

Abstract
Modulation of excitatory synaptic transmission by presynaptic metabotropic glutamate receptors (mGluRs) was examined in brain slices from control rats and rats with amygdala-kindled seizures. Using whole-cell voltage-clamp and current-clamp recordings, this study shows for the first time that in control and kindled basolateral amygdala neurons, two pharmacologically distinct presynaptic mGluRs mediate depression of synaptic transmission. Moreover, in kindled neurons, agonists at either group II- or group III-like mGluRs exhibit a 28- to 30-fold increase in potency and suppress synaptically evoked bursting. The group II mGluR agonist (2S,3S,4S)-2-(carboxycyclopropyl)glycine (l-CCG) dose-dependently depressed monosynaptic EPSCs evoked by stimulation in the lateral amygdala with EC50values of 36 nm (control) and 1.2 nm (kindled neurons). The group III mGluR agonistl-2-amino-4-phosphonobutyrate (l-AP4) was less potent, with EC50 values of 297 nm (control) and 10.8 nm (kindled neurons). The effects ofl-CCG and l-AP4 were fully reversible. Neitherl-CCG (0.0001–10 μm) nor l-AP4 (0.001–50 μm) caused membrane currents or changes in the current–voltage relationship. The novel mGluR antagonists (2S,3S,4S)-2-methyl-2-(carboxycyclopropyl)-glycine (MCCG; 100 μm) and (S)-2-methyl-2-amino-4-phos-phonobutyrate (MAP4; 100 μm) selectively reversed the inhibition byl-CCG and l-AP4 to 81.3 ± 12% and 65.3 ± 6.6% of predrug, respectively. MCCG and MAP4 (100–300 μm) themselves did not significantly affect synaptic transmission. The exquisite sensitivity of agonists in the kindling model of epilepsy and the lack of evidence for endogenous receptor activation suggest that presynaptic group II- and group III-like mGluRs might be useful targets for suppression of excessive synaptic activation in neurological disorders such as epilepsy.
Keywords: excitatory amino acids, metabotropic glutamate receptors, presynaptic, basolateral amygdala, synaptic transmission, neuromodulation, kindling, epilepsy, limbic seizures, anticonvulsant, l-CCG, l-AP4, MCCG, MAP4
Understanding the synaptic and cellular processes that cause or accompany the various forms of epilepsies may help not only to improve therapies but also to gain insights into brain functions (McNamara, 1994). One established animal model of human epilepsy is “kindling,” in which the repeated and focal applications of initially subconvulsive electrical stimuli to certain brain areas result in the progressive development of partial and generalized seizures (Goddard et al., 1969; Racine et al., 1972).
The amygdala is one of the brain areas most sensitive to kindling-induced neuroplasticity (Löscher et al., 1995). This lab has shown previously that for several weeks after the last amygdala-kindled seizure in vivo, spontaneous and evoked epileptiform bursting can be recorded in vitro (Gean et al., 1989; Asprodini et al., 1992a,b; Rainnie et al., 1992; Holmes et al., 1996). In basolateral amygdala (BLA) neurons, kindling reduces the GABA receptor-mediated inhibitory transmission (Gean et al., 1989; Asprodini et al., 1992b; Rainnie et al., 1992) and enhances NMDA and non-NMDA receptor-mediated glutamatergic transmission (Gean et al., 1989;Rainnie et al., 1992).
l-Glutamate activates not only ionotropic NMDA and non-NMDA receptors, both of which form ligand-gated ion channels, but also metabotropic glutamate receptors (mGluRs), which couple to second messengers through G-proteins (Schoepp and Conn, 1993; Hollmann and Heinemann, 1994; Nakanishi and Masu, 1994; Westbrook, 1994). To date, eight mGluR subtypes (mGluR1–mGluR8) have been cloned and are classified into three groups based on sequence homology, signal transduction mechanisms, and pharmacological profiles (Nakanishi, 1994;Knöpfel et al., 1995; Pin and Duvoisin, 1995): group I (mGluR1 and 5), group II (mGluR2 and 3), and group III (mGluR4, 6, 7, 8).
Although mGluRs play important roles in neuroplasticity and neuropathology (Schoepp and Conn, 1993; Nakanishi, 1994; Knöpfel et al., 1995; Miller et al., 1995; Pin and Duvoisin, 1995), their significance in epilepsy is not clear (Gallagher et al., 1994). Activation of mGluRs can induce/enhance or suppress epileptiform activity in vivo (Sacaan and Schoepp, 1992; McDonald et al., 1993; Thomsen et al., 1994; Attwell et al., 1995; Tanaka et al., 1995;Tizzano et al., 1995; Dalby and Thomsen, 1996; Suzuki et al., 1996) andin vitro (Taschenberger et al., 1992; Arvanov et al., 1995;Burke and Hablitz, 1995; Merlin et al., 1995). This heterogeneity of effects may reflect the diversity of mGluRs.
Enhancement and/or depression of neuronal excitability and neurotransmission is observed on mGluR activation in control neurons (Knöpfel et al., 1995; Pin and Duvoisin, 1995). Recent evidence suggests that the synaptic inhibition involves multiple presynaptic mGluRs (Gereau and Conn, 1995; Lovinger and McCool, 1995; Manzoni and Bockaert, 1995; Vignes et al., 1995). In BLA neurons, only one study showed presynaptic inhibition by group III-like mGluRs (Rainnie and Shinnick-Gallagher, 1992). In amygdala-kindled neurons, postsynaptic mGluR-mediated effects change (Holmes et al., 1996). The role of presynaptic mGluRs, however, has not been determined. Here we examined the modulation of neurotransmission by presynaptic mGluRs in control BLA neurons, possible changes induced by amygdala-kindling, and the endogenous activation of presynaptic mGluRs.
MATERIALS AND METHODS
Amygdala slice preparation. Brain slices containing the BLA and lateral amygdalae (LA) were obtained from control and kindled male Sprague Dawley rats (70–240 gm) as described previously (Rainnie et al., 1994; Holmes et al., 1996). Rats were decapitated, and the brains were quickly dissected out and blocked in cold (4°C) artificial cerebrospinal fluid (ACSF) containing (in mm): 117 NaCl, 4.7 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, and 11 glucose. ACSF was oxygenated and equilibrated to pH 7.4 with a mixture of 95% O2/5% CO2. Coronal brain slices (500 μm) were prepared using a vibroslice (Campden Instruments, London, UK). After incubation in ACSF at room temperature (21°C) for at least 1 hr, a single brain slice was transferred to the recording chamber and submerged in ACSF (31 ± 1°C), which perfused the slice at ∼2 ml/min.
Whole-cell patch-clamp recording. “Blind” whole-cell recordings (Blanton et al., 1989) were obtained from BLA neurons using patch electrodes made from 1.5 mm borosilicate glass capillaries (1.5 mm outer diameter, 1.12 mm inner diameter; Drummond, Broomall, PA) pulled on a Flaming-Brown micropipette puller (P-80/PC, Sutter Instrument, Novato, CA). The recording electrodes had tip resistances of 3–5 MΩ when filled with one of two internal solutions (compounds in mm): (1) 122 potassium gluconate, 5 NaCl, 0.3 CaCl2, 2 MgCl2, 1 EGTA, 10 HEPES, 5 Na2-ATP, 0.4 Na3-GTP; (2) 140 KMeSO4, 10 HEPES, 2 Na2-ATP, 0.3 Na3-GTP. Internal solutions were adjusted to pH 7.2–7.3 with KOH and to osmolality of 280 mmol/kg with sucrose. Because no difference of drug effects was found using either of these internal solutions, the data were pooled.
Recordings were performed in both current and voltage clamp. After tight seals (2–4 GΩ) were formed and the whole-cell configuration was obtained, neurons then were included in the sample if the resting membrane potential was more negative than −60 mV and action potentials overshooting 0 mV could be evoked by direct cathodal stimulation. Voltage and current signals were low-pass-filtered at 1 kHz with a 4-pole Bessel filter (Warner Instrument, Hamden, CT), digitized at 5 Hz (Digidata 1200 interface, Axon Instruments, Foster City, CA), and stored on a computer (4DX2-66V, Gateway 2000). Data were also continuously recorded on a pen chart recorder (Gould 2400, Gould Instruments, Valley View, OH) and a four-channel videotape recorder (Model 420C; A. R. Vetter, Rebersburg, PA). Evoked potential and evoked current data were acquired and analyzed using pCLAMP software (Axon Instruments). Discontinuous single-electrode voltage-clamp recordings were acquired using an Axoclamp-2A amplifier (Axon Instruments) with a switching frequency of 5–6 kHz (30% duty cycle), gain of 3–8 nA/mV, time constant 20 msec. Phase-shift and anti-alias filter were optimized. The headstage voltage was monitored on an oscilloscope (Tektronix, Pittsfield, MA) to ensure precise performance of the amplifier.
Synaptic stimulation. Monosynaptic EPSPs and EPSCs were evoked in BLA neurons by electrical stimulation (using a Grass S88 stimulator; Grass Instruments, Quincy, MA) of afferents from the LA with a concentric bipolar stimulating electrode (SNE-100, Kopf Instruments). Electrical stimuli (150 μsec square-wave pulses) were delivered at frequencies below 0.25 Hz. Thresholds for EPSPs, EPSCs, spiking, and burst-firing were defined as the respective intensity that evoked a response in at least 5 of 10 trials. Input–output relations were obtained by increasing the stimulus intensity in 0.5 V steps. For evaluation of a drug effect on EPSPs and EPSCs the stimulus intensities were adjusted to just subthreshold for orthodromic spike generation.
Kindling. Rats were anesthetized with Equithesin (35 mg/kg sodium pentobarbital and 145 mg/kg chloral hydrate) and implanted with tripolar electrodes (Plastics One, Roanoke, VA) into the right BLA, as described previously (Rainnie et al., 1992; Holmes et al., 1996). Using the coordinates from Paxinos and Watson (1986), the tips of the two leads were positioned 2.0 mm posterior and 4.5 mm lateral to bregma at a depth of 7.3 mm from the dura surface; the third lead served as a ground for monitoring and/or recording afterdischarges (ADs). Electrodes were fixed to the skull with dental acrylic (Plastics One). The kindling stimulation of the BLA was started after a postimplantation recovery period of 5 d. The stimulation consisted of a 2 sec train of 60 Hz monophasic square waves, each 2 msec in duration, administered twice daily at least 8 hr apart. On the first kindling stimulation, threshold for evoking ADs was determined (200–400 μA). Subsequent stimulations were give n at 50–100 μA above the AD threshold and were monitored on a storage oscilloscope (Tektronix, Pittsfield, MA). Behavioral seizure severity was rated according to the ranking scale of Racine (1972). Animals received stimulations until three consecutive stage 5 (fully kindled) seizures were evoked. Animals were sacrificed 3–7 d later, and brain slices were prepared for the electrophysiological experiments. Control slices were obtained from both unoperated and unstimulated-implanted rats.
Drugs. The following mGluR agonists and antagonists (purchased from Tocris Cookson, Bristol, UK) were used: (2S,3S,4S)-2-(carboxycyclopropyl)-glycine (l-CCG),l-2-amino-4-phosphonobutyrate (l-AP4), (2S,3S,4S)-2-methyl-2-(carboxycyclopropyl)glycine (MCCG), and (S)-2-amino-2-methyl-4-phosphonobutyrate (MAP4). All drugs were applied by gravity-driven superfusion in the ACSF. Solution flow into the recording chamber (1 ml volume) was controlled with a three-way stopcock. All applications were at least 10 min (usually 12–14 min) in duration to establish equilibrium in the tissue.
Data analysis and statistics. Concentration–response relationships and input–output relationships were compared using the two-way ANOVA with post hoc tests. The paired ttest was used to compare data before and during drug application and to evaluate agonist effects in the presence and absence of antagonists. All averaged values are given as the mean ± SEM. Statistical significance was accepted at the level p < 0.05. EC50 values were calculated from sigmoid curves fitted to the concentration–response data by nonlinear regression using the formula y = A + (B −A)/ [1 + (10C/10X)D], whereA = bottom plateau, B = top plateau,C = log (EC50), D = slope coefficient (Inplot, GraphPad Software, San Diego, CA) (Tallarida and Jacob, 1979). Using the linear curve fit function of pCLAMP software (Axon Instruments), slope conductances (in nS) in the absence and presence of agonists and antagonists were calculated from the linear portion of the current–voltage (I–V) relationships recorded in voltage-clamp mode.
RESULTS
l-CCG and l-AP4 depress synaptic transmission in control BLA neurons through a presynaptic action
Modulation of synaptic transmission by mGluRs at the LA-BLA synapse was examined using the mGluR agonists l-CCG, which at low concentrations discriminates between groups II and III mGluRs, and l-AP4, which is highly selective and the most potent agonist at group III mGluRs (Hayashi et al., 1994; Suzdak et al., 1994;Burke and Hablitz, 1995; Gereau and Conn, 1995; Lovinger and McCool, 1995; Pin and Duvoisin, 1995). For each agonist we obtained complete concentration–response curves on synaptic transmission and postsynaptic cell membrane properties.
Superfusing the brain slice with l-CCG reduced the amplitude of monosynaptic EPSCs in a concentration-dependent manner in 22 neurons recorded in the BLA. A typical example is shown in Figure1A,B. The depressive effect of l-CCG was slowly reversible, usually within 15–20 min of washing with control ACSF. Analysis of the sigmoid concentration–response curve (Fig. 3A) revealed an EC50 of 36 nm for the inhibition of EPSC peak amplitude by l-CCG (see Materials and Methods).
Fig. 1.

The group II mGluR agonist l-CCG depresses synaptic transmission in the BLA in a concentration-dependent manner without affecting postsynaptic membrane properties. A, B, Monosynaptic EPSCs were evoked by electrical stimulation of afferents from the LA (13.5 V; 150 μsec) before, during, and after superfusion of the brain slice with l-CCG. Each trace is an average of 8–10 EPSCs recorded immediately before drug addition (control), after 10 min in l-CCG, and after 20 min of washout. The traces shown in A are superimposed in B. C, D, l-CCG did not affect the I–V relationship of the neuron. C, Whole-cell currents were elicited by a series of 400 msec voltage steps (−120 to −50 mV) from a holding potential of −60 mV in control ACSF and after 11 min in l-CCG.D, I–V curves show steady-state currents from voltage-clamp records like those inC, plotted against membrane potential in the absence and presence of l-CCG at the indicated concentrations. The data in A–D were obtained from one BLA neuron voltage-clamped at −60 mV.
Fig. 3.

The concentration-dependent depression of synaptic transmission by mGluR agonists is enhanced in kindled neurons.A, The peak amplitudes of the EPSCs obtained with each concentration of l-CCG in control neurons (n = 22) and kindled neurons (n= 5) were averaged and expressed as percentage of predrug control values (100%). The two-way ANOVA revealed significant differences between control and kindled neurons (p < 0.0001) and between the different concentrations (p < 0.0001) but no significant interaction (p > 0.5), indicating a parallel shift. The EC50 (see Materials and Methods) is 30 × lower in kindled (1.2 nm) than in control neurons (36 nm). B, l-AP4 was less potent than l-CCG in depressing synaptic transmission. Effects ofl-AP4 on EPSC amplitudes in control (n= 27) and kindled neurons (n = 6) are displayed as in A. The two-way ANOVA detected significant differences between control and kindled neurons (p < 0.0001) and between the different concentrations (p < 0.0001) but no significant interaction (p > 0.5), thus indicating a parallel shift. The EC50 is 28 × lower in kindled (10.8 nm) than in control neurons (297 nm). Symbols and error bars represent mean ± SEM.
Concentrations of l-CCG up to 10 μm neither induced detectable postsynaptic membrane currents nor altered membrane conductance as reflected in the I–V relationship (Fig. 1C,D), effects that are consistent with a presynaptic site of action. For each neuron (n = 21), the slope conductance of the I–V relation was calculated (see Materials and Methods) in the absence and presence of different concentrations of l-CCG. Averaging the difference in slope conductance for each concentration showed thatl-CCG had no significant effect at concentrations of 0.1 nm to 10 μm (Fig. 4A).l-CCG (100 μm), however, increased the slope conductance significantly (paired t test, p< 0.01) and induced an outward current of 22 ± 8 pA (n = 5). Furthermore, l-CCG (1 μm) did not alter action potential threshold or accommodation (n = 3) (Fig. 5A). In current-clamp mode, the frequency of action potentials generated by transient (500 msec) depolarizing current injections of increasing amplitude (steps of 100 pA) was not different in the presence and absence of l-CCG (paired t test,p > 0.1).
Fig. 4.

Low concentrations of l-CCG (A) and l-AP4 (B) do not have postsynaptic membrane effects in control and in kindled neurons. For each neuron the slope conductance was calculated from theI–V relationships (see Figs. 1, 2) in the presence and absence of the different agonist concentrations. The averaged differences are shown as mean ± SEM. A, In control (n = 21) and kindled neurons (n = 5), the slope conductances in the presence of up to 10 μml-CCG were not different from their respective predrug controls (paired t test,p > 0.5). At 100 μm, however, there was a significant increase of slope conductance (pairedt test, n = 5; tested in control neurons only). B, In control neurons (n = 23) the slope conductances in the presence of 1 nm to 50 μml-AP4 were not different from their respective predrug controls (pairedt test, p > 0.5). l-AP4 (100 and 200 μm) significantly increased the slope conductance (paired t test, n = 5 and n = 4, respectively). In kindled neurons (n = 6), no effects of 10 nm to 100 μml-AP4 were observed on the slope conductance (paired t test, p > 0.1). *p < 0.05, **p < 0.01.
Fig. 5.

Action-potential firing patterns are not changed by l-CCG (A, C) andl-AP4 (B, D) in control (A, B) and kindled neurons (C, D). Current-clamp recordings from four different BLA neurons at membrane potential −60 mV. Action potentials were generated by a transient (500 msec) depolarizing current pulse of 400 pA in the predrug period (left traces in A–D) and after 13 min in the indicated drug (middle traces in A–D).Graphs on the right inA–D show action-potential frequency calculated from the current-clamp recordings as a function of depolarizing currents injected at increasing intensities (100 pA steps) in the absence and presence of l-CCG (1.0 μm inA, C) and l-AP4 (10 μm in B; 1.0 μm inD). Data from different neurons are averaged:A, n = 3; B,n = 4; C, n = 3;D, n = 3. Symbols and error bars represent mean ± SEM. The paired t test did not indicate significant effects of either agonist (p > 0.1).
l-AP4 was 8.3 times less potent than l-CCG in depressing the amplitude of monosynaptic EPSCs in control neurons (n = 27). A typical example is shown in Figure2A,B. The reduction in EPSC amplitude was readily reversible, usually within 10 min. Analysis of the sigmoid concentration–response curve (Fig.3B) showed an EC50 of 297 nm for the effect of l-AP4 on EPSC peak amplitude (see Materials and Methods).
Fig. 2.

The group III mGluR agonist l-AP4 produces a concentration-dependent inhibition of synaptic transmission in the BLA without affecting postsynaptic membrane properties.A, B, Monosynaptic EPSCs were evoked by electrical stimulation in the LA (15 V; 150 μsec) before, during, and after superfusion of the brain slice with l-AP4. Each trace is an average of 8–10 EPSCs recorded immediately before drug addition (control), after 10 min in l-AP4, and after 8 min of washout. The traces shown inA are superimposed in B. C, D,l-AP4 did not affect the I–Vrelationship of the neuron. C, Whole-cell currents were elicited by a series of 400 msec voltage steps (−120 to −50 mV) from a holding potential of −60 mV in control ACSF and after 11 min inl-AP4. D, I–Vcurves show steady-state currents from voltage-clamp records like those in C, plotted as a function of membrane potential before and after 11 min in l-AP4 at the indicated concentrations. The data in A–D are from one BLA neuron held at −60 mV.
l-AP4 neither evoked detectable membrane currents nor caused changes in the I–V relationship at concentrations up to 50 μm (Fig.2C,D), which are findings consistent with a presynaptic site of action. For each neuron (n = 23), the slope conductance was calculated (see Materials and Methods) in the absence and presence of different concentrations of l-AP4. When the resulting differences in slope conductance were averaged for each concentration, no significant effect of l-AP4 was measured at concentrations of 1 nm to 50 μm(Fig. 4B). At the 100 μm(n = 5) and 200 μm (n = 4) concentrations, however, l-AP4 decreased slope conductance significantly (paired t test, p< 0.05) and evoked inward currents that averaged to 19 ± 5 and 27 ± 9 pA, respectively. Action potential threshold and accommodation were not altered by l-AP4 (10 μm; n = 4) (Fig.5B) either. In current-clamp mode, the frequency of action potentials in response to transient (500 msec) depolarizing current injections of increasing amplitude (100 pA steps) was not different in the presence and absence of l-AP4 (paired t test, p > 0.1).
These data show that in control neurons the group II mGluR agonistl-CCG is more potent than the group III agonistl-AP4 in depressing synaptic transmission and there is a threshold concentration for each agonist below which a postsynaptic action on resting membrane conductance and action potential firing properties is not recorded.
l-CCG and l-AP4 presynaptically depress evoked bursting and synaptic transmission in kindled BLA neurons
The effects of l-CCG and l-AP4 on synaptic transmission and postsynaptic membrane conductance were studied in brain slices from kindled animals 3–7 d (mean 5.1 ± 0.3;n = 15) after the last of three consecutive stage 5 seizures (Racine, 1972). Because no obvious difference of the agonist effects was observed in neurons from animals at different times post-kindling, the data were pooled. BLA neurons were recorded in both voltage-clamp and current-clamp mode. Confirming our previous studies (Rainnie et al., 1992; Holmes et al., 1996), kindled neurons showed two characteristics that distinguished them from control neurons: synaptically evoked bursting (Figs.6A, 7A) and a steep input–output relationship for synaptically evoked responses (Figs. 6B, 7B).
Fig. 6.

l-CCG blocks evoked bursting and depresses evoked EPSPs without changes in input resistance.A, Current-clamp recordings (at −60 mV) from a kindled BLA neuron. Electrical stimulation (8 V; 150 μsec) in the LA evoked bursting before (predrug, A1) superfusion of l-CCG. After 10 min in l-CCG, evoked bursting was blocked; only a small EPSP could be elicited (A2). Increasing stimulus intensity overcame the block (A3). Bursting was evoked again with the lower stimulus intensity after 10 min of washout (A4).l-CCG did not change the input resistance measured as the electrotonic potential recorded in response to a hyperpolarizing current pulse (50 pA, 100 μsec; bottom traces inA1–A4). B, Input–output relationships of the same neuron show that l-CCG blocks bursting and depresses EPSPs. C, l-CCG increased the burst threshold (Tburst) more potently than the threshold for EPSPs (TEPSP) (two-way ANOVA,p < 0.05; n = 5). Threshold was defined as the stimulus intensity required to evoke the respective response in at least 5 out of 10 trials. The thresholds obtained with each dose of l-CCG were averaged and expressed as percentage of predrug control values (100%).
Fig. 7.

l-AP4 blocks evoked bursting and depresses evoked EPSPs without changes in input resistance.A, Current-clamp recordings (at −60 mV) from a kindled BLA neuron. Electrical stimulation (7 V; 150 μsec) in the LA evoked bursting before (predrug, A1) superfusion of l-AP4. After 10 min in l-AP4, evoked bursting was blocked; only a small EPSP could be elicited (A2). Increasing stimulus intensity overcame the block (A3). Bursting was evoked again with the lower stimulus intensity after 10 min of washout (A4).l-AP4 did not change the input resistance measured as the electrotonic potential recorded in response to a hyperpolarizing current pulse (100 pA, 150 μsec; bottom traces inA1–A4). B, Input–output relationships of the same neuron show that l-AP4 blocks bursting and depresses EPSPs. C, l-AP4 increased the burst threshold (Tburst) and threshold for EPSPs (TEPSP) in a similar way (two-way ANOVA, p > 0.1; n = 6). Threshold was defined as the stimulus intensity required to evoke EPSPs and bursting, respectively, in at least 5 out of 10 trials. The thresholds obtained with each dose of l-CCG were averaged and expressed as percentage of predrug control values, which were set to 100%.
Superfusion of l-CCG (0.1–10 μm) reversibly blocked synaptically evoked bursting. Figure 6A shows a typical example. In Figure 6A1, suprathreshold burst response is shown; the burst response is blocked, revealing an EPSP. An increase in the intensity of synaptic stimulation (Fig.6A3) could overcome the block by l-CCG.l-CCG (1 μm) did not alter input resistance (control, 94.2 MΩ; l-CCG, 95 MΩ) calculated from the electrotonic potential recorded in response to a hyperpolarizing current pulse (bottom traces in Fig.6A1–A4). Analysis of the input–output relation plotting EPSP amplitude and epileptiform bursting as a function of stimulus intensity in the same neuron (Fig.6B) showed that the presynaptic inhibitory effect ofl-CCG (1 μm) was significant (two-way ANOVA,p < 0.0001). Interestingly, in the population of kindled neurons tested (n = 5) (Fig. 6C),l-CCG affected the burst threshold more potently than the threshold for EPSPs (two-way ANOVA, p < 0.05). This difference became significant at concentrations of 1 and 10 μm (post hoc t test,p < 0.05). Although l-CCG (1 nm to 10 μm) did not significantly affect the slope conductances in the whole sample of kindled neurons (see below and Fig. 4A), it cannot be ruled out that the differential effects on burst threshold and EPSP threshold are not entirely attributable to presynaptic receptor activation, becausel-CCG (10 μm) caused a slight but inconsistent change of slope conductance in two of five kindled neurons.
Using voltage-clamp recordings, we compared the presynaptic depressant action of l-CCG in control and kindled neurons and measured its effects on membrane conductance. In kindled neurons,l-CCG produced concentration-dependent inhibition of the amplitude of monosynaptic EPSCs (n = 5) (Fig.3A). The EC50 determined from the sigmoid concentration–response curve (see Materials and Methods) was shifted 30-fold to the left in kindled (1.2 nm) compared to control (36 nm) neurons. Significant differences were found (two-way ANOVA) between control and kindled neurons (p < 0.0001) and between the different concentrations (p < 0.0001), but there was no significant interaction between treatment and concentration (p > 0.5), indicating a parallel shift. The slope coefficient of the curve fitted to the concentration–response data points (see Materials and Methods) was −0.62 ± 0.25 (mean ± SEM) in control neurons and −0.87 ± 0.15 in kindled neurons.
l-CCG did not alter postsynaptic membrane properties in kindled neurons (Fig. 4A). For each neuron, the slope conductance was calculated from the I–Vrelationship (Figs. 1, 2) in the absence and presence of the different agonist concentrations. In kindled neurons (n = 5), the slope conductances in the presence of l-CCG (1 nm to 10 μm) were not different from their respective predrug controls (paired t test,p > 0.5). Action potential threshold and accommodation recorded in current-clamp mode were also not altered byl-CCG (1.0 μm; n = 3) (Fig.5C). The frequency of action potentials generated by transient (500 msec) depolarizing current injections of increasing amplitude (steps of 100 pA) was not different in the presence and absence of l-CCG (paired t test,p > 0.1).
l-AP4 (1–100 μm) reversibly blocked synaptically evoked bursting as shown in Figure 7A. An increase in the intensity of synaptic stimulation overcame the depression of epileptiform bursting by l-AP4 (Fig.7A3). Input resistance as monitored from the elec-trotonic potential recorded in response to a hyperpolarizing current pulse (bottom traces in Fig.7A1–A4) was not altered by l-AP4 (1 μm; control, 100.2 MΩ; l-AP4, 100.8 MΩ). Analysis of the input–output relation of this neuron (Fig.7B) showed that the depression of EPSP amplitude by 1 μml-AP4 was significant (two-way ANOVA,p < 0.0001). In the population of kindled neurons tested (n = 6) (Fig. 7C), l-AP4 increased both the burst threshold and the threshold for EPSPs in a concentration-dependent fashion (two-way ANOVA, p < 0.0001). In contrast to l-CCG, l-AP4 was equally potent in affecting EPSP and burst threshold (two-way ANOVA,p > 0.1).
We compared the depression of synaptic transmission byl-AP4 in control and kindled neurons and analyzed effects on postsynaptic membrane conductance in voltage-clamp recordings. In kindled neurons, l-AP4 reduced the amplitude of monosynaptic EPSCs (n = 6) concentration-dependently (Fig. 3B). The concentration–response curve forl-AP4 (see Materials and Methods) had an EC50of 10.8 nm in kindled neurons, which represents a 28-fold increase in potency compared to control neurons (297 nm). Analysis of the EC50 values for l-AP4 andl-CCG showed that l-AP4 was nine times less potent than l-CCG in kindled neurons. The differences in the effects of l-AP4 between control and kindled neurons and between the different concentrations were significant (two-way ANOVA, p < 0.0001), and there was no significant interaction between treatment and concentration (p > 0.5), indicating a parallel shift. The slope of the linear portion of the curve fitted to the concentration–response data points (see Materials and Methods) was −0.46 ± 0.14 (mean ± SEM) in control neurons and −0.62 ± 0.11 in kindled neurons.
Measurements of membrane conductance showed that l-AP4 did not affect postsynaptic resting membrane properties in kindled neurons (Fig. 4B). For each neuron the slope conductance was calculated from the linear portion of the I–Vrelationship (Figs. 1, 2) in the absence and presence of the different agonist concentrations. Slope conductances in kindled neurons (n = 6) in the presence of l-AP4 (10 nm to 100 μm) were not different from their respective predrug controls (paired t test,p > 0.1). Furthermore, l-AP4 (1.0 μm; n = 3) (Fig. 5D) did not change action potential threshold and accommodation recorded in current-clamp mode. The frequency of action potentials in response to transient (500 msec) depolarizing current injections of increasing amplitude (100 pA steps) was not different in the presence and absence of l-AP4 (paired t test, p > 0.1).
Effects of the mGluR antagonists MCCG and MAP4 in control and kindled neurons
Because both l-CCG and l-AP4 depress synaptic transmission through a presynaptic action and both agonists have a ∼30-fold increase in potency in kindled neurons, we analyzed pharmacologically whether l-CCG andl-AP4 may be acting on two different receptors. If two different receptors are involved, different antagonists may have differential effects on the actions of each agonist. We used the novel second generation antagonists MCCG and MAP4, which have been shown to be specific for group II and group III mGluRs, respectively (Watkins and Collingridge, 1994; Knöpfel et al., 1995; Pin and Duvoisin, 1995; Roberts, 1995; Vignes et al., 1995).
MCCG itself did not significantly influence synaptic transmission but selectively antagonized the effect of l-CCG on EPSC amplitude. Although in individual neurons MCCG (100 μm) slightly reduced the monosynaptic EPSCs (Fig. 8A,B), in the control (79 ± 12%, n = 5) and kindled (86 ± 9%, n = 4) neuronal populations tested, MCCG (100 μm) was without significant effects on EPSC amplitude (paired t test, p > 0.1) (Fig.10C). MCCG (100 μm) also did not significantly change the threshold for synaptically evoked bursting in kindled neurons (96 ± 5%, n = 4; paired ttest, p > 0.1). Even higher concentrations (300 μm) of MCCG did not alter EPSC amplitude in control (n = 2) or kindled (n = 2) neurons (Fig. 10C).
Fig. 8.

MCCG but not MAP4 antagonizes the inhibition of synaptic transmission produced by l-CCG.A1–A4, MCCG (superfused for 14 min) only slightly affected monosynaptic EPSCs evoked by electrical stimulation in the LA (17 V; 150 μsec). A5–A8, When coapplied withl-CCG, MCCG (A7) but not MAP4 (A6) reversed the depression of synaptic transmission by l-CCG (A5).B, C, Superimposed traces taken fromA1–A8. The data in A–C are from one BLA neuron held at −60 mV. Each trace inA–C is an average of 7–10 EPSCs recorded after 10 min in the indicated drug(s).
Fig. 10.
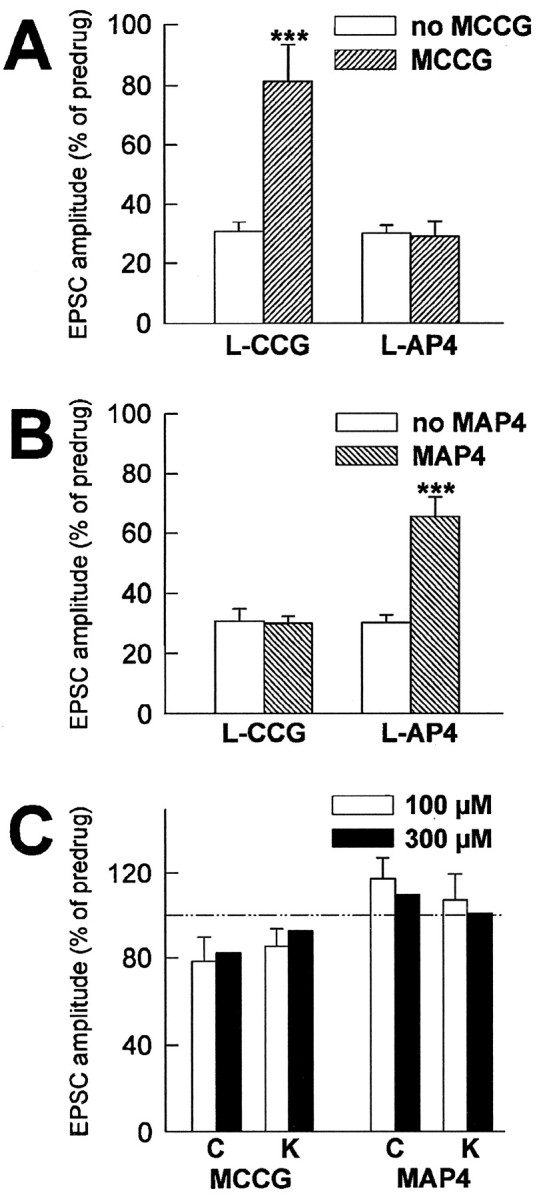
MCCG and MAP4 are selective and potent antagonists at l-CCG- and l-AP4-activated mGluRs, respectively. A, MCCG (100 μm) reversed the synaptic depression by l-CCG (1.0 μm) but not by l-AP4 (10 μm).B, MAP4 (100 μm) antagonized the synaptic depression by l-AP4 (10 μm) but not byl-CCG (1.0 μm). C, MCCG and MAP4 did not significantly change synaptic transmission in control (C) and in kindled (K) neurons. ***p < 0.001, paired t test. The EPSC peak amplitudes obtained for each neuron after 10 min in the indicated drug(s) were averaged and expressed as percentage of predrug control values (100%).
MCCG (100 μm) did antagonize the inhibition of EPSC amplitudes produced by l-CCG (1 μm), and this effect was not mimicked by MAP4 (100 μm) (Fig.8A,C). Figure 10Asummarizes the agonist-specific effects of MCCG in control neurons: MCCG (100 μm) reversed the depression of synaptic transmission produced by l-CCG (1 μm; pairedt test, p < 0.001; n = 8) but not that caused by l-AP4 (10 μm;p > 0.1; n = 4). Similarly, in one kindled neuron, MCCG (100 μm) reversed the inhibition byl-CCG (1.0 μm) to 78% of predrug value (data not shown). Importantly, MCCG (100 μm) did not change postsynaptic membrane properties of control and kindled neurons. The differences in slope conductances in the presence and absence of MCCG (100 μm) were 0.13 ± 0.42 nS (n = 5) in control neurons and 0.17 ± 0.56 nS (n = 4) in kindled neurons, an insignificant change (paired t test,p > 0.5). At higher concentrations, MCCG (300 μm) had inconsistent effects on the slope conductance in control (n = 2) and kindled neurons (n= 2), i.e., both increases and decreases were observed.
The presumed group III mGluR antagonist MAP4 itself did not significantly influence synaptic transmission but selectively antagonized the depression by l-AP4. Although in individual neurons MAP4 (100 μm) slightly potentiated the amplitude of monosynaptic EPSCs (Fig.9A,B), in the population of control (117 ± 11%, n = 5) and kindled (107 ± 12%, n = 4) neurons tested, MAP4 (100 μm) had no significant effects on synaptic transmission (paired t test, p > 0.1) (Fig.10C). MAP4 (100 μm) also did not significantly change burst threshold in kindled neurons (103 ± 4%, n = 4; paired t test,p > 0.1). Higher concentrations of MAP4 (300 μm) tested in control (n = 2) and kindled (n = 2) neurons still did not affect synaptic transmission (Fig. 10C).
Fig. 9.

MAP4 but not MCCG antagonizes the inhibitory effect of l-AP4 on EPSC amplitude. A, Superfusion of MAP4 for 15 min only slightly increased monosynaptic EPSCs evoked by electrical stimulation of afferents from the LA (7.2 V; 150 μsec). B, Superimposed traces taken from A. C, When coapplied with l-AP4, MAP4 but not MCCG reversed the inhibition of synaptic transmission by l-AP4. Monosynaptic EPSCs were evoked by electrical stimulation in the LA (15 V; 150 μsec). D, Superimposed traces taken fromC. The data in A, B and inC, D are from two different BLA neurons held at −60 mV. Each trace in A–D is an average of 7–10 EPSCs recorded after 10 min in the indicated drug(s).
MAP4 (100 μm) but not MCCG (100 μm) antagonized the depression of synaptic transmission byl-AP4 (10 μm) (Fig.9C,D). In Figure 10B, the agonist-specific effect of MAP4 in control neurons is shown: MAP4 (100 μm) antagonized the inhibitory effect ofl-AP4 (10 μm; paired t test,p < 0.0001; n = 8) but not that ofl-CCG (1.0 μm; p > 0.5;n = 5). Similarly, in one kindled neuron, MAP4 (100 μm) reversed the depression of synaptic transmission byl-AP4 (10 μm) to 73% of predrug value (data not shown). MAP4 (100 μm) did not affect postsynaptic membrane properties of control and kindled neurons. The changes in slope conductances in the presence and absence of MAP4 (100 μm) were 0.22 ± 0.42 nS (n = 5) in control neurons and 0.08 ± 0.51 nS (n = 4) in kindled neurons. The effects of MAP4 were not statistically significant in either control or kindled neurons (paired t test,p > 0.5). At a higher concentration, however, MAP4 (300 μm) decreased slope conductances by 2.41 ± 0.55 nS and 2.56 ± 0.78 nS in control (n = 2) and kindled neurons (n = 2), respectively, and induced inward currents of 10–40 pA.
These data show that MCCG and MAP4 are selective antagonists at thel-CCG-and l-AP4-activated mGluRs, respectively. Conversely, l-CCG and l-AP4 exhibit differential sensitivities toward these antagonists. Furthermore, the data of this study do not provide any apparent evidence for intrinsic activation of these presynaptic mGluRs, because the antagonists MCCG and MAP4 alone did not significantly influence synaptic transmission.
DISCUSSION
The main findings of this study are that (1) activation of two distinct group II- and group III-like mGluRs depresses synaptic transmission in the BLA nucleus presynaptically; (2) in amygdala-kindled BLA neurons, presynaptic mGluR agonists depress synaptic transmission with a 28- to 30-fold increased potency and block evoked epileptiform bursting; and (3) there is lack of evidence for intrinsic activation of presynaptic group II and group III mGluRs in either control or kindled BLA neurons.
The present study shows that inhibition of synaptic transmission in the BLA is mediated by two pharmacologically distinct presynaptic group II- and group III-like mGluRs. Evidence for two different mGluR subgroups is as follows. l-CCG and l-AP4 at the low nanomolar concentrations used in this study can discriminate between group II and group III mGluRs, respectively (Hayashi et al., 1994;Suzdak et al., 1994; Knöpfel et al., 1995; Pin and Duvoisin, 1995; Roberts, 1995). The inhibition of EPSC amplitude produced byl-CCG and l-AP4 was selectively antagonized by MCCG and MAP4, novel second generation group II and group III mGluR antagonists, respectively (Watkins and Collingridge, 1994;Knöpfel et al., 1995; Pin and Duvoisin, 1995; Roberts, 1995). Furthermore, in kindled neurons, l-AP4 affected EPSP and burst threshold equally, whereas l-CCG raised the threshold for evoked bursting more potently than for EPSPs (Figs.6, 7). These differences in agonist actions occurred in the absence of effects on action potential or firing properties of BLA neurons (Fig.5).
Importantly, although they clearly depressed monosynaptic EPSCs and EPSPs, l-CCG (<10 μm) and l-AP4 (<50 μm) did not affect postsynaptic membrane properties. There was no change in either the holding current when the neurons were voltage-clamped at −60 mV or in postsynaptic conductance, as evidenced by the unchanged slope of the I–Vrelationship; these observations are consistent with a presynaptic action. Furthermore, for each agonist we found a threshold concentration (10 μm for l-CCG; 50 μm for l-AP4) above which postsynaptic membrane effects occurred. These data are in agreement with the presynaptic receptor-specific agonist concentrations reported in the literature (Hayashi et al., 1994; Suzdak et al., 1994; Knöpfel et al., 1995; Pin and Duvoisin, 1995; Roberts, 1995). Furthermore, neitherl-CCG nor l-AP4 altered action potential threshold or accommodation in control and kindled BLA neurons. Additionally, a previous study in this laboratory (Rainnie and Shinnick-Gallagher, 1992) showed that 1SR,3RS-ACPD did not affect responses to exogenously applied ionotropic glutamate receptor agonists AMPA and NMDA, suggesting that the effects of l-AP4 andl-CCG are presynaptic.
The involvement of both group II and group III mGluRs in the presynaptic depression of neurotransmission in the BLA is similar to the pattern described for mossy fiber synapses in the hippocampal area CA3 (Manzoni et al., 1995; but see Lanthorn et al., 1984) and lateral perforant path synapses in the dentate gyrus (Lovinger and McCool, 1995), where group II and group III mGluRs also mediate presynaptic depression. Presynaptic group I and group III but not group II mGluRs depress transmission at the Schaffer collateral–CA1 synapse (Gereau and Conn, 1995) and the CA1–CA3 pyramidal cell synapse (Manzoni and Bockaert, 1995). Surprisingly, group II but not group III mGluRs depress transmission at corticostriatal synapses (Lovinger and McCool, 1995; but see Calabresi et al., 1993). These data suggest pathway-specific patterns for presynaptic mGluR subgroups involved in inhibition of synaptic transmission.
Variations between different brain areas and synapses also exist for mGluR agonist potencies. Based on complete concentration–response relationships in this study, l-CCG and l-AP4 are both extremely potent in depressing synaptic transmission at the LA-BLA synapse, the EC50 values being 36 nm and 297 nm, respectively. In other brain areas, the EC50 values for presynaptic inhibition by l-CCG and l-AP4 are in the micromolar range. EC50values of l-CCG are 6.1 μm at corticostriatal synapses (Lovinger and McCool, 1995) and 1.1 μm at the mossy fiber–CA3 synapse (Manzoni et al., 1995). EC50values of l-AP4 are ∼500 μm at the Schaffer collateral–CA1 synapse (Gereau and Conn, 1995), 112 μmat the CA3–CA1 pyramidal cell synapse (Manzoni and Bockaert, 1995), ∼50 μm at the corticostriatal synapse (Calabresi et al., 1993), 2.5 μm at the lateral perforant path in the dentate gyrus (Koerner and Cotman, 1981), and 1.1 μm at the mossy fiber–CA3 synapse (Manzoni et al., 1995). Even at the stria terminalis–BLA synapse, the EC50 of l-AP4 is at least 50 μm (Rainnie and Shinnick-Gallagher, 1992), suggesting the nanomolar EC50 values measured at the LA-BLA synapse are pathway-specific.
The nanomolar EC50 values of l-CCG andl-AP4 correlate well with the concentrations for specific binding to the cloned group II and group III mGluRs (Knöpfel et al., 1995; Pin and Duvoisin, 1995) and for inhibition of forskolin-stimulated cAMP production but not group I-mediated phosphoinositol hydrolysis (Ambrosini et al., 1995; Johansen et al., 1995; Roberts, 1995). Inhibition of cAMP accumulation in the hippocampus, however, requires higher agonist concentrations (Wright and Schoepp, 1996). Although it is not known whether inhibition of cAMP formation by mGluRs results in inhibition of synaptic potentials, presynaptic group II and group III mGluRs can depress neurotransmission by inhibition of high voltage-activated Ca2+-channels (Trombley and Westbrook, 1992; Sahara and Westbrook, 1993; Chavis et al., 1994; Glaum and Miller, 1994; Ikeda et al., 1995; Choi and Lovinger, 1996).
The role of mGluRs in the epilepsies is not clear (Gallagher et al., 1994). In different in vivo and in vitro models, activation of group I-like mGluRs evokes seizures or epileptiform bursting (Sacaan and Schoepp, 1992; McDonald et al., 1993; Burke and Hablitz, 1995; Merlin et al., 1995; Tizzano et al., 1995; Suzuki et al., 1996). In behavioral studies, group II mGluR agonists likel-CCG prevented or inhibited audiogenic seizures (Thomsen et al., 1994; Dalby and Thomsen, 1996), amygdala-kindled seizures (Attwell et al., 1995), and group I mGluR agonist-induced seizures (Tizzano et al., 1995), although higher concentrations ofl-CCG induced seizures (Tizzano et al., 1995). The group III mGluR agonist l-AP4 suppressed group I mGluR agonist-induced (Tizzano et al., 1995) and amygdala-kindled seizures (Suzuki et al., 1996).
In electrophysiological studies, l-CCG induced burst-firing in DLSN neurons (Zheng and Gallagher, 1995) and increased picrotoxin-induced bursting in hippocampal neurons (Merlin et al., 1995), whereas l-CCG and l-AP4 suppressed bicuculline-evoked paroxysmal depolarizations in immature neocortical neurons (Burke and Hablitz, 1995). A recent study from this lab showed that in amygdala-kindled BLA neurons postsynaptic mGluR-mediated membrane depolarization is enhanced, whereas hyperpolarization is downregulated (Holmes et al., 1996). The various effects of mGluR agonists may depend on the different subgroups, brain areas, and epilepsy models.
The present study shows for the first time that activation of presynaptic group II and group III mGluRs depresses synaptic activity and bursting in amygdala-kindled BLA neurons, suggesting that selective mGluR agonists may be anticonvulsive. Importantly, the potency of each agonist is increased 28- to 30-fold in kindled compared with control neurons. The parallel shift of the concentration–response curves is consistent with enhanced receptor affinities. Alterations in second messenger systems could also account for the enhanced agonist potency. Because group II and group III mGluRs couple to the cAMP cascade (Knöpfel et al., 1995; Pin et al., 1995), it is of interest that cAMP induces or enhances epileptiform activity (Yokoyama et al., 1989; Boulton et al., 1993), and elevations of cAMP levels are measured in amygdala-kindled animals (Mori, 1983). Because in our study the potencies of l-CCG and l-AP4 increased equally with an unchanged potency ratio of 10:1, it may be speculated that the increase in potency involves a mechanism that is shared by both group II and group III mGluRs, e.g., inhibition of cAMP production, whereas their effects on Ca2+-channel inhibition are less homogenous (Glaum and Miller, 1994; Choi and Lovinger, 1996).
Although presynaptic mGluRs in the BLA exhibited high agonist sensitivity, there was no apparent evidence for endogenous activation of these receptors in either control or kindled neurons. Endogenous activation of presynaptic mGluRs has not been shown in any study using specific group II and III mGluR antagonists (Jane et al., 1994; Bushell et al., 1995; Gereau and Conn, 1995; Manzoni et al., 1995; Salt and Eaton, 1995; Vignes et al., 1995). Reasonable concentrations (100 μm) of MCCG and MAP4 clearly antagonized the effects ofl-CCG (1 μm) and l-AP4 (10 μm), respectively, but their potency at receptors activated by the endogenous transmitter glutamate is not known. It is possible that more potent antagonists might reveal endogenous activation.
In conclusion, presynaptic group II and III mGluRs can mediate depression of synaptic transmission in control and amygdala-kindled neurons, but evidence for endogenous activation is lacking. The nanomolar EC50 values for the agonists and their 30-fold decrease in kindled neurons suggest that presynaptic group II and group III mGluR agonists may be useful anticonvulsants in epileptiform disorders.
Footnotes
This work was supported by National Institutes of Health Grant NS 24643 and the Deutsche Forschungsgemeinschaft. We thank Dr. Joel P. Gallagher for critical reading of and helpful comments on this manuscript.
Correspondence should be addressed to Patricia Shinnick-Gallagher, Ph.D., Department of Pharmacology, The University of Texas Medical Branch, Galveston, TX 77555-1031.
REFERENCES
- 1.Ambrosini A, Bresciani L, Fracchia S, Brunello N, Racagni G. Metabotropic glutamate receptors negatively coupled to adenylate cyclase inhibit N-methyl-d-aspartate receptor activity and prevent neurotoxicity in mesencephalic neurons in vitro. Mol Pharmacol. 1995;47:1057–1064. [PubMed] [Google Scholar]
- 2.Arvanov VL, Holmes KH, Keele NB, Shinnick-Gallagher P. The functional role of metabotropic glutamate receptors in epileptiform activity induced by 4-aminopyridine in the rat amygdala slice. Brain Res. 1995;669:140–144. doi: 10.1016/0006-8993(94)01243-b. [DOI] [PubMed] [Google Scholar]
- 3.Asprodini EK, Rainnie DG, Anderson AC, Shinnick-Gallagher P. In vivo kindling does not alter afterhyperpolarizations (AHPs) following action potential firing in vitro in basolateral amygdala neurons. Brain Res. 1992a;588:329–334. doi: 10.1016/0006-8993(92)91595-6. [DOI] [PubMed] [Google Scholar]
- 4.Asprodini EK, Rainnie DG, Shinnick-Gallagher P. Epileptogenesis reduces the sensitivity of presynaptic γ-aminobutyric acidB receptors on glutamatergic afferents in the amygdala. J Pharmacol Exp Ther. 1992b;262:1011–1021. [PubMed] [Google Scholar]
- 5.Attwell PJE, Kaura S, Sigala G, Bradford HF, Croucher MJ, Jane DE, Watkins JC. Blockade of both epileptogenesis and glutamate release by (1S,3S)-ACPD, a presynaptic glutamate receptor agonist. Brain Res. 1995;698:155–162. doi: 10.1016/0006-8993(95)00886-u. [DOI] [PubMed] [Google Scholar]
- 6.Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 7.Boulton CL, McCrohan CR, O’Shoughnessy CT. Cyclic AMP analogues increase excitability and enhance epileptiform activity in rat neocortex in vitro. Eur J Pharmacol. 1993;236:131–136. doi: 10.1016/0014-2999(93)90235-a. [DOI] [PubMed] [Google Scholar]
- 8.Burke JP, Hablitz JJ. Modulation of epileptiform activity by metabotropic glutamate receptors in immature rat neocortex. J Neurophysiol. 1995;73:205–217. doi: 10.1152/jn.1995.73.1.205. [DOI] [PubMed] [Google Scholar]
- 9.Bushell TJ, Jane DE, Tse H-W, Watkins JC, Davies CH, Garthwaite J, Collingridge GL. Antagonism of the synaptic depressant actions of l-AP4 in the lateral perforant path by MAP4. Neuropharmacology. 1995;34:239–241. doi: 10.1016/0028-3908(95)00012-u. [DOI] [PubMed] [Google Scholar]
- 10.Calabresi P, Pisani A, Mercuri NB, Bernardi G. Heterogeneity of metabotropic glutamate receptors in the striatum: electrophysiological evidence. Eur J Neurosci. 1993;5:1370–1377. doi: 10.1111/j.1460-9568.1993.tb00923.x. [DOI] [PubMed] [Google Scholar]
- 11.Chavis P, Shinozaki H, Bockaert J, Fagni L. The metabotropic glutamate receptor types 2/3 inhibit L-type calcium channels via a pertussis toxin-sensitive G-protein in cultured granule cells. J Neurosci. 1994;14:7067–7076. doi: 10.1523/JNEUROSCI.14-11-07067.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi S, Lovinger DM. Metabotropic glutamate receptor modulation of voltage-gated Ca2+ channels involves multiple receptor subtypes in cortical neurons. J Neurosci. 1996;16:36–45. doi: 10.1523/JNEUROSCI.16-01-00036.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalby NO, Thomsen C. Modulation of seizure activity in mice by metabotropic glutamate receptor ligands. J Pharmacol Exp Ther. 1996;276:516–522. [PubMed] [Google Scholar]
- 14.Gallagher JP, Zheng F, Shinnick-Gallagher P. Long-lasting modulation of synaptic transmission by metabotropic glutamate receptors. In: Conn PJ, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 173–193. [Google Scholar]
- 15.Gean P-W, Shinnick-Gallagher P, Anderson AC. Spontaneous epileptiform activity and alteration of GABA- and NMDA-mediated neurotransmission in amygdala neurons kindled in vivo. Brain Res. 1989;494:177–181. doi: 10.1016/0006-8993(89)90160-1. [DOI] [PubMed] [Google Scholar]
- 16.Gereau RW, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glaum SR, Miller RJ. Acute regulation of synaptic transmission by metabotropic glutamate receptors. In: Conn PJ, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 147–172. [Google Scholar]
- 18.Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. 1969;25:295–330. doi: 10.1016/0014-4886(69)90128-9. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi Y, Sekiyama N, Nakanishi S, Jane DE, Sunter DC, Birse EF, Udvarhelyi PM, Watkins JC. Analysis of agonist and antagonist activities of phenylglycine derivatives for different cloned metabotropic glutamate receptor subtypes. J Neurosci. 1994;14:3370–3377. doi: 10.1523/JNEUROSCI.14-05-03370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 21.Holmes KH, Keele NB, Shinnick-Gallagher P. Loss of metabotropic glutamate receptor (mGluR)-mediated hyperpolarizations and increase in mGluR depolarizations in basolateral amygdala neurons in kindling-induced epilepsy. J Neurophysiol. 1996;76:1208–1212. doi: 10.1152/jn.1996.76.4.2808. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda SR, Lovinger DM, McCool BA, Lewis DL. Heterologous expression of metabotropic glutamate receptors in adult sympathetic neurons: subtype-specific coupling to ion channels. Neuron. 1995;14:1029–1038. doi: 10.1016/0896-6273(95)90341-0. [DOI] [PubMed] [Google Scholar]
- 23.Jane DE, Jones PLSJ, Pook PC-K, Tse H-W, Watkins JC. Actions of two new antagonists showing selectivity for different sub-types of metabotropic glutamate receptor in the neonatal rat spinal cord. Br J Pharmacol. 1994;112:809–816. doi: 10.1111/j.1476-5381.1994.tb13151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansen PA, Chase LA, Sinor AD, Koerner JF, Johnson RL, Robinson MB. Type 4a metabotropic glutamate receptor: identification of new potent agonists and differentiation from the l-(+)-2-amino-4-phosphonobutanoic acid-sensitive receptor in the lateral perforant pathway in rats. Mol Pharmacol. 1995;48:140–149. [PubMed] [Google Scholar]
- 25.Knöpfel T, Kuhn R, Allgeier H. Metabotropic glutamate receptors: novel targets for drug development. J Med Chem. 1995;38:1417–1426. doi: 10.1021/jm00009a001. [DOI] [PubMed] [Google Scholar]
- 26.Koerner JF, Cotman CW. Micromolar l-2-amino-4-phosphonobutyric acid selectively inhibits perforant path synapse from lateral entorhinal cortex. Brain Res. 1981;216:192–198. doi: 10.1016/0006-8993(81)91288-9. [DOI] [PubMed] [Google Scholar]
- 27.Lanthorn TH, Ganong AH, Cotman CW. 2-Amino-4-phosphonobutyrate selectively blocks mossy fiber-CA3 responses in guinea pig but not rat hippocampus. Brain Res. 1984;290:174–178. doi: 10.1016/0006-8993(84)90750-9. [DOI] [PubMed] [Google Scholar]
- 28.Löscher W, Ebert U, Wahnschaffe U, Rundfeldt C (1995) Susceptibility of different cell layers of the anterior and posterior part of the piriform cortex to electrical stimulation and kindling: comparison with the basolateral amygdala and “area tempestas.” Neuroscience 66:265–276. [DOI] [PubMed]
- 29.Lovinger DM, McCool BA. Metabotropic glutamate receptor-mediated presynaptic depression at corticostriatal synapses involves mGluR2 or 3. J Neurophysiol. 1995;73:1076–1083. doi: 10.1152/jn.1995.73.3.1076. [DOI] [PubMed] [Google Scholar]
- 30.Manzoni O, Bockaert J. Metabotropic glutamate receptors inhibiting excitatory synapses in the CA1 area of rat hippocampus. Eur J Neurosci. 1995;7:2518–2523. doi: 10.1111/j.1460-9568.1995.tb01051.x. [DOI] [PubMed] [Google Scholar]
- 31.Manzoni OJ, Castillo PE, Nicoll RA. Pharmacology of metabotropic glutamate receptors at the mossy fiber synapses of the guinea pig hippocampus. Neuropharmacology. 1995;34:965–971. doi: 10.1016/0028-3908(95)00060-j. [DOI] [PubMed] [Google Scholar]
- 32.McDonald JW, Fix AS, Tizzano JP, Schoepp DD. Seizures and brain injury in neonatal rats induced by 1S,3R-ACPD, a metabotropic glutamate receptor agonist. J Neurosci. 1993;13:4445–4455. doi: 10.1523/JNEUROSCI.13-10-04445.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNamara JO. Cellular and molecular basis of epilepsy. J Neurosci. 1994;14:3413–3425. doi: 10.1523/JNEUROSCI.14-06-03413.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merlin LR, Taylor GW, Wong RKS. Role of metabotropic glutamate receptor subtypes in the patterning of epileptiform activities in vitro. J Neurophysiol. 1995;74:896–900. doi: 10.1152/jn.1995.74.2.896. [DOI] [PubMed] [Google Scholar]
- 35.Miller S, Kesslak JP, Romano C, Cotman CW. Roles of metabotropic glutamate receptors in brain plasticity and pathology. Ann NY Acad Sci. 1995;757:460–474. doi: 10.1111/j.1749-6632.1995.tb17506.x. [DOI] [PubMed] [Google Scholar]
- 36.Mori N. Long-lasting increase of regional cAMP and cGMP in amygdaloid kindled rat brain. J Jpn Epil Soc. 1983;1:79–87. [Google Scholar]
- 37.Nakanishi S. Metabotropic glutamate receptors: synaptic transmission, modulation and plasticity. Neuron. 1994;13:1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi S, Masu M. Molecular diversity and functions of glutamate receptors. Annu Rev Biophys Biomol Struct. 1994;23:319–348. doi: 10.1146/annurev.bb.23.060194.001535. [DOI] [PubMed] [Google Scholar]
- 39.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 40.Pin J-P, Duvoisin R. The metabotropic glutamate receptors: structure and function. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 41.Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 42.Racine RJ, Gartner JG, Burnham WM. Epileptiform activity and neural plasticity in limbic structures. Brain Res. 1972;47:262–268. doi: 10.1016/0006-8993(72)90268-5. [DOI] [PubMed] [Google Scholar]
- 43.Rainnie DG, Shinnick-Gallagher P. Trans-ACPD and L-APB presynaptically inhibit excitatory glutamatergic transmission in the basolateral amygdala (BLA). Neurosci Lett. 1992;139:87–91. doi: 10.1016/0304-3940(92)90864-4. [DOI] [PubMed] [Google Scholar]
- 44.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Kindling-induced long-lasting changes in synaptic transmission in the basolateral amygdala. J Neurophysiol. 1992;67:443–454. doi: 10.1152/jn.1992.67.2.443. [DOI] [PubMed] [Google Scholar]
- 45.Rainnie DG, Holmes KH, Shinnick-Gallagher P. Activation of postsynaptic metabotropic glutamate receptors by trans-ACPD hyperpolarizes neurons of the basolateral amygdala. J Neurosci. 1994;14:7208–7220. doi: 10.1523/JNEUROSCI.14-11-07208.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts PJ. Pharmacological tools for the investigation of metabotropic glutamate receptors (mGluRs): phenylglycine derivatives and other selective antagonists—an update. Neuropharmacology. 1995;34:813–819. doi: 10.1016/0028-3908(95)00094-m. [DOI] [PubMed] [Google Scholar]
- 47.Sacaan AI, Schoepp DD. Activation of hippocampal metabotropic excitatory amino acid receptors leads to seizures and neuronal damage. Neurosci Lett. 1992;139:77–82. doi: 10.1016/0304-3940(92)90862-2. [DOI] [PubMed] [Google Scholar]
- 48.Sahara Y, Westbrook GL. Modulation of calcium currents by a metabotropic glutamate receptor involves fast and slow kinetic components in cultured hippocampal neurons. J Neurosci. 1993;13:3041–3050. doi: 10.1523/JNEUROSCI.13-07-03041.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salt TE, Eaton SA. Modulation of sensory neurone excitatory and inhibitory responses in the ventrobasal thalamus by activation of metabotropic excitatory amino acid receptors. Neuropharmacology. 1995;34:1043–1051. doi: 10.1016/0028-3908(95)00052-8. [DOI] [PubMed] [Google Scholar]
- 50.Schoepp DD, Conn PJ. Metabotropic glutamate receptors in brain function and pathology. Trends Pharmacol Sci. 1993;14:13–20. doi: 10.1016/0165-6147(93)90107-u. [DOI] [PubMed] [Google Scholar]
- 51.Suzdak PD, Thomsen C, Mulvihill E, Kristensen P. Molecular cloning, expression, and characterization of metabotropic glutamate receptor subtypes. In: Conn PJ, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 1–30. [Google Scholar]
- 52.Suzuki K, Mori N, Kittaka H, Iwata Y, Yamada Y, Osonoe K, Niwa S-I. Anticonvulsant action of metabotropic glutamate receptor agonists in kindled amygdala of rats. Neurosci Lett. 1996;204:41–44. doi: 10.1016/0304-3940(96)12311-9. [DOI] [PubMed] [Google Scholar]
- 53.Tallarida RJ, Jacob LS. The dose-response relation in pharmacology. Springer; New York: 1979. [Google Scholar]
- 54.Tanaka K, Hirayama K, Murata R, Matsuura S. Relation of the enhancement of entorhinal tetanic responses by 50-Hz amygdala stimulation to the progression of kindling in the rat. Neurosci Res. 1995;23:249–255. doi: 10.1016/0168-0102(95)00949-3. [DOI] [PubMed] [Google Scholar]
- 55.Taschenberger H, Roy BL, Lowe DA. Effects of a metabotropic glutamate agonist, trans-ACPD, on cortical epileptiform activity. NeuroReport. 1992;3:629–632. doi: 10.1097/00001756-199207000-00022. [DOI] [PubMed] [Google Scholar]
- 56.Thomsen C, Klitgaard H, Sheardown M, Jackson HC, Eskesen K, Jacobsen P, Treppendahl S, Suzdak PD. (S)-4-carboxy-3-hydroxyphenylglycine, an antagonist of metabotropic glutamate receptor (mGluR)1a and an agonist of mGluR2, protects against audiogenic seizures in DBA/2 mice. J Neurochem. 1994;62:2492–2495. doi: 10.1046/j.1471-4159.1994.62062492.x. [DOI] [PubMed] [Google Scholar]
- 57.Tizzano JP, Griffey KI, Schoepp DD. Induction or protection of limbic seizures in mice by mGluR subtype selective agonists. Neuropharmacology. 1995;34:1063–1067. doi: 10.1016/0028-3908(95)00083-i. [DOI] [PubMed] [Google Scholar]
- 58.Trombley PQ, Westbrook GL. l-AP4 inhibits calcium currents and synaptic transmission via a G-protein-coupled glutamate receptor. J Neurosci. 1992;12:2043–2050. doi: 10.1523/JNEUROSCI.12-06-02043.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vignes M, Clarke VRJ, Davies CH, Chambers A, Jane DE, Watkins JC, Collingridge GL. Pharmacological evidence for an involvement of group II and group III mGluRs in the presynaptic regulation of excitatory synaptic responses in the CA1 region of rat hippocampal slices. Neuropharmacology. 1995;34:973–982. doi: 10.1016/0028-3908(95)00093-l. [DOI] [PubMed] [Google Scholar]
- 60.Watkins J, Collingridge G. Phenylglycine derivatives as antagonists of metabotropic glutamate receptors. Trends Pharmacol Sci. 1994;15:333–342. doi: 10.1016/0165-6147(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 61.Westbrook GL. Glutamate receptor update. Curr Opin Neurobiol. 1994;4:337–346. doi: 10.1016/0959-4388(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 62.Wright RA, Schoepp DD. Differentiation of group 2 and group 3 metabotropic glutamate receptor cAMP responses in the rat hippocampus. Eur J Pharmacol. 1996;297:275–282. doi: 10.1016/0014-2999(95)00747-4. [DOI] [PubMed] [Google Scholar]
- 63.Yokoyama N, Mori N, Kumashiro H. Chemical kindling induced by cAMP and transfer to electrical kindling. Brain Res. 1989;492:158–162. doi: 10.1016/0006-8993(89)90898-6. [DOI] [PubMed] [Google Scholar]
- 64.Zheng F, Gallagher JP. (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid-induced burst firing is mediated by a native pertussis toxin-sensitive metabotropic receptor at rat dorsolateral septal nucleus neurons. Neuroscience. 1995;68:423–434. doi: 10.1016/0306-4522(95)00128-6. [DOI] [PubMed] [Google Scholar]