

Abstract
Electrophysiological and pharmacological studies of a cloned human dopamine transporter (hDAT) were undertaken to investigate the mechanisms of transporter function and the actions of drugs at this target. Using two-electrode voltage-clamp techniques with hDAT-expressing Xenopus laevis oocytes, we show that hDAT can be considered electrogenic by two criteria. (1) Uptake of hDAT substrates gives rise to a pharmacologically appropriate “transport-associated” current. (2) The velocity of DA uptake measured in oocytes clamped at various membrane potentials was voltage-dependent, increasing with hyperpolarization. Concurrent measurement of transport-associated current and substrate flux in individual oocytes revealed that charge movement during substrate translocation was greater than would be expected for a transport mechanism with fixed stoichiometry of 2 Na+ and 1 Cl− per DA+ molecule. In addition to the transport-associated current, hDAT also mediates a constitutive leak current, the voltage and ionic dependencies of which differ markedly from those of the transport-associated current. Ion substitution experiments suggest that alkali cations and protons are carried by the hDAT leak conductance. In contrast to the transport-associated functions, the leak does not require Na+ or Cl−, and DAT ligands readily interact with the transporter even in the absence of these ions. The currents that hDAT mediates provide a functional assay that readily distinguishes the modes of action of amphetamine-like “DA-releasing” drugs from cocaine-like translocation blockers. In addition, the voltage dependence of DA uptake suggests a mechanism through which presynaptic DA autoreceptor activation may accelerate the termination of dopaminergic neurotransmission in vivo.
Keywords: Na+/Cl−-dependent, carrier, cocaine, amphetamine, methamphetamine, methylphenidate, MPP+, uptake, release, Xenopus oocyte, psychomotor stimulant
The dopamine transporter (DAT) is thought to be a principal site of action for several psychomotor stimulants, including cocaine, amphetamine, methamphetamine, and methylphenidate—drugs used widely for both therapeutic and nontherapeutic purposes. After release of the neurotransmitter dopamine (DA), its extracellular concentrations are regulated primarily by reaccumulation into dopaminergic neurons through the action of DAT and by diffusion. Accordingly, pharmacological inhibition of DA uptake may both prolong the duration of DA action at its receptors and expand the spatial domain of its actions in a manner comparable to that reported for GABA uptake inhibition (Isaacson et al., 1993). Numerous in vivomicrodialysis and electrochemical studies have shown that psychomotor stimulants raise extracellular DA concentrations (for review, see Wise, 1996). Similarly, in vitro and in vivoelectrophysiological experiments demonstrate that stimulant actions are consistent with elevation of DA concentrations (for review, see Lacey, 1993; White, 1996). Although cocaine, amphetamine, and many of their congeners display comparable actions at serotonin and norepinephrine transporters (SERT and NET), DAT is the pharmacological target best correlated with their reinforcing properties and abuse potential (Ritz et al., 1987; Nestler et al., 1993; Pulvirenti and Koob, 1994; Wise, 1996). Gene disruption experiments demonstrate that DAT may be the principal mediator of the locomotor stimulatory effects of cocaine and amphetamine, and that DAT plays a critical role setting dopaminergic “tone” in the murine CNS (Giros et al., 1996). Thus, there is considerable interest in understanding the molecular mechanisms by which DAT functions.
DAT belongs to a recently cloned family of neurotransmitter and amino acid transporters that are functionally related by their requirement for extracellular Na+ and Cl− (Amara and Kuhar, 1993). These transporters operate by coupling the transmembrane translocation of organic substrates to the movement of driving ions down preestablished electrochemical gradients (Kanner and Schuldiner, 1987; Rudnick and Clark, 1993). Biochemical studies of the dependence of DA uptake on Na+ and Cl− concentrations suggest that two Na+ ions and one Cl− ion are cotransported with each DA molecule (Krueger, 1990; McElvain and Schenk, 1992; Kilty, 1993; Gu et al., 1994). These results predict that the transport of each DA+ molecule will be accompanied by the movement of two net positive charges (because DA is positively charged at physiological pH), and will thereby generate an inward current.
A number of neurotransmitter/ion cotransporters generate detectable electrical currents during the process of substrate translocation bothin situ and in reconstituted systems. Certain transporters, furthermore, exhibit ion channel-like electrical activities that would not be predicted from classical alternating-access models of carrier function (for review, see Lester et al., 1994; DeFelice and Blakely, 1996; Sonders and Amara, 1996). To investigate the electrogenic properties of DAT, we have studied a cloned human DAT with two-electrode voltage-clamp methods. By expressing hDAT inXenopus laevis oocytes and applying a combination of electrophysiological, pharmacological, and biochemical techniques, we can assess its translocation activity in real time and examine in greater detail the voltage dependence, ionic coupling, and channel-like properties of this carrier. These studies also provide insights into the actions of an important class of neuropharmacological agents.
MATERIALS AND METHODS
hDAT cloning and expression. Total RNA was extracted according to the method of Chomczynski and Sacchi (1987) from a single human midbrain sample and reverse-transcribed into cDNA using SuperScript II (Life Technologies, Grand Island, NY) and the oligonucleotide GTCTTCGTCTCTGCTCCC complementary to the sense strand of hDAT subsequent to the termination codon (Giros et al., 1992;Vandenbergh et al., 1992). hDAT cDNA was amplified by PCR using Vent DNA polymerase (New England Biolabs, Beverly, MA) and primers overlapping 23 and 21 nucleotides of the 5′ and 3′ ends of the 1860 nucleotide coding region. The PCR transcript was digested and directionally ligated into the oocyte transcription vector pOTV (Arriza et al., 1994). The insert was sequenced in its entirety and found to be identical at the nucleotide level to the hDAT allele reported by Vandenbergh et al. (1992) (GenBank accession no. M95167). Capped cRNA was transcribed from linearized DAT plasmid using T7 polymerase (mMessage mMachine, Ambion), diluted with water, and injected into defolliculated stage V or VI oocytes (∼10 ng/oocyte). Oocytes were prepared as described by Quick and Lester (1994) and maintained at 17 or 21°C for up to 3 weeks. The presence of hDAT in cRNA-injected oocytes was confirmed by Western blot analyses, which displayed immunoreactive bands not detected in water-injected oocytes.
Uptake assays. Transport of DA into individual oocytes was quantitated either by liquid scintillation spectroscopy of [3H]DA or HPLC-coupled electrochemical detection (HPLC-EC) of nonradiolabeled DA. All uptake experiments were performed at ambient temperature using frog Ringer’s solution, containing (in mm): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES-NaOH, pH 7.4–7.5, or ion-substituted versions, as specified. Uptake kinetic experiments were initiated with the addition of tritiated or unlabeled DA to groups of oocytes (3–6) in a final volume of 500 μl and terminated by transferring the oocytes through three sequential 5 ml baths of ice-cold Ringer’s (total transfer time, <20 sec). Radiolabeled DA was quantitated by liquid scintillation spectroscopy after dissolving individual oocytes in 250 μl of 0.2% SDS. Intracellular DA concentration determination by HPLC-EC (Gerhardt et al., 1989) was performed by sonicating individual oocytes in 0.8 ml of 2 mm perchloric acid and chromatographing aliquots equivalent to 1/50-1/160 of the centrifugation supernatant (16,000 × g, 4°C, 15 min). Retention time of standards was used to identify peaks, and peak heights were used to calculate absolute amounts of DA. Nominal detection limits for DA and dihydroxyphenylacetic acid were 0.5 and 0.25 pg per injection, respectively.
The velocity of DA uptake was essentially constant over incubation periods between 0.5 and 60 min using 75–200 nm[3H]DA. Accordingly, incubation periods between 100 sec and 30 min were used in uptake studies. Saturation experiments used at least eight DA concentrations between 10 nm and 1 mm. Nonspecific uptake was determined by performing parallel incubations of water-injected oocytes or of cRNA-injected oocytes in the presence of saturating concentrations of uptake inhibitors mazindol, cocaine, RTI-55, or GBR 12909. Nonspecific uptake of radioligand was always <3% of total uptake, and for incubations ≥5 min, it was typically <1% of total uptake. Oocytes were preincubated in drug/buffer solutions for at least 5 min before addition of [3H]DA.
In ion substitution experiments, Na+ or Cl−ions in frog Ringer’s solution were either partially or fully replaced. LiCl and KCl were used to replace NaCl, and in most cases, LiOH and KOH were used to adjust the pH of the buffers. Cl− substitution experiments were performed with morpholinoethylsulfonate (MES)-NaOH and MES-LiOH in place of NaCl at a concentration of 96 mm, either with 6.6 or 0 mm Cl− remaining. In the latter case, KNO3, Ca(NO3)2, and MgSO4 were used in place of the corresponding chloride salts.
For uptake and binding data, nonlinear regression analyses were performed with KaleidaGraph 3.0 or GraphPAD software to generate estimates of kinetic constants KT (the apparent affinity of transport), Vmax,Ki, IC50,KD, and Bmax.Ki values were calculated from IC50 using the Cheng–Prusoff equation. Geometric means were determined for the apparent affinity valuesKT, Ki,KD, and IC50. Error values are given as SEMs or, where appropriate, 95 or 99% confidence intervals (CI95 or CI99).
Drugs were obtained from National Institute on Drug Abuse Research Technology Branch or were purchased from Research Biochemicals International (Natick, MA) or Sigma (St. Louis, MO). Radiochemicals were purchased from DuPont NEN (Boston, MA) and Amersham (Arlington Heights, IL).
Determination of hDAT density and turnover rate. The turnover rate of hDAT was estimated from the ratioVmax:Bmax. In each of three batches of oocytes, parallel measurements were made of the velocity of DA uptake (Vmax) and the density of hDAT sites (Bmax). Vmaxvalues were calculated from saturation analyses of assays using either liquid scintillation spectrometry or HPLC-EC to quantitate accumulation of tritiated or unlabeled DA, respectively (8 concentrations, triplicate determinations). Saturation binding assays were performed on intact ooctyes using 5 nm [3H]mazindol and six concentrations of unlabeled mazindol (1 nm–1 μm, triplicate determinations) in frog Ringer’s solution. Oocytes were incubated in 1 ml of [3H]mazindol solutions for 45–60 min on ice, washed for 5–10 sec in 4 ml of ice-cold buffer, and then solubilized individually in 200 μl of 1% SDS. Radioactivity was determined with liquid scintillation counting. Nonspecific binding was determined with 1 μm GBR 12909.
Two-electrode voltage-clamp electrophysiology.Two-microelectrode voltage-clamp recordings from oocytes were performed at room temperature using glass microelectrodes filled with 3m KCl solutions (resistance, <1 MΩ) and an Ag/AgCl-pellet bath ground or an active bath probe. Dagan TV-200, Axon GeneClamp 500, or Warner OC-725B amplifiers were used with AxoLab-1 or DigiData1200 interfaces. The pClamp suite of programs (Axon Instruments, Foster City, CA) was used to control stimulation parameters, for data acquisition, and for analysis. MacLab data acquisition software and a MacLab/2e interface (ADInstruments, Milford, MA) were used simultaneously to monitor and record electrophysiological experiments. Currents were low-pass filtered between 10 Hz and 2 kHz, and digitized at rates between 1 and 5 kHz. Frog Ringer’s buffer and ion-substituted versions (described above) were superfused over voltage-clamped oocytes at a rate of ∼4 ml/min (bath volume, 0.5 ml). During Cl− substitution experiments, 3 mKCl/agar bridges were used to avoid voltage offsets associated with buffer changes.
The voltage dependence of hDAT-mediated currents was studied using two voltage excursion protocols. In one protocol, membrane potentials were ramped between −130 mV and +80 mV over a 750 msec interval. The second protocol used a sequence of jumps in membrane potential in 10 mV increments to measure steady-state currents at potentials between −120 mV and +40 mV. Oocytes were held at −60 mV (600–750 msec) before jumps to each test potential (duration 250–400 msec). Before each application of drug, two voltage excursions were executed to measure the control currents and establish that they were stable. For voltage jump protocols, current values were measured and averaged during the final 80 msec of the test interval when they had reached steady state.
Currents attributable to the actions of drugs were determined by performing off-line subtraction of currents recorded during buffer perfusion (control) and currents recorded during drug perfusion. Substrate-elicited currents were measured by subtracting the control currents from those recorded during substrate application (IDrug − IControl). However, changes in membrane current brought about by nonsubstrate transport antagonists were determined by a subtraction using a reversed order (IControl −IDrug), because these drugs appear only to block membrane conductances. This convention is obeyed in all figures except Figure 3, in which the aim is to compare the actions of DA and cocaine, and all data are plotted as IDrug −IControl. The sole exception to the pattern of DA-elicited currents being plotted as IDA −IControl is found in Figure 7: under Na+-free conditions, all DAT ligands only block a membrane conductance; hence, the current–voltage (I–V) of the conductances blocked by DA and cocaine are both plotted asIControl − IDrug.
Fig. 3.

Antagonism of DA transport-associated current by cocaine. I–V plots of currents elicited during consecutive applications to a single oocyte of 3 μmcocaine (Coc; - - -), 2 μm DA (——), and 3 μm cocaine plus 2 μm DA (— —). Currents were measured during voltage ramps (−130 to +80 mV in 750 msec), and subtractions all used the IDrug− IControl convention. Comparison of the DAI–V with the DA + Cocaine I–V reveals the transport-associated component that is susceptible to blockade by cocaine.
Fig. 7.

Effects of Na+ substitution on leak conductance I–V relations. A, Sequential application to a single oocyte of 3 μm cocaine in Na+ Ringer’s buffer (▪), and then after a change to 96 mm K+ (2 mmNa+) Ringer’s buffer, of 10 μm DA (▵) and 3 μm cocaine (□). Subtractive currents (IControl −IDrug) determined during voltage jump protocols represent the I–V of the leak conductance, which is blocked by all three drug treatments. CocaineI–V plots display identical reversal potentials in the two buffers (−16 mV in this cell). In low Na+ buffer, the DA I–V displays the same reversal potential. Moreover, DA elicits no inward transport-associated current at negative potentials. B, In 96 mm Li+ (2 mm Na+) Ringer’s buffer, voltage jumps during sequential applications of 10 μm DA (▿) and 10 μm cocaine (⊞) reveal that both drugs block a conductance with the same reversal potential (−4 mV). Comparable results were observed when Cl− was replaced with MES in Li+ Ringer’s (not shown). These data are representative of four repetitions.
Concentration–response data were collected by measuring subtracted currents (IDA −IControl) during voltage jump protocols. To six oocytes, concentrations of DA were superfused at least twice (in randomized order), and the two responses for each concentration were averaged. The concentrations studied were 0.1, 0.3, 1.0, 3.0, 10.0, and 30.0 μm, although the 30 μm concentration was examined in only five oocytes. To control for variation in expression levels between oocytes, the concentration dependence and voltage dependence of DA-evoked currents were analyzed by normalizing current responses in an individual oocyte to that evoked by 10 μm DA at −120 mV. The DA concentrations evoking a half-maximal current (K0.5) at single membrane potentials were determined by nonlinear regression using the Michaelis–Menten equation. At potentials greater than −20 mV, responses evoked by 30 μm DA were omitted from the regressions because at high concentrations, several phenethylamines appeared to block endogenous channels. The voltage dependence of mean normalized currents evoked at each DA concentration was studied by nonlinear regression analysis. Comparable experiments were performed using S(+)amphetamine over the concentration range 0.03–30 μm.
DA uptake under voltage-clamp conditions. The dependence of DA uptake velocity on membrane potential was studied by measuring substrate accumulation in oocytes held in voltage-clamp at various potentials (+10, 0, −30, −60, −90, or −120 mV). DA uptake was measured in six batches of oocytes either by HPLC-EC after 3 or 5 min perfusions of 20 μm DA, or by liquid scintillation spectroscopy after 100 sec perfusions with 10.1 μm[3H]DA (0.37 Ci/mmol). After incubations with DA or [3H]DA, oocytes were briefly superfused with frog Ringer’s, voltage clamps were shut off, electrodes were withdrawn, and the oocytes were transferred to either perchlorate or SDS solutions and treated as described above (≤30 sec). DA uptake velocities were calculated on a per-second basis. The voltage dependence of uptake velocity was quantified in each batch of oocytes by normalizing mean velocities at each potential to that measured at −30 mV. Net charge movement attributable to substrate translocation during DA/[3H]DA perfusion was calculated off-line by graphically integrating the DA-elicited current in each oocyte with MacLab (5 of 6 oocyte batches). For each oocyte, an estimate of the net charge:DA flux ratio was calculated as follows: Net Charges Translocated/(Moles of Accumulated DA × Faraday Constant).
Ion substitution experiments. Reversal potentials were determined by visual inspection of I–V plots generated with Clampfit. After each cocaine perfusion, oocytes were perfused for at least 8 min before subsequent drug applications to allow adequate drug washout. Ion permeability ratios were calculated from shifts in the reversal potential of cocaine-elicited currents in different ion-substituted buffers using the Goldman–Hodgkin–Katz voltage equation (Hille, 1992).
RESULTS
DA uptake studies
To establish the validity of the Xenopus oocyte expression system for investigating the electrophysiological properties of hDAT, the kinetic and pharmacological properties of [3H]DA uptake were studied. Levels of hDAT expression were fairly consistent among oocytes of a single injection batch; however, they varied widely between batches of oocytes. The time of maximal hDAT expression also varied between batches, peaking between 4 and 14 d after injection. Oocytes expressing hDAT accumulated [3H]DA in a time- and ion-dependent manner. For instance, when oocytes from four separate batches were incubated in [3H]DA (30–150 nm) for 30 min, they accumulated radioligand to levels >40-fold above external concentrations, assuming an intracellular aqueous volume of 0.5 μl (n = 12 assays). By contrast, control oocytes from these and other batches did not concentrate [3H]DA whatsoever, and excluded more than three-fourths the radioactivity found in a comparable volume of incubation bath (n > 60 assays). Control oocytes included uninjected and water-injected oocytes, or cRNA-injected oocytes that were coincubated with ≥3 μm mazindol, ≥20 μm cocaine, ≥1 μm RTI-55, or 1 μm GBR 12909. [3H]DA uptake displayed strong Na+ and Cl− dependence: complete replacement of Na+with Li+, K+, Cs+, or choline reduced [3H]DA uptake by 97–98% (n ≥ 2 assays for each substitute). Complete replacement of external Cl− diminished [3H]DA uptake by 96.5%; in comparison, parallel water-injected oocytes accumulated 98.9% less than hDAT cRNA-injected oocytes (n = 2 assays).
Kinetic parameters for DA translocation by hDAT were similar in uptake assays using either liquid scintillation spectroscopy to detect accumulated [3H]DA or HPLC-EC to measure intracellular DA. Saturation uptake experiments performed at 21°C with six different oocyte batches yielded a mean KT value of 1.7 μm (CI95 0.7–4.1 μm). Greater batch-to-batch variability was observed in DA uptake velocity than in apparent affinity, as would be expected to result from differing levels of hDAT expression. (Vmaxvalues ranged between approximately 15 and 400 fmol/sec (21°C) over all experiments.) Cocaine and S(+)amphetamine inhibited uptake of [3H]DA in a concentration-dependent manner with meanKi values of 206 nm(CI95 102–414 nm; n = 3) and 297 nm (CI95 137–647 nm;n = 3), respectively. Cocaine exhibited a Schild slope (Kenakin, 1987) of 0.94 (1 experiment), which is compatible with a competitive interaction with DA at hDAT.
Estimation of hDAT turnover rate
To determine the turnover rate of hDAT, parallel measurements of DA uptake velocity and hDAT density were made in three oocyte batches to calculate theVmax:Bmax ratio (Table1). Maximal DA uptake velocities were measured at room temperature using radiolabeled flux or HPLC-EC. Catabolism of accumulated DA to dihydroxyphenylacetic acid in hDAT oocytes was not detected by HPLC-EC analysis, indicating that accumulated tritium likely represents authentic [3H]DA. Endogenous DA was not detected by HPLC-EC in uninjected or water-injected oocytes. TheBmax of [3H]mazindol, which labels DAT in rat striatum with nanomolar affinity (Javitch et al., 1984), was determined on intact oocytes to estimate the number of transporters expressed per oocyte. A single class of binding sites with an affinity similar to that in rat brain was revealed. Assuming that each mazindol binding site represents a functional DAT molecule on the oocyte surface, the meanVmax:Bmax ratio estimates the turnover rate to be 0.47 DA molecules/(sec × transporter molecule), suggesting that each transport cycle requires ∼2 sec at 21°C.
Table 1.
hDAT turnover rate determined from saturation analyses of DA uptake velocity and [3H]mazindol binding to intact oocytes
| DA uptake1-a | [3H]mazindol binding | Turnover rate | ||
|---|---|---|---|---|
| Vmax | KT | Bmax | KD | Vmax/Bmax |
| (fmol/(oocyte × sec)) | (μm) | (fmol/oocyte) | (nm) | (sec−1) |
| 66.9 ± 6.3 | 2.71-b | 152.5 ± 7.0 | 5.21-c | 0.47 ± 0.06 |
Mean values (±SEM) for Vmax andBmax were determined in three experiments using different oocyte batches (not voltage-clamped). In each experiment, parallel pools of oocytes were used for Vmax and for Bmax measurements.
DA uptake was measured solely with [3H]DA in one experiment and solely by HPLC-EC in a second experiment. Both methods were used in a third experiment and yielded Vmax values that differed by 7%.
CI95 0.2–36 μm.
CI95 2.9–9.2 nm.
Two-electrode voltage-clamp studies of hDAT currents
Preliminary calculations based on the Vmaxof DA uptake by hDAT-expressing oocytes indicated that if DA translocation were accompanied by a net charge movement, then an inward current attributable to hDAT transport activity should be detectable by two-electrode voltage-clamp methods. Figure1A shows the changes in membrane current elicited by drug application to a hDAT-expressing oocyte voltage-clamped at −60 mV. Superfusion with 20 μm DA evoked a downward displacement in the current trace, consistent with a net inward current that occurs as a result of translocation of DA+ and Na+ ions. In contrast, 10 μm cocaine evoked outward currents (that only slowly washed out) both on initial application and on reapplication after DA superfusion of the same oocyte. The drug-elicited currents are attributable to hDAT because they were not observed in uninjected or water-injected oocytes (Fig. 1B).
Fig. 1.

Cocaine and DA applications to a voltage-clamped hDAT oocyte evoke opposite changes in membrane current. Drug applications (solid bars) to oocytes voltage-clamped at −60 mV. A, An hDAT-expressing oocyte was initially superfused with 10 μm cocaine (Coc), which elicited a small outward current that slowly returned to baseline after 10 min of washout (flow rate 4 ml/min, chamber volume 0.5 ml). Superfusion of 20 μm DA induced an inward current that rapidly returned to baseline. Reapplication of cocaine caused an outward current comparable in magnitude and kinetics to that evoked by its initial application. B, No responses were evoked by drug application to a water-injected control oocyte.
That cocaine could elicit any change in the holding current of an hDAT-expressing oocyte in the absence of DA was somewhat unexpected, because cocaine is generally viewed as a nontransported uptake inhibitor that interacts with DA in an apparently competitive manner. It was also intriguing that the cocaine-evoked current was opposite in polarity to the DA-elicited current. Even though cocaine has occasionally been reported to cause DA release (Baumann and Maitre, 1976; Pifl et al., 1995), the outward current elicited by cocaine does not arise from DA extrusion because the outward current could be evoked by cocaine before the initial presentation of extracellular DA (Fig.1A), and oocytes were found by HPLC-EC to have little or no endogenous DA (≤50 nm, data not shown). The opposite DA- and cocaine-elicited currents suggest that hDAT mediates at least two distinct steady-state ionic conductances, one of which is associated with substrate transport, whereas the other can be observed in the absence of translocation activity. Mager and colleagues (1994) noted analogous currents manifested by rat SERT and termed them transport-associated and leak currents. The following experiments illustrate that the steady-state ionic conductances that give rise to the hDAT transport-associated and leak currents are distinguishable based on voltage dependence, ionic selectivity, and mode of drug action.
I–V relations of transport-associated and leak currents
To better understand the changes in ionic conductance that underlie the currents mediated by hDAT, drug-evoked currents were studied across a voltage range, using both ramp and jump protocols to control membrane potentials. The actions of drugs in changing membrane conductance were identified by comparing membrane currents measured during drug superfusion and during buffer superfusion (control conditions). Subtraction of currents measured in the presence and absence of hDAT ligands yielded I–V plots for the drugs.
Figure 2 shows results from a voltage ramp experiment in which DA and cocaine were applied sequentially to a single oocyte. At both negative and positive potentials, 3 μm cocaine superfusion diminished the magnitude of the membrane current compared with that measured during the control voltage ramp. This result is consistent with the cocaine-evoked outward current observed in an oocyte clamped at −60 mV (Fig. 1A) and suggests that the cocaine-elicited outward current likely reflects the action of the drug in blocking a constitutive membrane conductance, rather than an ability to activate either K+ ion efflux or Cl− ion influx. This point is made more obvious by inspection of the difference between cocaine and control voltage ramps.
Fig. 2.

Membrane conductance changes evoked by DA and cocaine. Currents measured in a single voltage-clamped oocyte during voltage ramps (−130 to +80 mV in 750 msec). A, Membrane currents measured during buffer (- - -) and 3 μm cocaine (——) superfusion. B, I–V plot describing current that is blocked by cocaine, determined by subtraction of currents (IControl −ICocaine) plotted in A.C, Membrane currents measured during buffer (- - -) and 20 μm DA (——) superfusion. D,I–V of current elicited by DA, determined by subtraction of currents (IDA −IControl) plotted inC.
The I–V curve describing the tonic leak conductance that is susceptible to blockade by cocaine is derived according to convention by taking the difference of IControl −ICocaine (Fig. 2B). This current was voltage-dependent, outwardly rectifying, and reversed at approximately −10 mV. The slope and reversal potential of this current both argue against an action of cocaine to increase membrane permeability to K+ or Cl− ions because: (1) the amplitude of currents carried by either ion would be expected to increase with depolarization between −60 mV and −10 mV, whereas the observed current decreases over this range; and (2) the reversal potential of approximately −10 mV is more positive than the Nernst potentials for K+ or Cl− in oocytes (−95 mV and −28 mV, respectively) (Dascal, 1987; Costa et al., 1989). That the observed reversal potential is also more negative than the Nernst potential for Na+ (61 mV) (Costa et al., 1989) suggests that the tonic leak conductance may be nonselective for cations, an assertion that is more directly supported by ion substitution experiments presented below.
The effect of DA superfusion on hDAT oocyte membrane conductance is more complex than that of cocaine because, at extremes of the potential range, DA clearly evokes two opposite effects. At positive potentials, DA acted to reduce the membrane conductance compared with control in a manner analogous to cocaine; in contrast, at negative potentials, DA superfusion increased the membrane conductance (Fig. 2C). Because DA is believed to increase transport-associated ionic flux, the DA-elicited current was determined by subtraction using the convention of IDA − IControl (Fig.2D). The resulting DA I–V curve exhibited an asymmetric inverted U-shape, with a positive slope conductance (at hyperpolarized potentials) representing the transport-associated current and a negative slope conductance (at depolarized potentials) representing a cocaine-like blocking action on a tonic leak conductance. As a result of the opposing conventions used to describe DA-elicited and cocaine-inhibitable I–V relationships, the similar inhibitory actions of the two drugs on a tonic leak conductance are manifested as I–V plots with negative slope conductance for DA but with positive slope conductance for cocaine over the range of positive potentials (Fig. 2B,D). Despite this artificial disparity in plotting, the suggestion that both drugs may block an identical leak conductance is explored further in ion substitution experiments presented below.
Over the range of negative potentials, Figures 1 and 2 suggest that when applied separately, DA and cocaine may act on distinct hDAT ionic conductances. However, because cocaine is a well established inhibitor of DA translocation, it would be expected that cocaine should be able to inhibit ionic flux (i.e., current) that accompanies substrate transport. To test for the possibility of such an interaction, voltage ramps were executed during superfusion with DA and cocaine in combination. Figure 3 displays the I–Vvalues elicited by sequential application to a single representative oocyte of 3 μm cocaine (∼10 ×Ki), 2 μm DA (approximately KT), and the two drugs together. (To facilitate comparison, only the IDrug(s) −IControl convention was used for current subtractions.) Coapplication of 2 μm DA and 3 μm cocaine gave rise to a voltage-sensitive current that was not the sum of the individually elicited currents but that virtually overlapped the cocaine I–V trace. Comparison of the DA I–V values in the absence and presence of cocaine makes clear that cocaine fully antagonized the inward DA current otherwise seen at negative potentials. That the DA-elicited current observed at negative potentials was abolished by a near-saturating concentration of cocaine supports the contention that this current is closely associated with substrate translocation by hDAT.
From these data, it appears that the electrophysiological action of cocaine on hDAT is limited to blocking ionic conductances—either the tonically active leak conductance when cocaine is applied alone, or the transport-associated conductance when it is coapplied with DA. DA, on the other hand, increases the transport-associated conductance, yet also blocks the tonic leak conductance in a cocaine-like manner. Hence, the observed substrate I–V plots likely represent the net effect of substrates acting simultaneously on the two different hDAT conductances: at negative potentials, activation of the transport-associated conductance is the predominating effect, whereas at positive potentials, blockade of the tonic leak conductance is most apparent.
Concentration and voltage dependence of DA-elicited currents
To further explore the concentration, voltage, and ionic dependence of the transport and leak conductances, as well as their pharmacological sensitivity, steady-state currents were measured in oocytes during voltage jump protocols. Figure4A displays the mean normalized responses elicited by application of varying DA concentrations to six oocytes. To control for variations in expression levels between cells, the currents elicited in each oocyte were normalized to that observed at −120 mV in response to 10 μm DA. For all DA concentrations, the I–V relations took the shape of an inverted U, the peak of which was approximately −20 mV and which resembled the subtracted I–V plots generated in non-steady-state ramp protocols (Fig. 2D). The amplitudes of DA-elicited currents increased in a dose-related manner in all oocytes. Comparable concentration-dependent responses were observed in two oocytes treated with a range of S(+)amphetamine concentrations (data not shown).
Fig. 4.
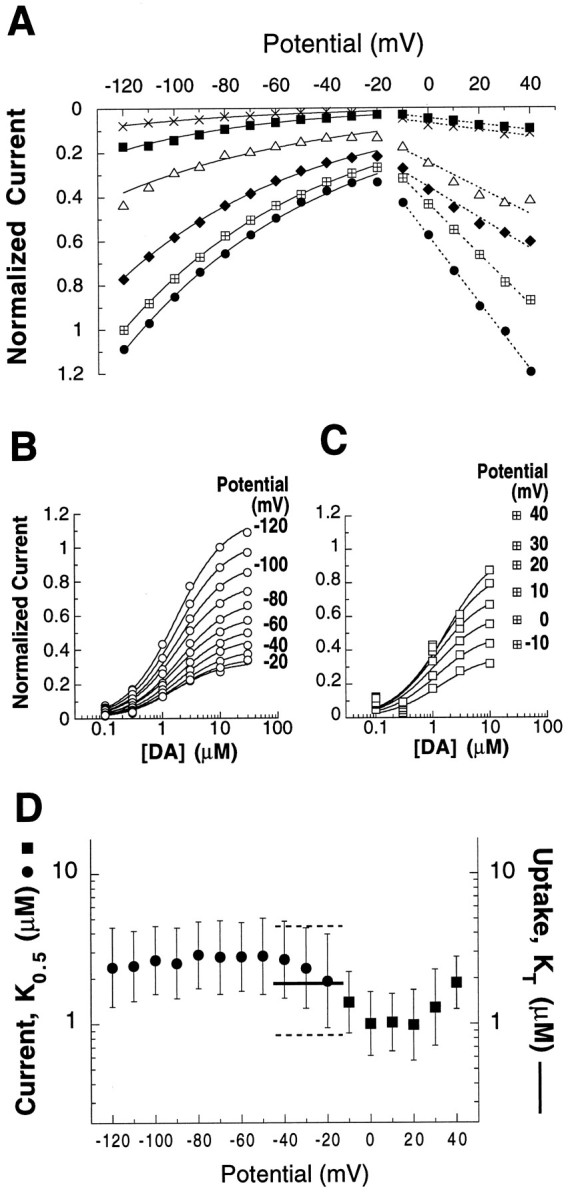
Concentration and voltage dependence of DA-evoked steady-state currents. Concentration-dependent current responses were measured in six oocytes using a voltage jump protocol (see Materials and Methods). To control for differing levels of hDAT expression between oocytes, IDA −IControl subtractions for each DA concentration and potential were normalized to the current elicited by 10 μm DA at −120 mV in the same cell (mean, −17.9 nA; range, −7.9 to −43.5 nA). A, Nonlinear curve fitting of mean current amplitudes for each DA concentration (x, 0.1 μm; ▪, 0.3 μm; ▵, 1.0 μm; ♦, 3.0 μm; ⊞, 10 μm; •, 30 μm). Over the potential range −120 to −20 mV, DA-elicited currents displayed an exponential dependence on voltage (mean, e-fold per 67 mV, solid lines; range, e-fold per 55–75 mV). At more positive potentials, however, current amplitudes were better described as having a linear relation to membrane potential (broken lines).B, The DA concentration dependence of mean-normalized currents (○) was well fit by a simple Michaelis–Menten equation for individual potentials over the range −120 to −20 mV. Currents appeared to saturate with increasing [DA] and displayed K0.5 values of 1.9–2.9 μm. C, At potentials greater than −20 mV, mean normalized current amplitudes (▪) were more poorly described by Michaelis–Menten kinetics as a function of concentration. In some oocyte batches, high DA concentrations affected an endogenous conductance (see Results), and therefore the 30 μm points (⊞) were omitted from the curve fits. D, At each membrane potential tested, geometric means ofK0.5 and CI95 values (error bars) were determined from affinity values derived individually for each of six oocytes (see Materials and Methods). The apparent affinities (K0.5) of DA for eliciting transport-associated current (•) and for blocking a leak current (▪) are displayed. The K0.5 for the transport-associated current (in the range of −120 to −20 mV) demonstrates little voltage sensitivity. For comparison, also plotted is the DA apparent substrate affinity (KT, ——; CI95 - - -) determined in uptake assays using six different batches of oocytes. These oocytes were not voltage-clamped, although other oocytes from the same batches displayed resting membrane potentials in the range of −15 to −45 mV.
Nonlinear curve fitting of the normalized I–V data suggested that the two limbs of I–V plot divided at −20 mV are best described by different functions. DA transport-associated inward currents exhibited an exponential dependence on membrane potential, increasing with hyperpolarization approximatelye-fold per 67 mV between potentials −20 mV and −120 mV. In distinction, at voltages more depolarized than −20 mV, DA-evoked currents displayed a dependence on membrane potential that was better described by a linear than by an exponential function. At neither extreme of the range studied did current magnitudes appear to saturate as a function of potential.
The concentration–response relations of DA-evoked currents were studied at individual potentials. Figure 4, B andC, graphically represents the concentration dependence of normalized DA-evoked currents at membrane potentials −120 to −20 mV and −10 mV to 40 mV, respectively. For the transport-associated currents, amplitudes appeared to saturate with increasing DA concentration and were well described by Michaelis–Menten kinetics. The DA concentrations that elicited half-maximal currents (K0.5) were calculated in each oocyte for every potential using the Michaelis function. The plot of meanK0.5 versus membrane potential (Fig.4D) illustrates that voltage has little influence on the apparent affinity of DA between −120 mV and −20 mV, where the mean values fluctuated only between 1.9 and 2.9 μm. TheseK0.5 values are in good agreement with the apparent affinity for DA transport (KT = 1.7 μm) determined in uptake assays using oocytes that were not voltage-clamped (typical resting membrane potentials of hDAT oocytes were −15 to −45 mV). The resemblance in apparent affinities of DA for uptake and for eliciting transport-associated currents supports the assertion that the currents are a manifestation of the translocation process. The functional link between translocation and the transport-associated current is reinforced by studies with S(+)amphetamine. Normalized S(+)amphetamine concentration–response curves generated in two oocytes yielded a meanK0.5 of 0.5 μm over hyperpolarized potentials (data not shown). This value closely corresponds to its potency (Ki = 0.3 μm) for inhibiting [3H]DA uptake, as would be expected for a DA uptake inhibitor that is itself a transport substrate (Ross, 1976).
Although the Michaelis–Menten function provided a less satisfactory fit to the DA concentration dependence of current at voltages more positive than −20 mV (Fig. 4C), the estimates of mean DAK0.5 values (1.0–2.0 μm) at depolarized potentials were in the same range as those found for hyperpolarized potentials (Fig. 4D) and for theKT. A number of factors may have contributed to the poorer fits of the DA currents to Michaelis–Menten kinetics at potentials more positive than −20 mV, one of which is that in certain batches of uninjected and water-injected oocytes, high concentrations (≥30 μm) of DA and other phenethylamine congeners were observed to block an endogenous ionic conductance (data not shown). In particular, such a phenomenon might account for why the largest divergences from the Michaelis–Menten model were observed at the highest DA concentrations. Accordingly, only concentrations up to 10 μm were used to estimate the apparent affinity of DA for blocking the leak conductance (Fig. 4C,D). Although there is some uncertainty in determining the apparent affinity of DA in blocking the leak conductance, the results presented in Figure4D argue that DA has quite comparable potencies as a transport substrate (KT), in evoking a transport-associated current, and in blocking a leak current (K0.5). In turn, the comparable affinities of DA for influencing the three functions support the assertion that all three are mediated by hDAT.
Dependence of DA transport velocity on membrane potential
The finding that DA uptake by hDAT is closely associated with an inward current is consistent with the hypothesis that the translocation process itself is electrogenic, because it couples the movement of some net charge with DA flux, as has been proposed for rat DAT by Krueger (1990), McElvain and Schenk (1992), Kilty (1993), and Gu et al. (1994). If transport-associated currents arise from a fixed stoichiometric coupling of driving ions to DA during translocation, two corollaries should follow, i.e., that the net charge:DA flux ratio should not vary with membrane potential, and that DA transport velocity should increase with membrane hyperpolarization in an exponential manner akin to the steady-state, transport-associated current (Fig.4A).
To examine the voltage dependence of DA uptake velocity and the hypothesis of fixed stoichiometric coupling, the rates of DA accumulation were quantified in individual oocytes held under voltage clamp and superfused (≤300 sec) with 20 μm DA or 10.1 μm [3H]DA. In each of six batches of oocytes, mean DA uptake velocity increased with membrane hyperpolarization (3–8 oocytes/potential). Pooled results from six batches (each normalized to its −30 mV value) are presented in Figure5A, and these demonstrated a clear voltage dependence of DA uptake velocity. The mean uptake velocity in oocytes clamped at −120 mV was 85% faster than that observed in those clamped at −30 mV. Although uptake velocities increased with hyperpolarization in all experiments, marked batch-to-batch differences in the magnitudes of increase were observed. In the different batches, uptake velocities observed in oocytes held at −90 were between 12 and 108% higher than the corresponding velocities measured at −30 mV. Moreover, these variations did not correlate with variations in levels of hDAT expression.
Fig. 5.

Voltage dependence of DA uptake velocity and of charge:DA flux ratios determined in voltage-clamped uptake experiments. Oocytes were voltage-clamped and superfused with DA or [3H]DA for 100–300 sec periods, after which the DA accumulated by each oocyte was quantitated using HPLC-EC or scintillation counting (see Materials and Methods). A, For each of six oocyte batches, uptake velocities at each potential were normalized to that seen at −30 mV (dotted line). The normalized values were pooled, and the mean values (±SEM) are plotted. The number of oocyte batches and the total number of oocytes studied at each potential are 1:3 at +10 mV, 5:25 at 0 mV, 6:28 at −30 mV, 6:31 at −60 mV, 6:28 at −90 mV, and 4:17 at −120 mV.B, From current records available for five of the six oocyte batches, net charge:DA flux ratios were calculated for each oocyte from the time integral of currents elicited during periods of DA perfusion and the corresponding measurements of accumulated DA. Mean ratios (±SEM) for oocytes tested at each potential are graphed. Means were compared with that determined at −30 mV (3.1 ± 0.26) using one-way ANOVA with a post hoc Dunnett’s multiple comparisons test, and they differ with p < 0.01 (asterisk). The number of oocyte batches and the total number of oocytes studied at each potential are 5:25 at 0 mV, 5:24 at −30 mV, 5:26 at −60 mV, 5:24 at −90 mV, and 3:10 at −120 mV. Thedashed line at 2.0 represents the net charge:DA flux ratio predicted for a fixed transport stoichiometry of 1 DA+/2 Na+/1 Cl−.
Correlation of charge and substrate fluxes
Time integrals of the DA-elicited currents were calculated for oocytes in five of the six batches used for clamped uptake studies. These measures of charge movement were correlated with the quantity of DA concurrently accumulated by the same oocytes, yielding a net charge:DA flux ratio for each of the 84 oocytes studied. Because this ratio eliminates the effect of differing expression levels between oocytes, net charge:DA flux determinations could be pooled for all oocytes held at each membrane potential. As Figure 5Billustrates, the charge:DA flux ratio displayed a minimum at −30 mV of 3.1 ± 0.26 e0/DA molecule (mean ± SEM; n = 24), and the ratio increased with either hyperpolarization or depolarization from that potential (−60 mV, 3.8 ± 0.4, n = 26; −90 mV, 4.1 ± 0.3, n = 24; −120 mV, 6.6 ± 0.9,n = 10; 0 mV, 6.8 ± 0.9, n = 25).
The change with voltage in the ratio of net charge:DA flux indicated that not all DA-elicited current was stoichiometrically coupled to DA translocation. This point has already been suggested for voltages above the leak conductance reversal potential (approximately −18 mV), where the inhibitory action of DA on the outward cation leak inflates the estimate of charges moving inward. In contrast, the rise in the net charge:DA flux ratio with hyperpolarization from −30 mV cannot be attributed to the same mechanism: at more negative potentials, inhibition of a tonic cationic leak current by DA would tend to diminish the net inward current. Hence, if DA elicits an inward transport-associated current and simultaneously blocks a tonic inward leak, the observed current will actually underestimate the magnitude of the transport-associated charge movement and the net charge:DA flux ratio. The elevation in the net charge:DA flux ratio attributable to hyperpolarization supports the existence of a second ionic conductance that is associated with DA transport (see Discussion).
Differentiation of pharmacological agents byI–V plots
To examine whether the hDAT-mediated currents elicited by DA and cocaine are also elicited by other DA uptake inhibitors (or releasers), a spectrum of pharmacological agents was assayed using voltage-jump protocols with hDAT-expressing oocytes. Examination of drug-elicited steady-state currents revealed that the ligands could be classified as either DA-like, cocaine-like, or electrically inactive based on theirI–V plots. The DA-like group all displayed inverted U-shaped I–V curves with peaks at approximately −20 mV (Fig. 6A, compare Fig.4A) and include the psychomotor stimulants S(+)amphetamine and S(+)methamphetamine, the neurotoxin MPP+ (1-methyl-4-phenylpyridinium), as well as (−)norepinephrine and the indirect sympathomimetic agentsp-tyramine, m-tyramine, β-phenethylamine, and (−)metaraminol. The catecholaminergic neurotoxin 6-hydroxydopamine elicited no transport-associated current at 10 μm, and at a 30 μm concentration, the transport-associated current (at −120 mV) was less than one-tenth the amplitude of that elicited by 10 μm S(+)amphetamine in the same oocytes.
Fig. 6.

I–V plots readily distinguish two classes of pharmacological agents that act at hDAT. Nineteen hDAT ligands that were studied in voltage jump protocols could be resolved into two groups, displaying either DA-like or cocaine-like voltage dependence of their subtractive currents. A, Substrates for hDAT such as dopamine (•), p-tyramine (□), and amphetamine (▿) elicit a conductance increase at potentials below −20 mV, and their subtractive currents are plotted asIDrug −IControl. These and other compounds (listedbelow I–V plot) thought to be substrates for hDAT displayed a characteristic inverted U-shaped I–V curve (see also Fig. 3A). B, In contrast, nontransported uptake inhibitors such as cocaine (•), GBR12909 (□), methylphenidate (▿), and other listed drugs all block an inward current at potentials below −20 mV and are plotted asIControl −IDrug. The reversal potential of the conductance blocked by these drugs was approximately −20 mV and was independent of drug concentration (≤30 μm). TheI–V plots represent data obtained from several different batches of oocytes, and each drug was tested in at least two hDAT oocytes. At the concentrations tested, none of the drugs affected ionic conductances of control oocytes.
In contrast, a number of drugs manifested cocaine-like I–Vcurves and appeared to reduce membrane conductance over the entire range of membrane potentials. Their I–V plots (IControl − IDrug) reversed at approximately −20 mV and grew steeper at positive potentials (Fig. 6B, compare Fig.2B). Drugs falling in this category include the stimulant/anorectic drugs methylphenidate, pemoline, amfonelic acid, phendimetrazine, aminorex, and mazindol, as well as GBR12909, indatraline (Lu19-005), and the cocaine analog RTI-55. Drugs that produced no detectable electrical responses in hDAT-expressing oocytes included the tyramine congeners with low potency for inhibiting [3H]DA uptake—l-tyrosine (100 μm) and 3-methoxytyramine (30 μm)—and the anti-parkinsonian drug amantadine (100 μm). At the concentrations tested, none of the drugs produced responses in water-injected or uninjected oocytes.
Comparison of DA and cocaine actions on the steady-state leak currents
It is clear from the I–V plots of the hDAT ligands examined, that like cocaine and DA at depolarized potentials, they too block a tonic leak conductance (Fig. 6). Therefore, the question arises whether substrates and nonsubstrate ligands inhibit an identical leak conductance or whether they affect distinct conductances. One criterion for determining whether two ligands affect the same conductance is whether their I–V curves reverse at the same potential. Because I–V plots of substrates do not cross the abscissa—a result of the drugs’ combined effects on the transport-associated and leak conductances—we reasoned that elimination of the transport-associated component from the total DA-evoked current might serve to isolate the membrane leak current that is blocked by DA. To abolish the transport-associated currents, buffer substitutions of K+ and Li+ were made for Na+, because substrate translocation by hDAT shows strict dependence on Na+ ions. By examining the DA and cocaineI–V values in ion-substituted buffers, their reversal potentials could be compared; furthermore, the relative permeability of ions through the leak conductance could be characterized by measuring the shifts in reversal potential brought about by ion substitutions.
As expected, substitution of K+ for Na+abolished the transport-associated component of the DA I–V, altering its inverted U-shape to a more ohmic current, which does indeed reverse and which now resembles the cocaine I–V. Figure 7A illustrates that in 96 mm K+ (2 mm Na+) Ringer’s buffer, IControl −IDrug subtractions for both DA and cocaine generated I–V plots that had comparable slope conductances over the entire voltage range (contrast Fig. 3). The DA- and cocaine-inhibited conductances both reversed at −16 mV in 96K+ buffer—the same potential at which the cocaineI–V reversed when assayed in Na+-Ringer’s buffer during a previous trial on the same oocyte. This reversal potential is not different from the mean reversal potential (−18.4 ± 2.4 mV, mean ± SEM) for cocaine I–Vvalues in Na+ Ringer’s buffer measured in 26 oocytes. When cocaine reversal potentials were measured successively in individual oocytes using both Na+ Ringer’s and 48 mmK+/50 mm Na+ Ringer’s buffers, these too showed that, despite a 24-fold increase in the extracellular K+ concentration, the reversal potentials were not appreciably different: respectively, −14.3 ± 5.7 mV and −15.4 ± 3.2 mV (n = 4 oocytes).
Substitution of Li+ for Na+ also eliminated substrate-elicited transport-associated currents and revealed that the leak currents blocked by substrates and nonsubstrate hDAT inhibitors had indistinguishable reversal potentials. Successive applications of DA and cocaine, both in 98 mmLi+ (0 Na+) buffer, blocked conductances with reversal potentials of 0.0 ± 3.1 mV and −0.8 ± 3.2 mV, respectively (Fig. 7B, representative of 4 oocytes). Overall, the mean reversal potential for cocaine in Li+buffer was 4.2 ± 3.1 mV (n = 10 oocytes). Comparable results were observed using Li+-substituted buffers with the substrates S(+)amphetamine, p-tyramine, and β-phenethylamine, and the nonsubstrate blocker GBR12909 (data not shown). Furthermore, similar experiments have been performed with DA and cocaine in buffer lacking both Na+ and Cl−(Li+ and MES substitutions, respectively), and the results are qualitatively the same as those in Figure 7B (data from 3 experiments not shown). That Cl− substitutions did not shift the cocaine reversal potentials strongly suggests that Cl− ions are not carried by the hDAT leak conductance.
The Goldman–Hodgkin–Katz voltage equation was used to characterize the ionic selectivity of the leak conductance from the shifts in the cocaine reversal potential observed in ion-substituted buffers. Assuming that the intracellular concentrations of Na+, K+, and Li+ do not change appreciably, the relative permeabilities of these ions are 1, 0.96, and 2.4 for the hDAT leak conductance. That removal of all external Cl− did not shift the cocaine reversal potential suggests that the tonic hDAT leak conductance is Cl−-impermeant, and yet it is fairly nonselective for alkali cations. This ionic selectivity of the leak conductance differs markedly from the comparatively strict dependence on Na+ and Cl− of the hDAT transport functions, i.e., substrate flux and transport-associated currents.
The ionic selectivity of the leak conductance was explored further with experiments in which reversal potentials were successively determined in individual oocytes from cocaine I–V plots executed in Ringer’s buffer and in choline-substituted buffer. Complete replacement of Na+ with choline caused leftward shifts in the I–V curves. The mean reversal potentials shifted from −15.4 ± 3.8 mV in Ringer’s to −48.4 ± 4.0 mV in choline (n = 7 oocytes), a value substantially more positive than the predicted Nernst potential for K+ in oocytes (−95 mV) (Dascal, 1987; Costa et al., 1989). These results raise the possibility that choline or some other ion may permeate the leak conductance aside from Na+ and K+. Although removal of Ca2+ or Mg2+ from the Ringer’s had no effect, manipulating the buffer between pH 6.5 and 8.5 strongly affected the cocaine reversal potentials, shifting them approximately −38.5 ± 3.5 mV per unit increase in pH (n = 10 oocytes). These data suggest that protons may be carried by the leak conductance or, alternatively, that pH can alter the ionic selectivity of the leak. Because a 10-fold change in [H+]o had a greater effect than an approximately 50-fold change in [Na+ + K+]o and that [Na+ + K+]o/[H+]o is ≥104, the Goldman–Hodgkin–Katz voltage equation suggests that if protons do permeate through the leak conductance, their relative permeability is more than four orders of magnitude greater than that of alkali cations.
DISCUSSION
The Xenopus oocyte expression system has proven an exceedingly useful method for investigating the function of cloned Na+-coupled transport proteins, in part because both biochemical and electrical indices of transport can be studied. To characterize the electrogenic properties of a DAT cloned from human midbrain RNA, we have measured substrate uptake, radioligand binding, and both ramp and steady-state currents evoked under voltage-clamp conditions in oocytes injected with hDAT cRNA. Expression of the hDAT in oocytes gives rise to DA transport activity, the pharmacological properties of which are consistent with those displayed in heterologous mammalian cell lines transfected with hDAT; the apparent affinity of DA uptake in oocytes, the inhibition constants for cocaine and S(+)amphetamine, as well as the potencies of mazindol, GBR12935, and RTI-55, were within the ranges determined previously in cell lines expressing hDAT (Giros et al., 1992; Pifl et al., 1993; Giros et al., 1994; Pristupa et al., 1994; Eshleman et al., 1995). As would be expected for a member of the Na+/Cl−-dependent neurotransmitter transporter family, removal of either extracellular Na+ or Cl− ions virtually abolished the transport of [3H]DA into hDAT-expressing oocytes.
DA uptake and transport-associated current
Using two-electrode voltage-clamp techniques, we have found that application of substrates to hDAT-expressing oocytes reliably elicited voltage-dependent steady-state currents that were not observed in control oocytes. When studied under conditions that precluded substrate translocation, the electrophysiological actions of substrates and nonsubstrate transport inhibitors revealed that hDAT mediates a cationic leak conductance that is constitutive and uncoupled from the uptake process. The two currents that hDAT expression confers on oocytes are designated the transport-associated and the leak currents.
Several lines of evidence suggest that the inward transport-associated current observed at hyperpolarized potentials after application of DA (or other hDAT substrates) is closely linked to substrate translocation by hDAT. The transport-associated current was caused by a conductance increase (Fig. 2C), consistent with the coupled movement of a positively charged DA molecule and Na+ ions. Numerous pharmacological attributes of the transport-associated current were essentially identical to those of [3H]DA uptake. The concentration dependence of [3H]DA uptake velocity and of DA-evoked transport-associated current amplitude were both well fit by rectangular hyperbolic functions with similar kinetic constants (KT = 1.7 μm andK0.5 = 1.9–2.9 μm). The set of pharmacological agents that evoked transport-associated currents includes compounds previously determined to be substrates by biochemical assay (Fig. 6A); conversely, nonsubstrate uptake blockers (e.g., cocaine and methylphenidate) were unable to elicit transport-associated currents (Fig. 6B). Furthermore, the apparent affinities of S(+)amphetamine were nearly identical for inhibiting [3H]DA uptake (Ki = 0.3 μm) and for evoking transport currents (K0.5 = 0.5 μm), as would be expected for a competitive inhibitor of DA uptake that is an alternative transport substrate (Ross, 1976). Finally, treatments that prevented uptake of substrate, such as coincubation with pharmacologically appropriate concentrations of cocaine or replacement of extracellular Na+ or Cl−, also abolished transport-associated currents evoked by DA (Figs. 3, 7).
Dependence of DA uptake on membrane potential
The close association of substrate-evoked voltage-dependent currents with substrate translocation strongly supports the assertion that hDAT is an electrogenic transporter, a hypothesis developed from studies of the apparent ionic coupling of rodent DATs (Krueger, 1990;McElvain and Schenk, 1992; Kilty, 1993; Gu et al., 1994). Our results provide direct evidence of the electrogenic nature of hDAT in their demonstration that the velocity of DA uptake increased with hyperpolarization (Fig. 5A). Several studies have demonstrated that the ability of striatal preparations to accumulate [3H]DA uptake could be diminished by agents that alter neuronal membrane potentials, such as ouabain, elevated external K+ concentrations, veratridine, batrachotoxin, and metabolic inhibitors (Baldessarini and Vogt, 1971; Holz and Coyle, 1974; Liang and Rutledge, 1982; Krueger, 1990). One ambiguity in these results, however, is that the effects of these agents on membrane potential could not be separated from their effects on the ionic gradients that drive substrate translocation.
hDAT-expressing oocytes, on the other hand, offered the opportunity to directly assess the influence of membrane potential on DA uptake velocity, because the voltage could be manipulated independently of ionic gradients, and the expression levels were sufficient to allow the biochemical quantitation of DA accumulated by single cells during periods of voltage clamp. The increase in uptake velocity observed with membrane hyperpolarization is consistent with the expectation that the thermodynamic driving force for an electrogenic transporter includes transmembrane electrical potential, as well as chemical gradients for substrates and cotransported ions. Furthermore, the data indicate that the effect of membrane potential on DA uptake is mediated primarily through alterations in the turnover rate (Vmax) of hDAT rather than its apparent affinity for substrate, because DA concentrations used in the clamped uptake experiments were near saturating. An effect of voltage on turnover rate, but not on substrate affinity, is also supported by the finding that theK0.5 of DA for eliciting transport-associated currents was largely independent of membrane potential (Fig.4D). The voltage-dependent and rate-limiting step in the hDAT translocation reaction, therefore, is likely to occur subsequent to substrate binding.
The clamped uptake experiments, apart from providing some biophysical insight into hDAT translocation, also suggest a physiological context in which the voltage dependence of DA uptake may help regulate intercellular signaling by dopaminergic neurons. Recent in vitro and in vivo studies have suggested that drugs acting at D2-like autoreceptors modulate the velocity of DA uptake (Meiergerd et al., 1993; Parsons et al., 1993; Cass and Gerhardt, 1994), and the voltage dependence of hDAT function demonstrated in our studies could provide the mechanism through which autoreceptor activation influences DA reuptake. In situhybridization of rat DAT mRNA indicates that DAT is expressed exclusively in dopaminergic neurons (Cerruti et al., 1993; Lorang et al., 1994), and immunocytochemical studies demonstrate that terminal and somatodendritic zones of rat midbrain DA neurons are rich in both DAT (Ciliax et al., 1995; Nirenberg et al., 1996) and D2-like autoreceptors (Sesack et al., 1994; Yung et al., 1995). Ample electrophysiological evidence shows that activation of autoreceptors in dopaminergic cells in the substantia nigra and ventral tegmental area causes the cells to hyperpolarize through the G-protein-mediated opening of K+ channels (for review, seeLacey, 1993; White, 1996). Our data predict that in regions of dopaminergic neurons in which DAT and D2 receptors are colocalized, hyperpolarization attributable to autoreceptor activation by DA will increase the DAT turnover rate and thus accelerate the clearance of extracellular DA. Faster removal of extracellular DA complements the autoreceptor-mediated inhibition of DA release and may sharpen the temporal response of DA signaling in these important cell groups. A second consequence predicted from the colocalization of autoreceptors and DAT is that the DA-activated K+conductance would act to offset the depolarizing action of the transport-associated current, and thereby cancel the small, positive feedback effect that DA uptake might contribute toward promoting Ca2+-dependent vesicular DA release.
Two components of transport-associated currents
Despite the numerous common properties that appear to unite transport-associated currents and substrate translocation in hDAT-expressing oocytes, clamped uptake experiments suggested that the transport-associated currents may be comprised of two components, only one of which is stoichiometrically tied to substrate translocation. Concurrent measurement of DA-elicited currents and DA uptake in voltage-clamped oocytes provides strong evidence that not all of the DA-elicited transport current arises from stoichiometric coupling of 2 Na+ and 1 Cl− ions per DA+molecule, as predicted from biochemical studies of ionic activation of rat DAT uptake activity (Krueger, 1990; McElvain and Schenk, 1992;Kilty, 1993; Gu et al., 1994) (but see Turner, 1985, regarding putative transport stoichiometry). The mean net charge:DA ratio displayed a minimum value of 3.1 in oocytes clamped at −30 mV, a value that significantly exceeds the predicted ratio of 2 net charges per DA (CI99 2.4–3.8; n = 24; Fig.5B). This divergence between charge and substrate flux is further reflected in Figure 5B, in which the mean net charge:DA flux ratio rises with hyperpolarization from 3.1 at −30 mV to 4.1 at −90 mV and 6.6 at −120 mV. Simply put, the DA transport-associated currents are too large to be accounted for solely by a translocation mechanism that operates with a fixed stoichiometry of 2 net charges per DA molecule.
Two different mechanistic hypotheses might account for the disparities between DA-elicited currents and uptake velocities. One is that the DA transporter operates with variable coupling of substrate and driving ions, and that hyperpolarization from −30 mV alters the ionic coupling such that more positive charges accompany each translocated DA molecule. Alternatively, one might posit that DA transport activates a distinct ionic conductance that is thermodynamically “uncoupled” from substrate translocation. (Both explanations would be consistent with transport-associated currents showing the same kinetics, ionic, and pharmacological sensitivities as uptake activity.) Although our data do not rule out either possibility, we favor the hypothesis that substrate transport by hDAT can regulate an uncoupled ionic conductance because several other electrogenic neurotransmitter transporters have recently been shown to mediate ion fluxes that are modulated by, but uncoupled from, substrate fluxes (Cammack et al., 1994; Mager et al., 1994; Fairman et al., 1995; Galli et al., 1995; Wadiche et al., 1995;Cammack and Schwartz, 1996; Eliasof and Jahr, 1996; Larsson et al., 1996; Risso et al., 1996) (for review, see Sonders and Amara, 1996). In particular, it is interesting to compare the hDAT results with those obtained by Mager and colleagues (1994), who studied oocytes expressing rat SERT. They found that although hyperpolarization from −30 mV to −80 mV had virtually no effect on the velocity of serotonin uptake, it markedly increased the transport-associated current elicited by serotonin. In combination with the finding that the mean net charge:serotonin flux ratio at −40 mV was 8.0 ± 1.0, these results suggest that a thermodynamically uncoupled component makes an even greater contribution to the rSERT than to the hDAT transport-associated current.
If a portion of the hDAT transport-associated current is indeed stoichiometrically uncoupled from substrate movement, this would suggest that hDAT mediates three discernible ionic conductances: the putative stoichiometrically coupled movement of DA+/2 Na+/Cl−, a transport-activated uncoupled conductance, and the tonic leak conductance. The two uncoupled conductances can be distinguished from each other because hDAT substrates have opposing actions on these two uncoupled conductances. The disparity is most obvious at hyperpolarized potentials, where the inferred action of the DA-activated uncoupled conductance is toincrease the inward current, whereas the action of DA on the tonic leak conductance is, in all likelihood, to block an inward current.
Characterization of the leak conductance
The action of DA on the tonic leak conductance at hyperpolarized potentials is stated in a tentative manner, because it is ordinarily masked by the transport-associated current under those ionic conditions that permit substrate translocation. It was possible, however, to isolate the action of DA on the tonic leak conductance from its action on transport-associated conductances by replacing external Na+ in the superfusing Ringer’s buffer. When K+, Li+, or choline was substituted for Na+, both specific [3H]DA uptake into oocytes and the transport-associated inward current elicited by DA were abolished (Fig. 7). Nevertheless, the ability of drugs, including DA and cocaine, to block the leak conductance in these buffers remained intact, and they did so at concentrations essentially the same as those effective in normal Na+ Ringer’s. In contrast to normal Na+ conditions, however, the I–V plots of DA and cocaine were comparable across the entire voltage range; furthermore, the I–V values reversed at the same potentials (Fig. 7), indicating that it is highly probable that both substrate and nonsubstrate translocation inhibitors block an identical hDAT leak conductance. Because ion substitution experiments revealed that the leak conductance blockade by DA (and other substrates) occurred at negative as well as positive potentials, it seems reasonable to infer that substrates also block the leak conductance at negative potentials in Na+ Ringer’s despite the overwhelming effect of the transport-associated currents they evoke.
The tonic leak conductance blocked by both DA and cocaine also appears to be susceptible to inhibition by virtually all DAT ligands tested regardless of their effect on the transport-associated current. The inhibition can be seen in subtractive currents for all drugs in the voltage range above −18 mV, where I–V curves for substrates displayed a negative slope conductance (Fig.6A) and those for translocation inhibitors displayed a positive slope conductance (Figs. 6B, 7). The ability of substrates to block transporter leak currents is a relatively uncommon phenomenon among Na+-dependent cotransporters. Although several members of this transporter superfamily display leak currents that can be inhibited by nonsubstrate translocation blockers (Umbach et al., 1990; Parent et al., 1992;Cammack et al., 1994; Mager et al., 1994; Chen et al., 1995; Galli et al., 1995; Vandenberg et al., 1995), only hDAT, rSERT (Mager et al., 1994), and perhaps a rat GABA transporter (GAT1) (Cammack and Schwartz, 1996) appear to have leak conductances that are also inhibited by substrates.
The leak conductances of hDAT and rSERT also share a rather uncommon ionic selectivity. Analysis of the shifts in the cocaine reversal potential attributable to ion substitution for Na+ indicate that Li+ and K+ are, respectively, 2.4-fold and 0.96-fold as permeant as Na+ through the hDAT leak conductance. These values are reasonably close to the estimates of Mager and colleagues (1994), who report that the magnitude of the rSERT leak current is increased threefold by substitution of Li+for Na+, but reduced by 27% by K+substitution. We found no evidence suggesting that Cl−anions are carried in the leak conductance; however, the robust shifts in the cocaine reversal potential observed with alterations in the buffer pH suggested that protons may also permeate the hDAT leak conductance—a phenomenon currently being investigated. It is somewhat surprising that, in contrast to the stringent Na+ and Cl− requirements for substrate translocation, the tonic leak conductance of hDAT readily carries Li+ and K+, and perhaps H+, ions. Like hDAT and rSERT, uncoupled conductances mediated by GAT1 are also supported by several alkali cations (Cammack and Schwartz, 1993, 1996; Cammack et al., 1994;Mager et al., 1996). In contrast, the leak “modes” of many other transporters display comparable ionic selectivities to their transport modes (for review, see Maloney, 1994; Sonders and Amara, 1996). Thus, it is an open question for hDAT and other members of this family whether a single permeation pathway mediates both the net vectorial movement of substrate/cosubstrate ions and also the bidirectional flux of leak ions or whether the transporters contain separate pathways that have markedly different ionic selectivities.
Similarly, it is not currently known whether the site at which substrates bind to block the leak conductance is in the permeation pathway or outside of it. Our ion substitution results indicate, however, that the binding of cosubstrate ions is not a prerequisite for substrate binding and leak blockade, because DA readily blocks the leak conductance in the complete absence of external Na+ and Cl−. Should this DA binding site be located within the substrate permeation pathway, the ion substitution experiments would suggest that the strict dependence of the hDAT transport function on Na+ and Cl− is not attributable to the ion-dependent binding of substrates, as has been suggested for GAT1 (Mager et al., 1996). Although hDAT and GAT1 belong to the same family of Na+/Cl−-dependent transporters, they can also be mechanistically distinguished by the finding that theK0.5 for DA-elicited transport-associated currents is independent of voltage for hDAT (Fig.4D), whereas the K0.5 for GABA-elicited current at GAT1 rises with hyperpolarization (Mager et al., 1993). It is possible that the voltage dependence of the GABAK0.5 may arise from the initial voltage-dependent binding of Na+, which was recently described by Mager and coworkers (1996).
hDAT currents as a pharmacological tool: implications for DAT-mediated DA release
The demonstration that the transport-associated and leak currents are intrinsic to hDAT function helps to highlight the possibility that the psychomotor stimulants, a class of drugs with broad societal relevance, may influence the electrical properties of neurons directly as well as indirectly through the regulation of extracellular DA levels. For instance, the observation that hDAT leak conductance can be inhibited by the same pharmacological agents and concentrations as those affecting DA translocation suggests that drugs such as cocaine, methylphenidate, pemoline, and mazindol modulate ionic flux as well as substrate uptake into dopaminergic cells in vivo. If protons are indeed carried by the hDAT leak conductance, our data suggest that the selectivity and voltage dependence of the leak is comparable to that of voltage-activated proton-selective conductances (DeCoursey and Cherny, 1994). One proposed function of these proton conductances is to enable the rapid alkalinization of the cytoplasm under conditions of heightened metabolic activity without further energy expenditure (for review, see Lukacs et al., 1993). If the hDAT leak serves a similar purpose, psychostimulants may directly influence the physiology of dopaminergic neurons by interfering with intracellular pH homeostasis.
Apart from the physiological consequences of the ionic currents of hDAT, the transport-associated and leak currents provide new tools with which to elucidate hDAT pharmacology and the mechanisms of action of psychomotor stimulants. Using the criterion of whether drugs elicited transport-associated currents, it was easy to discriminate hDAT substrates from nonsubstrate translocation inhibitors (Fig. 6). Our categorization accords well with and extends the current understanding of how drugs act on hDAT. Because both substrate and nonsubstrate ligands inhibit uptake of radiolabeled substrates by DAT, differentiation of the two classes of drugs has relied on two types of biochemical assays. By one approach, striatal preparations or transfected cells are used to measure the sequestration of putative substrates that have been radiolabeled or are detectable by other methods (e.g., electrochemistry, chromatography, spectrophotometry, or a combination). The second approach assesses the ability of putative substrates to promote the efflux of an authentic preloaded substrate, presumably through a mechanism akin to carrier-mediated facilitated diffusion (Stein, 1986). Nearly all of the compounds identified as substrates by electrophysiological assay have been shown previously to promote Ca2+-independent efflux of [3H]DA with a pharmacological profile appropriate to DAT (Heikkila et al., 1975; Raiteri et al., 1977, 1979; Fischer and Cho, 1979; Liang and Rutledge, 1982; Bonnet et al., 1984; Keller and Da Prada, 1985; Chang and Ramirez, 1986; Parker and Cubeddu, 1986; Rollema et al., 1986;Sirinathsinghji et al., 1988; Eshleman et al., 1995; Pifl et al., 1995;Wall et al., 1995). Metaraminol has not been studied previously as a promoter of [3H]DA efflux; however, it is an indirect sympathomimetic agent that promotes Ca2+-independent “release” of norepinephrine stores (Langeloh et al., 1987), underscoring the similarity among substrates for DAT and NET (Kilty, 1993; Buck and Amara, 1994). Conversely, cocaine and other drugs we have identified electrophysiologically as nonsubstrate translocation inhibitors have typically been reported to inhibit both releaser-elicited and spontaneous [3H]DA efflux at low concentrations.
Although assays of uptake and efflux have formed the basis of the contemporary classification of hDAT substrates and nonsubstrate inhibitors, they have not provided altogether unambiguous results, because the endpoints measured in such assays do not measure the activity of DAT alone but can also be influenced by agents that modify DA synthesis, degradation, and vesicular sequestration, as well as those that alter membrane potential and spontaneous release processes. Indeed, although it is well accepted that amphetamine, methamphetamine, and several β-phenethylamine congeners are excellent releasers of DA, the direct evidence indicating that these compounds can be taken up by DAT is scarce (Zaczek et al., 1991) compared with the data available for their less lipophilic, hydroxylated analogs (Steinberg and Smith, 1970; Baldessarini and Vogt, 1971; Dorris and Shore, 1971; Fischer and Cho, 1979). It has been proposed that the amphetamine-like releasing agents passively diffuse into the cell and increase DA efflux through their actions at an intracellular target (for review, see Seiden et al., 1993); by causing depletion of vesicular DA through a weak base mechanism, amphetamines elevate levels of cytosolic DA and promote its extrusion by DAT (Sulzer and Rayport, 1990; Sulzer et al., 1995). However, this model does not implicate any direct action of these releasers on DAT. In light of the several lines of evidence supporting the hypothesis that the transport-associated current is an indicator of ongoing hDAT translocation activity, the finding that S(+)amphetamine, S(+)methamphetamine, and β-phenethylamine all elicit transport-associated currents strongly suggests that they interact directly with hDAT and are substrates. To our knowledge, this is the first data, apart from biochemical release experiments, to indicate that S(+)methamphetamine is indeed a DAT substrate. These data support the idea that these drugs promote DA efflux through a sort of exchange mechanism involving hDAT, in addition to any intracellular actions they may have.
Conclusion
Data have been presented that indicate that hDAT mediates both a transport-associated current and a constitutive leak current inXenopus oocytes, and that these currents can be distinguished from each other by the criteria of ionic dependence and selectivity, voltage dependence, and the opposing actions substrates have on them. Furthermore, our evidence suggests that the transport-associated current itself is complex and may arise from two ionic conductances, both of which are activated by DA under conditions that permit substrate translocation (or a single transport conductance that operates with variable coupling). In contrast, the tonic leak conductance is inhibited by hDAT substrates and nontranslocated uptake blockers alike. All three putative conductances appear to be sensitive to individual hDAT ligands in the same concentration range, and therefore, all may be of potential relevance to the actions of hDAT agents. We do not know whether the three conductances are all intrinsic to a single transporter molecule, to hDAT oligomers, or to distinct subpopulations of modified hDAT molecules, or whether some ionic conductance attributed to hDAT is indirectly mediated through an extrinsic protein (Sonders and Amara, 1996).
Although our results predict that neuronal membrane potential will directly modulate hDAT uptake activity, at this stage it is not known whether hDAT-mediated currents contribute significantly to the electrical properties of dopaminergic neurons in vivo. Whole-cell currents attributable to neurotransmitter uptake have been demonstrated in a variety of neuronal and glial cells (for review, seeLester et al., 1994; Sonders and Amara, 1996). Estimation of the turnover rate for hDAT in oocytes, 0.47 DA molecules/sec at 21°C, indicates that the transporter is relatively slow and must reside at comparatively high density in terminal areas for it to effectively modulate extracellular DA concentrations, as has been suggested from work with rat striatal preparations (Boja et al., 1994). Accordingly, it is conceivable that the density of DATs may be high enough that even the small currents associated with hDAT expression in oocytes could affect the membrane potential in regions of dopaminergic neurons with high input resistance. Future experiments should reveal whether DAT influences the electrical signaling properties of dopaminergic neurons in a capacity beyond its function of regulating DA recycling.
Footnotes
These investigations have been supported by 5T32DA07262, DA07595, GM48709, DA04216, K02DA00174, F32DA05706, and HHMI. Tissue was obtained from the Oregon Brain Bank, which is partially supported by Grant P30A908017. We thank Geoffrey Murdoch, Wendy Fairman, and Weibin Zhang for multiple contributions to this work, and Jonathan Javitch for helpful editorial comments.
Correspondence should be addressed to Dr. Susan G. Amara, Vollum Institute, L474, Oregon Health Sciences University, Portland, OR 97201 (amaras@ohsu.edu) or to Dr. Mark S. Sonders (sondersm@ohsu.edu).
REFERENCES
- 1.Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- 2.Arriza JL, Fairman WA, Wadiche J, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldessarini RJ, Vogt M. The uptake and subcellular distribution of aromatic amines in the brain of the rat. J Neurochem. 1971;18:2519–2533. doi: 10.1111/j.1471-4159.1971.tb00208.x. [DOI] [PubMed] [Google Scholar]
- 4.Baumann PA, Maitre L. Is drug inhibition of dopamine uptake a misinterpretation of in vitro experiments? Nature. 1976;264:789–790. doi: 10.1038/264789a0. [DOI] [PubMed] [Google Scholar]
- 5.Boja JW, Vaughan R, Patel A, Shaya EK, Kuhar MJ. The dopamine transporter. In: Niznik HB, editor. Dopamine receptors and transporters. Marcel Dekker; New York: 1994. pp. 611–644. [Google Scholar]
- 6.Bonnet J-J, Lemasson M-H, Costentin J. Simultaneous evaluation by a double labelling method of drug-induced uptake inhibition and release of dopamine in synaptosomal preparation of rat striatum. Biochem Pharmacol. 1984;33:2129–2135. doi: 10.1016/0006-2952(84)90583-5. [DOI] [PubMed] [Google Scholar]
- 7.Buck KJ, Amara SG. Chimeric dopamine-norepinephrine transporters delineate structural domains influencing selectivity for catecholamines and 1-methyl-4-phenylpyridinium. Proc Natl Acad Sci USA. 1994;91:12584–12588. doi: 10.1073/pnas.91.26.12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cammack JN, Schwartz EA. Ions required for the electrogenic transport of GABA by horizontal cells of the catfish retina. J Physiol (Lond) 1993;472:81–102. doi: 10.1113/jphysiol.1993.sp019938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cammack JN, Schwartz EA. Channel behavior in a γ-aminobutyrate transporter. Proc Natl Acad Sci USA. 1996;93:723–727. doi: 10.1073/pnas.93.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cammack JN, Rakhilin SV, Schwartz EA. A GABA transporter operates asymmetrically and with variable stoichiometry. Neuron. 1994;13:949–960. doi: 10.1016/0896-6273(94)90260-7. [DOI] [PubMed] [Google Scholar]
- 11.Cass WA, Gerhardt GA. Direct in vivo evidence that D2 dopamine receptors can modulate dopamine uptake. Neurosci Lett. 1994;176:259–263. doi: 10.1016/0304-3940(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 12.Cerruti C, Walther DM, Kuhar MJ, Uhl GR. Dopamine transporter mRNA expression is intense in rat midbrain neurons and modest outside midbrain. Mol Brain Res. 1993;18:181–186. doi: 10.1016/0169-328x(93)90187-t. [DOI] [PubMed] [Google Scholar]
- 13.Chang GD, Ramirez VD. The mechanism of action of MPTP and MPP+ on endogenous dopamine release from the rat corpus striatum superfused in vitro. Brain Res. 1986;386:134–140. doi: 10.1016/0006-8993(86)91050-4. [DOI] [PubMed] [Google Scholar]
- 14.Chen X-Z, Coady MJ, Jackson F, Berteloot A, Lapointe J-Y. Thermodynamic determination of the Na+:glucose coupling ratio for the human SGLT1 cotransporter. Biophys J. 1995;69:2405–2414. doi: 10.1016/S0006-3495(95)80110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 16.Ciliax BJ, Heilman C, Demchyshyn LL, Pristupa ZB, Ince E, Hersch SM, Niznik HB, Levey AI. The dopamine transporter: immunochemical characterization and localization in brain. J Neurosci. 1995;15:1714–1723. doi: 10.1523/JNEUROSCI.15-03-01714.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costa PF, Emilio MG, Fernandes PL, Gil Ferreira H, Gil Ferreira K. Determination of ionic permeability coefficients of the plasma membrane of Xenopus laevis oocytes under voltage clamp. J Physiol (Lond) 1989;413:199–211. doi: 10.1113/jphysiol.1989.sp017649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dascal N. The use of Xenopus oocytes for the study of ion channels. CRC Crit Rev Biochem. 1987;22:317–387. doi: 10.3109/10409238709086960. [DOI] [PubMed] [Google Scholar]
- 19.DeCoursey TE, Cherny VV. Voltage-activated hydrogen ion currents. J Membr Biol. 1994;141:203–224. doi: 10.1007/BF00235130. [DOI] [PubMed] [Google Scholar]
- 20.DeFelice LJ, Blakely RD. Pore models for transporters? Biophys J. 1996;70:579–580. doi: 10.1016/S0006-3495(96)79604-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorris RL, Shore PA. Amine uptake and storage mechanisms in the corpus striatum of rat and rabbit. J Pharmacol Exp Ther. 1971;179:15–19. [PubMed] [Google Scholar]
- 22.Eliasof S, Jahr CE. Retinal glial cell glutamate transporter is coupled to an anionic conductance. Proc Natl Acad Sci USA. 1996;93:4153–4158. doi: 10.1073/pnas.93.9.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eshleman AJ, Neve RL, Janowsky A, Neve KA. Characterization of a recombinant human dopamine transporter in multiple cell lines. J Pharmacol Exp Ther. 1995;274:276–283. [PubMed] [Google Scholar]
- 24.Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- 25.Fischer JF, Cho AK. Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J Pharmacol Exp Ther. 1979;208:203–209. [PubMed] [Google Scholar]
- 26.Galli A, DeFelice LJ, Duke B-J, Moore KR, Blakely RD. Sodium-dependent norepinephrine-induced currents in norepinephrine transporter transfected HEK-293 cells blocked by cocaine and antidepressants. J Exp Biol. 1995;198:2197–2212. doi: 10.1242/jeb.198.10.2197. [DOI] [PubMed] [Google Scholar]
- 27.Gerhardt GA, Dwoskin LP, Zahniser NR. Outflow and overflow of picogram levels of endogenous dopamine and DOPAC from rat striatal slices: improved methodology for studies of stimulus-evoked release and metabolism. J Neurosci Methods. 1989;26:217–227. doi: 10.1016/0165-0270(89)90119-2. [DOI] [PubMed] [Google Scholar]
- 28.Giros B, Mestikawy SE, Godinot N, Zheng K, Han H, Yang-Feng T, Caron MG. Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter. Mol Pharmacol. 1992;42:383–390. [PubMed] [Google Scholar]
- 29.Giros B, Wang Y-M, Suter S, McLeskey SB, Pifl C, Caron MG. Delineation of discrete domains for substrate, cocaine, and tricyclic antidepressant interactions using chimeric dopamine-norepinephrine transporters. J Biol Chem. 1994;2169:15985–15988. [PubMed] [Google Scholar]
- 30.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 31.Gu H, Wall SC, Rudnick G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem. 1994;269:7124–7130. [PubMed] [Google Scholar]
- 32.Heikkila RE, Orlansky H, Cohen G. Studies on the distinction between uptake inhibition and release of [3H]dopamine in rat brain tissue slices. Biochem Pharmacol. 1975;24:847–852. doi: 10.1016/0006-2952(75)90152-5. [DOI] [PubMed] [Google Scholar]
- 33.Hille B. Ionic channels of excitable membranes. Sinauer; Sunderland, MA: 1992. [Google Scholar]
- 34.Holz RW, Coyle JT. The effects of various salts, temperature and the alkaloids veratridine and batrachotoxin on the uptake of [3H]-dopamine into synaptosomes from rat striatum. Mol Pharmacol. 1974;10:746–758. [Google Scholar]
- 35.Isaacson JS, Solís JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- 36.Javitch JA, Blaustein RO, Snyder SH. [3H]Mazindol binding associated with neuronal dopamine and norepinephrine uptake sites. Mol Pharmacol. 1984;26:35–44. [PubMed] [Google Scholar]
- 37.Kanner BI, Schuldiner S. Mechanism of transport and storage of neurotransmitters. CRC Crit Rev Biochem. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- 38.Keller HH, Da Prada M. Evidence for the release of 1-methyl-4-pyridinium (MPP+) from rat striatal neurons in vitro. Eur J Pharmacol. 1985;119:247–250. doi: 10.1016/0014-2999(85)90304-8. [DOI] [PubMed] [Google Scholar]
- 39.Kenakin TP. Pharmacologic analysis of drug-receptor interaction. Raven; New York: 1987. [Google Scholar]
- 40.Kilty JE (1993) Cloning and characterization of the rat dopamine transporter: the ionic-dependence and substrate selectivity of catecholamine transporters. Dissertation, Yale University.
- 41.Krueger BK. Kinetics and block of dopamine uptake in synaptosomes from rat caudate nucleus. J Neurochem. 1990;55:260–267. doi: 10.1111/j.1471-4159.1990.tb08847.x. [DOI] [PubMed] [Google Scholar]
- 42.Lacey MG. Neurotransmitter receptors and ionic conductances regulating the activity of neurones in the substantia nigra pars compacta and ventral tegmental area. Prog Brain Res. 1993;99:251–276. doi: 10.1016/s0079-6123(08)61351-5. [DOI] [PubMed] [Google Scholar]
- 43.Langeloh A, Bönisch H, Trendelenburg U. The mechanics of the 3H-noradrenaline releasing effect of various substrates of uptake1: multifactorial induction of outward transport. Naunyn Schmiedebergs Arch Pharmacol. 1987;336:602–610. doi: 10.1007/BF00165750. [DOI] [PubMed] [Google Scholar]
- 44.Larsson HP, Picaud SA, Werblin FS, Lecar H. Noise analysis of the glutamate-activated current in photoreceptors. Biophys J. 1996;70:733–742. doi: 10.1016/S0006-3495(96)79613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lester HA, Mager S, Quick MW, Corey JL. Permeation properties of neurotransmitter transporters. Annu Rev Pharmacol Toxicol. 1994;34:219–249. doi: 10.1146/annurev.pa.34.040194.001251. [DOI] [PubMed] [Google Scholar]
- 46.Liang NY, Rutledge CO. Evidence for carrier-mediated efflux of dopamine from corpus striatum. Biochem Pharmacol. 1982;31:2479–2484. doi: 10.1016/0006-2952(82)90057-0. [DOI] [PubMed] [Google Scholar]
- 47.Lorang D, Amara SG, Simerly RB. Cell-type-specific expression of catecholamine transporters in the rat brain. J Neurosci. 1994;14:4903–4914. doi: 10.1523/JNEUROSCI.14-08-04903.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lukacs GL, Kapus A, Nanda A, Romanek R, Grinstein S. Proton conductance of the plasma membrane: properties, regulation, and functional role. Am J Physiol. 1993;265:C3–C14. doi: 10.1152/ajpcell.1993.265.1.C3. [DOI] [PubMed] [Google Scholar]
- 49.Mager S, Min C, Henry DJ, Chavkin C, Hoffman BJ, Davidson N, Lester HA. Conducting states of a mammalian serotonin transporter. Neuron. 1994;12:845–859. doi: 10.1016/0896-6273(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 50.Mager S, Kleinberger-Doron N, Keshet GI, Davidson N, Kanner BI, Lester HA. Ion binding and permeation at the GABA transporter GAT1. J Neurosci. 1996;16:5405–5414. doi: 10.1523/JNEUROSCI.16-17-05405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maloney PC. Bacterial transporters. Curr Opin Cell Biol. 1994;6:571–582. doi: 10.1016/0955-0674(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 52.McElvain JS, Schenk JO. A multisubstrate mechanism of dopamine uptake and its inhibition by cocaine. Biochem Pharmacol. 1992;43:2189–2199. doi: 10.1016/0006-2952(92)90178-l. [DOI] [PubMed] [Google Scholar]
- 53.Meiergerd SM, Patterson TA, Schenk JO. D2 receptors may modulate the function of the striatal transporter for dopamine: kinetic evidence from studies in vitro and in vivo. J Neurochem. 1993;61:764–767. doi: 10.1111/j.1471-4159.1993.tb02185.x. [DOI] [PubMed] [Google Scholar]
- 54.Nestler EJ, Hope BT, Widnell KL. Drug addiction: a model for the molecular basis of neural plasticity. Neuron. 1993;11:995–1006. doi: 10.1016/0896-6273(93)90213-b. [DOI] [PubMed] [Google Scholar]
- 55.Nirenberg MJ, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. The dopamine transporter is localized to dendritic and axonal plasma membranes of nigrostriatal dopaminergic neurons. J Neurosci. 1996;16:436–447. doi: 10.1523/JNEUROSCI.16-02-00436.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parent L, Supplisson S, Loo DDF, Wright EM. Electrogenic properties of the cloned Na+/glucose cotransporter. I. Voltage-clamp studies. J Membr Biol. 1992;125:49–62. doi: 10.1007/BF00235797. [DOI] [PubMed] [Google Scholar]
- 57.Parker EM, Cubeddu LX. Effects of d-amphetamine and dopamine synthesis inhibitors on dopamine and acetylcholine neurotransmission in the striatum. I. Release in the absence of vesicular transmitter stores. J Pharmacol Exp Ther. 1986;237:179–192. [PubMed] [Google Scholar]
- 58.Parsons LH, Schad CA, Justice JB., Jr Coadministration of the D2 antagonist pimozide inhibits up-regulation of dopamine release and uptake induced by cocaine. J Neurochem. 1993;60:376–379. doi: 10.1111/j.1471-4159.1993.tb05864.x. [DOI] [PubMed] [Google Scholar]
- 59.Pifl C, Giros B, Caron MG. Dopamine transporter expression confers cytotoxicity to low doses of the parkinsonism-inducing neurotoxin 1-methyl-4-phenylpyridinium. J Neurosci. 1993;13:4246–4253. doi: 10.1523/JNEUROSCI.13-10-04246.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pifl C, Drobny H, Reither H, Hornykiewicz O, Singer EA. Mechanism of the dopamine-releasing actions of amphetamine and cocaine: plasmalemmal dopamine transporter versus vesicular monoamine transporter. Mol Pharmacol. 1995;47:368–373. [PubMed] [Google Scholar]
- 61.Pristupa ZB, Wilson JM, Hoffman BJ, Kish SJ, Niznik HB. Pharmacological heterogeneity of the cloned and native human dopamine transporter: disassociation of [3H]WIN 35,428 and [3H]GBR 12,935 binding. Mol Pharmacol. 1994;45:125–135. [PubMed] [Google Scholar]
- 62.Pulvirenti L, Koob GF. Dopamine receptor agonists, partial agonists and psychostimulant addiction. Trends Pharmacol Sci. 1994;15:374–379. doi: 10.1016/0165-6147(94)90158-9. [DOI] [PubMed] [Google Scholar]
- 63.Quick MW, Lester HA. Methods for expression of excitability proteins in Xenopus oocytes. Methods Neurosci. 1994;19:261–279. [Google Scholar]
- 64.Raiteri M, Del Carmine R, Bertollini A, Levi G. Effect of sympathomimetic amines on the synaptosomal transport of noradrenaline, dopamine and 5-hydroxytryptamine. Eur J Pharmacol. 1977;41:133–143. doi: 10.1016/0014-2999(77)90202-3. [DOI] [PubMed] [Google Scholar]
- 65.Raiteri M, Cerrito F, Cervoni AM, Levi G. Dopamine can be released by two mechanisms differentially affected by the dopamine transport inhibitor nomifensine. J Pharmacol Exp Ther. 1979;208:195–202. [PubMed] [Google Scholar]
- 66.Risso S, DeFelice LJ, Blakely RD. Sodium-dependent GABA-induced currents in GAT1-transfected HeLa cells. J Physiol (Lond) 1996;490:691–702. doi: 10.1113/jphysiol.1996.sp021178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 68.Rollema H, Damsma G, Horn AS, De Vries JB, Westerink BH. Brain dialysis in conscious rats reveals an instantaneous massive release of striatal dopamine in response to MPP+. Eur J Pharmacol. 1986;126:345–346. doi: 10.1016/0014-2999(86)90071-3. [DOI] [PubMed] [Google Scholar]
- 69.Ross SB. Structural requirements for uptake into catecholamine neurons. In: Paton DM, editor. The mechanism of neuronal and extraneuronal transport of catecholamines. Raven; New York: 1976. pp. 67–93. [Google Scholar]
- 70.Rudnick G, Clark J. From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim Biophys Acta. 1993;1144:249–263. doi: 10.1016/0005-2728(93)90109-s. [DOI] [PubMed] [Google Scholar]
- 71.Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;32:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- 72.Sesack SR, Aoki C, Pickel VM. Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci. 1994;14:88–106. doi: 10.1523/JNEUROSCI.14-01-00088.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sirinathsinghji DJ, Heavens RP, McBride CS. Dopamine-releasing action of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 1-methyl-4-phenylpyridine (MPP+) in the neostriatum of the rat as demonstrated in vivo by the push-pull perfusion technique: dependence on sodium but not calcium ions. Brain Res. 1988;443:101–116. doi: 10.1016/0006-8993(88)91603-4. [DOI] [PubMed] [Google Scholar]
- 74.Sonders MS, Amara SG. Channels in transporters. Curr Opin Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- 75.Stein WD. Transport and diffusion across cell membranes. Academic; San Diego: 1986. [Google Scholar]
- 76.Steinberg MI, Smith CB. Effects of desmethylimipramine and cocaine on the uptake, retention and metabolism of H3-tyramine in rat brain slices. J Pharmacol Exp Ther. 1970;173:176–192. [PubMed] [Google Scholar]
- 77.Sulzer D, Rayport S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron. 1990;5:797–808. doi: 10.1016/0896-6273(90)90339-h. [DOI] [PubMed] [Google Scholar]
- 78.Sulzer D, Chen T-K, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Turner RJ. Stoichiometry of cotransport systems. Ann NY Acad Sci. 1985;456:10–25. doi: 10.1111/j.1749-6632.1985.tb14839.x. [DOI] [PubMed] [Google Scholar]
- 80.Umbach JA, Coady MJ, Wright EM. Intestinal Na+/glucose transporter expressed in Xenopus oocytes is electrogenic. Biophys J. 1990;57:1217–1224. doi: 10.1016/S0006-3495(90)82640-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vandenberg RJ, Arriza JL, Amara SG, Kavanaugh MP. Constitutive ion fluxes and substrate binding domains of human glutamate transporters. J Biol Chem. 1995;270:17668–17671. doi: 10.1074/jbc.270.30.17668. [DOI] [PubMed] [Google Scholar]
- 82.Vandenbergh DJ, Persico AM, Uhl GR. A human dopamine transporter cDNA predicts reduced glycosylation, displays a novel repetitive element and provides racially-dimorphic TaqI RFLPs. Mol Brain Res. 1992;15:161–166. doi: 10.1016/0169-328x(92)90165-8. [DOI] [PubMed] [Google Scholar]
- 83.Wadiche JI, Amara SG, Kavanaugh MP. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 84.Wall SC, Gu H, Rudnick G. Biogenic amine flux mediated by cloned transporters stably expressed in cultured cell lines: amphetamine specificity for inhibition and efflux. Mol Pharmacol. 1995;47:544–550. [PubMed] [Google Scholar]
- 85.White FJ. Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci. 1996;19:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- 86.Wise RA. Addictive drugs and brain stimulation reward. Annu Rev Neurosci. 1996;19:319–340. doi: 10.1146/annurev.ne.19.030196.001535. [DOI] [PubMed] [Google Scholar]
- 87.Yung KKL, Bolam JP, Smith AD, Hersch SM, Ciliax BJ, Levey AI. Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience. 1995;65:709–730. doi: 10.1016/0306-4522(94)00536-e. [DOI] [PubMed] [Google Scholar]
- 88.Zaczek R, Culp S, De Souza EB. Interactions of [3H]amphetamine with rat brain synaptosomes. II. Active transport. J Pharmacol Exp Ther. 1991;257:830–835. [PubMed] [Google Scholar]