

Abstract
We show that three of the eleven genes of the nematodeCaenorhabditis elegans that mediate resistance to the nematocide levamisole and to other cholinergic agonists encode nicotinic acetylcholine receptor (nAChR) subunits.unc-38 encodes an α subunit while lev-1and unc-29 encode non-α subunits. The nematode nAChR subunits show conservation of many mammalian nAChR sequence features, implying an ancient evolutionary origin of nAChR proteins. Expression in Xenopus oocytes of combinations of these subunits that include the unc-38 α subunit results in levamisole-induced currents that are suppressed by the nAChR antagonists mecamylamine, neosurugatoxin, andd-tubocurarine but not α-bungarotoxin. The mutant phenotypes reveal that unc-38 and unc-29subunits are necessary for nAChR function, whereas thelev-1 subunit is not. An UNC-29–GFP fusion shows that UNC-29 is expressed in body and head muscles. Two dominant mutations oflev-1 result in a single amino acid substitution or addition in or near transmembrane domain 2, a region important to ion channel conductance and desensitization. The identification of viable nAChR mutants in C. elegans provides an advantageous system in which receptor expression and synaptic targeting can be manipulated and studied in vivo.
Keywords: acetylcholine receptor; levamisole resistance genes; receptor mutations; Caenorhabditis elegans; evolution; nematode, unc-29; unc-38; lev-1; transmembrane domain mutation; Xenopus oocyte expression; GFP; confocal microscopy; receptor localization
Nicotinic acetylcholine receptors (nAChRs) are the most thoroughly characterized receptors in the family of ligand-gated ion channels (Karlin, 1993; Unwin, 1993a,b, 1995). The vertebrate muscle nAChR is a pentamer of four subunit types in the stoichiometry α2, β, γ, and δ. Each subunit contains four hydrophobic, putative transmembrane regions (TM1–TM4), with TM2 in each subunit contributing to the lining of the channel (Devillers-Thiery et al., 1993). A main part of the binding sites for ACh has been localized to each of the two α subunits, close to a pair of vicinal cysteines (positions 192 and 193 in the Torpedoα subunit) that define α subunits in all nAChRs (Kao and Karlin, 1986). Residues of the γ and δ subunits also contribute to ACh binding (Karlin, 1993). The subunits of vertebrate neuronal nAChRs are encoded by a separate set of genes and fall into two major classes, either α or non-α, each with several isoforms (α2–α9 and β2–β5) (Sargent, 1993; Ortells and Lunt, 1995).
Beyond cloning the subunits of nAChRs, considerable effort has been expended both in vitro and in vivo toward understanding the precise contribution of each subunit and the identities and functions of proteins that interact with the nAChR (Gautam et al., 1995; Karlin and Akabas, 1995; Gautam et al., 1996). To aid in this endeavor, Caenorhabditis elegans offers a genetic facility for defining the molecules associated with nAChR function that would be difficult to achieve in more complex animals.
Nematodes possess nAChRs (Johnson and Stretton, 1980; Fleming et al., 1993; Ajuh and Egwang, 1994; Squire et al., 1995; Treinin and Chalfie, 1995; Ballivet et al., 1996; Wiley et al., 1996). Although >20 nAChR sequences have been uncovered in the C. elegans genome sequencing project, our attention has been focused on the levamisole receptor. Levamisole is a more potent agonist than acetylcholine at nematode muscle nAChRs (Lewis et al., 1980b; Harrow and Gration, 1985;Martin et al., 1991; Robertson and Martin, 1993). Mutants resistant to levamisole define 11 genes (Brenner, 1974; Lewis et al., 1980a). These mutants fall into three classes: uncs, pseudo-wild types, and twitchers (Table 1). Mutants in six genes with the unc phenotype exhibit extreme levamisole resistance, uncoordinated motor behavior, and resistance to other cholinergic agonists. Weakly resistant mutants (pseudo-wild types) move normally but have severalfold higher resistance to levamisole and other cholinergic agonists. All genes mutable to extreme resistance (exceptunc-50) also produce weaker mutant alleles having a partially resistant phenotype (Table 1). lev-1 is the only locus for which the predominant mutant phenotype is that of partial resistance but for which two rare unc extreme resistance alleles, x21 and x61, have also been found. These two alleles are the only extreme levamisole resistance mutations that show any dominance.
Table 1.
Major levamisole resistance loci of Caenorhabditis elegans
| Levamisole resistance phenotype | Gene | Phenotypes1-a | Number of alleles | [3H]MAL binding1-b | Gene product |
|---|---|---|---|---|---|
| Extremely resistantunc | lev-1 | s + w | 2 + 13 | Variable | Non-α-nAChR subunit1-c |
| unc-29 | s + w | 74 + 2 | − | Non-α-nAChR subunit1-c | |
| unc-38 | s + w | 44 + 3 | +/− | α-nAChR subunit1-c | |
| unc-63 | s + w | 58 + 1 | +/− | Unknown | |
| unc-74 | s + w | 27 + 1 | − | Unknown | |
| unc-50 | s | 5 | − | Not an nAChR subunit1-d | |
| Partially resistant pseudo-wild type | lev-8 | w | 1 | + | Unknown |
| lev-9 | w | 3 | + | Unknown | |
| lev-10 | w | 1 | + | Unknown | |
| Twitcher | unc-22 | w | 16 | + | Muscle-specific1-e |
| lev-11 | w | 1 | + | Muscle-specific1-f |
Phenotypes: s, strong; w, weak. Only a limited effort was made to isolate and characterize weak, partially levamisole-resistant alleles. The number of weak alleles isolated thus mainly reflects their relative ease of isolation. This table summarizes discussion of previously reported work described in the text (Lewis et al., 1980a, 1987b; work cited in other footnotes).
The [3H]MAL binding characteristic of the most levamisole-resistant alleles of each gene is indicated. +, Substantial specific [3H]MAL binding; −, little or no specific [3H]MAL binding.
This work.
M. O. Hengartner, N. Tsung, J. A. Lewis, and H. R. Horvitz, unpublished data.
Using [3H]meta-aminolevamisole (MAL), nAChR binding has been detected in nematode extracts (Lewis et al., 1987a,b). Mutants of the six loci that give rise to strong resistance have deficient or altered receptor binding. Such receptor mutants are only moderately impaired in motor behavior as adults but are severely incapacitated at early larval stages.
Here we show that three genes associated with levamisole resistance encode nAChR subunits, and that α and non-α subunit combinations of these genes generate functional nAChRs when co-expressed inXenopus oocytes. The eight other levamisole resistance genes are candidates for components of nAChR-mediated synaptic signaling. The ability to exploit C. elegans genetics offers the prospect of identifying additional novel molecular components of nicotinic cholinergic synapses.
MATERIALS AND METHODS
Nematode strains. The wild-type C. elegansused was Bristol strain N2 (Brenner, 1974). The Bergerac BO transposon mutator strain (Moerman and Waterston, 1984) was obtained from theCaenorhabditis Genetics Center, and the TR679 mutator strain was kindly provided by P. Anderson (University of Wisconsin) (Collins et al., 1987)
Mutant isolation. Mutants containing restriction fragment polymorphisms were obtained in the following ways. For the isolation of spontaneous transposon-induced mutations, 30 (BO strain) or 40 (TR679 strain) adult hermaphrodites were placed on a 100 mm diameter Petri dish spread with bacteria. After 4–5 d at 20°C, progeny worms were washed off the plate and placed to one side of a separate plate containing 1 mm levamisole, as described previously (Lewis et al., 1980a). Drug plates were then screened at daily intervals for extreme resistance to levamisole. To isolate γ-ray-induced mutants, worms were first irradiated for 6 min with 1500 rads from a60Co source, and 20 mutagenized adults were transferred to each Petri plate. Mutants were tested for genetic identity as described previously (Lewis et al., 1980a).
Identification of restriction fragment length polymorphisms in mutants. The mutator strains used contain high copy numbers of the transposon Tc1. Genomic probing with Tc1 DNA was used to identify a novel candidate Tc1 element generating a levamisole resistance mutation. Extraneous background Tc1 elements were eliminated by balancing the mutation against a Bristol chromosome containing left- and right-flanking markers and then back-crossing the mutation 12 times into a strain homozygous for the same genetic markers. The candidate mutation was then recombined with the left- and right-flanking markers, and, if it proved necessary to achieve adequate viability, the mutation was separated by further recombination from one or both flanking markers before isolating a strain homozygous for the back-crossed mutation. Control constructs homozygous for approximately the same high-copy Tc1 chromosomal region, but otherwise having a Bristol low-copy Tc1 genetic background, were generated by back-crossing the wild-type allele present in the parent mutator strain into a Bristol strain having an ethylmethane sulfonate (EMS)-induced mutation in the gene of interest, usually flanked by the same left and right genetic markers used in constructing the mutants. For producingunc-38 mutant constructs, unc-57(e406) anddpy-5(e61) marker mutations (0.5 map units to either side ofunc-38) were used. For control constructs,unc-11(e47) was used instead of unc-57. In the case of unc-29 constructs, unc-13(e450) andlin-11(n389), 1.1 and 1.6 map units to either side ofunc-29, were used. For lev-1,unc-30(e191) and dpy-4(e1166), 0.3 and 4.5 map units to either side of lev-1, were used.
lev-1 mapping. Several mistakes in the chromosomal placement of lev-1 were discovered by T.M.B. The needed corrections in its map position were the placement of lev-1 close tounc-30, the inversion of the unc-26, lev-1 gene order, and the finding that the deficiency sDf23 complementslev-1(x38) and therefore does not delete the gene. The correct position of lev-1 on the genetic map was determined as follows. First, the two-factor distance between lev-1 anddpy-4 was measured. From a strain that waslev-1(x21)tra-3(e1767)dpy-4(e1166)/+++, 74 Lev non-Dpy and 61 Dpy non-Lev recombinants were found among the 3014 progeny constituting 10 complete broods, giving a distance of 4.5 map units. For deficiency mapping, the recessive allele x38 was used.x38/+ males were mated into Df/nT1 strains, and outcross males were scored. The deficiencies sDf21 andnDf27 failed to complement x38, whereassDf22, sDf23, and sDf60 did complementx38. For three-factor mapping, Unc non-Dpy and Dpy non-Unc recombinants were picked from a strain that wasdpy-20(e1282) + unc-26(e205)/+lev-1(x21) +. Twenty of 22 Dpy recombinants and 2 of 21 Unc recombinants contained x21. In a further three-factor mapping experiment, Unc non-Dpy and Dpy non-Unc recombinants were selected from a strain that was unc-30(e191) +dpy-4(e1166)/+ lev-1 +, using mutator- or γ-ray-generated lev-1 alleles (x505,x508, x548, and x566) isolated in this study (see above). The cumulative data were that 4 of 64 Unc and 24 of 27 Dpy recombinants contained the lev-1 marker. In summary these data give a gene order of unc-30–lev-1–unc-26. unc-26 is genetically inseparable from the Bergerac polymorphismnP33 both from the left (Yuan et al., 1993) and to the right (T. M. Barnes and J. Hodgkin, unpublished data). Thus lev-1was inferred to lie physically between the unc-30-rescuing cosmid (Jin et al., 1994) and the nP33-detecting cosmid, a distance of about 120 kb.
Recombinant DNA techniques. Standard recombinant DNA techniques were used (Sambrook et al., 1989). The Tc1 transposable element used as a genomic probe was an EcoRV fragment prepared by D. Bird (North Carolina State University) from an isolate supplied by S. Emmons (Albert Einstein College of Medicine). The flanking genomic DNA associated with a novel Tc1-induced mutation was separated from the Tc1 DNA in any genomic subclone by excision withEcoRV and purification by gel electrophoresis. Most hybridizations and washes performed in the course of screening C. elegans genomic libraries were performed under high stringency conditions (washing at 65°C with 0.2× SSC).
Reduced stringency conditions were used for screening with an 800 bp ard probe (from position 500 to position 1350) (Hermans-Borgmeyer et al., 1986). The probe was labeled to a specific radioactivity of about 1 × 109 cpm/μg by random priming. Suitable hybridization conditions were chosen by probing a Southern blot of C. elegans genomic DNA with theard cDNA. Hybridization was performed at 30°C in 43% (w/v) formamide and 5× SSC for 48 hr. Filters were washed for 30 min each, twice at room temperature, twice at 37°C in 2× SSC and 0.1% SDS, and then for at least 45 min at 45°C in 1× SSC and 0.1% SDS before autoradiography with intensification at −70°C. Fifty thousand phage from a C. elegans genomic library in λ2001 (approximately eight genome equivalents) were screened.
Phage clones were fingerprinted by A. Coulson [Medical Research Council (MRC), Cambridge, UK] (Coulson et al., 1986). DNA sequencing followed the method of McCombie et al. (1991). Cosmids spanning theunc-38 and unc-29 loci were provided by A. Coulson. Cosmid DNA was microinjected after removal of RNA by LiCl precipitation and purification by repeated ethanol precipitation. For reverse transcription (RT)-PCR, except as noted below, poly(A+) RNA (Jacobson, 1987) was prepared from total nematode RNA or was obtained as a gift from D. Zarkower (MRC); 5′- or 3′-anchored PCR was performed using standard procedures (Frohman et al., 1988).
lev-1 cDNA. A full-length cDNA of lev-1 was obtained as follows. An oligonucleotide complementary to bp 2208–2237 of the genomic sequence was used to isolate a partial cDNA clone from a mixed stage C. elegans library (provided by I. Maruyama, MRC). The cDNA contained 232 bp of sequence upstream of the predicted start of translation but lacked the predicted 3′ end. The 3′-anchored PCR (Frohman et al., 1988) using an internal primer (position 3181–3200) upstream of the EcoRI site at 3306 was used to generate a fragment containing the 3′ end of the message. The available partial cDNA clone contained what appeared to be an unspliced 48 bp intron, but because there was no termination codon in this intron and the reading frame was maintained, it was not clear whether this sequence was present in the mature mRNA. Therefore we analyzed the splicing pattern across this region using RT-PCR. The data (not shown) were consistent with the quantitative removal of this intron. Therefore, a partial cDNA lacking the 48 bp intron was cleaved withEcoRI (which cleaves in the 3′ primer as well), and the resulting fragment was cloned into the EcoRI site of the original cDNA, thus regenerating a complete cDNA.
Cloning unc-29. The mutator-induced unc-29mutation x513 was back-crossed into the wild-type strain and recombined with the closely linked left and right flanking markersunc-13(e450) and lin-11(n389) to reduce the background of unrelated Tc1 elements. Hybridization of a Tc1 probe toEcoRI-digested DNA from the back-crossed strain showed a novel 3.7 kb EcoRI fragment not present in the wild-type strain. The Tc1 element was not separable from the x513mutation in 12 recombination events with either the unc-13or the lin-11 flanking markers. The x513 Tc1 element was cloned, and the genomic DNA flanking the insert was used as a probe to identify nine wild-type genomic clones in an EMBL4 phage library (supplied by C. Link and W. Wood, University of Colorado). The physical map position of these phage was the same as that of the putative nAChR homolog JF#WA33 isolated by cross-hybridization withard and was consistent with the genetic map location ofunc-29 on chromosome I (Brenner, 1974; Lewis et al., 1980a) (Fig. 1B). Southern blots of a γ-ray-induced mutant and five additional mutator-induced mutants all revealed chromosomal rearrangements when DNA from one of the EMBL4 phage (ZZ#1) was used as a probe (data not shown). Of the putative transposon-induced unc-29 mutants, four had insertions in either a 1.4 or 5.0 kb EcoRI fragment. TheseEcoRI fragments lie immediately downstream from the 2.0 kbEcoRI fragment in which both x513 and another insertion occurred (Fig. 1B). The insertions in two of these six mutants were lost in spontaneously occurring revertants to the wild-type phenotype, further indicating association of the restriction fragment length polymorphisms (RFLPs) with theunc-29 gene.
Fig. 1.

Structures of thelev-1, unc-29, and unc-38nAChR subunit genes. A, lev-1 structure. The position of lev-1 on the genetic map of chromosome IV is shown in relation to nearby genetic markers. The relative contig positions on the physical map of cosmids W07H6, C43C9, and B0564 and λ phage clones JF#WA10 and JF#WA18 derived from thelev-1 region are indicated. The genomic organization of the lev-1 gene is shown. Restriction enzyme sites are indicated to define possible limits of mutations: R,EcoRI; S, SalI;H, HindIII; X,XhoI; and B, BamHI. Mutant alleles found to have substantial DNA sequence rearrangements or Tc1 insertions are diagrammed. The open bar forx548 represents a deletion with a range of end points indicated. x416, x427, x438, andx566 represent complex rearrangements, mostly insertions of the sizes indicated, affecting the restriction fragments spanned by the bar shown for each mutation. Forx504, x508, and x562, the positions of 1.6 kb Tc1 insertions are indicated byarrows. For e211, e289,x21, x38, x61, x400, andx505, no DNA differences were detected. GenBank entryX98601 gives the DNA sequence of lev-1. B,unc-29 structure. The position of unc-29on the genetic map of chromosome I is shown in relation to nearby genetic markers. The relative contig positions on the physical map of cosmids C45D10, C11C3, and C34D2 and λ phage clones ZZ#1, ZZ#2, and JF#WA33 derived from the unc-29 region are indicated. The genomic organization of the unc-29 gene is shown. Restriction enzyme sites: R, EcoRI;S, SalI; and H,HindIII. The positions of a mutation caused by DNA rearrangement and of six mutations caused by apparent transposon insertion are shown relative to the EcoRI fragments that span the unc-29 gene. The sizes of the apparent inserts found were 1.6 kb each for x513, x522,x544, x545, and x554 and 2.5 kb for x520. The inserts of x520,x544, and x554 were lost in revertants, and the insert of x513 hybridized to Tc1. The exact nature and extent of the x415 mutation are unknown, but at least several hundred bases near the 3′ end of the coding seem to be involved (see Materials and Methods). For x401,x417, x429, and x433, noDNA differences were detected. The DNA sequence ofunc-29 is given by GenBank entry U81144.C, Structure of the unc-38 nAChR α subunit gene. The position of unc-38 on the genetic map of chromosome I is shown in relation to nearby genetic markers. The relative contig positions on the physical map of cosmids C09C3, B0241, and C04E4 and λ phage clones ZZ#11 and ZZ#15 are indicated. The genomic organization of the unc-38 gene is shown. Restriction enzyme sites: P, PstI; andH, HindIII. Mutations associated with Tc1 insertion or DNA rearrangements within the HindIII fragment spanning the unc-38 gene are indicated. Forx402, x404, x414, andx511, no DNA differences were found in this fragment. Other than causing a size alteration of the 3.2 kbHindIII fragment, the exact nature and extent of thex411 and x419 mutations are unknown. The DNA sequence of unc-38 is given by GenBank entryX98599.
A 6.0 kb HindIII fragment contained within the 8.7 kb of DNA spanned by the three neighboring EcoRI fragments associated with the unc-29 mutation was sequenced (GenBank accession number U81144). The subcloned 1.4 kb EcoRI fragment (Fig.1B) was used as a probe to isolate a full-length cDNA. The cDNA had an SL1 trans-spliced leader sequence at its 5′ end, as commonly found on many nematode mRNAs (Krause and Hirsh, 1987; Zorio et al., 1994), eight nucleotides upstream of the ATG start.
Cloning unc-38. A Southern blot containingHindIII-digested genomic DNA from three back-crossedunc-38 mutator-induced mutants, five nonmutant back-crossed control strains, and the Bristol wild-type strain was probed with Tc1. All three unc-38 mutants contained a 4.8 kbHindIII band not present in the wild-type strain and all nonmutant controls. The 4.8 kb fragment from unc-38(x525)was subcloned, and the genomic DNA flanking the Tc1 site was used as a probe on Southern blots of five mutator-induced and five γ-ray-induced unc-38 mutants. Four of the mutator-induced (including the Tc1 mutants probed above) and two of the γ-ray mutants showed a size distinct from that seen in the wild-type strain (3.2 kb). The subcloned DNA flanking the x525 Tc1 element was used as a hybridization probe to identify corresponding λ phage from a wild-type EMBL4 genomic library. The physical map position of the phage was consistent with the unc-38 genetic map position on chromosome I (Brenner, 1974; Lewis et al., 1980a) (Fig. 1C). The sequence of the 3′ end of the unc-38 gene, which was missing from the 3.2 kb HindIII fragment, was obtained directly from cosmid B0241 (Fig. 1C) by linear amplification sequencing (Craxton, 1991) using oligonucleotide primers derived from the cDNA sequence that was obtained.
The HindIII fragment was used to screen two C. elegans cDNA libraries (provided by S. Kim, Stanford University; and R. Barstead, Oklahoma Medical Research Foundation). Three cDNAs were isolated from 240,000 clones screened. One cDNA was sequenced and appeared to have the complete 3′ end, because it contained a polyA tail 65 bases downstream of the predicted stop codon. Based on the genomic sequence, the 5′ coding region was not present on any of the cDNA clones. To recover the missing 5′ end, a forward primer was made corresponding to the 5′ untranslated leader predicted from the genomic sequence (nucleotides 273–309, but containing an XbaI site: ATTCTC TCTAGAACACTTTCTTTCAAGGCTTTTCATA). This forward primer and a reverse primer (nucleotides 854–882) corresponding to the known partial cDNA sequence were used to amplify total first strand cDNA from a mixed stage population of C. elegans. The resulting PCR product was cleaved with XbaI and BamHI (uniqueBamHI site at position 837) and ligated to the partial, but mature, cDNA that itself was cleaved with BamHI. TheXbaI site was ligated to the vector polylinker cleaved withXbaI. The resulting hybrid PCR cDNA sequence corresponded to the predicted mature full-length cDNA sequence.
To search for null mutations of unc-38 andunc-29, cDNA clones were generated by RT-PCR using total RNA prepared according to the method of Chomczynski and Sacchi (1987) with Trizol (Life Technologies, Gaithersburg, MD) phenol–guanidine isothiocyanate reagent. The RNA (20 μg) was treated with 10 U of RQ1 RNase-free DNase and 56 U of RNasin (both from Promega, Madison, WI) in 85 μl of buffer for 20 min at 37°C. After phenol/CHCl3extraction and ethanol precipitation, 1 μg of the resuspended RNA was used in RT-PCR according to the protocols of the Perkin-Elmer (Norwalk, CT) GeneAmp RNA PCR kit in a Perkin-Elmer Thermocycler 480, using thick-walled GeneAmp tubes. RT-PCR reactions, after a hot start, were conducted for 45 cycles of 1 min denaturation at 94°, 1 min annealing at 59°, and 2 min extension at 72°C with a final extension of 5 min. To produce cDNA products from the 5′ ends of unc-38transcripts, a forward primer derived from positions 339–364 in the genomic sequence was used with a reverse primer from positions 2184–2209 antisense. To obtain an overlapping cDNA from the 3′-end ofunc-38 transcripts, forward and reverse primers from positions 1669–1691 and 3517–3541 antisense were used, respectively. The 5′ and 3′ RT-PCR products obtained for unc-38(x411) andunc-38(x419) were the same sizes as for wild type, 915 and 847 bp, respectively, as was the 3′ PCR product ofunc-38(x20). The 5′ RT-PCR product from x20 RNA was only about 760 bp in length in two independent amplifications done from the same RNA preparation. After purification with the QIAquick PCR Purification System (Qiagen, Chatsworth, CA), the 5′ x20products were ligated to pT7Blue (Novagen, Madison, WI) and electroporated into DH12S. Sequencing of the two independentx20 5′ clones showed that the base sequence of the third exon was precisely missing in both clones, accounting for the smaller size of the PCR products. Two independent genomic clones were generated using the Boehringer-Mannheim (Indianapolis, IN) EXPAND PCR kit with forward and reverse primers derived from positions 638–663 and 1120–1143 antisense, respectively. Each PCR amplification was done using the genomic DNA from two adult worms digested with protease K (Williams et al., 1992) using the shortened cycle times recommended (10 cycles without autoextension followed by 20 cycles with an additional 20 sec each). Unlike the cDNA products, the genomic products seemed to be of wild-type length (506 bp). In both clones the G found in the universal 3′ splice acceptor AG consensus sequence was found mutated to an A, accounting for the loss of the third exon in the RNA transcript (genomic position 764). The finding of apparently normal sizes found for 5′ and 3′ RT-PCR products prepared from unc-38(x411) andunc-38(x419) was surprising, because these mutants showed significant RFLP differences from the wild type on Southern blots (Fig.1C). To obtain 5′ and 3′ RT-PCR products ofunc-29 transcripts, sets of forward and reverse primers were used from positions 494 to 521 and 2996 to 2972 antisense and from positions 2632 to 2655 and 4147 to 4170 antisense, respectively. The 5′ and 3′ RT-PCR products produced from unc-29(x29) andunc-29(e1072) total RNA were wild type in size (835 and 915 bp, respectively), as was the 5′ product of unc-29(x415). No 3′ product was obtained for x415 RNA even when a primer 214 bases downstream from the initial reverse primer position was used (positions 4361–4381). Southern blots showed that the 5.0 kbEcoRI fragment containing the 3′ coding region ofunc-29 was about 200 bases shorter than the wild-type fragment, consistent with the PCR results and indicating thatx415 contains a rearrangement involving the 3′ end of theunc-29 coding region (Fig. 1B).
Mutant rescue. Germ line transformation of unc-29and unc-38 was accomplished using the methods developed byFire (1986) and Mello et al. (1991).
Construction of UNC-29:: GFP fusions. Two UNC-29:: GFP fusions were made for this work, and each was found to rescue the unc-29 mutant phenotypes (both uncoordinated movement and levamisole insensitivity). Rescue indicates that transgene expression is physiologically relevant. Details of transgene construction are as follows. For LJH5, a genomic clone carrying the unc-29 coding region and 1.5 kb upstream was fused after the C terminus to the coding region for Aequora victoria gfp (Chalfie et al., 1994). For these constructs, the Ser65→Thr variant of GFP (Heim et al., 1994) was used for its improved fluorescent properties. The gfp coding region in these constructs also incorporates several consensus nematode introns to facilitate nuclear export of RNA products (A. Fire, G. Seydoux, J. Ahnn, and S. Xu, unpublished observations). The UNC-29-GFP fusion junction is just at the C terminus of UNC-29, so that no UNC-29 amino acids are removed from the construct. The 3′ nontranslated region is from unc-54. Construct LJH9 is similar to LJH5, except that the unc-29 upstream regions have been replaced by the body muscle-specific myo-3 promoter (Okkema et al., 1993) to generate a myo-3:: unc-29:: gfp fusion. The entire coding sequence from unc-29 is in this construct. There are six introns from unc-29 (these are introns 6–11 from the 3′ end of the endogenous primary transcript), and one upstream intron is provided by the expression vector, whereas three artificial introns are present in gfp. The 3′ untranslated region is from unc-54. The unc-29 introns are not necessary for rescue by the myo-3:: unc-29 constructs; a simple fusion of the myo-3 promoter to unc-29cDNA was constructed and found to rescue the unc-29 movement defect. Injection of these constructs, derivation, and maintenance of transgenic lines were by standard protocols (Mello and Fire, 1995), using the selectable marker rol-6 (Mello et al., 1991). Strain PD9253 carries the gfp-tagged unc-29 clone LJH5, whereas strain PD9254 carries themyo-3:: unc-29:: gfp fusion construct LJH9, each as an unstable, extrachromosomal array. A control transgenic PD9258 strain carries just the rol-6 marker. Each of these transgenics was constructed by injection into a unc-29(e193)genetic background. To examine the unc-29:: gfpfusions in other genetic backgrounds, hermaphrodites of strains PD9253 and PD9254 were first crossed with N2 males to obtain the arrays in a wild-type background. The arrays were then crossed into the background of other unc-29 mutants and were found to rescue all otherunc-29 mutants tested (mutant alleles x29,x513, x520, x522, x545, ande1072).
Confocal microscopy on strains carrying gfpfusions. Worms were grown at 20°C on NGM plates. Young adult worms that rolled strongly were picked to a 5% agarose pad containing 1× M9 and 10 mm sodium azide (Bargmann and Avery, 1995) and covered with a number 1½ coverslip. Confocal microscopy was done with a Bio-Rad (Richmond, CA) MRC-1024 microscope equipped with a 60×, 1.4 numerical aperture oil immersion objective. Specimens were viewed with 488 nm excitation from a krypton–argon laser at 10% transmission. Emitted light passing through a 522DF32 filter was collected at a normal scan speed with an iris setting of 3.4, gain of 1113, and a black level of −2, using Lasersharp software. Each optical section shown represents the accumulation over a 3 μm vertical distance of images scanned every 0.5 μm.
Transient heterologous expression in Xenopusoocytes. In vitro RNA transcription was performed using a Riboprobe kit obtained from Promega. Oocytes were obtained from female Xenopus laevis from Blades Biological (Kent, UK). The oocytes were kept in standard oocyte saline (SOS) medium [containing (in mm) 100 NaCl, 5 HEPES, 1.8 CaCl2, 1 MgCl2, and 2 KCl, pH 7.6]. Under the dissecting microscope, individual oocytes were defolliculated manually (i.e., the outer thecal layer and the follicle cell layers were removed with forceps), leaving the innermost vitelline membrane intact. The oocytes were then transferred to 1 mg/ml collagenase (type 1A; Sigma, St. Louis, MO) in SOS and incubated for 20 min to ensure that any remains of the follicle layer were digested. After a short recovery period in SOS (10–30 min), the oocytes were ready for injection. Test oocytes were injected with 50 nl of RNA solution (1 ng/nl for each subunit transcript) in the vegetal hemisphere. Control oocytes were either left uninjected or were injected with sterile distilled water.
Recordings were performed using a standard two-electrode voltage clamp (Dascal et al., 1984). A single oocyte was placed on the Sylgard base in a 1 ml experimental chamber. The oocyte was impaled with two glass microelectrodes (Clarke Electromedical glass GC 150 GF-150), fabricated using an electrode puller (Scientific and Research Instruments Ltd; catalog number 2001), and filled with 1 m KCl. Electrodes with a resistance of 5 MΩ were used for the voltage electrode, and current electrodes of 1–2 MΩ were used. Oocytes were voltage-clamped using a GeneClamp 500 amplifier. Current responses were monitored on an oscilloscope (Nicolet 3091) and recorded on a Gould BS-272 pen recorder. In some experiments, responses were recorded and stored using pClamp5 software installed on an IBM Personal Computer. Medium (SOS) and drugs were applied by a perfusion pump (Pharmacia LKB pump P-1) at a rate of 4 ml/min. Levamisole, mecamylamine,d-tubocurarine, neosurugatoxin, and α-bungarotoxin were each dissolved in the standard SOS medium. Levamisole was bath-applied. In experiments with nAChR antagonists, oocytes were preexposed to either mecamylamine (at the concentration indicated),d-tubocurarine (1 × 10−5m), or neosurugatoxin (5 × 10−7m) for 5 min, and then mecamylamine,d-tubocurarine, or neosurugatoxin was co-applied with levamisole for 30 sec. For α-bungarotoxin (Mr ∼7800), preexposure was done with a 5.0 × 10−6m concentration for 30 min.
Construction of an evolutionary tree. An alignment of 23 receptor sequences was generated using the computer program CLUSTAL V (Higgins et al., 1992) and edited using Genetic Data Environment. The resulting data set was analyzed under maximum parsimony conditions using the program Phylogenetic Analysis Using Parsimony (PAUP) (Swofford, 1991). Briefly, a 1000 replicate heuristic search was performed using independent random number seeds and tree bisection reconnection. The resulting tree of length 2384 changes was evaluated using a 1000 replicate bootstrap analysis in PAUP (Swofford and Olsen, 1990; Felsenstein, 1992). The resulting tree was rooted using the rat GABAA α1 receptor subunit. The data set contains phylogenetic data as determined using the gl statistic bracket (gl = −0.869766; p < 0.01) (Huelsenbeck and Hillis, 1993). An essentially identical tree was generated from the same starting alignment by nearest neighbor-joining analysis using the program CLUSTAL W (Thompson et al., 1994), which uses the neighbor-joining algorithm of Saitou and Nei (1987). The alignment and the PAUP data file are available from J.T.F., D.B.S., or J.A.L.
The following protein sequences, available from the SWISS-PROT protein sequence database, were used in constructing Figures 2and 4. All except GAA1_RAT are nAChR subunits. Rat α subunits: ACHA_RAT, ACH2_RAT, ACH3_RAT, ACH4_RAT, ACH5_RAT, ACH6_RAT, and ACH7_RAT (muscle α and neuronal α2–7). Rat neuronal β subunits: ACHN_RAT, ACHO_RAT, and ACHP_RAT (β2–β4). Rat muscle non-α: ACHB_RAT, ACHD_RAT, ACHE_RAT, ACHG_RAT (β, δ, ε, and γ).Drosophila subunits: ACH1_DROME, ACH2_DROME, ACH3_DROME, ACH4_DROME (ALS, SAD, ARD, and SBD). Locust Schistocerca gregaria αL1: ACH1_SCHGR. Rat GABA α subunit: GAA1_RAT (GABAA α1).
Fig. 2.

Amino acid sequence alignment of theCaenorhabditis elegans presumptive mature nAChR subunit sequences UNC-38, UNC-29, and LEV-1 with locust (Schistocerca gregaria) αL1, Drosophila(Dros.) non-α ARD, and rat α2 and β2 nAChR subunit sequences. The alignment was constructed using MACAW, version 2.0.5 (Schuler et al., 1991). Regions of sequence identity or high similarity within blocks of homology are indicated by dark coloring. Regions of moderate similarity or regions at boundaries of homology blocks are indicated by light coloring. Sequences in regions with no significant similarities between subunits are given in lower case letters, and no effort was made to align the amino acids in these regions. The positions of the four transmembrane domains (TM1–TM4) and the extracellular dicysteine loop (CL) characteristic of nAChR subunits are indicated.Asterisks indicate the positions of the vicinal cysteines characteristic of α-acetylcholine-binding subunits. The percent identity and similarity of UNC-38 to the locust and rat α sequences are 48 and 58% and 42 and 55%, respectively, as determined by individual pairwise comparisons. The percent identity and similarity of UNC-29 to the LEV-1, ARD, and rat β sequences are 66 and 77%, 50 and 65%, and 39 and 56%, respectively, determined by pairwise comparisons.
Fig. 4.

Maximum parsimony phylogenetic tree showing the relationship between the nAChR subunits. The tree is shown rooted using the rat GABAA receptor α1 sequence. LEV-1 and UNC-29 are shown in a class of polypeptides that include mammalian muscle nAChR non-α subunits. UNC-38 is shown clearly related to other invertebrate α-like AChR subunits. The values over the branches represent the minimum number of times from 1000 random seeds in the bootstrap analysis that a particular branch is expected to appear (p < 0.01). Branches without numbers do not have significant probability of appearing at that exact point in the tree. Regions with little homology, such as the intracellular cytoplasmic loop, were not used in the comparison. A tree of the same shape was generated by nearest neighbor-joining analysis, and the bootstrap values for 1000 random seeds are shown for comparisonbelow the branches.
RESULTS
Deletion and transposon insertion alleles of the levamisole resistance genes
The isolation and characterization of nAChR subunit genes is an essential beginning to understanding the effects of levamisole resistance mutations on synaptic signaling. To clone the subunit genes, two different experimental approaches were adopted to maximize the chances of success. One approach was to search for C. elegans nAChR subunit homologs by cross-hybridization and placement of the clones on the physical map of the nematode genome (Coulson et al., 1986). The other approach was to generate levamisole-resistant mutants by transposon insertion followed by cloning of the insertion site (Greenwald, 1985; Moerman et al., 1986), allowing genes to be cloned regardless of their actual functions or homology to known nAChR subunit genes.
Putative transposon insertion mutants were obtained by screening the progeny of either the Bergerac BO or the TR679 mutator strains (Moerman and Waterston, 1984; Collins et al., 1987) for spontaneous resistance to 1 mm levamisole. Isolates showing strong levamisole resistance were tested by complementation against the six known extreme levamisole resistance loci (Table 1). To confirm the identity of any clone found by transposon tagging or by cross-hybridization, mutants that might represent DNA rearrangements in the same resistance genes were also isolated by a similar selection scheme using the progeny of γ-irradiated worms. New alleles of unc-29, unc-38, unc-63,unc-74, unc-50, and lev-1 were isolated. All showed extreme levamisole resistance, except for the lev-1isolates, which produced only partial resistance, consistent with partial resistance being the null phenotype for this gene (Lewis et al., 1980a).
Isolation and identification of λ phage containing nAChR subunit gene homologs
To isolate λ phage carrying nAChR subunit genes, a library ofC. elegans genomic DNA was screened as described in Materials and Methods with an ard cDNA probe, which encodes a Drosophila non-α subunit (Hermans-Borgmeyer et al., 1986). Fifty-eight positive hybridizing phage were isolated. DNA from the 14 strongest positives was isolated and placed on the physical map by fingerprinting (Coulson et al., 1986). Several of these nAChR homologs mapped to positions on the physical map that were very close to the known genetic locations of levamisole resistance genes or corresponded to clones simultaneously identified by transposon tagging (unc-38 and unc-29). After revision of the genetic map position of lev-1 by T.M.B., the map position of this gene on chromosome IV was also consistent with the physical map position of two phage clones (JF#WA10 and JF#WA18; Fig.1A and Materials and Methods).
Characterization of the lev-1 locus
The identity of lev-1 as a gene encoding a non-α-nAChR was established as follows. First, sequence analysis of the phage occupying the expected physical map position oflev-1 identified a 4.8 kb HindIII fragment that contained an almost intact nAChR subunit gene, organized into eight exons (GenBank accession number X98601). Second, a complete cDNA sequence recovered from this region was found to encode an open reading frame for a non-α-nAChR subunit of 507 amino acids clearly homologous to vertebrate and insect nAChR subunits (Fig. 2). Third, when the genomic phage clones were used to probe Southern blots of genomic DNA from 11 mutator- and γ-ray-induced lev-1 mutants and five EMS-induced mutants, allele-specific rearrangements were found by T.M.B. that defined the lev-1 locus (Fig.1A). Three of four γ-ray-induced alleles (x416, x427, and x438) and two of seven mutator-induced alleles (x548 andx566) were found to have rearrangements that included at least part of the 4.8 kb HindIII fragment identified byard hybridization or the adjacent 1.4 kb HindIII fragment (x416). For three other mutator-induced alleles, the 4.8 kb HindIII fragment was replaced by a 6.4 kb fragment, consistent with the insertion of a typical 1.6 kb Tc1 transposon element. By using two PCR primers, one an oligonucleotide corresponding to the ends of the Tc1 element together and the other an oligonucleotide corresponding to the sequence of either strand at various positions within the 4.8 kb HindIII fragment (Barnes, 1990), it was possible to locate precisely the transposon within the 4.8 kb HindIII fragment (Fig.1A). These results show that the lev-1gene and a nAChR homolog co-localize to the same 4.8 kbHindIII fragment. The null phenotype of lev-1 was defined by the finding that most, if not all, of the coding region of the gene is deleted by the x548 mutation and the homozygousx548 null mutants are viable, partially levamisole-resistant, pseudo-wild-type mutants.
Characterization of the unc-29 locus
unc-29 was identified as a non-α-nAChR subunit as follows. Genomic phage fingerprinting to the region ofunc-29 on chromosome I were isolated based on the identification of a novel Tc1 insertion associated with theunc-29(x513) mutation. These phage overlapped the phage JF#WA33 identified by cross-hybridization with the Drosophila ard probe (Fig. 1B). Hybridization of the phage to Southern blots of DNA from mutator- and γ-ray-inducedunc-29 mutant identified RFLPs through a region spanned by a 6.0 kb HindIII fragment (Fig. 1B). The sequence the 6.0 kb HindIII fragment contained the entire coding region, divided into 12 exons, of a non-α-nAChR subunit highly homologous to lev-1 (GenBank accession number X98601). A full-length 1.7 kb cDNA that contained an open reading frame of 493 amino acids (Fig. 2) was obtained from a C. elegans mixed stage library (provided by R. Barstead).
Mutant rescue experiments confirmed the cloning of unc-29. Three cosmids that encompass the λ clone JF#WA33, and thus include all the regions affected by the unc-29 transpositions described above (Fig. 1B), could rescue theunc-29 mutant phenotype in transgenic animals. The cosmids were able to rescue completely the unc phenotype and to restore levamisole sensitivity, as were the smaller λ phage clones ZZ#1 and JF#WA33. Even the 6.0 kb HindIII fragment that spans the unc-29 coding region and contains only 490 bp of DNA upstream from the translational start site (Fig.1B) is capable of rescuing unc-29 (S. Kim, personal communication) (this work).
Because the mutant phenotype can be rescued by a transgene sequence, it can be inferred that the transgene expression pattern includes those tissues in which the endogenous gene is required. To monitor transgene expression, we constructed a set of plasmids in which the coding region for GFP (A. victoria green fluorescent protein) (Chalfie et al., 1994) was fused to the C-terminal coding region for UNC-29. An activated form of GFP (S65T) (Heim et al., 1994) was used to maximize fluorescent signal. The resulting constructs were capable of rescuing the mutant phenotypes of unc-29 alleles e193,x29, x513, x520, x522,x545, and e1072. A major focus ofunc-29 promoter activity was seen in body muscles. To test the hypothesis that UNC-29 expression in body muscles was sufficient for its function, we produced a construct in which unc-29upstream sequences (“promoter”) were removed and UNC-29/GFP expression was driven by the body muscle-specific myo-3promoter. The myo-3:: unc-29:: gfp fusion was indeed capable of rescuing the mutant phenotypes ofunc-29 mutant animals. This was consistent with the hypothesis that the primary focus of unc-29 was in body muscle. The muscle staining is consistent with previous pharmacological data from studying C. elegans (Lewis et al., 1980b) and physiological data from Ascaris that levamisole-sensitive nAChRs are present on muscle (Harrow and Gration, 1985). Recent mosaic studies using a unc-29 clone provided by us also show a major focus of unc-29 expression to be muscle (Miller et al., 1996b). We observed a low level of neuronal fluorescence, both with the unc-29:: gfp fusion and with themyo-3:: unc-29:: gfp fusion. It is not clear whether this fluorescence represents bona fide activity in these cells.
The ability of GFP fusions to rescue the mutant phenotype allowed us to examine the intracellular localization of a biologically functionalunc-29 derivative in living cells. Intracellular localization in a wild-type background (i.e., in the presence of a wild-type chromosomal copy of the gene) was somewhat surprising, with activity predominantly internal to muscle cells. Because the presence of the natural UNC-29 product might be expected to affect assembly of the recombinant protein, we examined localization of the protein produced from an LJH5-derived extrachromosomal array in a variety ofunc-29 mutant genetic backgrounds. Several unc-29mutant backgrounds behaved similarly to wild type in these assays (Fig.3A–E), with the exogenous GFP fusion present primarily in the cell body (e193, e1072,x520, x522, and x545). However, when the fusions were crossed into the homozygous mutant background provided by unc-29(x29) (and to a lesser extent, x513), relative localization was closer to that expected for a neurotransmitter receptor (head region for x29 homozygote shown in Fig. 3F–J) In addition, the overall amount of staining within muscle appeared to decrease. Internal staining of body muscles almost disappeared with punctate staining found along the nerve cords. Because of the background of neuronal staining, it was difficult to say what fraction of the staining in the nerve cords arose from body muscles. Some staining in the unc-29(x29) mutant background was still found internally in head muscles, but the relative amount of staining localized to synaptic regions of the ring (the central neuropil) greatly increased. These observations are consistent with the idea that the UNC-29 protein produced from the chromosomal copy of the unc-29 gene competes in the assembly and localization of nAChR molecules with the UNC-29–GFP fusion protein. Paradoxically, from the decrease in overall staining observed in aunc-29(x29) background, the endogenous gene product may also help stabilize the GFP fusion protein in assembly intermediates, possibly suggesting that more than one UNC-29 subunit can be assembled into an nAChR molecule. The null phenotype of unc-29 has yet to be defined, but both homozygotes of the unc-29(x29) andunc-29(e1072) mutations have been shown previously to contain little or no detectable specific high-affinity [3H]MAL binding (Lewis et al., 1987b). Although the focus of unc-38 and lev-1 expression has not been defined in this study, the great similarity in mutant phenotypes of unc-38 mutants and lev-1 semidominants tounc-29 mutants makes it likely that muscle is also the major focus for the expression of these genes as well.
Fig. 3.
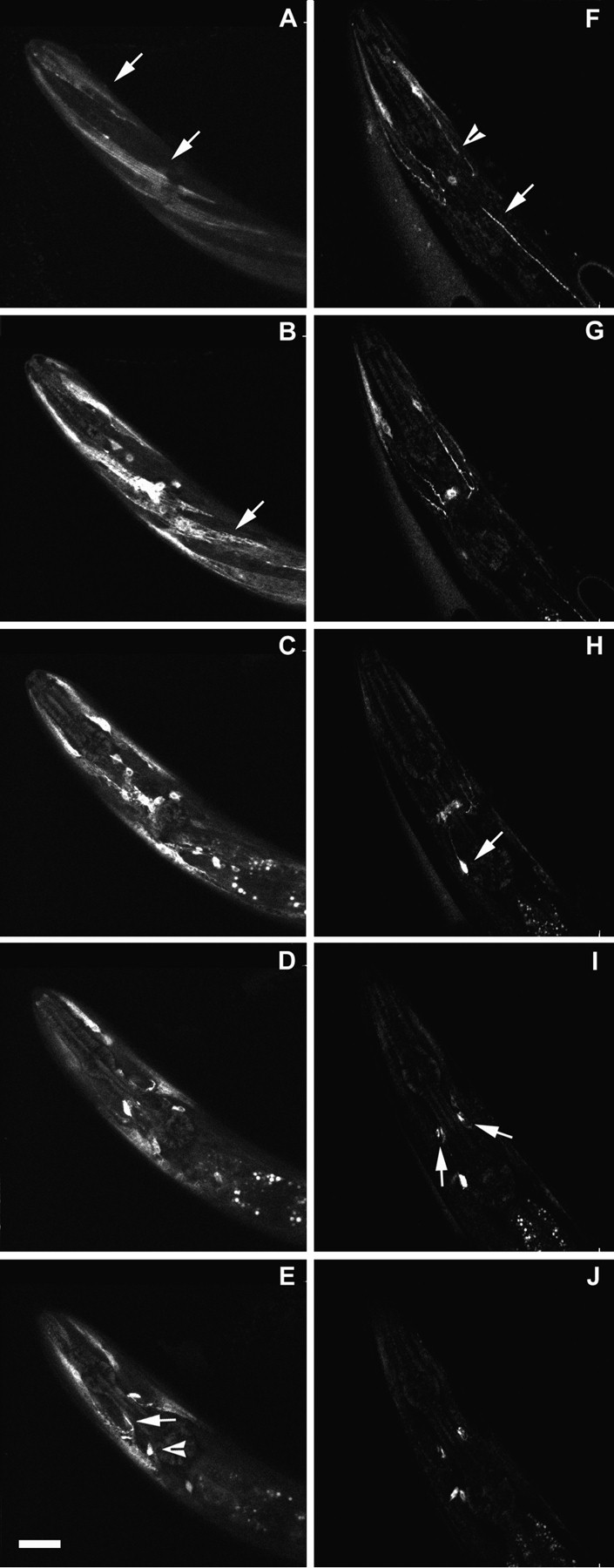
Serial optical sections showing expression of theunc-29 promoter-drivenunc-29:: gfp fusion LJH5 in N2 wild type and in the unc-29(x29) mutant strain. Confocal microscopic sections through the head region of a wild-type and anx29 mutant animal were accumulated as described in Materials and Methods. Each picture represents a succesive 3 μm thickness of the head. A–E, Wild type. Arrows in A point to fluorescence in head muscles. Fluorescence is seen more intensely inside the same muscles in B and also within a body muscle (arrow). In E, a muscle process entering the central neuropil next to the isthmus of the pharynx is stained (arrow). Stain is also accumulated in a neuronal cell body (arrowhead). F–J,unc-29(×29) mutant. Less staining is seen overall, and the stain is relatively concentrated in head muscle processes and nerve cords with continued neuronal staining. In F, process from anterior head muscles stain (arrowhead) and punctate staining is seen in a nerve cord (arrow). InH, the arrow points to a brightly staining neuronal cell body with a process running to the central neuropil, appearing as a hazy band in thecenter. In I, arrows point to brightly stained areas in the central neuropil immediately adjacent to the isthmus of the pharynx that appear to be associated with muscle processes running into the neuropil at this point. The same region is stained in the center in J, with two neuronal cell bodies below thecenter.
Characterization of the unc-38 locus
The unc-38 gene was identified by a novel Tc1-containing RFLP found in four of five spontaneously inducedunc-38 mutants (see Fig. 1C and Materials and Methods). Two of five γ-ray-induced unc-38 mutants also showed a restriction size difference from wild type when the subcloned genomic DNA flanking one of the Tc1 inserts was used as a hybridization probe on genomic Southern blots. DNA extending through and beyond the 3.2 kb HindIII fragment that was the site of sixunc-38 mutations was sequenced and found to encode a complete α subunit of an nAChR (GenBank accession X98599). A complete cDNA sequence reconstructed from this region encoded an open reading frame of 511 amino acids with strong homology to known α subunit sequences of vertebrates and insects (Fig. 2). The fingerprint of genomic phage containing this α subunit gene was consistent with the genetic map position of unc-38 to the left ofdpy-5 on chromosome I (Fig. 1C).
The cloning of unc-38 was confirmed by mutant rescue. The injection of either cosmid B0241 or C04E4 into the germ line ofunc-38 mutants completely restored normal movement in the L1 stage offspring, and the transgenic worms could now be killed by exposure to 1 mm levamisole. The B0241 and C04E4 cosmids completely encompass the λ phage ZZ#11 and ZZ#15 shown to contain the sites of the unc-38 mutations (Fig. 1C).
The null phenotype of unc-38 was defined to be that of an extremely levamisole-resistant unc. This finding was originally suggested by sequence analysis that showed that the Tc1 insertion site in unc-38(x525) disrupts a reading frame 15 amino acids upstream from the dicysteine loop (amino acids 128–142,Torpedo α numbering) that is characteristic of all nAChR subunits. Further mutant analysis showed that the EMS-induced mutationunc-38(x20) is an absolute splicing defect in which the third exon is skipped because the universal AG intron consensus sequence at the third exon splice acceptor site is mutated to AA. The mutation causes a transcript to be produced that is 155 bases shorter than the wild type when detected by RT-PCR. Homozygousunc-38(x20) mutants grow well and are among the most resistant of all levamisole-resistant mutants to the effects of levamisole and other cholinergic agonists (Lewis et al., 1980b). Because body muscles of unc-38 mutants are more resistant than head muscles to agonists, it is likely that there is at least one other α-nAChR subunit participating in the formation of other nAChR isotypes, and these isotypes differ in tissue distribution from the receptor formed with the unc-38 α subunit.
Sequence comparison of LEV-1, UNC-29, and UNC-38
In Figure 2, the deduced amino acid sequences of the LEV-1, UNC-29, and UNC-38 proteins are aligned and compared with vertebrate and insect nAChR subunit sequences. Comparison of the C. elegans sequences with database sequences shows that the nematode sequences are most similar to the Drosophila melanogasternAChR subunit sequences ARD and ALS (Hermans-Borgmeyer et al., 1986;Bossy et al., 1988) and to a partially sequenced putative nAChR subunit of the parasitic nematode Onchocerca volvulus (Ajuh and Egwang, 1994). The nematode non-α subunits UNC-29 and LEV-1 exhibit the highest amino acid sequence identity to ARD (50 and 46%), whereas the α subunit UNC-38 shows 49% identity to the ALS subunit. The predicted mature lengths of the three C. elegans nAChR subunits are 488 amino acids for UNC-38, 467 for UNC-29, and 473 for LEV-1. Whereas the α subunit of vertebrate muscle is the smallest muscle nAChR subunit, UNC-38 is the longest of the known nematode nAChR subunits, a relative size difference that holds for other invertebrate and for vertebrate neuronal α subunits compared with their non-α counterparts. The main structural features of the nAChR subunits of higher organisms are strikingly conserved in phylogeny down to the nematode, including the positions of the four transmembrane domains (TM1–4), the high sequence similarity found in each TM region, the variable long loop between TM3 and TM4, and the dicysteine loop, which is a hallmark of this superfamily and invariably found at the equivalent to positions 128–142 of the Torpedo α subunit (Barnard, 1992). There seem to be additional regions in all α subunits of invertebrates that have no counterpart in vertebrates. These insertions occur 25 amino acids N-terminal to the vicinal cysteines (equivalent to positions 192 and 193 of theTorpedo α subunit) and at the C terminus of the polypeptide. The functional significance of these domains is unknown. The insertion N-terminal to the vicinal cysteines is 13 amino acids longer for the nematode UNC-38 α subunit than for locust andDrosophila α subunits and is absent from invertebrate non-α subunits.
UNC-29 and LEV-1 are highly homologous: 66% amino acid identity or, with conservative substitutions, 77% similarity, a resemblance found for few other nAChR subunit pairs from the same species. They are categorized as non-α subunits by the absence of vicinal cysteines (at positions 192 and 193, Torpedo α numbering) and when compared with vertebrate nAChRs show the closest homology to neuronal subunits. Both UNC-29 and LEV-1 are about equally similar to neuronal α and non-α of the rat and chick (rat α2–α4 and β2–β4, chick α2 and α4 and β2 and β4) with UNC-29 showing slightly greater over all sequence similarity, ∼55 versus ∼50% for LEV-1. Consistent with these results, many aspects of the in situpharmacology of muscle nAChRs of C. elegans andAscaris resemble those of vertebrate neuronal nAChRs, including insensitivity to α-bungarotoxin, block by mecamylamine (Lewis et al., 1987a, 1980b; Fleming et al., 1993), and, in the case ofAscaris suum muscle nAChRs, block by κ-bungarotoxin (Colquhoun et al., 1993).
Intron–exon structure comparisons and phylogenetic analysis using accepted mutation parsimony trees
Between unc-29 and the γ, δ, and ε subunits of vertebrate muscle (Nef et al., 1984; Buonanno et al., 1989), the positions of 5 of the 11 introns are completely conserved, and an additional 3 intron positions are within four amino acids of the equivalent sites in the vertebrate muscle subunits. The inexact conservation of some intron–exon boundaries may be attributable to splice junction drift or de novo creation of introns at “permissible” sites. The locations of four of the sevenlev-1 introns and three of the seven unc-38introns are identical in unc-29. However, only one intron location, between exons 5 and 6 in unc-29, is shared betweenunc-38 and lev-1, and this site is present in all nAChR subunit genes. The flanking exons seem to form a calcium-binding domain (Godzik and Sander, 1989), although its significance has not been well defined. A unc-29–unc-38 common splice site occurring between exons 3 and 4 of unc-29 is conserved in all nAChR subunit genes examined except lev-1. These findings suggest that the conserved splice sites predate the divergence of nematodes, insects, and vertebrates, which occurred about 600 million years ago. Unique to the nematode receptors is an intron that interrupts the coding region between TM4 and the C terminus in each of the three nematode subunits. Although the gene structures ofunc-29 and lev-1 show considerable similarity,unc-38 is no more homologous to the other C. elegans subunits than any other insect or vertebrate nAChR subunit genes, suggesting that the divergence between α and non-α subunits is very ancient. The intron–exon structure of all three nematode genes is also more typical of the highly interrupted vertebrate muscle nAChR genes than it is of the vertebrate neuronal nAChR genes (Nef et al., 1988), in which there is generally only a single intron between the conserved splice sites in the extracellular domain and at the end of the protein.
A maximum parsimony analysis for nAChR polypeptides is shown in Figure4. The polypeptide sequences of 22 nAChRs and the rat GABAA α1 receptor subunit were aligned. Using PAUP, a single tree of length 2384 changes was found (Swofford, 1991). The significance of this tree was evaluated using bootstrap analysis (Felsenstein, 1992). The resulting tree shows that nAChRs fall into several distinct classes. The C. elegans subunits LEV-1 and UNC-29 are very closely related and, of the other sequences analyzed, are most similar to the Drosophila neuronal subunit ARD. UNC-38 represents an α subunit type closely related to theDrosophila α-like neuronal subunits. Although the distinctiveness of UNC-38 on the tree is not strongly supported by the bootstrap analysis, it is consistent with the unique pharmacological properties of nematode nAChRs as observed for C. elegans andAscaris.
Potential glycosylation and phosphorylation signals in UNC-29, UNC-38, and LEV-1
Sequence prediction of glycosylation sites for the vertebrate muscle nAChR subunits, now confirmed biochemically (Claudio et al., 1989), indicated a single glycosylation site for the α and β subunits, two for the γ, and three for the δ muscle subunits. The vertebrate neuronal sequences usually contain two potential glycosylation signals. The sequences of UNC-29, UNC-38, and LEV-1 predict two, three, and five asparagine-linked glycosylation sites, respectively. There is no glycosylation site common to all three subunits, and only one shared between UNC-29 and LEV-1 at asparagine 50 (UNC-29 numbering), a site that is also present in nearly all invertebrate and vertebrate neuronal sequences (but lacking in human β4 and goldfish non-α3). UNC-38 is the only invertebrate sequence to lack this site, and the three glycosylation signals UNC-38 contains are not in positions common to any other nAChR. An asparagine 109-linked glycosylation site in LEV-1 is conserved in α2 and α4 neuronal sequences and in the chick muscle δ subunit.
The Torpedo nAChR can be phosphorylated by at least three different protein kinases: cAMP-dependent protein kinase (PKA), protein kinase C (PKC), and a tyrosine kinase (TK) (Huganir and Greengard, 1983; Huganir et al., 1984; Huganir, 1988). The region of the Torpedo receptor that was shown to be phosphorylated is the intracellular loop between TM3 and TM4. In the equivalent regions, the C. elegans UNC-29 subunit has three PKC sites, one PKA site, and one TK site; UNC-38 has single PKC and TK sites; LEV-1 has only a single PKC site. Studies with vertebrate nAChR subunits have shown phosphorylation to be involved in desensitization, receptor turnover, and receptor assembly. The in vivo functions of the glycosylation and phosphorylation sites can be investigated inC. elegans by mutagenizing a given site and then returning the altered gene to a null mutant strain that cannot otherwise produce the targeted subunit.
Expression of unc-29, unc-38, and lev-1 inXenopus oocytes
In earlier studies, RNA isolated from mixed stage wild-typeC. elegans was injected into Xenopus oocytes with the result that a dose-dependent depolarization was detected in response to bath-applied levamisole (Fleming et al., 1991, 1993). The levamisole response was not blocked by α-bungarotoxin (Tornoe et al., 1995), in agreement with [3H]meta-aminolevamisole-binding studies using membrane fragments (Lewis et al., 1987a) and cut worm muscle contraction assays (Lewis et al., 1980b). Cytoplasmic co-injection of cRNAs encoding UNC-29, LEV-1, and UNC-38 resulted in inwardly directed currents (holding potential, −60 mV) in response to levamisole (1 × 10−4m), whereas oocytes injected separately with message encoding a single subunit or the equivalent volume of distilled water showed no such responses (Fig. 5A–E). Pairwise injection of all possible combinations yielded either no responses or inconsistent responses. In oocytes injected with all three cRNAs, the response to levamisole was mimicked by dimethylphenylpiperazinium (DMPP; 1.0 × 10−4m; n = 4; Fig.5F) and less efficiently by ACh (1.0 × 10−4m; n = 4; Fig.5G). The membrane potential of the amplitude of the levamisole-induced currents indicated the gating of a cation ion channel (Fig. 5H). Responses to levamisole were dose-dependent (Fig. 5I) and blocked in a dose-dependent manner by the potent antagonist of Ascarismuscle nAChRs mecamylamine (Fig. 5J). Neosurugatoxin (5 × 10−7m; Fig.5K), which blocks reversibly Ascarismuscle nAChRs, also reversibly blocked the response to levamisole (1.0 × 10−4m) in oocytes injected with UNC-29, LEV-1, and UNC-38 (n = 6), as didd-tubocurarine (1.0 × 10−5m; data not shown; n = 5). In contrast, α-bungarotoxin (5 × 10−6m; Fig. 5L), a weak antagonist of Ascaris muscle nAChRs, was ineffective on responses to levamisole even after 30 min exposure of oocytes injected with all three subunits. The observed currents are of small amplitude and do not require a rigorous subunit stoichiometry. Perhaps not all subunits needed for full expression have been identified, or alternatively, some factor useful in assembling the nematode receptor, e.g., one of the other gene functions identified by levamisole resistance mutation, may be lacking in the frog oocyte.
Fig. 5.

Transient expression of unc-29,unc-38, and lev-1 cRNAs inXenopus oocytes. A–E, Responses to levamisole (100 μm) of Xenopus oocytes injected cytoplasmically with one (A–C) or a combination (E) of cRNAs encoding C. elegans putative nAChR subunits or the equivalent volume (50 nl) of distilled water (D). Whereas, when injected separately (A–C) no response was obtained, and all pairwise combinations yielded either no expression or unreliable expression, when all three subunits were co-expressed, small amplitude inward currents were observed in response to levamisole (E), DMPP (F), and acetylcholine (G). Levamisole-induced currents recorded when all three subunits were co-expressed were membrane potential-dependent, and the estimated reversal potential suggested a cationic current (H). Such responses to levamisole were dose-dependent (I) and were blocked in a dose-dependent manner by the nicotinic antagonist mecamylamine (J), which also blocks native muscle nAChRs in Ascaris suum. As is also the case for nativeAscaris muscle nAChRs, on the expressed receptors, neosurugatoxin (0.5 μm, 10 min) was an effective blocker of levamisole responses (K), whereas α-bungarotoxin (5.0 μm, 30 min) was ineffective (L).
Sequence analysis of the lev-1 dominant alleles (x21 and x61)
We also determined the sequence alterations in the two unusual lev-1 semidominant alleles, x21 and x61. Thelev-1 cDNAs from these mutants were amplified by PCR and sequenced. To minimize the possibility of a cloning artifact, cDNAs were amplified and sequenced from two different preparations of each mutant. Both alleles contained mutations in or very near the TM2 membrane-spanning domain of the LEV-1 protein (Fig. 6). For x21, a glutamate to lysine (E to K) mutation was found at the hydrophilic site equivalent to the −1 position ofTorpedo TM2 (numbering from the N-terminal direction), a position known to influence the overall conductance and Ca2+ permeability of the ion channel. Mutations at this position in vertebrate nAChR subunits produce the largest changes in channel conductance and ion selectivity (Imoto et al., 1988; Konno et al., 1991; Galzi et al., 1992). For x61, a leucine insertion was found at a position equivalent to the +11 position ofTorpedo TM2, a position in TM2 demonstrated to affect ion conductance by in vitro mutagenesis of the rat neuronal α7 nAChR subunit (Galzi et al., 1992). The sites of the twolev-1 dominant mutations are thus 11 amino acids apart in the same functional domain of the protein, consistent with their similar mutant phenotypes. The mutations are about three turns of an α helix apart and are presumed to face or be close to the lining of the channel (Unwin, 1989; Revah et al., 1991).
Fig. 6.

Comparison of TM2 sequences from the twolev-1 dominant alleles with the chick α7 nAChR subunit mutations shown to convert cationic to anionic selectivity (Galzi et al., 1992) and a rat α7 nAChR subunit mutated in leu-247, resulting in altered desensitization (Revah et al., 1991). Mutated amino acids are shown in bold italic underlined.
DISCUSSION
We have shown that the lev-1, unc-29, andunc-38 genes of C. elegans associated with resistance to the cholinergic anthelmintic drug levamisole encode nAChR subunits. Furthermore, subunit combinations that include theunc-38 α subunit can be expressed in Xenopusoocytes to form ion channels activated by levamisole. The viability ofC. elegans nAChR mutants provides unique opportunities to study both molecular aspects of nAChR function and the interactions of nAChR molecules with other cellular components throughout development.
The non-α subunit UNC-29 is required for agonist binding
Earlier studies implicated UNC-29 as part of the levamisole binding site in nematode nAChRs. unc-29 mutants had little specific [3H]MAL binding, whereasunc-38 mutants retained detectable binding (Lewis et al., 1987b). Levamisole seems to act like nicotine, competing with acetylcholine for binding to the receptor (Lewis et al., 1980b; Harrow and Gration, 1985; Lewis et al., 1987a). The finding thatunc-38 rather than unc-29 encodes an nAChR α subunit suggests that the levamisole and acetylcholine binding site may be formed between α and non-α subunits, as occurs for vertebrate muscle nAChRs (Pedersen and Cohen, 1990; Czajkowski et al., 1993). Ten rare mutants of unc-29 and unc-38 retain normal motor behavior but are extremely resistant to levamisole, as might result from missense amino acid substitutions that interfere with the binding of levamisole but not of acetylcholine. Sequencing these rareunc-38 and unc-29 mutations should help further define the levamisole and acetylcholine binding site.
LEV-1 is highly homologous to UNC-29 but not required for nAChR function
LEV-1 and UNC-29 are much more homologous than almost all other nAChR subunit pairs from the same species. Mutation ofunc-29 is more functionally debilitating than mutation oflev-1 (Lewis et al., 1980b). unc-29 mutants have lost high-affinity specific [3H]MAL binding, are extremely uncoordinated, and are extremely resistant to muscle contraction induced by levamisole. lev-1 mutants have detectable levamisole binding, wild-type motility, and much weaker levamisole resistance. In vertebrate muscle, the γ and ε subunits can functionally replace one another while sharing 52% amino acid identity (65% similarity). UNC-29 with 66% identity and 77% similarity to LEV-1 might substitute for a missing LEV-1 subunit and allow the partial function seen in lev-1 mutants (including deletion and transposon mutants; Fig. 1A). The inability of LEV-1 to replace the UNC-29 subunit is consistent with UNC-29 but not LEV-1, being required for the function of most levamisole-sensitive nAChR molecules in C. elegans.
lev-1 semidominant mutations block nAChR function
Although the LEV-1 subunit is normally not essential, two rare semidominant (sd) mutations of lev-1, when homozygous, greatly reduce levamisole-sensitive nAChR function. Our results show that these mutations represent either an amino acid substitution (x21) or an amino acid addition (x61) within or very near the TM2 domain of the LEV-1 subunit. For several hundred other extreme levamisole resistance mutations that have been complemented, including unc-29 andunc-38 mutants, the single copy of the wild-type gene in a heterozygote is sufficient for wild-type sensitivity to levamisole, whereas a single copy of the wild-type lev-1 gene inlev-1(sd)/+ heterozygote results in partial resistance. Normal assembly of a receptor with a defective LEV-1 subunit that interferes with or alters ion conductance could produce thelev-1(sd) mutant phenotype, especially if the LEV-1 subunit is normally present in the great majority of levamisole receptor molecules. The hypothesis that most levamisole-sensitive nAChR molecules contain LEV-1 is consistent with the observations that mutation of lev-1 affects the major portion of high-affinity specific [3H]MAL binding, and that the twolev-1(sd) mutants as homozygotes have normal amounts of specific [3H]MAL binding trapped in an unusual high-affinity state (Lewis et al., 1987b). The semidominant phenotype could be explained if at least 50% of the assembled molecules in alev-1(sd)/+ heterozygote were poisoned by thelev-1(sd) subunit, whereas in all other unc/+ heterozygotes examined, in which the unc allele was selected for strong dysfunction in the homozygous state, the limited amount of receptor molecules assembled were preferentially drawn from the wild-type pool of receptor subunits for the gene in question.
The lev-1(x61) mutant phenotype is caused by insertion of an additional leucine into TM2 at the equivalent position of leucine 247 in the chick α7 neuronal nAChR. Replacement of the chick α7 Leu-247 with either a threonine or a serine results in a channel with no rectification, reduced desensitization, and increased affinity for ACh (Revah et al., 1991). The change in x61, adding an additional hydrophobic leucine residue next to this position, causes the opposite mutant phenotype with respect to agonist sensitivity: reduced sensitivity to agonist and an apparent desensitized, high-affinity binding state insensitive to the addition of the noncompetitive antagonist mecamylamine (Lewis et al., 1987b). Similarly, the charge reversal in x21 (E to K transition) at a position known to be critical to ion conductance in vertebrate nAChRs is consistent with the insensitivity of the x21 mutant to agonist and its similarly desensitized binding state (Lewis et al., 1987b). A series of engineered changes in the rat neuronal α7 subunit including an amino acid addition and substitution at the site of thex21 mutation can even convert the α7 channel to one conducting anions (Galzi et al., 1992). Because α7 forms a homo-oligomer, the α7 mutant phenotypes are the result of the same mutation being present in all five subunits of a receptor molecule. By contrast, the lev-1(sd) mutant phenotype is generated by mutation of a single non-α subunit that only forms a channel when combined with other subunits, as shown both by mutant phenotypes and by oocyte expression studies. The lev-1 mutations may act by causing desensitization of the entire receptor molecule rather than by simply blocking ionic conductance, consistent with the apparent importance of TM2 to desensitization as well as to ion passage (Unwin, 1995). The lack of similar dominant isolates for other subunit genes such as unc-29 and unc-38 may simply be caused by the rarity of dysfunctional subunits that assemble well yet poison the assembled receptor strongly.
The predicted UNC-38 amino acid sequence is consistent with the insensitivity to α-bungarotoxin and lophotoxin observed for nematode nAChRs
The potent neurotoxin lophotoxin is a cyclic diterpene isolated from the gorgonian coral Lophogorgia chilensis and has been shown to block all vertebrate muscle, neuronal, and invertebrate neuronal nAChRs tested (Abramson et al., 1988; Bai et al., 1993). This toxin binds covalently to the α subunit of the Torpedoreceptor, and protease digestion followed by sequence analysis has identified tyrosine 190 as the target amino acid residue. This residue is conserved in all α subunits sensitive to lophotoxin tested to date (Abramson et al., 1989) and is close to the vicinal cysteines that define a main part of the acetylcholine binding site on α subunits (Kao and Karlin, 1986). UNC-38, however, contains a proline at the position equivalent to Tyr-190 (Fig. 7), and lophotoxin is unable to block the levamisole-induced responses of Ascaris suum muscle (Tornoe et al., 1995). In general, this region of UNC-38, extending through and beyond the vicinal cysteines equivalent to residues 192 and 193 of Torpedo α, is more typical of a muscle-type α subunit than a neuronal α subunit (Fig. 7). In addition, UNC-38 has an alanine at position 197 (Torpedo α numbering). This position is occupied by a proline in all α subunits that are capable of binding α-bungarotoxin. This is consistent with our finding that the C. elegans nAChR is not blocked by α-bungarotoxin in living worms and cut worms, in binding assays, or on expressed receptors in Xenopus oocytes (Lewis et al., 1980b; Lewis et al., 1987a; Tornoe et al., 1995).
Fig. 7.

Amino acid sequence comparison of UNC-38 with neuronal α and vertebrate muscle α subunits in the region known to include the α-bungarotoxin and lophotoxin binding sites. In all species in which the sequence and pharmacology are known, a proline is present at position 197 (Torpedo α subunit numbering, indicated by asterisk) if the α subunit binds α-bungarotoxin. The muscle α subunits that do not bind α-bungarotoxin used in this alignment (muscle αR) are from two snakes (Neumann et al., 1989) and a mongoose (Barchan et al., 1992). The neuronal α subunits that bind bungarotoxin include chick α7 (Couturier et al., 1990), invertebrate locust α1 (Marshall et al., 1990), and the DrosophilaALS α-like sequence. The tyrosine present at the equivalent ofTorpedo α position 190 (✙) is found in subunits binding lophotoxin (see Discussion). Sequence positions important to the comparison are shown in upper case. The letterx is used to indicate positions not considered important to the comparison. R and S, α-Bungarotoxin-resistant and -sensitive, respectively.
Conclusions and future prospects
We have shown here that 3 of the 11 C. eleganslevamisole resistance loci encode nAChR subunits (see Table 1). The complete loss of levamisole sensitivity in mutants of eitherunc-29 or unc-38 suggests that these subunits co-exist in the same receptor molecules. The dominant effect of rarelev-1 mutations argues that the LEV-1 subunit is also an integral although normally dispensable part of most receptor molecules formed from UNC-29 and UNC-38. Two other loci (unc-22 andlev-11) encode muscle proteins (Moerman et al., 1988;Williams and Waterston, 1994). Of the remaining six levamisole resistance loci, some may encode proteins important to receptor function other than additional nAChR subunits. Two of these genes,unc-50 and unc-74, are essential for nAChR response and [3H]MAL binding (Lewis et al., 1987b). The gene for unc-50 has recently been cloned (M. O. Hengartner, N. Tsung, J. A. Lewis, and H. R. Horvitz, unpublished data), and it is not an nAChR subunit. The genes lev-8,lev-9, and lev-10 are probably not major structural subunits of the levamisole nAChR, because mutants of these genes have relatively normal levamisole binding (Lewis et al., 1987b).unc-63 is not predicted to encode an nAChR structural subunit because of the isolation of a rare allele (b404) that is markedly uncoordinated although still very sensitive to levamisole (Lewis et al., 1980b). The genetic loci identified so far have been discovered on the basis of uncoordination or levamisole resistance. Other mutant types that might identify additional genes important to receptor function, such as revertants or suppressors of the levamisole-resistant mutant phenotype, have yet to be sought extensively.
In conclusion, a number of genes affecting cholinergic neurotransmission have now been identified in C. elegans(Nonet et al., 1993; Alfonso et al., 1994; Arpagaus et al., 1994;Squire et al., 1995; Treinin and Chalfie, 1995; Miller et al., 1996a;Baylis et al., 1997) in addition to the novel nAChR subunit genes described in this work. The ease and rapidity of working with C. elegans make it well suited for the manipulation and expressionin vivo of genes that are important to cholinergic synapse formation and function. Receptors from other organisms that are difficult to study, such as parasitic nematodes (e.g., Onchocerca volvulus), might be rendered more amenable to analysis by transgenic expression in C. elegans in place of the native nicotinic acetylcholine receptor.
Footnotes
This work was supported National Institutes of Health Grants GM 08194 and GM 37706, National Science Foundation Grant HRD-9253024, the Medical Research Council (MRC) of the United Kingdom, the Isaac Newton Trust, DuPont Agricultural Products, the Association of Commonwealth Universities (for a Commonwealth Scholarship to T.M.B.), and the Korean Ministry of Education (for Genetic Engineering Grant GE96–192 to J.A.). We thank S. Kim for sharing unpublished observations, and H. Betz (Max-Planck-Institut für Hirnforschung, Frankfurt, Germany) and E. Gundelfinger (Federal Institute for Neurobiology, Magdeburg, Germany) for provision of the Drosophila ard cDNA. I. Maruyama, S. Kim, R. Barstead, C. Link, and A. Coulson kindly providedC. elegans cDNA and genomic libraries. We are indebted to C. Venter for generously providing facilities at the National Institutes of Health (Bethesda, MD) for automated DNA sequencing, D. Bird for expert advice on recombinant DNA techniques, L. F. Kolakowski for performing the maximum parsimony analyses and providing advice, S. Hardies for additional helpful advice on evolutionary comparisons, M. Nonet for help in analyzing the GFP fusion studies, and D. Zarkower for L2 RNA. Thanks to S. Hekimi, L. Avery, and N. Unwin for helpful discussions and A. Eisenstark and P. D. Gardner for aiding J.L. Thanks to the Caenorhabditis Genetics Center for providing strains and map data.
The GenBank accession numbers for the lev-1,unc-29, and unc-38 sequences reported in this paper are X98601, U81144, and X98599, respectively.
Correspondence should be addressed to Dr. James A. Lewis, Occupational and Safety Programs, University of Texas at San Antonio, San Antonio, TX 78249.
Dr. Barnes’ present address: Department of Biology, McGill University, Montreal, Quebec H3A 1B1, Canada.
Dr. Matsuda’s present address: Department of Agricultural Chemistry, Faculty of Agriculture, Kinki University, 3327-204 Nakamachi, Nara 631, Japan.
Dr. Ahnn’s present address: Department of Life Science, Kwangju Institute of Science and Technology, Kwangsan-Ku Sangam-Dong 572, Kwangju 506-712, Korea.
Dr. Sulston’s present address: Sanger Centre, Cambridge CB10 1RQ, UK.
Dr. Barnard’s present address: Molecular Neurobiology Unit, Royal Free Hospital, School of Medicine, London NW3 2PF, UK.
REFERENCES
- 1.Abramson SN, Culver P, Kline T, Li Y, Guest P, Gutman L, Taylor P. Lophotoxin and related coral toxins covalently label the α-subunit of the nicotinic acetylcholine receptor. J Biol Chem. 1988;263:18568–18573. [PubMed] [Google Scholar]
- 2.Abramson SN, Li Y, Culver P, Taylor P. An analog of lophotoxin reacts covalently with Tyr190 in the α-subunit of the nicotinic acetylcholine receptor. J Biol Chem. 1989;264:12666–12672. [PubMed] [Google Scholar]
- 3.Ajuh PM, Egwang TG. Cloning of a cDNA encoding a putative nicotinic acetylcholine receptor subunit of the human filarial parasite Onchocerca volvulus. Gene. 1994;144:127–129. doi: 10.1016/0378-1119(94)90216-x. [DOI] [PubMed] [Google Scholar]
- 4.Alfonso A, Grundahl K, McManus JR, Rand JB. Cloning and characterization of the choline acetyltransferase structural gene (cha-1) from C. elegans. J Neurosci. 1994;14:2290–2300. doi: 10.1523/JNEUROSCI.14-04-02290.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arpagaus M, Fedon Y, Cousin X, Chatonne A, Berge J-B, Fournier D, Toutant J-P. cDNA sequence, gene structure, and in vitro expression of ace-1, the gene encoding acetylcholinesterase of class A in the nematode Caenorhabditis elegans. J Biol Chem. 1994;269:9957–9965. [PubMed] [Google Scholar]
- 6.Bai D, Abramson SN, Sattelle DB. Actions of a coral toxin analogue (Bipinnatin-B) on an insect nicotinic acetylcholine receptor. Arch Insect Biochem Physiol. 1993;23:155–159. doi: 10.1002/arch.940230402. [DOI] [PubMed] [Google Scholar]
- 7.Ballivet M, Alliod C, Bertrand S, Bertrand D. Nicotinic acetylcholine receptors in the nematode Caenorhabditis elegans. J Mol Biol. 1996;258:261–269. doi: 10.1006/jmbi.1996.0248. [DOI] [PubMed] [Google Scholar]
- 8.Barchan D, Kachalsky S, Neumann D, Vogel Z, Ovadia M, Kochva E, Fuchs S. How the mongoose can fight the snake: the binding site of the mongoose acetylcholine receptor. Proc Natl Acad Sci USA. 1992;89:7717–7721. doi: 10.1073/pnas.89.16.7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bargmann CI, Avery L. Laser killing of cell in Caenorhabditis elegans. In: Epstein HF, Shakes DC, editors. Methods in cell biology, Vol 48, Caenorhabditis elegans: modern biological analysis of an organism. Academic; New York: 1995. pp. 225–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnard EA. Receptor classes and neurotransmitter-gated ion channels. Trends Biochem Sci. 1992;17:368–374. doi: 10.1016/0968-0004(92)90002-q. [DOI] [PubMed] [Google Scholar]
- 11.Barnes TM. Echolocation: a PCR-based strategy for reliably and rapidly mapping transposon insertion sites. Nucleic Acids Res. 1990;18:6741–6742. doi: 10.1093/nar/18.22.6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baylis HA, Matsuda K, Squire MD, Fleming JT, Harvey RJ, Darlison MG, Barnard EA, Sattelle DB (1997) ACR-3, a Caenorhabditis elegans nicotinic acetylcholine receptor subunit: molecular cloning and functional expression. Receptors Channels, in press. [PubMed]
- 13.Bossy B, Ballivet M, Spierer P. Conservation of neuronal nicotinic acetylcholine receptors from Drosophila to vertebrate central nervous systems. EMBO J. 1988;7:611–618. doi: 10.1002/j.1460-2075.1988.tb02854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buonanno A, Mudd J, Merlie JP. Isolation and characterization of the β and ε subunit genes of mouse muscle acetylcholine receptor. J Biol Chem. 1989;264:7611–7616. [PubMed] [Google Scholar]
- 16.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 17.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 18.Claudio T, Paulson HL, Green WN, Ross AF, Hartman DS, Hayden D. Fibroblasts transfected with Torpedo acetylcholine receptor β-, γ-, and δ-subunit cDNAs express functional receptors when infected with a retroviral α recombinant. J Cell Biol. 1989;108:2277–2290. doi: 10.1083/jcb.108.6.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins J, Saari B, Anderson P. Activation of a transposable element in the germ line but not the soma of Caenorhabditis elegans. Nature. 1987;328:726–728. doi: 10.1038/328726a0. [DOI] [PubMed] [Google Scholar]
- 20.Colquhoun LM, Holden-Dye L, Walker RJ. The action of nicotinic receptor specific toxins on the somatic muscle cells of the parasitic nematode Ascaris suum. Mol Neuropharmacol. 1993;3:11–16. [Google Scholar]
- 21.Coulson A, Sulston J, Brenner S, Karn J. Towards a physical map of the genome of the nematode Caenorhabditis elegans. Proc Natl Acad Sci USA. 1986;83:7821–7825. doi: 10.1073/pnas.83.20.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Couturier S, Bertrand D, Matter JM, Hernandez MC, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M. A neuronal nicotinic acetylcholine receptor subunit (α 7) is developmentally regulated and forms a homo-oligomeric channel blocked by α-BTX. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 23.Craxton M. Linear amplification sequencing, a powerful method for sequencing DNA. Methods Companion Methods Enzymol. 1991;3:20–26. [Google Scholar]
- 24.Czajkowski C, Kaufmann C, Karlin A. Negatively charged amino acid residues in the nicotinic receptor δ subunit that contribute to the binding of acetylcholine. Proc Natl Acad Sci USA. 1993;90:6285–6289. doi: 10.1073/pnas.90.13.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dascal N, Landau EM, Lass Y. Xenopus oocyte resting potential, muscarinic responses and the role of calcium and guanosine 3′,5′-cyclic monophosphate. J Physiol (Lond) 1984;352:551–574. doi: 10.1113/jphysiol.1984.sp015310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Devillers-Thiery A, Galzi JL, Eisele JL, Bertrand S, Bertrand D, Changeux JP. Functional architecture of the nicotinic acetylcholine receptor: a prototype of ligand-gated ion channels. J Membr Biol. 1993;136:97–112. doi: 10.1007/BF02505755. [DOI] [PubMed] [Google Scholar]
- 27.Felsenstein J. Estimating effective population size from samples of sequences: a bootstrap Monte Carlo integration method. Genet Res. 1992;60:209–220. doi: 10.1017/s0016672300030962. [DOI] [PubMed] [Google Scholar]
- 28.Fire A. Integrative transformation of Caenorhabditis elegans. EMBO J. 1986;5:2673–2680. doi: 10.1002/j.1460-2075.1986.tb04550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fleming JT, Riina HA, Sattelle DB. Acetylcholine and GABA receptors of Caenorhabditis elegans expressed in Xenopus oocytes. J Physiol (Lond) 1991;438:371p. [Google Scholar]
- 30.Fleming JT, Tornoe C, Riina HA, Coadwell J, Lewis JA, Sattelle DB. Acetylcholine receptor molecules of the nematode Caenorhabditis elegans. In: Pichon Y, editor. Comparative molecular neurobiology. Birkhauser Verlag; Basel: 1993. pp. 65–80. [DOI] [PubMed] [Google Scholar]
- 31.Frohman MA, Dush MK, Martin GR. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galzi J-L, Devillers-Thiery A, Hussy N, Bertrand S, Changeux J-P, Bertrand D. Mutations in the channel domain of a neuronal nicotinic receptor convert ion selectivity from cationic to anionic. Nature. 1992;359:500–505. doi: 10.1038/359500a0. [DOI] [PubMed] [Google Scholar]
- 33.Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- 34.Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- 35.Godzik A, Sander C. Conservation of residue interactions in a family of Ca-binding proteins. Protein Eng. 1989;2:589–596. doi: 10.1093/protein/2.8.589. [DOI] [PubMed] [Google Scholar]
- 36.Greenwald I. lin-12, a nematode homeotic gene, is homologous to a set of mammalian proteins that includes epidermal growth factor. Cell. 1985;43:583–590. doi: 10.1016/0092-8674(85)90230-2. [DOI] [PubMed] [Google Scholar]
- 37.Harrow ID, Gration KAF. Mode of action of the anthelmintics morantel, pyrantel and levamisole on muscle cell membrane of the nematode Ascaris suum. Pestic Sci. 1985;16:662–672. [Google Scholar]
- 38.Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hermans-Borgmeyer I, Zopf D, Ryseck R-P, Hovemann B, Betz H, Gundelfinger ED. Primary structure of a developmentally regulated nicotinic acetylcholine receptor protein from Drosophila. EMBO J. 1986;5:1503–1508. doi: 10.1002/j.1460-2075.1986.tb04389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Higgins DG, Bleasby AJ, Fuchs R. CLUSTALV: improved software for multiple sequence alignment. Comput Appl Biosci. 1992;8:189–191. doi: 10.1093/bioinformatics/8.2.189. [DOI] [PubMed] [Google Scholar]
- 41.Huelsenbeck JP, Hillis DM. Success of phylogenetic methods in the four-taxon case. J Systematic Biol. 1993;42:247–264. [Google Scholar]
- 42.Huganir RL. Regulation of the nicotinic acetylcholine receptor channel by protein phosphorylation. Curr Top Membr Transp. 1988;33:147–163. [Google Scholar]
- 43.Huganir RL, Greengard P. cAMP-dependent protein kinase phosphorylates the nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 1983;80:1130–1134. doi: 10.1073/pnas.80.4.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huganir RL, Miles K, Greengard P. Phosphorylation of the nicotinic acetylcholine receptor by an endogenous tyrosine-specific protein kinase. Proc Natl Acad Sci USA. 1984;81:6968–6972. doi: 10.1073/pnas.81.22.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imoto K, Busch C, Sakmann B, Mishina M, Konno T, Nakai J, Bujo H, Mori Y, Fukuda K, Numa S. Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature. 1988;335:645–648. doi: 10.1038/335645a0. [DOI] [PubMed] [Google Scholar]
- 46.Jacobson A. Purification and fractionation of poly(A)+ RNA. Methods Enzymol. 1987;152:254–261. doi: 10.1016/0076-6879(87)52028-6. [DOI] [PubMed] [Google Scholar]
- 47.Jin Y, Hoskins R, Horvitz HR. Control of type-D GABAergic neuron differentiation by C. elegans UNC-30 homeodomain protein. Nature. 1994;372:780–783. doi: 10.1038/372780a0. [DOI] [PubMed] [Google Scholar]
- 48.Johnson CD, Stretton AOW. Neural control of locomotion in Ascaris: anatomy, electrophysiology, and biochemistry. In: Zuckerman BM, editor. Nematodes as biological models, Vol 1, Behavioral and developmental models. Academic; New York: 1980. pp. 159–196. [Google Scholar]
- 49.Kao PN, Karlin A. Acetylcholine receptor binding site contains a disulfide crosslink between adjacent half-cystinyl residues. J Biol Chem. 1986;261:8085–8088. [PubMed] [Google Scholar]
- 50.Karlin A. Structure of nicotinic acetylcholine receptors. Curr Opin Neurobiol. 1993;3:299–309. doi: 10.1016/0959-4388(93)90121-e. [DOI] [PubMed] [Google Scholar]
- 51.Karlin A, Akabas MH. Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron. 1995;15:1231–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- 52.Konno T, Busch C, Von Kitzing E, Imoto K, Wang F, Nakai J, Mishina M, Numa S, Sakmann B. Rings of anionic amino acids as structural determinants of ion selectivity in the acetylcholine receptor channel. Proc R Soc Lond [Biol] 1991;244:69–79. doi: 10.1098/rspb.1991.0053. [DOI] [PubMed] [Google Scholar]
- 53.Krause M, Hirsh D. A trans-spliced leader sequence on actin mRNA in C. elegans. Cell. 1987;49:753–761. doi: 10.1016/0092-8674(87)90613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lewis JA, Wu C-H, Berg H, Levine JH. The genetics of levamisole resistance in the nematode Caenorhabditis elegans. Genetics. 1980a;95:905–928. doi: 10.1093/genetics/95.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lewis JA, Wu C-H, Levine JH, Berg H. Levamisole-resistant mutants of the nematode Caenorhabditis elegans appear to lack pharmacological acetylcholine receptors. Neuroscience. 1980b;5:967–989. doi: 10.1016/0306-4522(80)90180-3. [DOI] [PubMed] [Google Scholar]
- 56.Lewis JA, Fleming JT, McLafferty S, Murphy H, Wu C. The levamisole receptor, a cholinergic receptor of the nematode Caenorhabditis elegans. Mol Pharmacol. 1987a;31:185–193. [PubMed] [Google Scholar]
- 57.Lewis JA, Elmer JS, Skimming J, McLafferty S, Fleming J, McGee T. Cholinergic receptor mutants of the nematode Caenorhabditis elegans. J Neurosci. 1987b;7:3059–3071. doi: 10.1523/JNEUROSCI.07-10-03059.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marshall J, Buckingham SD, Shingai R, Lunt GG, Goosey MW, Darlison MG, Sattelle DB, Barnard EA. Sequence and functional expression of a single α subunit of an insect nicotinic acetylcholine receptor. EMBO J. 1990;9:4391–4398. doi: 10.1002/j.1460-2075.1990.tb07889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin RJ, Pennington AJ, Duittoz AH, Robertson S, Kusel JR. The physiology and pharmacology of neuromuscular transmission in the nematode parasite Ascaris suum. Parasitology. 1991;102:S41–S58. doi: 10.1017/s0031182000073285. [DOI] [PubMed] [Google Scholar]
- 60.McCombie WR, Kirkness E, Fleming JT, Kerlavage AR, Iovannisci DM, Martin-Gallardo A. The use of exonuclease III deletions in automated DNA sequencing. Methods Companion Methods Enzymol. 1991;3:33–40. [Google Scholar]
- 61.Mello C, Fire A. DNA transformation. In: Epstein HF, Shakes DC, editors. Methods in cell biology, Vol 48, Caenorhabditis elegans: modern biological analysis of an organism. Academic; New York: 1995. pp. 451–482. [PubMed] [Google Scholar]
- 62.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci USA. 1996a;93:12593–12598. doi: 10.1073/pnas.93.22.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller LM, Waring DA, Kim S. Mosaic analysis using a ncl-1 (+) extrachromosomal array reveals that lin-31 acts in the Pn.p cells during Caenorhabditis elegans vulval development. Genetics. 1996b;143:1181–1191. doi: 10.1093/genetics/143.3.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moerman DG, Waterston RH. Spontaneous unstable unc-22 IV mutations in C. elegans var. Bergerac. Genetics. 1984;108:859–877. doi: 10.1093/genetics/108.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moerman DG, Benian GM, Waterston RH. Molecular cloning of the muscle gene unc-22 in Caenorhabditis elegans by Tc1 transposon tagging. Proc Natl Acad Sci USA. 1986;83:2579–2583. doi: 10.1073/pnas.83.8.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moerman DG, Benian GM, Barstead RJ, Schriefer LA, Waterston RH. Identification and intracellular localization of the unc-22 gene product of Caenorhabditis elegans. Genes Dev. 1988;2:93–105. doi: 10.1101/gad.2.1.93. [DOI] [PubMed] [Google Scholar]
- 68.Nef P, Mauron A, Stalder R, Alliod C, Ballivet M. Structure, linkage, and sequence of the two genes encoding the δ and γ subunits of the nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 1984;81:7975–7979. doi: 10.1073/pnas.81.24.7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nef P, Oneyser C, Alliod C, Couturier S, Ballivet M. Genes expressed in the brain define three distinct neuronal nicotinic acetylcholine receptors. EMBO J. 1988;7:595–601. doi: 10.1002/j.1460-2075.1988.tb02852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Neumann D, Barchan D, Horowitz M, Kochva E, Fuchs S. Snake acetylcholine receptor: cloning of the domain containing the four extracellular cysteines of the α subunit. Proc Natl Acad Sci USA. 1989;86:7255–7259. doi: 10.1073/pnas.86.18.7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- 72.Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A. Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics. 1993;135:385–404. doi: 10.1093/genetics/135.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ortells MO, Lunt GG. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci. 1995;18:121–127. doi: 10.1016/0166-2236(95)93887-4. [DOI] [PubMed] [Google Scholar]
- 74.Pedersen SE, Cohen JB. d-Tubocurarine binding sites are located at α-γ and α-δ subunit interfaces of the nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 1990;87:2785–2789. doi: 10.1073/pnas.87.7.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Revah F, Bertrand D, Galzi J-L, Devillers-Thiery A, Hussy N, Mulle C, Bertrand S, Ballivet M, Changeux J-P. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 76.Robertson S, Martin RJ. Levamisole-activated single-channel currents from muscle of the nematode parasite Ascaris suum. Br J Pharmacol. 1993;108:170–178. doi: 10.1111/j.1476-5381.1993.tb13458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 78.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual, Ed 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 79.Sargent PB. The diversity of neural nicotinic acetylcholine receptors. Annu Rev Neurosci. 1993;16:403–443. doi: 10.1146/annurev.ne.16.030193.002155. [DOI] [PubMed] [Google Scholar]
- 80.Schuler GD, Altschul SF, Lipman DJ. A workbench for multiple alignment construction and analysis. Proteins. 1991;9:180–190. doi: 10.1002/prot.340090304. [DOI] [PubMed] [Google Scholar]
- 81.Squire MD, Tornoe C, Baylis HA, Fleming JT, Barnard EA, Sattelle DB. Molecular cloning and functional co-expression of a Caenorhabditis elegans nicotinic acetylcholine receptor subunit (acr-2). Receptors Channels. 1995;3:107–115. [PubMed] [Google Scholar]
- 82.Swofford DL. PAUP: phylogenetic analysis using parsimony, version 3.0 sec +3. Illinois Natural History Survey; Champaign, IL: 1991. [Google Scholar]
- 83.Swofford DL, Olsen GJ. Phylogeny reconstruction. In: Hillis DM, Moritz C, editors. Molecular systematics. Sinauer; Sunderland, MA: 1990. pp. 411–501. [Google Scholar]
- 84.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tornoe C, Bai D, Holden-Dye L, Abramson SN, Sattelle DB. Actions of neurotoxins (bungarotoxins, neosurugatoxin, and lophotoxins) on insect and nematode nicotinic acetylcholine receptors. Toxicon. 1995;33:411–424. doi: 10.1016/0041-0101(94)00163-3. [DOI] [PubMed] [Google Scholar]
- 86.Treinin M, Chalfie M. A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron. 1995;14:871–877. doi: 10.1016/0896-6273(95)90231-7. [DOI] [PubMed] [Google Scholar]
- 87.Unwin N. The structure of ion channels in membranes of excitable cells. Neuron. 1989;3:665–676. doi: 10.1016/0896-6273(89)90235-3. [DOI] [PubMed] [Google Scholar]
- 88.Unwin N. Neurotransmitter Action: opening of ligand-gated ion channels. Cell. 1993a;72:31–41. doi: 10.1016/s0092-8674(05)80026-1. [DOI] [PubMed] [Google Scholar]
- 89.Unwin N. Nicotinic acetylcholine receptor at 9Å resolution. J Mol Biol. 1993b;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 90.Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 91.Wiley LJ, Weiss AS, Sangster NC, Qun Li. Cloning and sequence analysis of the candidate nicotinic acetylcholine receptor alpha subunit gene tar1 from Trichostrongylus colubriformis. Gene. 1996;182:97–100. doi: 10.1016/s0378-1119(96)00520-3. [DOI] [PubMed] [Google Scholar]
- 92.Williams BD, Waterston RH. Genes critical for muscle development and function in Caenorhabditis elegans identified through lethal mutations. J Cell Biol. 1994;124:475–490. doi: 10.1083/jcb.124.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Williams BD, Schrank B, Huynh C, Shownkeen R, Waterston RH. A genetic mapping system in Caenorhabditis elegans based on polymorphic sequence-tagged sites. Genetics. 1992;131:609–624. doi: 10.1093/genetics/131.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1B-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 95.Zorio DAR, Cheng NSN, Blumenthal TE, Spieth J. Operons as a common form of chromosomal organization in C. elegans. Nature. 1994;372:270–272. doi: 10.1038/372270a0. [DOI] [PubMed] [Google Scholar]