of the staining intensity of the trypsin-treated versus nontreated sections was used to quantify the extent of digestion of the normal and abnormal NF-positive profiles.
RESULTS
Immunochemical profile of perikaryal NFHLacZ inclusions
The generation and expression of the NFHLacZ transgene in two lines of mice (44A and 44B) have been described (Eyer and Peterson, 1994). Briefly, the NFHLacZ fusion protein was detected initially at approximately embryonic day 15 in the spinal cord motoneurons by β-galactosidase histochemistry staining, but a high level of expression was achieved only after birth. Histochemical staining indicated that NFHLacZ expression increased in motoneurons progressively through the first few weeks of postnatal life during which time other populations of CNS neurons became NFHLacZ-positive. Although NFHLacZ was expressed in a neuron-specific manner, the abundance of this protein varied among different populations of CNS neurons. For example, NFHLacZ appeared to be more abundant in large neurons with long projections (e.g., cortical pyramidal neurons of layer V, Purkinje cells, neurons in cranial nerve nuclei, and spinal cord motoneurons) compared with smaller neurons (i.e., granule cells in hippocampus).
Immunohistochemical staining with a panel of anti-NF subunit-specific antibodies demonstrated perikaryal inclusions in the cell bodies of almost all neurons in the telencephalon and Purkinje cells in cerebellum of adult transgenic mice (Fig. 1). The size of these inclusions correlated with the regional expression level (see below) of the NFHLacZ fusion protein and with the size of the affected neurons. Furthermore, the numbers of affected neurons and the size of the inclusions in the transgenic mice increased from 1 week of age (Fig. 1A) to reach their maximum size at ∼2 months of age (data not shown). In addition to the NFHLacZ fusion protein, which was revealed by the X-gal staining (Fig. 1B), these inclusions also contained all three endogenous NF subunits (Fig.1C–F). Similar to NF-rich LBs in human neurodegenerative diseases, these inclusions were also stained by monoclonal antibodies (mAbs), which recognize heavily phosphorylated epitopes in the tail domains of NFM and NFH (NFMP+++ and NFHP+++, respectively), such as RMO32 and RMO24 (Fig.1E,F). Thus, like LBs, these perikaryal inclusions contain heavily phosphorylated isoforms of NFH and NFM. Because RMO32 is a sensitive and specific probe for detecting LBs in the brains of patients with PD and DLBD (Hill et al., 1991;Schmidt et al., 1991), the RMO32 immunoreactivity of these inclusions suggests that they share specific characteristics common to human LBs.
Fig. 1.
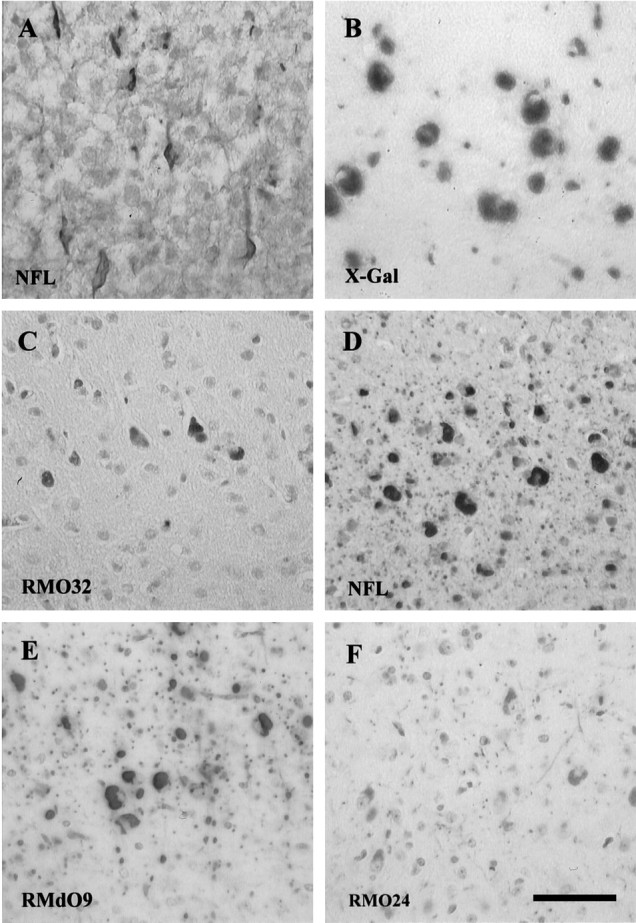
The NFHLacZ inclusions contain all three endogenous NF subunits and the NFHLacZ fusion protein. Ais a frozen section of the neocortex of a 1-week-old transgenic mouse stained with rabbit anti-NFL polyclonal antiserum (NFL). The NF-rich inclusions first appear in the perikarya of large pyramidal neurons of neocortex. B is a frozen section of the neocortex of a 6-month-old transgenic mouse stained with X-gal (X-Gal). C–F show paraffin sections of the neocortex of a 6-month-old transgenic mouse stained with RMO32 (an mAb specific for NFM), RMO24, and RMdO9 (mAbs specific for NFH) and rabbit anti-NFL antiserum. The sections inA and C–F were counterstained with hematoxylin. A–F are at the same magnification; scale bar: F, 50 μm.
The nascent NFHLacZ inclusions, which appeared in some large cortical pyramidal neurons of 1-week-old 44A mice (Fig. 1A), were flame-shaped, and they extended into proximal neurites. At this time, the processes of inclusion-bearing neurons were still strongly stained by antibodies to each of the three NF subunits, suggesting that abundant NFs were present in these neurites. However, with the growth of these perikaryal inclusions, most neuronal processes (except for some white matter axons) showed markedly diminished NF immunoreactivity, suggesting that dendrites and axons gradually become depleted of NFs as a function of age (Fig.1A,D).
Endogenous NF proteins are reduced in the NFHLacZ transgenic mice
In 6-month-old transgenic mice, the NFHLacZ fusion polypeptide was detected with RMdO9, an mAb to a poorly or nonphosphorylated epitope in NFH (NFHP−), as a 210 kDa band in all of the nervous system regions containing neuronal cell bodies, but not in sciatic nerve containing only axons and Schwann cells (Fig.2A). In contrast, this band was not identified by RMO24, an mAb specific for highly phosphorylated NFH (Fig. 2B). These data suggest that the NFHLacZ protein is not highly phosphorylated in vivo despite the fact that it retains 45 of the 51 KSP repeats normally present in the tail domain of mouse NFH. Further, retention of the NFHLacZ fusion protein in the somatodendritic compartment of neurons probably accounts for the fact that it was undetectable in sciatic nerve. However, endogenous NFH was detected with RMdO9 as a broad band in the transgenic mice, where it migrated faster than its counterpart in control mice (Figs. 2A, 3), suggesting that endogenous NFH is less phosphorylated in the NFHLacZ mice compared with control mice. In contrast, NFM appeared to be phosphorylated to the same extent in the transgenic and control mice (Figs. 2, 3).
Fig. 2.

NF proteins are reduced in 6-month-old 44A NFHLacZ mice. Two pairs of age-matched transgenic and control mice were examined. The transgenic NFHLacZ fusion protein (TGNFH) was detected in all samples containing neuronal cell bodies by RMdO9, an mAb specific for non- and poorly phosphorylated NFH (NFHP−) (A), but not by RMO24, an mAb specific for highly phosphorylated NFH (NFHP+++) (B), suggesting that the TGNFH is poorly or nonphosphorylated. The levels of NFM and NFL are decreased in all nervous system regions of the transgenic mice, as revealed by the decrease in the immunoreactivities of NFM (detected by the RMO189, a phosphorylation-independent NFM mAb) and NFL (detected by the rabbit anti-NFL antiserum). However, the increase in RMdO9 immunoreactivity (A) and the decrease in the RMO24 immunoreactivity (B) of the endogenous mouse NFH (MNFH) suggest that NFH levels may not diminish but that the phosphorylation state of NFH decreases.NEO, Neocortex; HIP, hippocampus;CERB, cerebellum; BS, brainstem;SC, spinal cord; SN, sciatic nerve.
Fig. 3.

The decrease in NF proteins in the neocortex of the 6-month-old NFHLacZ transgenic (TG) mice is selective. Whereas there is a decrease in NFM and NFL in the TG mouse, the levels of tubulin (TUB) and synaptophysin (SYN) are not reduced in the same TG mouse compared with the wild-type (WT) mouse. However, elevations in the levels of the microtubule-associated protein 2 (MAP2) and 5 (MAP5) are noted in the same TG mouse.
Notably, the levels of NFM were reduced by ∼50% in all CNS regions examined in the transgenic versus the control mice (Figs.2A,B, 3). Further, when the signals for the NFL and NFH immunobands were normalized to that of NFM in the same region, the relative ratios of NFL/NFM in the transgenic mice was ∼50% less than that of control mice, suggesting that NFL was decreased more than NFM in all CNS tissues of the transgenic mice. Thus, these data clearly demonstrate that the abundance of each NF subunit was reduced to a different extent in the transgenic mice, with NFL being the most markedly reduced component, and NFH being the least affected one. Therefore, the stoichiometry of the NFs in the NFHLacZ inclusions may be different from that of native NFs. Additionally, the NFHP+++ signal in the transgenic mice was dramatically decreased to ∼25% of that in control mice (Fig. 2B), but the signal for NFHP− remained unchanged in the transgenic hippocampus and cerebellum, whereas in the transgenic neocortex and spinal cord, it was twice that of the control mice (Fig. 2A). Because the phosphorylation of NFH at the multiphosphorylation repeat domain occurs primarily in the axon, it is not surprising that the formation of perikaryal NF inclusions results in a decrease in the phosphorylation state of NFH in the NFHLacZ mice.
As for the sciatic nerve, the amount of NF proteins dramatically decreased in the transgenic versus the control mice. This finding is consistent with the previous report that the caliber of axons in the transgenic sciatic nerves is smaller than that in control mice (Eyer and Peterson, 1994). However, all three endogenous NFH subunits were clearly identified in the transgenic nerves (Fig. 2,SN). Thus, at least some endogenous NF proteins escape entrapment in the perikaryal inclusions and enter the axons of sciatic nerves. Interestingly, the ratios of NFL/NFM, NFHP+++/NFM, and NFHP−/NFM in the transgenic mice were very similar to those of the control mice. These data are consistent with the notion that NF proteins are transported as subunits or in oligomers (Nixon, 1993;Terada et al., 1996) and that the normal stoichiometry and phosphorylation state of NFs are maintained in the transgenic sciatic nerves.
Other neuronal proteins are not reduced in transgenic mice
To understand whether the decrease in endogenous NF proteins occurred in concert with a generalized disruption of protein synthesis, we also examined the expression of other neuron-specific proteins. No significant reduction in synaptophysin or tubulin proteins (Fig. 3) was detected in the NFHLacZ mice. Surprisingly, with the antibodies we used, we observed a twofold increase in the levels of MAP2 and MAP5 in both the brain (Fig. 3) and the spinal cord (data not shown) of 6-month-old transgenic mice. Thus, these data demonstrate that the reduction in NF proteins in the transgenic mice is selective and that it does not reflect a generalized perturbation of protein synthesis or metabolism.
The NFHLacZ inclusions are more resistant to trypsin digestion than normal NFs
To determine whether the long-term persistence of the NFHLacZ inclusions might be attributable to a relative resistance to proteolysis, we compared the effect of trypsin digestion on these inclusions and normal NFs, using the method described by Yamada and Hazama (1993). NFs in the corpus callosum of control mice were compared with those in inclusions of neocortical neurons, because previous studies showed that the phosphorylated NFs in axons were more resistant to enzymatic digestion than the nonphosphorylated NFs in perikarya (Pant, 1988). Consistent with the findings of Pant (1988), after treatment with low concentrations (0.1–0.5 mg/ml) of trypsin, NF immunoreactivity persisted in axons of the corpus callosum but not in the somatodendritic compartment of neurons (data not shown). However, digestion with a high concentration (1 mg/ml) of trypsin completely eliminated NF immunoreactivity from both axons and dendrites in the control mice (Fig.4A,B), but did not eliminate, the NF staining intensity of the NFHLacZ inclusions (Fig.4C,D). For example, after treatment for 30 min at 37°C, the staining intensity of some inclusions with RMO189 was ∼43% of that before trypsin treatment in many NFHLacZ inclusions. Thus, this suggests that the NFs in the inclusions are more resistant to trypsin digestion than native axonal NFs. Interestingly, the staining of these inclusions with antibodies specific for epitopes in the tail domains of both NFM and NFH was eliminated after the same trypsin digestion (data not shown). Because NFH was less phosphorylated in transgenic versus control mice, these data suggest that the resistance of these inclusions to trypsin digestion may not be attributable to the phosphorylation state of the NF proteins in these inclusions.
Fig. 4.

NFHLacZ inclusions in neuronal perikarya are more resistant to trypsin digestion than native NFs in axons of the corpus callosum in control mice. A and Billustrate the corpus callosum of a wild-type (WT) mouse, whereas C andD show NF inclusions in the neocortex of a NFHLacZ (TG) mouse. The sections in A andC were stained with the mAb RMO189 before trypsin treatment (TS−), whereas the adjacent sections inB and D were stained with the same mAb after trypsin treatment (TS+). Although immunoreactive NFM in normal axons of the corpus callosum is completely eliminated by trypsin digestion in the wild-type mouse, the same does not eliminate NFM immunoreactivity in the NFHLacZ inclusions of the NFHLacZ transgenic mouse. The sections in A–Dwere counterstained with hematoxylin.A–D are at the same magnification; scale bar: D, 50 μm.
Two types of ultrastructural profiles exist in perikaryal NFHLacZ inclusions
Because the NFHLacZ inclusions were found in diverse neuronal populations, we asked whether all these inclusions were identical at the ultrastructural level, and we showed that these inclusions had two distinct types of ultrastructural morphologies by electron microscopy. Type I inclusions were composed of densely packed interwoven fascicles of long (10 nm in diameter) IFs, which, like native NFs, had clear sidearms protruding from the bodies of these filaments (Fig.5A). Type II inclusions also contained a loose network of apparently shorter (10 nm in diameter) IFs that were arranged in a haphazard pattern and did not form fascicles (Fig.5A). Notably, entrapped within this filamentous network were islands of cytoplasmic organelles, including polyribosomes, endoplasmic reticulum, mitochondria, lysosomes, and some undefined vesicles. In sharp contrast, type I inclusions contained similar masses of filaments, but they were nearly devoid of any cellular organelles. Quantitative regional analysis showed that type I inclusions predominated in hippocampus and spinal cord, whereas the Purkinje cells exclusively contained type II inclusions, and cortical neurons had a similar number of type I and type II inclusions (Fig. 5B). The ratios of type I/type II inclusions in these four neuronal populations remained quite constant over the life span of the mice examined here (data not shown). Thus, these data suggest that the ultrastructural morphology of these inclusions is a function of the type of neuron in which they arise, rather than the stage in the formation of these inclusions or the age of the mice.
Fig. 5.
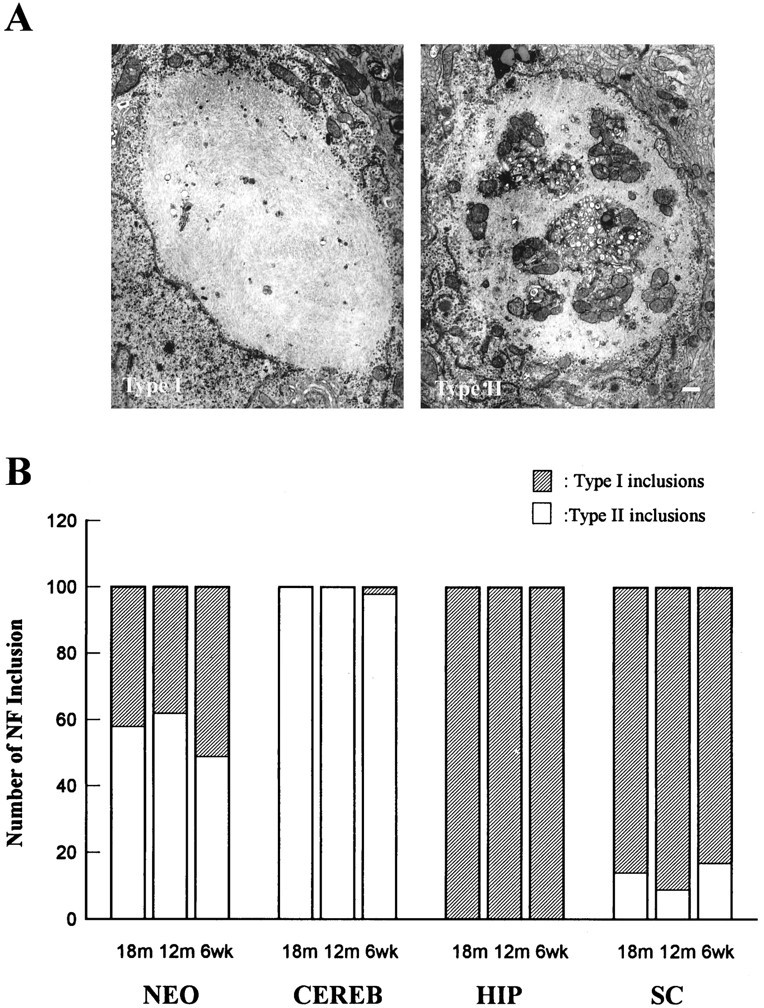
Differences in the ultrastructure of the NFHLacZ inclusions define two types of lesions. A shows representative electron photomicrographs of the two distinct types of ultrastructural morphologies of the NFHLacZ inclusions. The type I inclusion (left) is composed of compact filaments with few or no entrapped organelles. In contrast, the type II inclusion (right) entrap numerous different cellular organelles (e.g., mitochondria, endoplasmic reticulum, polyribosomes) within a tangled mass of filaments. The images in A are at the same magnification; scale bar: right, 500 nm.B shows the number of neurons with type I or type II inclusions in four different nervous regions of the TG mice at 6 weeks, 12 months, and 18 months of age. Fifty neurons from each CNS region were counted, and two animals were examined for each age group. The type I/II ratios remain quite constant in all four CNS regions throughout the life span of these mice, suggesting that the ultrastructure of the NFHLacZ inclusions is a function of the class of neuron in which it forms. NEO, Neocortical pyramidal neurons; HIP, hippocampal CA1 neurons;CERB, cerebellar Purkinje cells; SC, spinal cord motoneurons.
Additional analysis of other neuronal compartments revealed that the normal NF network was not evident by electron microscopy in either dendrites or axons of the transgenic mice. However, both type I and type II inclusions were identified in dendrites (data not shown), and some organelles (e.g., mitochondria, polyribosomes) frequently clustered around these inclusions.
Age-dependent, selective loss of Purkinje cells in the aged NFHLacZ transgenic mice
Because the role of NF inclusions in the death of neurons remains poorly defined in human neurodegenerative diseases, we scrutinized the brain and cerebellum of NFHLacZ mice for evidence of neuronal loss as a function of age. Remarkably, we detected a dramatic and selective loss of Purkinje cells beginning at ∼1 year of age in the NFHLacZ mice. Accordingly, we counted Purkinje cells and hippocampal CA1 neurons in control and NFHLacZ mice at different ages. We focused our studies on these two classes of neurons, because nearly all of them accumulated either type I (CA1 neurons) or type II (Purkinje cells) NFHLacZ inclusions, and because both populations of neurons have been observed to be selectively vulnerable to degeneration after different injuries. For the quantitative studies, we randomly chose 100-unit-lengths of the Purkinje cell layer and CA1 from matched coronal levels of transgenic and control mice and counted ∼2500 Purkinje cells and 6500 hippocampal neurons per animal. There was a selective loss (>60%) of Purkinje cells (Figs. 6A,7I,M), but not hippocampal CA1 neurons (Fig. 6B), in the 18-month-old 44A transgenic versus the age-matched control mice. At this age, the average number of Purkinje cells per unit length was 8 in the transgenic versus 22 in the control mice. Although the precise onset of Purkinje cell loss was difficult to determine, there was a significant decrease (23%) in the number of Purkinje cells in the 12-month-old transgenic mice, but there was no evidence of Purkinje cell loss in these mice at 6 months (see also Plummer et al., 1995). Degenerating neurons with pyknotic nuclei were easily identified in the cerebellar Purkinje cell layer of the aged transgenic mice (Fig.7I, arrowhead), but none were seen in the hippocampus. A similar loss of Purkinje cells also was detected in the aged 44B transgenic mice (data not shown). Taken together, these data suggest that the loss of Purkinje cells results from a highly selective, age-dependent detrimental effect of the NFHLacZ inclusions, rather than from any harmful effect of the insertion site of the transgenes. Interestingly, a mild ataxia gradually appeared at ∼1 year in these transgenic mice, and the severity of the ataxia progressed as the loss of Purkinje cells became more prominent (Table1).
Fig. 6.
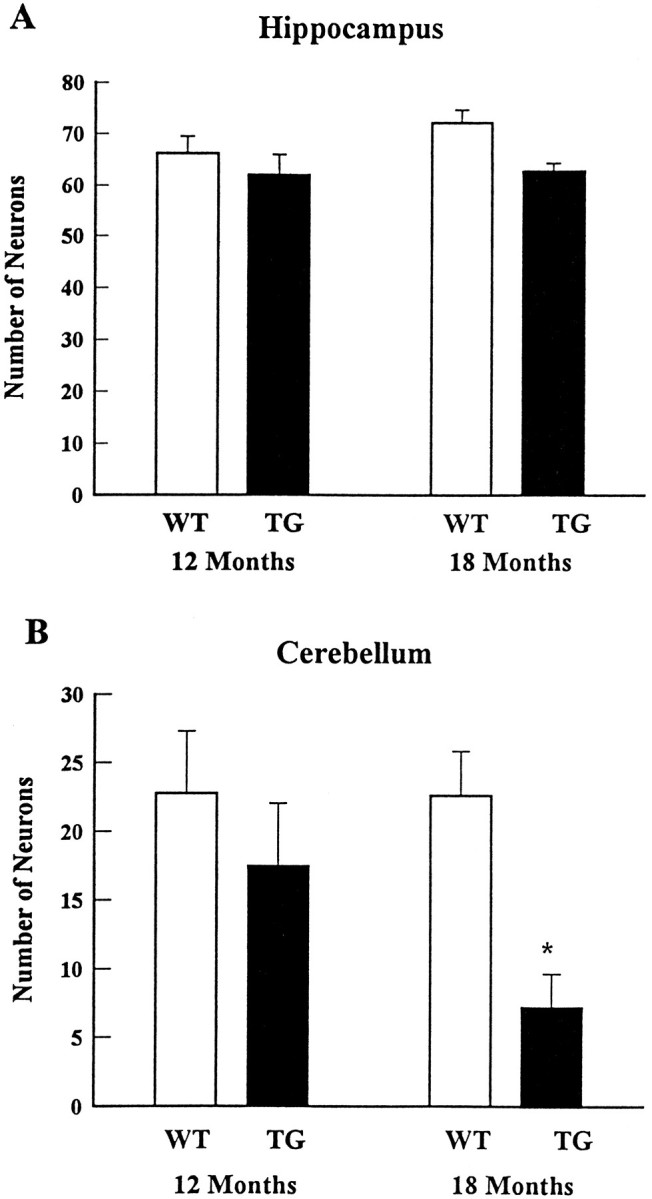
Selective loss of Purkinje cells, but not the CA1 neurons of hippocampus, is noted in the aged NFHLacZ (TG) mice compared with age-matched wild-type (WT) mice. Three pairs of age-matched NFHLacZ transgenic and wild-type mice from each age group are examined. The number of Purkinje cells per unit length slightly decreases (∼20%) in the 12-month-old NFHLacZ transgenic mice compared with the age-matched wild-type mice, but a 60% decrease in the number of Purkinje cells/unit length is noted in the 18-month-old NFHLacZ transgenic mice (8 cells/unit length) compared with the age-matched wild-type mice (22 cells/unit length; *p < 0.0001 by Student’s t test) (B). In contrast to the cerebellum, the number of CA1 hippocampal neurons per unit length does not decrease either in the 12-month-old or the 18-month-old mice compared with the age-matched wild-type mice (A).
Fig. 7.
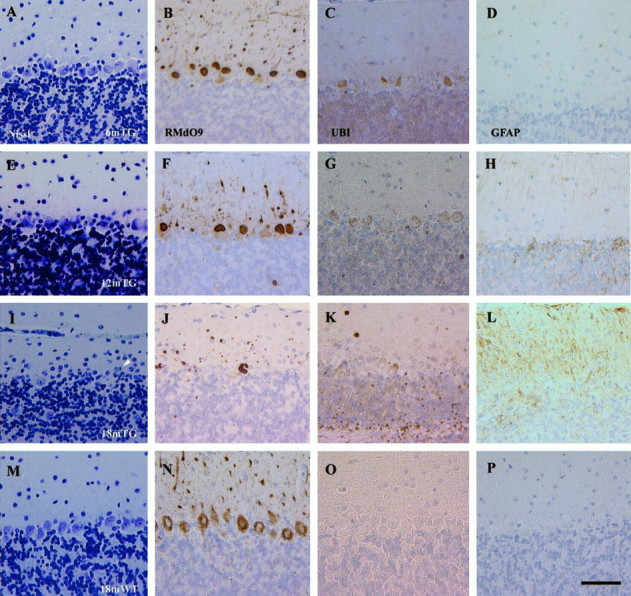
A progressive loss of Purkinje cells is detected in the NFHLacZ transgenic (TG) mice.A–D, Six-month-old NFHLacZ transgenic mouse. E–H, Twelve-month-old NFHLacZ transgenic mouse. I–L, Eighteen-month-old NFHLacZ transgenic mouse. M–P, Eighteen-month-old wild-type (WT) mouse.A, E, I, M, Nissl-stained sections. B, F,J, N, RMdO9-stained sections.C, G, K, O, Anti-ubiquitin (UBI)- stained sections.D, H, L, P, Anti-GFAP (GFAP)- stained sections. The loss of Purkinje cells starts in the 12-month-old TG mice (panels Eand F) and a prominent loss is identified in the 18-month-old NFHLacZ transgenic mice with the Nissl stain and RMdO9 (I, J). A degenerating Purkinje cell in the 18-month-old transgenic mice is shown in I(arrowhead). A concomitant increase in the immunoreactivity of anti-ubiquitin and GFAP is observed in the cerebellum of the 18-month-old NFHLacZ transgenic mice but not in the age-matched wild-type mice. The antibody-labeled sections were counterstained with hematoxylin. A–P are at same magnification; scale bar: P, 50 μm.
In addition to the loss of Purkinje cells in the NFHLacZ mice, there was a prominent increase in Bergmann glial cells in the molecular layer and in the cerebellar white matter, as demonstrated by increased immunoreactivity for glial fibrillary acidic protein (Fig.7L). No gliosis in any other CNS region (i.e., neocortex, hippocampus) was detected (data not shown). Thus, the onset of fibrillary gliosis beginning at ∼1 year of age exclusively in the cerebellum of the NFHLacZ mice is consistent with the view that Purkinje cells are selectively vulnerable to death in the brains of these transgenic mice.
The ubiquitination of cytoskeletal inclusions, including LBs, is another pathological hallmark associated with neuronal loss in a variety of neurodegenerative diseases (Lowe et al., 1993), and the NFHLacZ inclusions also were stained by an antibody to ubiquitin (Fig.7C,G,K). Because other molecules such as the microtubule-associated proteins 2 and 5 (MAP2 and MAP5) and tubulin were not found in these inclusions (data not shown), one or more NF proteins in these inclusions may be ubiquitinated. However, as in other neurodegenerative diseases with abundant NF-rich inclusions, ubiquitin immunoreactivity was not completely localized to these inclusions, i.e., it also was seen in some regions of the white matter (Fig. 7K).
Features of Purkinje cell death
To determine whether an apoptotic cell death mechanism might play a role in the massive degeneration of Purkinje cells, TUNEL staining was conducted, but it failed to label any neuronal nuclei (data not shown). Further, no apoptotic bodies were seen by light or electron microscopy (data not shown). Thus, both the TUNEL results and the morphological data favor the notion that Purkinje cells do not degenerate by a classic apoptotic pathway.
DISCUSSION
NF-rich inclusions are hallmark brain lesions of a variety of late-life neurodegenerative diseases, but the precise role these lesions play in the pathogenesis of these disorders remains controversial (Calne et al., 1992). However, the development of transgenic mice that overexpress one or more NF subunits has made it possible to address this issue experimentally (Côté et al., 1993; Xu et al., 1993; Eyer and Peterson, 1994; Lee et al., 1994;Vickers et al., 1994; Tu et al., 1995; Wong et al., 1995). For example, some of these transgenic mice develop NF inclusions in the perikarya and proximal axons of spinal cord motoneurons as well as clinical (motor weakness) and pathological (motoneuron loss) findings similar to those seen in human ALS (Côté et al., 1993; Xu et al., 1993; Lee et al., 1994). In fact, recent studies suggest that the degeneration of spinal cord motoneurons in these mice may result from a blockage of axonal transport by the accumulation of large NF inclusions in the proximal axons of these neurons (Collard et al., 1995). However, the present study suggests another mechanism whereby NF inclusions lead to the demise of affected neurons. Specifically, our data suggest that the selective degeneration of Purkinje cells in the NFHLacZ transgenic mice could result from the entrapment of vital cellular organelles in type II NF-rich inclusions, located in perikarya and dendrites, leading to the functional isolation of these organelles, thereby compromising the long-term viability of Purkinje cells. Indeed, similar to human neurodegenerative diseases such as PD and DLBD, the loss of Purkinje cells in the NFHLacZ mice was an age-dependent and protracted process.
Compared with previously described lines of NF transgenic mice, the NFHLacZ mice are unique in several aspects. First, in other transgenic mice, the development of NF inclusions was limited to specific groups of neurons such as spinal cord motoneurons (Côté et al., 1993; Xu et al., 1993; Lee et al., 1994; Wong et al., 1995), large pyramidal neurons of the cortex (Vickers et al., 1994), or thalamic neurons (Mathieu et al., 1995). In contrast, almost all CNS neurons in the NFHLacZ mice developed NF inclusions, presumably because the transgene is regulated by the NFH promoter and is expressed in most neurons, and only small quantities of the fusion protein are necessary to effectively cross-link NFs. Second, although subsets of neurons were selectively vulnerable to degenerate or become functionally impaired in several previously described lines of NF transgenic mice, this selectivity was determined by the neurons in which the NF inclusions developed. However, in the NFHLacZ mice, only a subset of CNS neurons harboring inclusions (i.e., Purkinje cells) died prematurely. Third, the clinical phenotypes (i.e., tremor and muscular weakness) seen in previously described NF transgenic mouse lines appeared early in life (i.e., a few months after birth), but the NFHLacZ mice did not develop ataxia until they were ∼1-year-old, and this coincided with the massive loss of Purkinje cells. Thus, the delayed loss of Purkinje cells in aged NFHLacZ transgenic mice closely simulates the loss of neurons in authentic human neurodegenerative diseases such as PD and DLBD.
Although the progressive loss of Purkinje cells in the NFHLacZ mice was age-dependent and highly selective, the mechanisms that account for the selective vulnerability of aging Purkinje cells to degenerate remains to be clarified. Purkinje cells are prone to degenerate as a result of a variety of insults including genetic mutations such as those seen in “lurcher” and “pcd” mice (Phillips, 1960; Caddy and Biscoe, 1979; Roffler-Tarlov et al., 1979), the lack of prion proteins (Sakaguchi et al., 1996), chemical intoxication (West et al., 1990;Brorson et al., 1995), global ischemia (Cervos-Navarro and Diemer, 1991), ectopic oncogene expression (Feddersen et al., 1992), paraneoplastic syndromes (Mizutani et al., 1988), and some neurodegenerative diseases (Ferrer et al., 1994). However, the manner in which these insults induce neuron death remains enigmatic. Here, we present data suggesting that Purkinje cells die through a nonapoptotic pathway, and these Purkinje cells exclusively contain the type II inclusions that entrap cellular organelles. Interestingly, organelles such as mitochondria, secretory vesicles, and granular material also are found in the NF-rich inclusions of neurons that selectively degenerate in human neurodegenerative diseases (Forno, 1986; Murayama et al., 1989, 1991; Sasaki and Maruyama, 1991). Thus, it is plausible that type II inclusions are more harmful to neurons than type I inclusions, because they entrap cellular organelles that may functionally isolate them or interfere with their normal functions. Although the reason(s) why Purkinje cells only accumulate type II inclusions is not clear, this could reflect heterogeneity in the post-translational modifications of NF proteins in different classes of neurons (Sternberger and Sternberger, 1983; Trojanowski et al., 1985;Fujii et al., 1993; Archer et al., 1994; Soussan et al., 1994). Notably, these post-translational modifications are thought to play important roles in the assembly and degradation of NF proteins as well as in the interactions of NFs with other organelles (Pant, 1988;Hisanaga et al., 1991). Differences in the ultrastructural profiles of NF inclusions also were demonstrated in human neurodegenerative diseases, and this may be attributable to unique properties of the affected neurons (Forno, 1986; Murayama et al., 1989, 1991; Sasaki and Maruyama, 1991). Our studies suggest that the two distinct ultrastructural profiles of the NFHLacZ inclusions also may be determined by the class of neuron in which they accumulate.
The selective loss of Purkinje cells in the aged, but not in the young, mice, nor in other neuronal populations may imply the existence of one or more compensatory mechanisms that counterbalance the potentially deleterious effects of NF inclusions. What kind of compensatory mechanisms might these be? First, the selective decrease in the levels of NF proteins in the brains of the NFHLacZ mice may limit the progressive aggregation of NFs into the NFHLacZ inclusions. Because a reduction in NF mRNAs or subunits has been described after nerve transection (Goldstein et al., 1988) and acute aluminum neurotoxicity (Strong et al., 1994) and in human neurodegenerative diseases (Hill et al., 1993; Bergeron et al., 1994), downregulation of NF protein synthesis may be a common mechanism that enables such injured neurons to prevent the formation of NF inclusions. Second, because the polyubiquitination of aberrant proteins targets these proteins for extralysosomal degradation (Ciechanover, 1994), and we detected ubiquitin immunoreactivity in the NFHLacZ inclusions, it is possible that the ubiquitin-mediated degradation of proteins in the NFHLacZ inclusions serves as a mechanism to limit the size of the inclusions. Third, an augmentation of the microtubule network has been suggested to compensate for the depletion of NFs in the Quiver quail (Yamasaki et al., 1992; Ohara et al., 1993; Zhao et al., 1994). A similar increase in microtubule density in axons occurs in the NFHLacZ mice (Eyer and Peterson, 1994), and we found that the levels of MAP2 and MAP5 also were increased in these mice. Thus, augmentation of the microtubule network may delay some of the deleterious effects of the NF inclusions in the brains of the NFHLacZ mice.
Based on the present studies of the NFHLacZ transgenic mice as well as on earlier studies of previous lines of NF transgenic mice that develop intra-axonal NF inclusions, impaired axonal transport and an ALS-like phenotype (Côté et al., 1993; Collard et al., 1995), we propose that NF inclusions may induce neuronal dysfunction and degeneration by at least two alternative mechanisms, depending on the composition and the location of these inclusions within a neuron. First, the formation of NF inclusions in the initial segment of axons appears to occlude the axon leading to impaired axonal transport and the demise of the affected neurons (Côté et al., 1993;Collard et al., 1995). Second, in the NFHLacZ mice, the entrapment of cellular organelles in type II perikaryal NF inclusions in Purkinje cells (as in Purkinje cells of the NFHLacZ mice) is likely to compromise neuronal viability by sequestering these vital organelles such that they no longer support the survival of Purkinje cells as they age. Accordingly, NF inclusions may not be “toxic” unless they are positioned or formed in such a manner that they compromise vital processes in neurons, as in the case of the type II inclusions in the NFHLacZ mice or the NF transgenic mice with impaired axonal transport (Côté et al., 1993; Collard et al., 1995). Although these hypothetical mechanisms of selective neuron death induced by NF inclusions must be tested further, the NFHLacZ mice provide a unique opportunity to gain insights into any mechanistic role that NF inclusions may play in human neurodegenerative conditions such as ALS, PD, and DLBD.
Footnotes
This study was supported by grants from National Institutes of Health (V.M.-Y.L. and J.Q.T), the Association Francaise contre les Myopathies (J.E.), and the Medical Research Council and Muscular Dystrophy Association of Canada (A.P.); and support from the Penn Award, the Jan Kornelis de Cock Foundation, the Stimuleringsprogramma voor Internationalisering van het hoger onderwijs Foundation, and the Groningen University Foundation (F.d.S.). We gratefully acknowledge Dr. M. L. Schmidt for thoughtful discussions, and we also thank Ms. M. Minda and Mr. T.-H. Chiu for their assistance with electron microscopy and photography. F.d.S. thanks Dr. W. M. Molenaar for encouragement.
Correspondence should be addressed to Dr. John Q. Trojanowski, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, Hospital of the University of Pennsylvania, Room A009, 3400 Spruce Street, Philadelphia, PA 19104.
REFERENCES
- 1.Archer DR, Watson DF, Griffin JW. Phosphorylation-dependent immunoreactivity of neurofilaments and the rate of slow axonal transport in the central and peripheral axons of the rat dorsal root ganglion. J Neurochem. 1994;62:1119–1125. doi: 10.1046/j.1471-4159.1994.62031119.x. [DOI] [PubMed] [Google Scholar]
- 2.Arnold SE, Lee VMY, Gurney RE, Trojanowski JQ. Abnormal expression of two microtubule-associated proteins (MAP2 and MAP5) in specific subfields of the hippocampal formation in schizophrenia. Proc Natl Acad Sci USA. 1991;88:10850–10854. doi: 10.1073/pnas.88.23.10850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergeron C, Beric-Maskarel K, Muntasser S, Weyer L, Somerville MJ, Percy ME. Neurofilament light and polyadenylated mRNA levels are decreased in amyotrophic lateral sclerosis motor neurons. J Neuropathol Exp Neurol. 1994;53:221–230. doi: 10.1097/00005072-199405000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VMY. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 5.Brorson JR, Manzolillo PA, Gibbons SJ, Miller RJ (1995) AMPA receptor desensitization predicts the selective vulnerability of cerebellar Purkinje cells to excitotoxicity. 15:4515–4524. [DOI] [PMC free article] [PubMed]
- 6.Caddy KWT, Biscoe TJ. Structural and quantitative studies on the normal C3H and lurcher mutant mouse. Philos Trans R Soc Lond [Biol] 1979;287:167–201. doi: 10.1098/rstb.1979.0055. [DOI] [PubMed] [Google Scholar]
- 7.Calne DB, Hochberg FH, Snow BJ, Nygaard T. Theories of neurodegeneration. Ann NY Acad Sci. 1992;648:1–5. doi: 10.1111/j.1749-6632.1992.tb24518.x. [DOI] [PubMed] [Google Scholar]
- 8.Carden MJ, Trojanowski JQ, Schlaepfer WW, Lee VMY. Two-stage expression of neurofilament polypeptides during rat neurogenesis with early establishment of adult phosphorylation patterns. J Neurosci. 1987;7:3489–3504. doi: 10.1523/JNEUROSCI.07-11-03489.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cervos-Navarro J, Diemer NH. Selective vulnerability in brain hypoxia. Crit Rev Neurobiol. 1991;6:149–182. [PubMed] [Google Scholar]
- 10.Ching GY, Liem RKH. Assembly of type IV neuronal intermediate filaments in nonneuronal cells in the absence of preexisting cytoplasmic intermediate filaments. J Cell Biol. 1993;122:1323–1335. doi: 10.1083/jcb.122.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 12.Cleeter MWJ, Cooper JM, Schapira AHV. Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: evidence for free radical involvement. J Neurochem. 1992;58:786–789. doi: 10.1111/j.1471-4159.1992.tb09789.x. [DOI] [PubMed] [Google Scholar]
- 13.Cole JS, Messing A, Trojanowski JQ, Lee VMY. Modulation of axon diameter and neurofilaments by hypomyelinating Schwann cells in transgenic mice. J Neurosci. 1994;14:6956–6966. doi: 10.1523/JNEUROSCI.14-11-06956.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collard JF, Côté F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- 15.Côté F, Collard JF, Julien JP. Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell. 1993;73:35–46. doi: 10.1016/0092-8674(93)90158-m. [DOI] [PubMed] [Google Scholar]
- 16.de Waegh S, Lee VMY, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- 17.Eyer J, Peterson A. Neurofilament-deficient axons and perikaryal aggregates in viable transgenic mice expressing a neurofilament-β-galactosidase fusion protein. Neuron. 1994;12:389–405. doi: 10.1016/0896-6273(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 18.Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT. Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron. 1992;9:955–966. doi: 10.1016/0896-6273(92)90247-b. [DOI] [PubMed] [Google Scholar]
- 19.Ferrer I, Genist D, Bernado L, Sant F, Serrano The Purkinje cell in olivopontocerebellar atrophy. A Golgi and immunohistochemical study. Neuropathol Appl Neurobiol. 1994;20:38–46. doi: 10.1111/j.1365-2990.1994.tb00955.x. [DOI] [PubMed] [Google Scholar]
- 20.Forno LS. The Lewy body in Parkinson’s disease. Adv Neurol. 1986;45:35–43. [PubMed] [Google Scholar]
- 21.Fujii H, Nakamura K, Takeo K, Kawai S. Heterogeneity of carbonic anhydrase and 68 kDa neurofilament in nerve roots analyzed by two-dimensional electrophoresis. Electrophoresis. 1993;14:1074–1078. doi: 10.1002/elps.11501401171. [DOI] [PubMed] [Google Scholar]
- 22.Gavrieli Y, Sherman Y, Ben-Sasson S. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gold BG, Austin DR. Regulation of aberrant neurofilament phosphorylation in neuronal perikarya. I. Production following colchicine application to the sciatic nerve. J Neuropathol Exp Neurol. 1991;50:615–626. doi: 10.1097/00005072-199109000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Goldman JE, Yen SH. Cytoskeletal protein abnormalities in neurodegenerative diseases. Ann Neurol. 1987;19:209–223. doi: 10.1002/ana.410190302. [DOI] [PubMed] [Google Scholar]
- 25.Goldstein ME, Cooper HS, Carden MJ, Lee VMY, Schlaepfer WW. Phosphorylation of neurofilament proteins and chromatolysis following transection of rat sciatic nerve. J Neurosci. 1987;7:1586–1594. doi: 10.1523/JNEUROSCI.07-05-01586.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldstein ME, Weiss SR, Lazzarini RA, Shneidman PS, Lees JF, Schlaepfer WW. mRNA levels of all three neurofilament proteins decline following nerve transection. Mol Brain Res. 1988;3:287–292. doi: 10.1016/0169-328x(88)90051-4. [DOI] [PubMed] [Google Scholar]
- 27.Gurney ME, Pu H, Chiu AY, Canto MCD, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 28.Hill WD, Lee VMY, Hurtig H, Murray JM, Trojanowski JQ. Epitopes located in spatially separate domains of each neurofilament subunit are present in Parkinson’s disease Lewy body. J Comp Neurol. 1991;309:150–160. doi: 10.1002/cne.903090111. [DOI] [PubMed] [Google Scholar]
- 29.Hill WD, Arai M, Cohen JA, Trojanowski JQ. Neurofilament mRNA is reduced in Parkinson’s disease substantia nigra pars compacta. J Comp Neurol. 1993;329:328–336. doi: 10.1002/cne.903290304. [DOI] [PubMed] [Google Scholar]
- 30.Hisanaga S, Kusubata M, Okumura E, Kishimoto T. Phosphorylation of neurofilament H subunit at the tail domain by cdc2 kinase dissociates the association to microtubules. J Biol Chem. 1991;266:21798–21803. [PubMed] [Google Scholar]
- 31.Holtzman DM, Li Y, DeArmond SJ, McKinley MP, Gage FH, Epstein CJ, Mobley WC. Mouse model of neurodegeneration: atrophy of basal forebrain cholinergic neurons in trisomy 16 transplants. Proc Natl Acad Sci USA. 1992;89:1383–1387. doi: 10.1073/pnas.89.4.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong K, Driscoll M. A transmembrane domain of the putative channel subunit MEC-4 influences mechanotransduction and neurodegeneration in C. elegans. Nature. 1994;367:470–473. doi: 10.1038/367470a0. [DOI] [PubMed] [Google Scholar]
- 33.Huang M, Chalfie M. Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans. Nature. 1994;367:467–470. doi: 10.1038/367467a0. [DOI] [PubMed] [Google Scholar]
- 34.Itoh T, Sobue G, Ken E, Mitsuma T, Takahashi A, Trojanowski JQ. Phosphorylated high molecular weight neurofilament protein in the peripheral motor, sensory and sympathetic neuronal perikarya: system-dependent normal variations and changes in amyotrophic lateral sclerosis and multiple system atrophy. Acta Neuropathol (Berl) 1992;83:240–245. doi: 10.1007/BF00296785. [DOI] [PubMed] [Google Scholar]
- 35.Jenner P, Dexter DT, Sian J, Schapira AHV, Marsden CD. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. Ann Neurol. 1992;32:S82–S87. doi: 10.1002/ana.410320714. [DOI] [PubMed] [Google Scholar]
- 36.Johnson WG (1991) Genetic susceptibility to Parkinson’s disease. Neurol 41[Suppl 2]:82–87. [DOI] [PubMed]
- 37.Lee MK, Xu Z, Wong PC, Cleveland DW. Neurofilaments are obligate heteropolymers in vivo. J Cell Biol. 1993;122:1337–1350. doi: 10.1083/jcb.122.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee MK, Marszalek JR, Cleveland DW. A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron. 1994;13:975–988. doi: 10.1016/0896-6273(94)90263-1. [DOI] [PubMed] [Google Scholar]
- 39.Lee VMY, Carden MJ, Schlaepfer WW, Trojanowski JQ, Otvos L, Jr, Hollosi M, Dietzschold B, Lazzarini RA. Identification of the major multiphosphorylation site in mammalian neurofilaments. Proc Natl Acad Sci USA. 1988;85:1998–2002. doi: 10.1073/pnas.85.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lew J, Wang JH. Neuronal cdc2-like kinase. Trends Biochem Sci. 1995;20:33–37. doi: 10.1016/s0968-0004(00)88948-3. [DOI] [PubMed] [Google Scholar]
- 41.Li YT, Woodruff-Pak DS, Trojanowski JQ. Amyloid plaques in cerebellar cortex and the integrity of Purkinje cell dendrites. Neurobiol Aging. 1994;15:1–9. doi: 10.1016/0197-4580(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 42.Lo AC, Houenou LJ, Oppenheim RW. Apoptosis in the nervous system: morphological features, methods, pathology, and prevention. Arch Histol Cytol. 1995;58:139–149. doi: 10.1679/aohc.58.139. [DOI] [PubMed] [Google Scholar]
- 43.Lowe J, Mayer RJ, Landon M. Ubiquitin in neurodegenerative diseases. Brain Pathol. 1993;3:55–65. doi: 10.1111/j.1750-3639.1993.tb00726.x. [DOI] [PubMed] [Google Scholar]
- 44.Mathieu JF, Ma D, Descarries L, Vallee A, Parent A, Julien JP, Doucet G. CNS distribution and overexpression of neurofilament light proteins (NF-L) in mice transgenic for the human NF-L: aberrant accumulation in thalamic perikarya. Exp Neurol. 1995;132:134–146. doi: 10.1016/0014-4886(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 45.Mizutani T, Maeda S, Hayakawa K, Tanaka U, Hirahata S, Kamoshita H, Taketani T, Morimatsu Y. Paraneoplastic cortical cerebellar degeneration. A neuropathological study of an autopsy case in comparison with cortical cerebellar degeneration in alcoholics. Acta Neuropathol. 1988;77:206–212. [PubMed] [Google Scholar]
- 46.Murayama S, Ookawa Y, Mori H, Nakano I, Ihara Y, Kuzuhara S, Tomonaga M. Immunocytochemical and ultrastructural study of Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis. Acta Neuropathol. 1989;78:143–152. doi: 10.1007/BF00688202. [DOI] [PubMed] [Google Scholar]
- 47.Murayama S, Bouldin TW, Suzuki K. Immunocytochemical and ultrastructural studies of Werdnig-Hoffmann disease. Acta Neuropathol. 1991;81:408–417. doi: 10.1007/BF00293462. [DOI] [PubMed] [Google Scholar]
- 48.Nakazato Y, Junko H, Yoichi I, Hoshi S, Masatoshi H, Fukuda T. Swollen cortical neurons in Creutzfeldt-Jakob disease contain a phosphorylated neurofilament epitope. J Neuropathol Exp Neurol. 1990;49:197–205. doi: 10.1097/00005072-199005000-00001. [DOI] [PubMed] [Google Scholar]
- 49.Nixon RA. The regulation of neurofilament protein dynamics by phosphorylation: clues to neurofibrillary pathobiology. Brain Pathol. 1993;3:29–38. doi: 10.1111/j.1750-3639.1993.tb00723.x. [DOI] [PubMed] [Google Scholar]
- 50.Ohara O, Gahara Y, Miyake T, Teraoka H, Kitamura T. Neurofilament deficiency in quail caused by nonsense mutation in neurofilament-L gene. J Cell Biol. 1993;121:387–395. doi: 10.1083/jcb.121.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olanow CW. A radical hypothesis for neurodegeneration. Trends Neurosci. 1993;16:439–444. doi: 10.1016/0166-2236(93)90070-3. [DOI] [PubMed] [Google Scholar]
- 52.Pant HC (1988) Dephosphorylation of neurofilament proteins enhances their susceptibility to degradation by calpain. Biochem J 256(2):665–668. [DOI] [PMC free article] [PubMed]
- 53.Pendlebury WW, Munoz-Garcia D, Perl DP. Cytoskeletal pathology in neurodegenerative diseases. Adv Exp Med Biol. 1987;221:427–442. doi: 10.1007/978-1-4684-7618-7_31. [DOI] [PubMed] [Google Scholar]
- 54.Phillips RJS. “Lurcher,” a new gene in linkage group XI of the house mouse. J Genet. 1960;57:35–42. [Google Scholar]
- 55.Pleasure SJ, Page C, Lee VMY. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J Neurosci. 1992;12:1802–1815. doi: 10.1523/JNEUROSCI.12-05-01802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plummer J, Peterson A, Messer A. Accelerated and widespread neuronal loss occurs in motor neuron degeneration (MND) mice expressing a neurofilament-disrupting transgene. Mol Cell Neurosci. 1995;6:532–543. doi: 10.1006/mcne.1995.0005. [DOI] [PubMed] [Google Scholar]
- 57.Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol. 1993;52:183–191. doi: 10.1097/00005072-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 58.Roffler-Tarlov S, Beart PM, O’Gorman S, Sidman RL. Neurochemical and morphological consequences of axon terminal degeneration in cerebellar deep nuclei of mice with inherited Purkinje cell degeneration. Brain Res. 1979;168:75–95. doi: 10.1016/0006-8993(79)90129-x. [DOI] [PubMed] [Google Scholar]
- 59.Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Houtani T, Shirabe S, Okada H, Hasegawa S, Miyamoto T, Noda T. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature. 1996;380:528–531. doi: 10.1038/380528a0. [DOI] [PubMed] [Google Scholar]
- 60.Sasaki S, Maruyama S. Immunocytochemical and ultrastructural studies of hyaline inclusions in sporadic motor neuron disease. Acta Neuropathol. 1991;82:295–301. doi: 10.1007/BF00308815. [DOI] [PubMed] [Google Scholar]
- 61.Schapira AHV, Cooper JM (1992) Mitochondrial function in neurodegeneration and aging. Mutat Res 275(N3–6):133–143. [DOI] [PubMed]
- 62.Schmidt ML, Carden MJ, Lee VMY, Trojanowski JQ. Phosphate dependent and independent neurofilament epitopes in the axonal swellings of patients with motor neuron disease and controls. Lab Invest. 1987;56:282–294. [PubMed] [Google Scholar]
- 63.Schmidt ML, Murray J, Lee VMY, Hill WD, Trojanowski JQ. Epitope map of neurofilament protein domains in cortical and peripheral nervous system Lewy bodies. Am J Pathol. 1991;139:53–65. [PMC free article] [PubMed] [Google Scholar]
- 64.Shetty KT, Link WT, Pant HC. cdc2-Like kinase form rat spinal cord specifically phosphorylates KSPXK motifs in neurofilament proteins: isolation and characterization. Proc Natl Acad Sci USA. 1993;90:6844–6848. doi: 10.1073/pnas.90.14.6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soussan L, Barzilai A, Michaelson DM. Distinctly phosphorylated neurofilaments in different classes of neurons. J Neurochem. 1994;62:770–776. doi: 10.1046/j.1471-4159.1994.62020770.x. [DOI] [PubMed] [Google Scholar]
- 66.Sternberger LA, Sternberger NH. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci USA. 1983;80:6126–6130. doi: 10.1073/pnas.80.19.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strong MJ, Mao K, Nerurkar VR, Wakayama I, Yanagihara R, Garruto RM. Dose-dependent selective suppression of light (NFL) and medium (NFM) but not high (NFH) molecular weight neurofilament gene transcription in acute aluminum neurotoxicity. Mol Cell Neurosci. 1994;5:319–326. doi: 10.1006/mcne.1994.1038. [DOI] [PubMed] [Google Scholar]
- 68.Terada S, Nakata T, Peterson AC, Hirokawa N. Visualization of slow axonal transport in vivo. Science. 1996;273:784–788. doi: 10.1126/science.273.5276.784. [DOI] [PubMed] [Google Scholar]
- 69.Tokutake S. On the assembly mechanism of neurofilaments. Int J Biochem. 1990;22:1–6. doi: 10.1016/0020-711x(90)90068-e. [DOI] [PubMed] [Google Scholar]
- 70.Trojanowski JQ, Lee VMY. Phosphorylation of neuronal cytoskeletal proteins in Alzheimer’s disease and Lewy body dementias. Ann NY Acad Sci. 1994;747:92–109. doi: 10.1111/j.1749-6632.1994.tb44403.x. [DOI] [PubMed] [Google Scholar]
- 71.Trojanowski JQ, Obrocka MA, Lee VM. Distribution of neurofilament subunits in neurons and neuronal processes; immunohistochemical studies of bovine cerebellum with subunit-specific monoclonal antibodies. J Histochem Cytochem. 1985;33:557–563. doi: 10.1177/33.6.3889140. [DOI] [PubMed] [Google Scholar]
- 72.Trojanowski JQ, Schuck T, Schmidt ML, Lee VMY. Distribution of tau proteins in normal human central and peripheral nervous system. J Histochem Cytochem. 1989;37:209–215. doi: 10.1177/37.2.2492045. [DOI] [PubMed] [Google Scholar]
- 73.Trojanowski JQ, Schmidt ML, Shin RW, Bramblett GT, Rao D, Lee VMY. Altered tau and neurofilament proteins in neurodegenerative diseases: diagnostic implications for Alzheimer’s disease and Lewy body dementias. Brain Pathol. 1993;3:45–54. doi: 10.1111/j.1750-3639.1993.tb00725.x. [DOI] [PubMed] [Google Scholar]
- 74.Tu PH, Lee VMY (1996) Neurofilaments: structure, function and involvement in neurodegeneration. Adv Mol Cell Biol, in press.
- 75.Tu PH, Elder G, Lazzarini RA, Nelson D, Trojanowski JQ, Lee VMY. Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J Cell Biol. 1995;129:1629–1640. doi: 10.1083/jcb.129.6.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VMY. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vaux DL. Toward an understanding of the molecular mechanisms of physiological cell death. Proc Natl Acad Sci USA. 1993;90:786–789. doi: 10.1073/pnas.90.3.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vickers JC, Morrison JH, Friedrich VL, Elder GA, Perl DP, Katz RN, Lazzarini RA. Age-associated and cell-type-specific neurofibrillary pathology in transgenic mice expressing the human midsized neurofilament subunit. J Neurosci. 1994;14:5603–5612. doi: 10.1523/JNEUROSCI.14-09-05603.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.West JR, Goodlett CR, Bonthius DJ, Hamre KM, Marcussen BL. Cell population depletion associated with fetal alcohol brain damage: mechanisms of BAC-dependent cell loss. Alcohol Clin Exp Res. 1990;14:813–818. doi: 10.1111/j.1530-0277.1990.tb01820.x. [DOI] [PubMed] [Google Scholar]
- 80.Wong PC, Marszalek J, Crawford TO, Xu Z, Hsieh ST, Griffin JW, Cleveland DW. Increasing neurofilament subunit NF-M expression reduces axonal NF-H, inhibits radial growth, and results in neurofilamentous accumulation in motor neurons. J Cell Biol. 1995;130:1413–1422. doi: 10.1083/jcb.130.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu ZH, Cork LC, Griffin JW, Cleveland DW. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell. 1993;73:23–33. doi: 10.1016/0092-8674(93)90157-l. [DOI] [PubMed] [Google Scholar]
- 82.Yamada E, Hazama F. Different stability of neurofilaments for trypsin treatment after axotomy in the dorsal motor nucleus of the vagal nerve and the hypoglossal nucleus. Brain Res. 1993;612:210–215. doi: 10.1016/0006-8993(93)91662-c. [DOI] [PubMed] [Google Scholar]
- 83.Yamasaki H, Bennett GS, Itakura C, Mizutani M. Defective expression of neurofilament protein subunits in hereditary hypotrophic axonopathy of quail. Lab Invest. 1992;66:734–743. [PubMed] [Google Scholar]
- 84.Zhao JX, Ohnishi A, Itakura C, Mizutani M, Yamamoto T, Hayashi H, Murai Y. Greater number of microtubules per axon of unmyelinated fibers of mutant quails deficient in neurofilaments: possible compensation for the absence of neurofilaments. Acta Neuropathol. 1994;87:332–336. doi: 10.1007/BF00313600. [DOI] [PubMed] [Google Scholar]