

Abstract
The capacity of the proinflammatory cytokines, tumor necrosis factor α (TNFα) and interleukin 1β (IL-1β), to modulate the sensitivity of isolated sensory neurons grown in culture to the excitatory chemical agent capsaicin was examined. Alterations in capsaicin sensitivity were assessed by quantifying the number of neurons labeled with cobalt after exposure to capsaicin and by recording the whole-cell response from a single neuron to the focal application of capsaicin. A 24 hr pretreatment of the neuronal cultures with TNFα (10 or 50 ng/ml), but not IL-1β (10 or 50 ng/ml), produced a concentration-dependent increase in the number of cobalt-labeled neurons after exposure to 100 nm capsaicin. The peak increase in the number of labeled neurons was attained after a 4 hr treatment with 10 ng/ml TNFα. Similarly, pretreatment with TNFα (10 ng/ml for 4, 12, and 24 hr) produced a greater than twofold increase in the average peak amplitude of the inward current evoked by 100 nm capsaicin. Both the TNFα-induced increase in labeling and current amplitude were blocked by treating the neuronal cultures with indomethacin before the addition of TNFα. Enhancement of the capsaicin-evoked current also was blocked by the specific cyclo-oxygenase-2 inhibitor SC-236. These results indicate that TNFα can enhance the sensitivity of sensory neurons to the excitation produced by capsaicin and that this enhancement likely is mediated by the neuronal production of prostaglandins. Isolated sensory neurons grown in culture may prove to be a useful model system in which to explore how prolonged exposure to mediators associated with chronic inflammation alter the regulatory pathways that modulate the excitability of the nervous system.
Keywords: tumor necrosis factor α, interleukin 1β, capsaicin, sensitization, prostaglandins, cyclo-oxygenase-2, membrane excitability
Tumor necrosis factor α and interleukin 1β serve as potent intermediaries between tissue injury and the resulting physiological indices of inflammation, such as neutrophil activation, plasma extravasation, and vasodilatation (Dinarello, 1987, 1991; Le and Vilc̆ek, 1987; Kimball, 1991). In rheumatoid arthritis, a well characterized pathology exemplifying the effects of chronic inflammation, the levels of tumor necrosis factor α (TNFα) and interleukin 1β (IL-1β) are elevated in synovial fluid (Dayer and Demczuk, 1984; Arend and Dayer, 1990; Feldman et al., 1990). In animal models of synovitis, injection of TNFα or IL-1β into the joints produces many of the symptoms observed with rheumatoid arthritis (Arend and Dayer, 1990). The responses associated with inflammation include a heightened sensitivity to painful stimuli, a condition known as hyperalgesia (Treede et al., 1992). Indeed, in animal models of peripheral hyperalgesia, the injection of IL-1β or TNFα lowers the response threshold to noxious stimulation (Ferreira et al., 1988;Schweizer et al., 1988; Follenfant et al., 1989; Cunha et al., 1992).
Although the cellular mechanisms whereby TNFα and IL-1β enhance the sensitivity to noxious stimuli are unknown, this sensitization may involve prostaglandins. Both TNFα and IL-1β enhance the release of arachidonic acid and the synthesis of eicosanoids, especially prostaglandin E2 (PGE2; Dayer et al., 1985), via the induction of phospholipase A2 and cyclo-oxygenase activities, respectively (Chang et al., 1986; Burch et al., 1988; Raz et al., 1988; Burch and Tiffany, 1989). The cytokine-induced release of PGE2 may sensitize cells and thus potentiate the response to other inflammatory agents, such as bradykinin (Higgs et al., 1984;Salmon and Higgs, 1987; Smith, 1992). Indeed, previous injection of PGE2 into a rat’s hind paw decreases the withdrawal time in response to noxious stimulation (Ferreira et al., 1978). Similarly, in single unit recordings from sensory nerves, pretreatment with PGE2 increases the firing activity evoked by either bradykinin (Handwerker, 1976; Mense, 1981) or mechanical stimulation (Pateromichelakis and Rood, 1982; Heppelmann et al., 1985). Furthermore, in isolated sensory neurons grown in culture, the number of action potentials elicited by either elevated potassium concentration or focally applied bradykinin is increased after pretreatment with PGE2 (Baccaglini and Hogan, 1983; Nicol and Cui, 1994). Thus, the sensitizing actions of PGE2 are directly on the sensory neuron.
Therefore, to determine whether the enhanced sensitivity produced by the proinflammatory cytokines TNFα or IL-1β results from a direct action on the sensory neurons or via a secondary mediator, we examined the excitation produced by capsaicin in embryonic rat sensory neurons grown in culture as a measure of neuronal sensitivity. Capsaicin was used because it selectively stimulates most, but not all, C-fibers and some Aδ fibers, the neurons associated with nociceptive signaling and neurogenic inflammation (Holzer, 1991). Capsaicin activates a nonselective cationic channel that gives rise to an inward current (Heyman and Rang, 1985; Marsh et al., 1987; Bevan and Forbes, 1988;Wood et al., 1988). Activation of this channel by capsaicin in the presence of extracellular cobalt results in the labeling of sensory neurons; therefore, putative nociceptive neurons can be distinguished from other neurons in a given population (Wood et al., 1988; Hingtgen and Nicol, 1994). In this report we demonstrate that pretreatment with TNFα, but not IL-1β, increased the number of neurons labeled with cobalt after exposure to capsaicin. Furthermore, the peak amplitude of the capsaicin-evoked current produced by submaximal concentrations was enhanced after treatment with TNFα. These findings suggest that TNFα can directly enhance the sensitivity of sensory neurons to excitatory chemical agents.
MATERIALS AND METHODS
Isolation and culture of embryonic rat sensory neurons.The procedures for isolation and culture of rat sensory neurons have been described previously (Vasko et al., 1994). Briefly, the dorsal root ganglia (DRG) from E15–E17 fetal rats were dissected free and placed in a dish containing sterile calcium-free, magnesium-free HBSS at 4°C. The DRGs were incubated in HBSS containing 0.025% trypsin for 25 min at 37°C. The digestion was terminated with the addition of 0.25% trypsin inhibitor; then the cells were washed and centrifuged. Ganglia were washed once with HBSS and then resuspended in DMEM (Life Technologies, Grand Island, NY) supplemented with 2 mm glutamine, 50 μg/ml penicillin and streptomycin, 10% heat-inactivated fetal bovine serum, 50 μm5-fluoro-2′-deoxyuridine, 150 μm uridine, and 250 ng/ml 7S-nerve growth factor (Harlan Bioproducts for Science, Indianapolis, IN). Individual cells were obtained by mechanical agitation with a fire-polished pipette until a cloudy suspension was observed. For the cobalt-labeling studies, ∼300,000 cells were plated into each well (35 mm diameter) of a six well Falcon culture dish coated with poly-d-lysine (100 μg/ml in sterile water). For the electrophysiological recordings, ∼300,000 cells were plated in a collagen-coated culture dish containing small plastic coverslips. Cells were grown at 37°C in a 5% CO2 atmosphere, and the medium was changed every 2 d. These procedures have been approved by the Animal Care and Use Committee of Indiana University School of Medicine.
Cobalt labeling of sensory neurons. These experiments were performed via methods that have been described previously (Hingtgen and Nicol, 1994). Briefly, after 5 d in culture, cells were washed with normal Ringer’s solution of the following composition (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH at 7.4 with NaOH. Then cells were exposed to Ringer’s solution containing 100 nmcapsaicin and 10 mm CoCl2 for 8 min. The capsaicin–cobalt Ringer’s solution was removed, the cells were washed with Ringer’s solution, and then the cells were exposed to Ringer’s solution containing 1% ammonium sulfide for 5 min. After the cobalt precipitation, cells were washed with PBS (100 mmNaH2PO4 and 155 mm NaCl, pH 7.4) and fixed with 4% paraformaldehyde in PBS for 20 min. The cobalt staining was enhanced by incubating the cells in a developer solution (15 mm hydroquinone, 38 mm citric acid, and 280 mm sucrose) for 15 min at 60°C. Then the cells were exposed to developer containing 0.1% (w/v) AgNO3 for ∼30 min at 60°C. The intensity of the staining was inspected visually, and enhancement was stopped by washing the cells with fresh developer. Neuronal staining was determined by counting the number of positive (dark-brown staining) and negative (clear or yellowish) cells in five random fields per well (∼80–100 cells per field) from dishes of neuronal cultures established on at least three different days.
To examine the effects of TNFα or IL-1β on the capsaicin sensitivity of these sensory neurons, we pretreated cell cultures with various concentrations of these agents for various times before exposure to capsaicin (day 4 in culture). Untreated cultures obtained from the same neuronal harvest served as the control cells for both the labeling and electrophysiological studies. In those experiments examining the effects of cyclo-oxygenase inhibition on the actions of TNFα, neuronal cultures were treated with either indomethacin or SC-236 (a specific cyclo-oxygenase-2 inhibitor) for 1 hr before the addition of TNFα. To explore the effects of carba prostacyclin (CPGI2, a nonhydrolyzable analog of prostacyclin or prostaglandin I2) alone or in combination with TNFα on the capsaicin–cobalt loading, cells were exposed to 10 nmCPGI2 or 10 nm CPGI2 and 10 ng/ml TNFα for 24 hr before exposure to capsaicin and cobalt.
Electrophysiology. The procedures for whole-cell patch-clamp recordings of rat sensory neurons have been described in detail previously (Nicol and Cui, 1994). Briefly, a coverslip with the DRG neurons (5 d in culture) was placed in a recording chamber, where the neurons were superfused with normal Ringer’s solution. Using the whole-cell patch-clamp technique (Hamill et al., 1981), we recorded membrane currents from a holding potential of −60 mV with a List EPC-7 (List Electronic, Darmstadt, Germany) patch-clamp amplifier. Recording pipettes were pulled from borosilicate disposable pipettes and had resistances of 3–5 MΩ when filled with the following solution (in mm): 140 KCl, 5 MgCl2, 4 ATP, 0.3 GTP, 2.5 CaCl2, 5 EGTA (calculated free Ca2+concentration of 100 nm), and 10 HEPES, at pH 7.2 with KOH. The cell capacitance was compensated by the nulling circuitry of the recording amplifier.
Capsaicin was applied focally to the neuron by placing a pipette (5–10 μm in diameter) filled with Ringer’s solution containing 100 nm capsaicin and 1 mm trypan blue within 10–30 μm of the cell body. Then positive pressure was applied to the pipette to eject the solution for which trypan blue served to visualize this process. The focal application of trypan blue alone had no effect on these sensory neurons (Nicol and Cui, 1994). Capsaicin was diluted to a concentration of 100 nm, because this value is slightly less than the EC50 obtained for the concentration–response relation for capsaicin in embryonic sensory neurons (J. C. Lopshire and G. D. Nicol, unpublished observations). While continuously superfusing the bath with Ringer’s solution, we obtained two responses, separated by ∼2 min, to the focal application of 100 nm capsaicin. After obtaining the test responses, we changed the superfusate to a Ringer’s solution containing 1 μm capsaicin to determine the maximal response. Responses from only a single neuron were obtained from each coverslip to avoid any possible desensitization that might occur after the application of 1 μm capsaicin. All experiments were done at room temperature (∼23°C).
Only the results obtained from neurons that satisfied the following criteria are presented in this report. First, after establishing the whole-cell configuration, neurons had to maintain zero-current potentials more hyperpolarized than −45 mV for at least 4–5 min before setting the holding potential to −60 mV. Second, the amplitudes of the two responses obtained to the focal application of 100 nm capsaicin had to be within ± 10% of their average value.
Analysis. Statistical differences among the numbers of neurons labeled with cobalt for the various experimental treatments were determined by a χ2 analysis. In this set of studies, one or two wells of cells from each neuronal harvest served as a control group for each experimental treatment. To determine significance for each experimental treatment relative to their respective controls, we obtained a χ2 value for each contingency table. The amplitudes of the inward currents elicited by two applications of 100 nm capsaicin were averaged to obtain the mean response to capsaicin. To account for the varying sensitivity of different sensory neurons to capsaicin, we normalized the mean response to the maximal response obtained with the bath application of 1 μm capsaicin. Statistical differences between the control recordings and those obtained under various treatment conditions were determined by one-way ANOVA. When a significant difference was obtained, a Student–Newman–Keuls or Dunnett’s post hoc test was performed. Values ofp < 0.05 were judged to be statistically significant.
Chemicals. Carba prostacyclin was obtained from Cayman Chemical (Ann Arbor, MI); tumor necrosis factor-α and interleukin-1β (recombinant murine) were obtained from R & D Systems (Minneapolis, MN). The cyclo-oxygenase-2 inhibitor SC-236 was a generous gift of Dr. Peter Isakson (G. D. Searle, St. Louis, MO). All other chemicals were obtained from Sigma (St. Louis, MO). Carba prostacyclin, capsaicin, and SC-236 were dissolved in 1-methyl-2-pyrrolidinone (HPLC grade, Aldrich, Milwaukee, WI) to obtain concentrated stock solutions. Then these stock solutions were diluted with Ringer’s solution to yield the appropriate concentration.
RESULTS
Tumor necrosis factor enhances the number of capsaicin-sensitive neurons
Previously, we demonstrated that capsaicin selectively labeled with cobalt the small-diameter sensory neurons isolated from the dorsal root ganglia of embryonic rats (Hingtgen and Nicol, 1994). In the present study, the capacity of the proinflammatory cytokines, tumor necrosis factor α (TNFα) and interleukin-1β (IL-1β), to modulate the sensitivity of isolated sensory neurons to the excitatory agent capsaicin was examined by quantifying the number of neurons labeled with cobalt after treatment with capsaicin. Under control conditions, the average percentage of cells labeled by 100 nm capsaicin was 15.5 ± 1.2% (range 10–26%;n = 14 neuronal harvests), indicating that, from preparation to preparation, 100 nm capsaicin labeled a relatively stable number of cells from the total neuronal population. As illustrated in Figure 1, pretreatment with TNFα enhanced the number of capsaicin-sensitive neurons (Fig.1A), whereas IL-1β was without effect (Fig.1C). After a 24 hr pretreatment with TNFα, the number of capsaicin-sensitive neurons was increased in a concentration-dependent manner. TNFα at 1 ng/ml did not enhance the number of positively stained neurons (36 of 292 neurons, 12.3% positively labeled), whereas the number was increased significantly by twofold for 10 ng/ml (883 of 2556 neurons, 34.5% labeled) and 50 ng/ml TNFα (448 of 970 neurons, 46.2% labeled). In another series of experiments, the time course of the TNFα-induced sensitization was determined. As illustrated in Figure 1B, it appeared that the maximal increase in the number of labeled neurons (27.3%, 163 of 597 neurons) was attained after a 4 hr treatment with 10 ng/ml TNFα. Similar values for the number of labeled neurons were observed after 12 hr (25.7%, 177 of 689 neurons) and 24 hr exposures (26.7%, 172 of 645 neurons). In contrast to the sensitization produced by TNFα, only 17.7% (91 of 514 neurons) and 12.6% (101 of 799 neurons) of the neurons were labeled with cobalt after a 24 hr exposure to 10 and 50 ng/ml IL-1β, respectively (Fig. 1C). In control experiments, sensory neurons exposed to either 100 nm capsaicin in the absence of cobalt or 10 mm CoCl2 in the absence of capsaicin produced no labeling (data not shown).
Fig. 1.
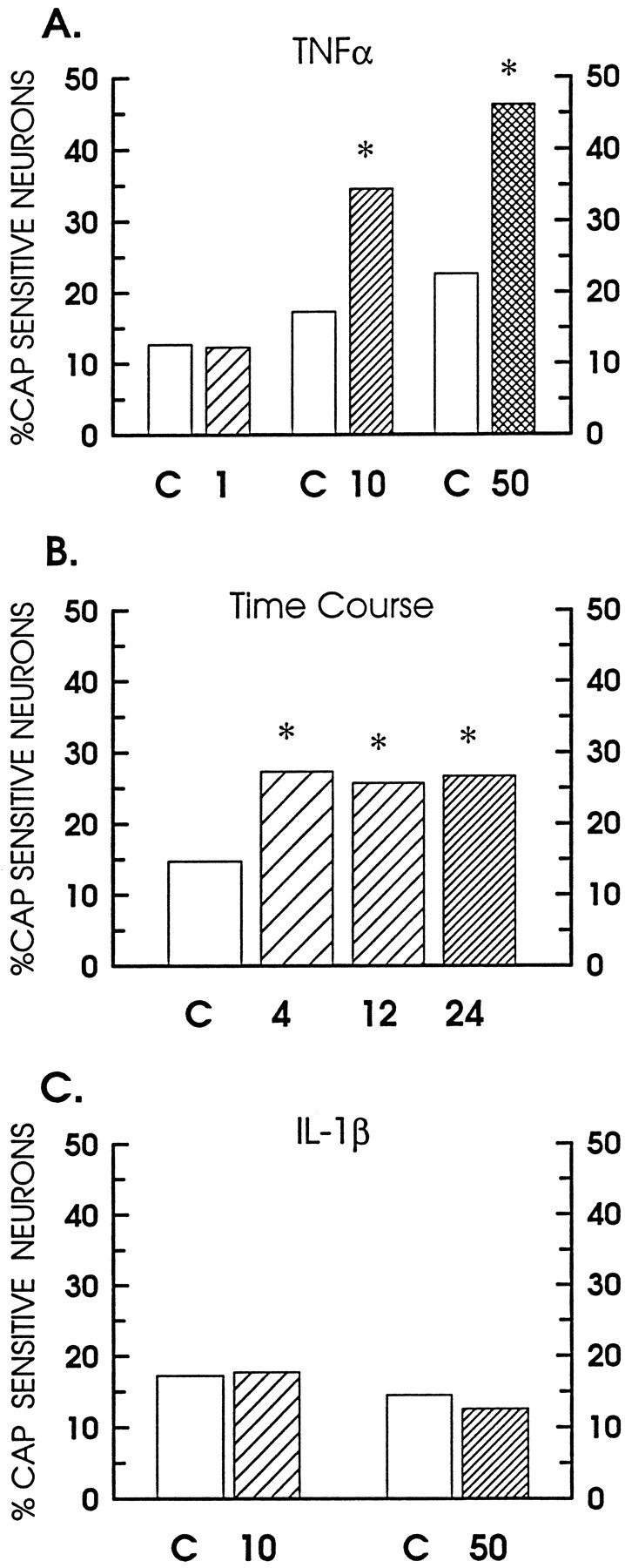
TNFα increases the number of neurons labeled with cobalt after exposure to 100 nm capsaicin. The percentage of labeled neurons for each control condition is represented by the open bars. A, The percentage of neurons positively labeled with cobalt after a 24 hr pretreatment with TNFα. The hatched bars represent the percentages obtained for the following concentrations of TNFα: 1 ng/ml (coarse), 10 ng/ml (medium), or 50 ng/ml (crossed). B, Time course for the sensitization produced by treatment with 10 ng/ml TNFα for the indicated times in hours. These experiments were performed in a set of neurons that differed from those shown in A.C, The percentage of neurons that were positively labeled after a 24 hr pretreatment with 10 and 50 ng/ml of IL 1β, shown as the coarse- and fine-hatched bars, respectively. The asterisks indicate a statistical difference at p < 0.05.
In many instances, the physiological actions of cytokines are believed to be intertwined with prostaglandins. This interdependency may result from either a cytokine-mediated activation of the cyclo-oxygenase pathway, i.e., the production of prostaglandins (Dayer et al., 1985; Burch et al., 1988; Burch and Tiffany, 1989), or prostaglandins may act synergistically to enhance the actions of the cytokine (Burch and Tiffany, 1989). To investigate the possibility that prostaglandins mediated the TNFα-induced increase in labeled neurons, we pretreated cell cultures with 30 μm indomethacin, a nonselective blocker of cyclo-oxygenase activity, for 1 hr before the addition of TNFα to the cultures. As shown in Figure2A, indomethacin blocked the sensitization produced by TNFα. The number of labeled neurons was reduced significantly from a value of 28.6% after treatment with TNFα to 17% after indomethacin and TNFα (189 of 1114 neurons). Exposure to 30 μm indomethacin alone had no significant effect on the number of labeled neurons (125 of 937 neurons, 13.3% positive).
Fig. 2.
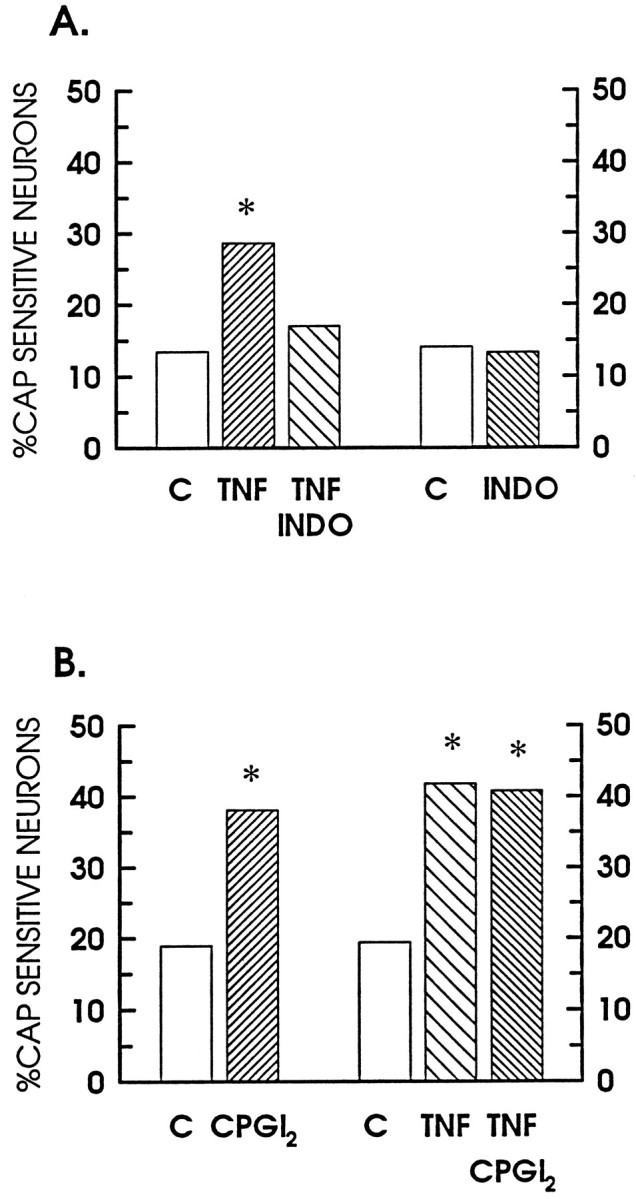
Indomethacin blocks the TNFα-induced increase in labeled neurons. These data represent the percentage of neurons labeled with cobalt after exposure to 100 nm capsaicin. Theopen bars show the percentage of neurons obtained for the various control conditions. A, Suppression of the TNFα-mediated enhancement of labeling by indomethacin. Thefine-hatched bar, labeled TNF, represents the percentage after a 24 pretreatment with 10 ng/ml TNFα; thecoarse-hatched bar, labeled TNF INDO, demonstrates the effects of 30 μm indomethacin in combination with 10 ng/ml TNFα; and the fine-hatched bar, labeled INDO, represents the percentage obtained after a 24 hr treatment with 30 μm indomethacin alone. B, The enhancement of labeling by CPGI2, TNF, and a combination of CPGI2 and TNF. The fine-hatched bar, labeledCPGI2, represents the percentage after a 24 hr pretreatment with 10 nm CPGI2; the coarse-hatched bar, labeled TNF, shows the percentage after a 24 treatment with 10 ng/ml TNFα; and thefine-hatched bar, labeled TNF CPGI2, illustrates the percentage obtained for the combined treatment with 10 nmCPGI2 and 10 ng/ml TNFα. The asterisksindicate significant differences between the control and experimental treatments (p < 0.05).
Another series of experiments examined the possibility that the TNFα-induced sensitization might be enhanced further by cotreatment with a prostaglandin. Neuronal cultures were treated with 10 nm CPGI2 alone or in combination with 10 ng/ml TNFα for 24 hr. This concentration was chosen because a previous study demonstrated that 10 nm CPGI2 produced a two- to threefold enhancement in the release of substance P or calcitonin gene-related peptide (CGRP) when sensory neurons grown in culture were stimulated by capsaicin (Hingtgen and Vasko, 1994). As illustrated in Figure 2B, CPGI2 alone significantly increased the number of labeled neurons to 38.1% (463 of 1216 neurons). The combination of CPGI2 and TNFα had no additional effect on the increase in neuronal number (510 of 1251 neurons, 40.8% positive), as compared with TNFα alone (41.8% positive). It does not seem likely that values between 30 and 40% were maximal, because previous work in adult, neonatal, or embryonic sensory neurons demonstrated that, with a higher concentration of capsaicin (1 μm), >50% of the neurons can be labeled with cobalt (Winter, 1987; Wood et al., 1988; Hingtgen and Nicol, 1994).
Tumor necrosis factor sensitizes the neuronal response to capsaicin
The results presented above demonstrate that TNFα increased the number of neurons that were labeled by cobalt after exposure to capsaicin. To further investigate the possibility that the increased number of positively labeled neurons resulted from a TNFα-mediated increase in the sensitivity of these neurons to capsaicin, we obtained the response to focally applied capsaicin from a single neuron via the whole-cell patch-clamp recording technique (Hamill et al., 1981). A representative response to capsaicin obtained under control conditions is illustrated in Figure 3 (top panel). At a holding potential of −60 mV, the focal application of 100 nm capsaicin (2 sec duration) elicited a peak inward current of 850 pA that slowly recovered to baseline. At the peak of the response there was an increase in the membrane current noise, suggesting that this inward current resulted from the opening of capsaicin-gated ion channels. Then the cell was superfused for another 2 min with Ringer’s solution to ensure complete recovery. At this point, the superfusate was changed to Ringer’s containing 1 μm capsaicin, for which a maximal inward current of 2300 pA was elicited. In recordings from eight neurons under control conditions, the inward current elicited by 100 nm capsaicin had an average value of 610 ± 163 pA (range 65–1238 pA). Exposure of these neurons to 1 μm capsaicin produced an average maximal response of 1701 ± 311 pA (range 700–3000 pA). In comparison with the control recordings, the neuronal response to capsaicin was enhanced greatly after a 24 hr treatment with 10 ng/ml TNFα (Fig. 3, bottom panel). In this representative neuron, a 2 sec pulse of 100 nm capsaicin applied focally elicited an inward current of 1725 pA; the bath application of 1 μm capsaicin produced a maximal response of 2450 pA. After a 24 hr treatment with TNFα, the average response to 100 nm capsaicin was 1446 ± 339 pA (range 400–4000 pA,n = 10), whereas the average maximal response was 1898 ± 387 pA (range 600–4800 pA). This large increase in the inward current evoked by the focal application of capsaicin after TNFα treatment did not result from an overall enhancement of the capsaicin response, because the average maximum response to 1 μm capsaicin obtained under control conditions was not significantly different from that after TNFα treatment.
Fig. 3.

TNFα enhances the amplitude of the capsaicin response. The top panel illustrates a representative response to the focal application of 100 nm capsaicin obtained under control conditions. The bottom panelshows the response from a different neuron to the focal application of capsaicin after a 24 hr treatment with 10 ng/ml TNFα. Thebars labeled CAP indicate the timing and duration of the applications of capsaicin. Both neurons were held at −60 mV; inward currents are shown as downward.
The notion that TNFα enhanced the capsaicin sensitivity of sensory neurons is supported further by examining the response amplitudes to the focal application of 100 nm capsaicin as a function of the response amplitudes to the bath application of 1 μmcapsaicin (i.e., the maximum response). As shown in Figure4, the amplitudes of the responses obtained under control conditions are fit by a linear regression line having a slope of 0.47 and a Pearson’s correlation coefficient of 0.90. In contrast, after treatment with TNFα the response amplitudes are larger and are now fit by a linear regression line having a slope of 0.77 and a correlation coefficient of 0.88. Therefore, these results indicate that treatment with TNFα heightened the sensitivity of sensory neurons to this excitatory agent without altering the magnitude of the maximal response.
Fig. 4.

TNFα enhances the response to capsaicin without altering the maximal response. The amplitude of the response to the focal application of 100 nm capsaicin is plotted as a function of the maximal response obtained in that same neuron for the bath application of 1 μm capsaicin. The filled circles represent those responses obtained under control conditions (n = 8), whereas the filled triangles illustrate the responses after a 24 hr treatment with 10 ng/ml TNFα (n = 10). The linesthrough the points are the linear regressions in which the fitting parameters are listed in the text. The broken lines represent the 95% confidence limits for each regression line.
The time course for sensitization of the capsaicin-evoked current by TNFα was very similar to that observed for the cobalt-labeling studies. As shown in Figure 5, the capsaicin response was enhanced by nearly twofold after a 4 hr treatment with 10 ng/ml TNFα. Likewise, after 12 and 24 hr treatments with 10 ng/ml TNFα, the relative amplitudes of the capsaicin-elicited current were doubled. However, an acute 10 min exposure to TNFα had no significant effect on the capsaicin response. In our previous studies, a 10 min exposure to PGE2 was sufficient to produce a maximal enhancement in the number of action potentials evoked by bradykinin (Nicol and Cui, 1994; Cui and Nicol, 1995). Therefore, these results suggest that TNFα does not have an immediate action on the sensitivity of these neurons to capsaicin, but, rather, the processes mediating this enhancement require a period of hours to become effective.
Fig. 5.

Time course for the TNFα-induced sensitization of the capsaicin response. The bars represent the current elicited by the focal application of 100 nmcapsaicin that was normalized to the maximal response obtained to the bath application of 1 μm capsaicin for different experimental treatments. The open bar represents untreated neurons under control conditions (n = 9). The hatched bar, labeled 10 M, represents the currents obtained after a 10 min exposure to 10 ng/ml TNFα (n = 5). The coarse-,medium-, and fine-hatched bars denote those currents recorded after 4 (n = 5), 12 (n = 5), and 24 (n = 5) hr treatments, respectively, with 10 ng/ml TNFα.
The neuronal responses to capsaicin obtained under control conditions and after TNFα treatment from two different sets of neurons are summarized in Figure 6A. After normalization, the application of 100 nm capsaicin under control conditions produced an average response that was 0.32 ± 0.05 (n = 8) of the maximal response. In those sensory neurons treated with 10 ng/ml TNFα for 24 hr, the average response to 100 nm capsaicin was increased significantly to 0.77 ± 0.07 (n = 10) of the maximal response. Thus, TNFα produced a greater than twofold enhancement in the sensitivity of sensory neurons to the focal application of capsaicin.
Fig. 6.
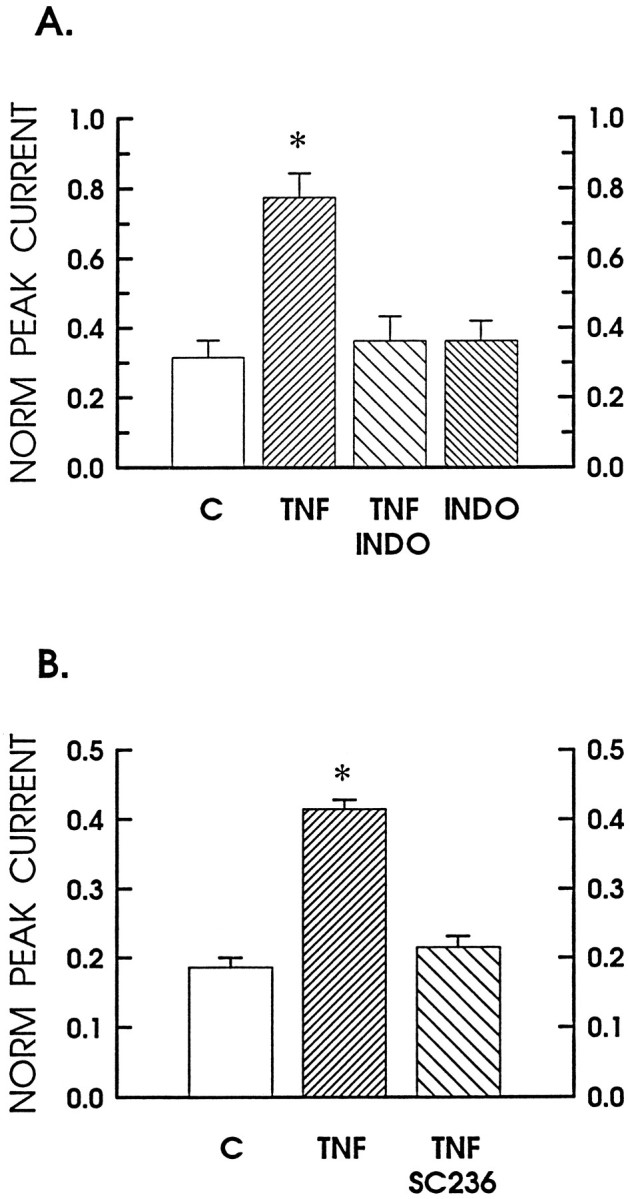
TNFα enhancement of the capsaicin response is blocked by inhibition of cyclo-oxygenase. A, The sensitizing effects of TNFα on the normalized response to capsaicin and its inhibition by indomethacin. The focal response to 100 nm capsaicin is expressed as the fraction of the maximal response obtained with the bath application of 1 μmcapsaicin. The bars represent the following experimental conditions: the control is shown as the open bar(n = 8); the fine-hatched bar, labeled TNF, is after a 24 hr treatment with 10 ng/ml TNFα (n = 10); the coarse-hatched bar is after treatment with 30 μm indomethacin and 10 ng/ml TNFα (n = 4); and thefine-hatched bar, labeled INDO, is after a 24 hr treatment with 30 μm indomethacin alone (n = 4). B demonstrates in another set of sensory neurons inhibition of the TNFα-induced sensitization by treatment with the selective COX-2 inhibitor SC-236. The open bar represents the untreated control neurons (n = 3). The fine-hatched barrepresents results obtained after a 24 hr treatment with 10 ng/ml TNFα (n = 5); the coarse-hatched bar is after treatment with 300 nm SC-236 and 10 ng/ml TNFα (n = 5). The asterisksindicate statistical significance at p < 0.05.
As with the cobalt-labeling studies, the possible contribution of cyclo-oxygenase products to the TNFα sensitization was examined by pretreating the neuronal cultures with 30 μm indomethacin before the addition of 10 ng/ml TNFα. As shown in Figure6A, indomethacin blocked the TNFα-induced increase in the response to 100 nm capsaicin such that, after exposure to indomethacin and TNFα, the average fractional response was only 0.36 ± 0.07 (n = 4) of the maximum and was similar to the control value of 0.32. A 24 hr treatment with 30 μm indomethacin alone had no significant effect on the average fractional response to 100 nm capsaicin (0.36 ± 0.06, n = 4). At this concentration, indomethacin inhibits both the constitutively active cyclo-oxygenase-1 (COX-1) and the inducible cyclo-oxygenase-2 [COX-2; Masferrer et al. (1994);Seibert et al. (1994b); but see Meade et al. (1993)]. In a separate series of experiments, a selective inhibitor of COX-2, SC-236 (Peter Isakson, personal communication), was used to distinguish which isoform mediated the TNFα-induced sensitization. This inhibitor of COX-2 has an activity on human enzymes with an IC50 of 0.01 μm for COX-2 and 18 μm for COX-1 (T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Zhang, P. C. Isakson, unpublished data). In the presence of 300 nm SC-236, the twofold increase in the capsaicin sensitivity produced by TNFα was blocked completely (Fig.6B). In another series of experiments, 10 μm SC-236 also produced a complete suppression of the TNFα-mediated enhancement (from 0.49 ± 0.03 to 0.21 ± 0.02), whereas SC-236 alone had no effect on the response to capsaicin (0.26 ± 0.03 vs 0.22 ± 0.02 for the control; data not shown). Therefore, using both population studies involving cobalt-labeling as well as electrophysiological studies examining the response obtained from a single neuron, we show results that indicate that pretreatment with TNFα enhanced the sensitivity of sensory neurons to the excitation elicited by capsaicin. Furthermore, the delayed time course of sensitization likely results from a TNFα-mediated induction of COX-2 that ultimately leads to the production of prostaglandins.
DISCUSSION
The results presented in this study demonstrate that TNFα, but not IL-1β, can sensitize isolated sensory neurons to the excitation produced by capsaicin. With the use of two different assays of neuronal sensitivity, TNFα increased the number of neurons labeled by cobalt after exposure to capsaicin as well as the amplitude of the inward current evoked by the focal application of capsaicin to a single neuron. Although the cellular pathways mediating this sensitization are not well characterized, it seems unlikely that the increased responsiveness results from an increase in the total number of capsaicin receptors, because the maximal inward current elicited by 1 μm capsaicin was similar in the absence or presence of TNFα.
The TNFα-induced sensitization likely is mediated by prostaglandins, because enhancement of the capsaicin response was blocked by both indomethacin, a nonselective inhibitor of cyclo-oxygenase, and SC-236, a specific inhibitor of COX-2. These results suggest that TNFα somehow leads to the induction and activation of COX-2 to release prostaglandins and, thus, enhances the response to capsaicin. Indeed, sensory neurons grown in culture can synthesize, from labeled arachidonic acid, many different prostanoids, most prominently PGE2, with smaller levels of 6-keto PGF1α (a breakdown product of PGI2), PGD2, and PGF2α (Vasko et al., 1994). Also, these sensory neurons were labeled heavily by a nonspecific antibody to cyclo-oxygenase (Vasko et al., 1994). Thus, sensory neurons express cyclo-oxygenase that is capable of generating prostaglandins. It is well documented that prostaglandins, especially PGE2 and PGI2(prostacyclin), play critical roles in the initiation of the heightened sensitivity to stimulation in both in vivo and in vitro models of neurogenic inflammation (Davies et al., 1984;Higgs et al., 1984). This notion is supported further by our observations that the number of cobalt-labeled neurons was increased by approximately twofold after a 24 hr pretreatment with the proinflammatory prostaglandin CPGI2. Likewise, exposure to CPGI2 on a much shorter time scale sensitizes isolated sensory neurons. Previously, we reported that pretreatment of sensory neurons with a higher concentration of CPGI2 (1 μm) for only 20 min produced a threefold increase the number of cobalt-labeled cells by capsaicin (Hingtgen and Nicol, 1994).
Currently, the intracellular transduction cascades linking the TNFα receptor and activation of cyclo-oxygenase are unknown. Recent evidence indicates that exposure to inflammatory mediators stimulates cyclo-oxygenase activity; however, the increased activity is limited to the inducible isoform COX-2 (Fu et al., 1990; Kujubu et al., 1991;O’Banion et al., 1991). Treatment with proinflammatory agents (Crofford et al., 1994; Masferrer et al., 1994; Seibert et al., 1994a,b; Feng et al., 1995) or tissue injury (Pritchard et al., 1994) induces the synthesis of COX-2 (prostaglandin G/H synthase 2) but has no effect on the constitutively active isoform COX-1 (prostaglandin G/H synthase 1). These observations indicate that it is the induction of COX-2, rather than increased activity of COX-1, which mediates the release of prostaglandins during the inflammatory response. Indeed, COX-2 is expressed in synovial tissues isolated from patients with rheumatoid arthritis, and when explants of these synovial tissues are grown in culture, IL-1β significantly increases the mRNA level of COX-2, but not COX-1 (Crofford et al., 1994). The notion that TNFα promotes the induction of COX-2 in sensory neurons is supported by two of our findings. First, our results demonstrate that selective inhibition of COX-2 with the compound SC-236 prevents the TNFα-induced sensitization of the capsaicin response. Second, TNFα does not have an immediate action on the capsaicin sensitivity of these neurons but, rather, requires ∼4 hr to become effective. This time course of cytokine action is supported by recent observations in which a significant increase in the relative expression of COX-2 mRNA was found 3 hr after producing carrageenan-induced inflammation in the paw of a rat (Seibert et al., 1994b). Also, in the isolated rat trachea, the capsaicin-evoked release of CGRP was potentiated after a 5 hr exposure to either TNFα or IL-1β; however, there was no effect after 2 hr (Hua et al., 1996). When taken together, experimental observations indicate that proinflammatory mediators have no significant effect on the levels or activity of COX-1; therefore, it seems highly likely that the enhanced sensitivity to capsaicin results from the ability of TNFα to induce the synthesis of COX-2.
In many ways the physiological actions of TNFα and IL-1β are believed to be quite similar (Dinarello, 1987; Le and Vilc̆ek, 1987; Arend and Dayer, 1990). For example, studies using behavioral measures for the perception of noxious stimuli in intact animals report that injection of either TNFα or IL-1β produces a hyperalgesic response (Ferreira et al., 1988; Schweizer et al., 1988; Follenfant et al., 1989; Cunha et al., 1992). In the intact animals, the cytokine-induced sensitization was blocked by pretreatment with indomethacin, also indicating a role for cyclo-oxygenase products. Similarly, in the isolated and perfused rat trachea, lipopolysaccharide, IL-1β, or TNFα facilitated the release of CGRP that was evoked by stimulation with capsaicin (Hua et al., 1996). These results, then, suggest that the threshold had been lowered, wherein TNFα or IL-1β had a sensitizing effect on the sensory afferent nerves, responding to the noxious or chemical stimulation. It is, however, curious that TNFα, but not IL-1β, sensitizes the isolated sensory neurons grown in culture to the excitation produced by capsaicin. Compared with TNFα, IL-1β had no significant effect on the number of cobalt-labeled neurons, even at a relatively high concentration (50 ng/ml). The lack of an IL-1β effect on the excitability of sensory neurons is supported by a similar finding in which, in recordings from cultured sympathetic neurons isolated from rat superior cervical ganglia, the calcium current was potentiated by long-term exposures (>4 hr) to TNFα, but not to IL-1β (Soliven and Albert, 1992). Therefore, our observations are consistent with the notion that the sensitizing effects of IL-1β observed in the intact animal or isolated tissue might result from an IL-1β-induced production of a secondary mediator released from other cell types rather than from a direct action of IL-1β on the sensory neuron.
The intracellular transduction cascades that mediate the prostaglandin-induced sensitization are poorly understood. Our previous observations suggest that these isolated sensory neurons grown in culture are an excellent model system in which to investigate the regulatory pathways activated by proinflammatory prostaglandins. Acute treatment with PGE2 enhances the excitability of isolated sensory neurons grown in culture in a manner that is similar to that observed in both in vivo and in vitro animal models of pain and neurogenic inflammation. For example, a 10 min pretreatment with 1 μm PGE2 produced a threefold increase in the number of action potentials elicited by the focal application of bradykinin (Nicol and Cui, 1994). Similarly, in the intact animal, the frequency of action potentials recorded from C-fibers in response to bradykinin was increased after the application of PGE2 (Handwerker, 1976) or PGE1 (Chahl and Iggo, 1977). Furthermore, acute treatment with either PGE2or CPGI2 enhanced the bradykinin-evoked release of substance P and calcitonin gene-related peptide from isolated sensory neurons grown in culture (Hingtgen and Vasko, 1994;Vasko et al., 1994). These neuroactive peptides have been demonstrated to be important mediators of neurogenic inflammation (Cuello, 1987; Foreman, 1987).
In a manner analogous to short-term applications, our current findings describing the effects of sustained treatment (24 hr) with TNFα and CPGI2 suggest that isolated sensory neurons grown in culture may prove to be a useful model system in which to explore how prolonged exposure to mediators associated with chronic inflammation alters the pathways controlling cellular sensitivity and excitability. Investigation of the cellular mechanisms whereby proinflammatory cytokines, such as TNFα, produce transcriptional changes that lead to either the induction or downregulation of various enzyme systems, such as COX-2, would enhance our understanding of the role played by sensory neurons in the maintenance of neurogenic inflammation. Indeed, patients afflicted with rheumatoid arthritis (a condition associated with chronic inflammation of the joints) have elevated levels of inflammatory cytokines and prostaglandins in their synovial fluids as well as a tendency for chronic pain in those joints (Arend and Dayer, 1990; Feldman et al., 1990; Bhoola et al., 1992; Konttinen et al., 1994).
In conclusion, the proinflammatory cytokine, TNFα, directly enhances the sensitivity of sensory neurons to the chemical excitatory agent capsaicin via a cyclo-oxygenase-dependent pathway. It is possible that this cytokine-mediated sensitization may be part of a more global mechanism whereby regulatory pathways associated with the immune system modulate the excitability of the nervous system.
Footnotes
This work was supported by National Institutes of Health Grant RO1-NS30527 (G.D.N.). J.C.L. was supported by the Indiana University Purdue University, Indianapolis (IUPUI) Research Investment Fund; C.M.P. was supported by the IUPUI Research Investment Fund and an Indiana Medical Scholar Fellowship. We are grateful to Drs. Michael Vasko and Angela Evans for discussions about signal transduction in sensory neurons. We thank Dr. Peter Isakson for the generous gift of SC-236.
Correspondence should be addressed to Dr. G. D. Nicol, Department of Pharmacology and Toxicology, School of Medicine, Indiana University, 635 Barnhill Drive, Indianapolis, IN 46202-5120.
REFERENCES
- 1.Arend WP, Dayer J-M. Cytokines and cytokine inhibitors or antagonists in rheumatoid arthritis. Arthritis Rheum. 1990;33:305–315. doi: 10.1002/art.1780330302. [DOI] [PubMed] [Google Scholar]
- 2.Baccaglini PI, Hogan PG. Some rat sensory neurons in culture express characteristics of differentiated pain sensory cells. Proc Natl Acad Sci USA. 1983;80:594–598. doi: 10.1073/pnas.80.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bevan S, Forbes CA. Membrane effects of capsaicin on rat dorsal root ganglion neurones in cell culture. J Physiol (Lond) 1988;398:28. [Google Scholar]
- 4.Bhoola KD, Elson CJ, Dieppe PA. Kinins—key mediators in inflammatory arthritis. Br J Rheumatol. 1992;31:509–518. doi: 10.1093/rheumatology/31.8.509. [DOI] [PubMed] [Google Scholar]
- 5.Burch RM, Tiffany CW. Tumor necrosis factor causes amplification of arachidonic acid metabolism in response to interleukin 1, bradykinin, and other agonists. J Cell Physiol. 1989;141:85–89. doi: 10.1002/jcp.1041410113. [DOI] [PubMed] [Google Scholar]
- 6.Burch RM, Connor JR, Axelrod J. Interleukin 1 amplifies receptor-mediated activation of phospholipase A2 in 3T3 fibroblasts. Proc Natl Acad Sci USA. 1988;85:6306–6309. doi: 10.1073/pnas.85.17.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang J, Gilman SC, Lewis AJ. Interleukin 1 activates phospholipase A2 in rabbit chondrocytes: a possible signal for IL-1 action. J Immunol. 1986;136:1283–1287. [PubMed] [Google Scholar]
- 8.Chahl LA, Iggo A. The effects of bradykinin and prostaglandin E1 on rat cutaneous afferent nerve activity. Br J Pharmacol. 1977;59:343–347. doi: 10.1111/j.1476-5381.1977.tb07498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crofford LJ, Wilder RL, Ristimäki AP, Sano H, Remmers EF, Epps HR, Hla T. Cyclo-oxygenase-1 and -2 expression in rheumatoid synovial tissues. Effects of interleukin-1β: phorbol ester and corticosteroids. J Clin Invest. 1994;93:1095–1101. doi: 10.1172/JCI117060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuello AC. Peptides as modulators in primary sensory neurons. Neuropharmacology. 1987;26:971–979. doi: 10.1016/0028-3908(87)90075-x. [DOI] [PubMed] [Google Scholar]
- 11.Cui M, Nicol GD. Cyclic AMP mediates the prostaglandin E2-induced potentiation of bradykinin excitation in rat sensory neurons. Neuroscience. 1995;66:459–466. doi: 10.1016/0306-4522(94)00567-o. [DOI] [PubMed] [Google Scholar]
- 12.Cunha FQ, Poole S, Lorenzetti BB, Ferreira SH. The pivotal role of tumour necrosis factor α in the development of inflammatory hyperalgesia. Br J Pharmacol. 1992;107:660–664. doi: 10.1111/j.1476-5381.1992.tb14503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies P, Bailey PJ, Goldenberg MM, Ford-Hutchinson AW. The role of arachidonic acid oxygenation products in pain and inflammation. Annu Rev Immunol. 1984;2:335–357. doi: 10.1146/annurev.iy.02.040184.002003. [DOI] [PubMed] [Google Scholar]
- 14.Dayer J-M, Demczuk S. Cytokines and other mediators in rheumatoid arthritis. Springer Semin Immunopathol. 1984;7:387–413. doi: 10.1007/BF00201968. [DOI] [PubMed] [Google Scholar]
- 15.Dayer J-M, Beutler B, Cerami A. Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med. 1985;162:2163–2168. doi: 10.1084/jem.162.6.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinarello CA. The biology of interleukin 1 and comparison to tumor necrosis factor. Immunol Lett. 1987;16:227–232. doi: 10.1016/0165-2478(87)90151-9. [DOI] [PubMed] [Google Scholar]
- 17.Dinarello CA. The proinflammatory cytokines interleukin 1 and tumor necrosis factor and treatment of the septic shock syndrome. J Infect Dis. 1991;163:1177–1184. doi: 10.1093/infdis/163.6.1177. [DOI] [PubMed] [Google Scholar]
- 18.Feldman M, Brennan FM, Chantry D, Haworth C, Turner M, Abney E, Buchan G, Barrett K, Barkley D, Chu A, Field M, Maini RN. Cytokine production in the rheumatoid joint: implications for treatment. Ann Rheum Dis. 1990;49:480–486. [PubMed] [Google Scholar]
- 19.Feng L, Xia Y, Garcia GE, Hwang D, Wilson CB. Involvement of reactive oxygen intermediates in cyclo-oxygenase-2 expression induced by interleukin-1, tumor necrosis factor-α, and lipopolysaccharide. J Clin Invest. 1995;95:1669–1675. doi: 10.1172/JCI117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira SH, Nakamura M, Salete de Abreu Castro M. The hyperalgesic effects of prostacyclin and prostaglandin E2. Prostaglandins. 1978;16:31–37. doi: 10.1016/0090-6980(78)90199-5. [DOI] [PubMed] [Google Scholar]
- 21.Ferreira SH, Lorenzetti BB, Bristow AF, Poole S. Interleukin 1β as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature. 1988;334:698–700. doi: 10.1038/334698a0. [DOI] [PubMed] [Google Scholar]
- 22.Follenfant RL, Nakamura-Craig M, Henderson B, Higgs GA. Inhibition by neuropeptides of interleukin-1β-induced prostaglandin-independent hyperalgesia. Br J Pharmacol. 1989;98:41–43. doi: 10.1111/j.1476-5381.1989.tb16860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foreman JC. Peptides and neurogenic inflammation. Br Med Bull. 1987;43:386–400. doi: 10.1093/oxfordjournals.bmb.a072189. [DOI] [PubMed] [Google Scholar]
- 24.Fu J-Y, Masferrer JL, Seibert K, Raz A, Needleman P. The induction and suppression of prostaglandin H2 synthase (cyclo-oxygenase) in human monocytes. J Biol Chem. 1990;265:16737–16740. [PubMed] [Google Scholar]
- 25.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 26.Handwerker HO. Influences of algogenic substances and prostaglandins on the discharges of unmyelinated cutaneous nerve fibers identified as nociceptors. Adv Pain Res Ther. 1976;1:41–45. [Google Scholar]
- 27.Heppelmann B, Schaible H-G, Schmidt RF. Effects of prostaglandins E1 and E2 on the mechanosensitivity of group III afferents from normal and inflamed cat knee joints. Adv Pain Res Ther. 1985;9:91–101. [Google Scholar]
- 28.Heyman I, Rang HP. Depolarizing responses to capsaicin in a subpopulation of rat dorsal root ganglion cells. Neurosci Lett. 1985;56:69–75. doi: 10.1016/0304-3940(85)90442-2. [DOI] [PubMed] [Google Scholar]
- 29.Higgs GA, Moncada S, Vane JR. Eicosanoids in inflammation. Ann Clin Res. 1984;16:287–299. [PubMed] [Google Scholar]
- 30.Hingtgen CM, Nicol GD. Carba prostacyclin enhances the capsaicin-induced cobalt loading of rat sensory neurons grown in culture. Neurosci Lett. 1994;173:99–102. doi: 10.1016/0304-3940(94)90158-9. [DOI] [PubMed] [Google Scholar]
- 31.Hingtgen CM, Vasko MR. Prostacyclin enhances the evoked-release of substance P and calcitonin gene-related peptide from rat sensory neurons. Brain Res. 1994;655:51–60. doi: 10.1016/0006-8993(94)91596-2. [DOI] [PubMed] [Google Scholar]
- 32.Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev. 1991;43:143–201. [PubMed] [Google Scholar]
- 33.Hua X-Y, Chen P, Fox A, Myers RR. Involvement of cytokines in lipopolysaccharide-induced facilitation of CGRP release from capsaicin-sensitive nerves in the trachea: studies with interleukin-1β and tumor necrosis factor-α. J Neurosci. 1996;16:4742–4748. doi: 10.1523/JNEUROSCI.16-15-04742.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimball ES. Involvement of cytokines in neurogenic inflammation. In: Kimball ES, editor. Cytokines and inflammation. CRC; Boston: 1991. pp. 169–189. [Google Scholar]
- 35.Konttinen YT, Kemppinen P, Segerberg M, Hukkanen M, Rees R, Santavirta S, Sorsa T, Pertovaara A, Polak JM. Peripheral and spinal neural mechanisms in arthritis, with particular reference to treatment of inflammation and pain. Arthritis Rheum. 1994;37:965–982. doi: 10.1002/art.1780370701. [DOI] [PubMed] [Google Scholar]
- 36.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclo-oxygenase homologue. J Biol Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- 37.Le J, Vilc̆ek J. Tumor necrosis factor and interleukin 1: cytokines with multiple overlapping biological activities. Lab Invest. 1987;56:234–248. [PubMed] [Google Scholar]
- 38.Marsh SJ, Stansfeld CE, Brown DA, Davey R, McCarthy D. The mechanism of action of capsaicin on sensory C-type neurons and their axons in vitro. Neuroscience. 1987;23:275–289. doi: 10.1016/0306-4522(87)90289-2. [DOI] [PubMed] [Google Scholar]
- 39.Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclo-oxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meade EA, Smith WL, DeWitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclo-oxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1993;268:6610–6614. [PubMed] [Google Scholar]
- 41.Mense S. Sensitization of group IV muscle receptors to bradykinin by 5-hydroxytryptamine and prostaglandin E2. Brain Res. 1981;225:95–105. doi: 10.1016/0006-8993(81)90320-6. [DOI] [PubMed] [Google Scholar]
- 42.Nicol GD, Cui M. Prostaglandin E2 enhances bradykinin activation of embryonic rat sensory neurones. J Physiol (Lond) 1994;480:485–492. doi: 10.1113/jphysiol.1994.sp020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Banion MK, Sadowski HB, Winn V, Young DA. A serum- and glucocorticoid-regulated 4-kilobase mRNA encodes a cyclo-oxygenase-related protein. J Biol Chem. 1991;266:23261–23267. [PubMed] [Google Scholar]
- 44.Pateromichelakis S, Rood JP. Prostaglandin E1-induced sensitization of Aδ moderate pressure mechanoreceptors. Brain Res. 1982;232:89–96. doi: 10.1016/0006-8993(82)90612-6. [DOI] [PubMed] [Google Scholar]
- 45.Pritchard KA, Jr, O’Banion MK, Miano JM, Vlasic N, Bhatia UG, Young DA, Stemerman MB. Induction of cyclo-oxygenase-2 in rat vascular smooth muscle cells in vitro and in vivo. J Biol Chem. 1994;269:8504–8509. [PubMed] [Google Scholar]
- 46.Raz A, Wyche AW, Siegel N, Needleman P. Regulation of fibroblast cyclo-oxygenase synthesis by interleukin-1. J Biol Chem. 1988;263:3022–3028. [PubMed] [Google Scholar]
- 47.Salmon JA, Higgs GA. Prostaglandins and leukotrienes as inflammatory mediators. Br Med Bull. 1987;43:285–296. doi: 10.1093/oxfordjournals.bmb.a072183. [DOI] [PubMed] [Google Scholar]
- 48.Schweizer A, Feige U, Fontana A, Müller K, Dinarello CA. Interleukin-1 enhances pain reflexes. Mediations through increased prostaglandin E2 levels. Agents Actions. 1988;25:246–251. doi: 10.1007/BF01965025. [DOI] [PubMed] [Google Scholar]
- 49.Seibert K, Masferrer J, Zhang Y, Gregory S, Olson G, Hauser S, Leahy K, Perkins W, Lee L, Isakson P. Mediation of inflammation by cyclo-oxygenase-2. Agents Actions Suppl. 1994a;46:41–50. doi: 10.1007/978-3-0348-7276-8_5. [DOI] [PubMed] [Google Scholar]
- 50.Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Pharmacological and biochemical demonstration of the role of cyclo-oxygenase 2 in inflammation and pain. Proc Natl Acad Sci USA. 1994b;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith WL. Prostanoid biosynthesis and mechanisms of action. Am J Physiol. 1992;263:F181–F191. doi: 10.1152/ajprenal.1992.263.2.F181. [DOI] [PubMed] [Google Scholar]
- 52.Soliven B, Albert J. Tumor necrosis factor modulates Ca2+ currents in cultured sympathetic neurons. J Neurosci. 1992;12:2665–2671. doi: 10.1523/JNEUROSCI.12-07-02665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Treede R-F, Meyer RA, Raja SN, Campbell JN. Peripheral and central mechanisms of cutaneous hyperalgesia. Prog Neurobiol. 1992;38:397–421. doi: 10.1016/0301-0082(92)90027-c. [DOI] [PubMed] [Google Scholar]
- 54.Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winter J. Characterization of capsaicin-sensitive neurones in adult rat dorsal root ganglion cultures. Neurosci Lett. 1987;80:134–140. doi: 10.1016/0304-3940(87)90642-2. [DOI] [PubMed] [Google Scholar]
- 56.Wood JN, Winter J, James IF, Rang HP, Yeats J, Bevan S. Capsaicin-induced ion fluxes in dorsal root ganglion cells in culture. J Neurosci. 1988;8:3208–3220. doi: 10.1523/JNEUROSCI.08-09-03208.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]