

Abstract
We describe a novel genetic and kinetic defect in a slow-channel congenital myasthenic syndrome. The severely disabled propositus has advanced endplate myopathy, prolonged and biexponentially decaying endplate currents, and prolonged acetylcholine receptor (AChR) channel openings. Genetic analysis reveals the heterozygous mutation αV249F in the propositus and mosaicism for αV249F in the asymptomatic father. Unlike mutations described previously in the M2 transmembrane domain, αV249F is located N-terminal to the conserved leucines and is not predicted to face the channel lumen. Expression of the αV249F AChR in HEK fibroblasts demonstrates increased channel openings in the absence of ACh, prolonged openings in its presence, enhanced steady-state desensitization, and nanomolar rather than micromolar affinity of one of the two binding sites in the resting activatable state. Thus, neuromuscular transmission is compromised because cationic overloading leads to degenerating junctional folds and loss of AChR, because an increased fraction of AChR is desensitized in the resting state, and because physiological rates of stimulation elicit additional desensitization and depolarization block of transmission.
Keywords: slow-channel congenital myasthenic syndrome, neuromuscular transmission, endplate myopathy, acetylcholine receptor α subunit gene, mutation analysis, single-channel recording, desensitization, agonist binding affinity
Recent studies of congenital myasthenic syndromes (CMS) revealed mutations in acetylcholine receptor (AChR) subunit genes that either reduce expression of AChR (Engel et al., 1996a) or alter its kinetic properties to decrease (Ohno et al., 1996) or increase (Ohno et al., 1995; Sine et al., 1995; Engel et al., 1996b; Gomez et al., 1996) the response to ACh. Mutations that increase response to ACh prolong elementary activation episodes, and these disorders are referred to as slow-channel CMS (SCCMS). Mutations underlying SCCMS have been identified in different AChR subunits, and in different domains of the subunits, and affect AChR function through different mechanisms. The mutation αG153S is in the major extracellular domain and near residues that contribute to ligand binding; it increases affinity of ACh for the resting closed state and prolongs activation episodes by allowing multiple reopenings before ACh can dissociate (Sine et al., 1995). The mutation αN217K is in the M1 transmembrane domain and both slows the rate of channel closing and allows multiple reopenings per activation episode (Engel et al., 1996b). Mutations in the M2 domain, εT264P, εL269F, and βV266M (Ohno et al., 1995;Engel et al., 1996b), increase spontaneous opening of the channel, increase apparent affinity for ACh, and slow the rate of channel closing.
Here we describe and functionally characterize the mutation αV249F in the M2 domain that causes severe SCCMS through a novel combination of mechanisms. Our mechanistic studies reveal functional consequences attributable to local perturbation of the M2 domain, including increased opening in the absence and prolonged opening in the presence of ACh, and enhanced steady-state desensitization. Moreover, the perturbation in M2 spreads to the binding site to increase affinity of the resting closed state from the micromolar to the nanomolar range. High affinity of the resting state predicts occupancy of sufficient duration to allow desensitization at physiological rates of stimulation. By enhancing both activation and desensitization, αV249F severely compromises the safety margin of neuromuscular transmission.
MATERIALS AND METHODS
Muscle specimens
Intercostal muscle specimens were obtained intact from origin to insertion from the patient and from control subjects without muscle disease undergoing thoracic surgery. All human studies were in accord with the guidelines of the Institutional Review Board of the Mayo Clinic.
Endplate studies
Morphology and counts of AChR per endplate. For electron microscopy, endplates (EPs) were localized and analyzed by established methods (Engel, 1994a). Peroxidase-labeled α-bungarotoxin (α-bgt) was used for the ultrastructural localization of AChR (Engel et al., 1977b). The number of AChRs per EP was measured with125I-labeled α-bgt, as described previously (Engel et al., 1993).
Intracellular microelectrode studies. Miniature EP potential (MEPP), miniature EP current (MEPC) and EP potential (EPP) recordings, estimates of the number of transmitter quanta released by nerve impulse, and analysis of the ACh-induced current noise were performed as described previously (Engel et al., 1993; Uchitel et al., 1993).
Patch-clamp recordings from EP AChRs. These were performed in the cell-attached mode by a slight modification of the methods described previously (Milone et al., 1994; Ohno et al., 1995). For all patches, the membrane potential was set to −80 mV; when possible, recordings were also obtained at −40, −120, and −160 mV. These membrane potentials were achieved by applying a corresponding potential to the patch pipette and assuming a resting potential of 0 mV. A null resting potential was confirmed by the absence of detectable channel events with 0 mV applied to the pipette. Channel currents were recorded using an Axopatch 200A amplifier (Axon Instruments, Foster City, CA) and analyzed at a final bandwidth of 5.8 kHz using the program pClamp 6 (Axon Instruments). Burst durations were determined by grouping openings separated by a specified closed time that misclassifies equal proportions of long closed times within bursts and short intervals between bursts (Colquhoun and Sakmann, 1985). Dwell time histograms were plotted on logarithmic abscissa and fitted to the sum of exponentials by maximum likelihood (Sigworth and Sine, 1987).
Mutation analysis
Single-strand conformation polymorphism (SSCP) and sequencing. mRNA, first-strand cDNA, and genomic DNA were obtained as described previously (Ohno et al., 1996). PCR primers were designed to amplify exons with their flanking regions from each AChR subunit as previously described (Ohno et al., 1995). Published cDNA sequences of the human α (Noda et al., 1983), β (Beeson et al., 1989), δ (Luther et al., 1989), and ε (Beeson et al., 1993) subunits were used to design cDNA primers. The “cold” SSCP procedure was used as described previously (Ohno et al., 1996). PCR-amplified fragments of genomic DNA or cDNA were purified by Wizard PCR Preps (Promega, Madison, WI). Plasmids were purified by QIAwell 8 Plus Plasmid kit (Qiagen, Santa Clarita, CA). DNA fragments and plasmids were sequenced with an Applied Biosystems model 377 DNA sequencer using fluorescently labeled dideoxy terminators.
Allele-specific PCR. We used allele-specific PCR to search for the αV249F mutation in the patient’s relatives and normal controls. The respective wild-type and mutant sense primers were 5′-AAGATGACTCTGAGCATCTgTG-3′ and 5′-AAGATGACTCTGAGCATCTgTT-3′. Mismatched nucleotide “g” was deliberately introduced two nucleotides upstream of the 3′ end of the primer to avoid misannealing of the primer to the opposite allele. The antisense primer was 5′-GTTGATGACGATGACAGTGA-3′.
Mutagenic PCR plus restriction analysis. We also synthesized a mutagenic PCR primer to check for mosaicism for the αV249F mutation in the patient’s family. A 157 bp fragment of genomic DNA spanning the αV249F mutation was amplified with primers 5′-AGGACTCAGGACTTCCACATA-3′ in α intron 6 and 5′-AGAAGGAACACAGTCAAAGACAGTgAGA-3′ in α exon 7. The mismatched nucleotide “g,” which introduces a mutation four nucleotides downstream from the αG745T (αV249F) mutant site, enablesBsmAI to differentiate among PCR products with or without the αV249F: BsmAI yields 134 and 23 bp fragments from the PCR product lacking αV249F mutation, and a 157 bp fragment from the PCR product harboring αV249F. The PCR product was digested withBsmAI (New England Biolabs, Beverly, MA).
Cloning of PCR products to detect mosaicism. A 157 bp fragment spanning the αV249F mutation was amplified from genomic DNA from the patient and his father and was ligated into a vector using the TA cloning kit (Invitrogen, Carlsbad, CA). White colonies were picked up from Luria-Bertani plates, boiled for 10 min in 50 μl of distilled water, and immediately cooled on ice. Five microliters of boiled sample was mixed with three PCR primers, 5′-GTAAAACGACGGCCAGT-3′ (forward generic primer for M13), 5′-GGAAACAGCTATGACCATG-3′ (reverse generic primer for M13), and 5′-AAGATGACTCTGAGCATCTgTT-3′ (mutant-allele-specific primer, as shown in the section for allele-specific PCR) in 25 μl of reaction mixture. All clones with inserts yield a 355 bp fragment, clones with no inserts produce a 198 bp fragment, and clones harboring αV249F additionally produce a 166 or 137 bp fragment, depending on the direction of the insert. Thus, we could characterize the inserts by size fractionation of the PCR products. Presence of αV249F in the 161 and 137 bp fragments was verified by BsmAI digestion, as described above.
Expression studies
Construction of human wild-type and mutant AChR cDNAs and expression in 293 HEK cells. Human α and δ subunit cDNAs were generously provided by Dr. Jon Lindstrom (Schoepfer et al., 1988;Luther et al., 1989). β and ε subunit cDNAs were cloned from normal human skeletal muscle as described previously (Ohno et al., 1996). All four cDNAs were subcloned into the CMV-based expression vector pRBG4 (Lee et al., 1991) for expression in 293 HEK cells.
The αV249F mutation was constructed using QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer’s recommendations. The presence of the desired mutation and the absence of unwanted mutations were confirmed by sequencing the entire insert.
HEK cells were transfected with mutant or wild-type AChR subunit cDNAs using calcium phosphate precipitation as described (Bouzat et al., 1994). For patch recordings, plasmid-encoding green fluorescent protein (Life Technologies, Grand Island, NY) was included for identification of transfected cells.
Patch-clamp recordings from AChRs expressed in HEK cells.Recordings were obtained in the cell-attached configuration, with a membrane potential of −70 mV, a temperature of 22°C, and with bath and pipette solutions containing (in mm): KCl 142, NaCl 5.4, CaCl2 1.8, MgCl2 1.7, HEPES 10, pH 7.4 (Bouzat et al., 1994; Ohno et al., 1996). Single-channel currents were recorded using an Axopatch 200A at a bandwidth of 50 kHz, digitized at 100 kHz, transferred to a Macintosh computer using the program Acquire (Instrutech, Great Neck, NY), and detected by the half-amplitude threshold criterion using the program MacTac (Instrutech) at a final bandwidth of 10 kHz. For αV249F receptors, infrequent subconductance states before or after an opening were excluded from the measured open durations, whereas those interrupting an opening were included. Open- and closed-duration histograms were constructed using a logarithmic abscissa and square root ordinate (Sigworth and Sine, 1987) and fitted to the sum of exponentials by maximum likelihood. The resulting time constants, relative areas, and total numbers of events were used to determine opening, closing, and agonist dissociation rate constants as described (Sine and Steinbach, 1986). For openings interrupted by closings approaching the instrumentation dead time (18 μsec), we determined the true mean open duration from the total open duration in the recording (apparent mean open duration multiplied by the total number of openings) divided by the total number of brief closings (determined from the area of the brief component of closings) as described (Sine et al., 1990).
To confirm our analysis method, we simulated single-channel events according to Scheme FS1 (see text) using the parameters obtained from the experimental data. We simulated open and closed intervals as described by Clay and deFelice (1983) and constructed bursts using our experimental closed time criterion (1 msec). After compiling the bursts into histograms, we fitted the histograms by the sum of exponentials, as described above for the experimental data. We computed the probability of openings in the three types of burst events (P1, P2, P3) by multiplying the relative area by the corresponding mean open duration.
Fig. FS1.

α-Bgt and ACh binding measurements. The total number of125I-α-bgt sites and ACh competition against the initial rate of 125I-α-bgt binding were determined as described previously (Ohno et al., 1996). ACh competition measurements were analyzed according to either the monophasic Hill equation (Eq. 1) or the sum of two distinct binding sites (Eq. 2): 1 − Y = 1/(1 + ([ACh]/Kov)n), (1)1 − Y = fractA(1/(1 + [ACh]/KA)) + (1 − fractA)(1/(1 + [ACh]/KB)), (2)
where Y is fractional occupancy by ACh,n is the Hill coefficient, Kov is an overall dissociation constant for a monophasic binding profile,KA and KB are intrinsic dissociation constants for two binding sites, andfractA is the fraction of sites with dissociation constant KA.
RESULTS
Clinical data
The patient, a 12-year-old male, had myasthenic symptoms involving the ocular, bulbar, trunkal, and limb muscles since early infancy. Between the ages of 3 and 8 years, he could walk only short distance before having to rest and subsequently he became wheelchair-dependent. He had negative tests for anti-AChR antibodies, a decremental electromyographic response on stimulation of motor nerves, and a repetitive action potential response to single nerve stimuli, as seen in the SCCMS (Engel et al., 1982) or in EP acetylcholinesterase deficiency (Engel et al., 1977a).
Morphological observations and counts of AChR per EP
Acetylcholinesterase activity was preserved at all EPs. Ultrastructural studies revealed an EP myopathy with honeycomb networks in junctional folds (Fig. 1A), degeneration of junctional folds with widening of the synaptic space (Fig. 1B) and loss of AChR (Fig.1D), degenerating organelles in the junctional sarcoplasm (Fig. 1C), and apoptosis of some junctional nuclei (Fig. 1E). The number of125I-α-bgt binding sites/EP was 5.02 × 106, or 39% of control.
Fig. 1.
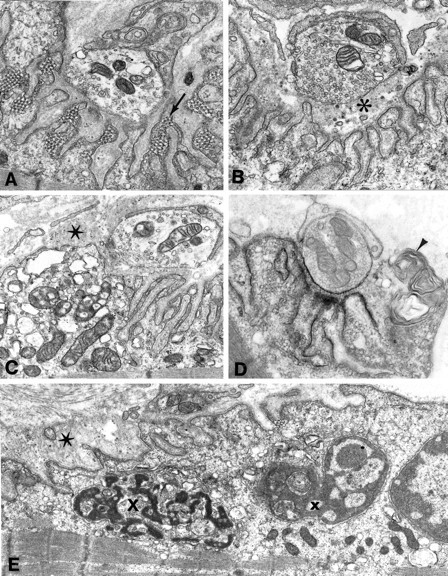
EP fine structure. A, Many junctional folds are honeycombed by membranous networks. This is a common ultrastructural reaction of the EP in states of cholinergic overactivity. Magnification, 18,100×. B, Degeneration of the junctional folds leaves globular debris in the widened synaptic space (asterisk). Magnification, 21,500×.C, The junctional sarcoplasm at the leftis filled with degenerating organelles; the starindicates remnants of degenerated junctional folds. Magnification, 17,500×. D, Localization of EP AChR with peroxidase labeled α-bungarotoxin. Note loss of AChR from degenerating junctional folds (arrowhead). Magnification, 23,800×.E, The junctional sarcoplasm harbors nuclei in early (x) and advanced (X) stages of apoptosis. The star indicates remnants of degenerated junctional folds. Magnification, 14,800×.
Intracellular microelectrode studies
Quantal release by nerve impulse was normal. MEPPs and MEPCs had a reduced amplitude, consistent with EP AChR deficiency. The MEPCs decayed biexponentially, suggesting two populations of channel openings, with one τMEPC close to normal and one markedly prolonged (Fig. 2, top panels, and Table1). Spectral analysis of the ACh-induced current noise indicated two channel open times, with one τnoise normal and one markedly prolonged (Table 1). These findings pointed to a kinetic abnormality of at least some of the EP AChRs.
Fig. 2.

MEPCs and channel events recorded from control and patient EPs. Top traces, MEPCs; bottom traces, channel events; left, control EP;right, patient EP. Note markedly prolonged MEPC and some highly prolonged channel events in the SCCMS. The MEPC decay is best fitted by two exponentials. The vertical arrows indicate decay time constants. MEPCs filtered at 500 Hz and channel currents at 5.8 kHz; −80 mV; temperature, 22°C.
Table 1.
Conventional microelectrode recordings and noise analysis
| Patient (No. of EPs) | Controls (No. of EPs) | |
|---|---|---|
| EPP quantal content (1 Hz)1-a | 36 ± 2.0 | 31 ± 1 |
| (10) | (190) | |
| MEPP amplitude (mV)b | 0.41 | 1.00 ± 0.03 |
| (10) | (164) | |
| MEPC amplitude (nA)c | 1.72 | 3.92 ± 0.14 |
| (15) | (78) | |
| τMEPC(msec)c | (i) 2.42 ± 0.21 | 3.23 ± 0.06 |
| (ii) 43.9 ± 2.31 | (78) | |
| (15) | ||
| τnoise(msec)c | (i) 1.40 ± 0.12 | 2.30 ± 0.04 |
| (ii) 15.1 ± 0.03 | (52) | |
| (5) |
Values indicate mean ± SEM.
EPP, EP potential; MEPP, miniature EP potential; MEPC, miniature EP current.
Corrected for membrane potential of −80 mV, nonlinear summation, and non-Poisson release; 29EC.bCorrected for membrane potential of −80 mV and a fiber diameter of 50 μm; 29°C. c−80 mV, 22°C.
Patch-clamp analysis of EP AChR
To gain additional insight into the kinetic defect of AChR, we recorded single-channel currents from EPs of the patient in the presence of 1 μm ACh and compared the findings with those at control EPs. Simple inspection of the recordings from the patient EPs revealed a mixture of apparently normal and markedly prolonged channel events (Fig. 2, bottom panels). On formal analysis, at control EPs both the open intervals and the bursts had a minor, brief (τ1) and a major, longer component (τ2), as described previously (Milone et al., 1994). At the patient EPs, the channel open intervals and bursts had two components, τ1 and τ2, similar to those observed at the control EPs, and a third component, τ3, that was five- to sevenfold longer than the respective τ2 (Table 2). The above channel events had a conductance of 60–66 pS, similar or up to 10% greater than the conductance of channel events at control EPs. Approximately 12% of all channel events at the patient’s EPs had a reduced conductance of 45 pS. The open interval and burst distributions of these channels showed two components, a brief, minor component, τ1, similar to that of the 60–66 pS channels, and a second, prolonged component (τ2 = 5.88 ± 0.58 msec for open intervals, and 10.86 ± 1.93 msec for bursts). The conductance and open durations of these channels were typical of AChRs containing the γ instead of the ε subunit (γ-AChR) (Mishina et al., 1986). In addition, at two EPs the 45 pS channels had an additional longer component accounting for 15–20% of the 45 pS channel events (τ3 = 11.27 and 17.4 msec for open intervals, and 16.74 and 41.68 msec for bursts). The patch-clamp data were compatible with different species of EP AChRs: adult-type AChR composed of wild-type subunits, adult-type AChR harboring one or more mutant subunits, γ-AChR composed of wild-type subunits, and γ-AChR harboring one or more mutant subunits. To identify the structural basis of the abnormal channel kinetics, we searched for mutations in AChR subunit genes.
Table 2.
Patch-clamp analysis of EP channels
| Open intervals | Bursts | |||
|---|---|---|---|---|
| Controls | Patient | Controls | Patient | |
| τ1(msec) | 0.20 ± 0.022 | 0.20 ± 0.082 | 0.12 ± 0.012 | 0.13 ± 0.06 |
| Area | 0.16 ± 0.015 | 0.12 ± 0.045 | 0.16 ± 0.014 | 0.17 ± 0.027 |
| Number of EPs | 34 | 5 | 34 | 5 |
| τ2 (msec) | 1.90 ± 0.085 | 2.07 ± 0.22 | 3.04 ± 0.17 | 2.95 ± 0.25 |
| Area | 0.85 ± 0.016 | 0.73 ± 0.066 | 0.85 ± 0.014 | 0.75 ± 0.064 |
| Number of EPs | 34 | 6 | 34 | 6 |
| τ3 (msec) | ND | 10.11 ± 2.86 | ND | 19.71 ± 5.89 |
| Area | ND | 0.33 ± 0.15 | 0.28 ± 0.15 | |
| Number of EPs | 6 | 6 | ||
Values indicate mean ± SEM. Recordings were obtained from 7 SCCMS EPs; the τ1 component of open intervals and bursts was absent at EPs 4 and 7, the τ2 component at EP 4, and the τ3 component at EP 7. ND, not detected. Recordings were done in the presence of 1 μm ACh. Bandwidth = 5.8 kHz; potential = −80 mV; T = 22 ± 0.5°C.
Mutation analysis
SSCP analysis of PCR-amplified fragments of genomic DNA encoding the α, β, δ, and ε subunits revealed seven aberrant conformers, all commonly observed in controls (Table 3), but no mutations. Sequencing of all exons and flanking intronic regions of genomic DNA for the α, β, δ, and ε subunit genes as well as overlapping cDNA fragments of the entire coding regions for the four AChR subunit genes revealed one additional polymorphism (Table 3) and a heterozygous G→T transversion in α exon 7 at nucleotide 745 (αG745T) that converted a valine to a phenylalanine codon at position 249 (αV249F) (Fig. 3A). The altered valine is located in the M2 transmembrane domain that lines the channel pore and is conserved across the α subunit of all species and the human γ, δ, and ε subunits (Fig. 3C).
Table 3.
Identified polymorphisms in AChR subunit genes and their allelic frequencies in humans
| Polymorphism | Allelic frequency |
|---|---|
| αG − 18 + 59T | 11 /28 |
| α130 − 13insT | 29 /70 |
| βA26G [βE9G] | 40 /124 |
| δA − 52G | 32 /64 |
| δA1543G | 10 /38 |
| ɛC − 60 − 70T | 7 /98 |
| ɛC285 − 7T | 5 /98 |
| ɛA1245G3-150 | 6 /108 |
Nucleotides and codons are numbered from the beginning of the mature peptide. Negative nucleotide numbers are in the signal peptide region. Plus (+) or minus (−) symbol after nucleotide numbers indicates position in an intron relative to the nearest last or first base of an exon. Codon changes, if any, are shown in brackets. All polymorphisms are heterozygous.
F3-150: Not detected by SSCP. Nomenclature for designating polymorphisms is according to Beaudet and Tsui (1993).
Fig. 3.
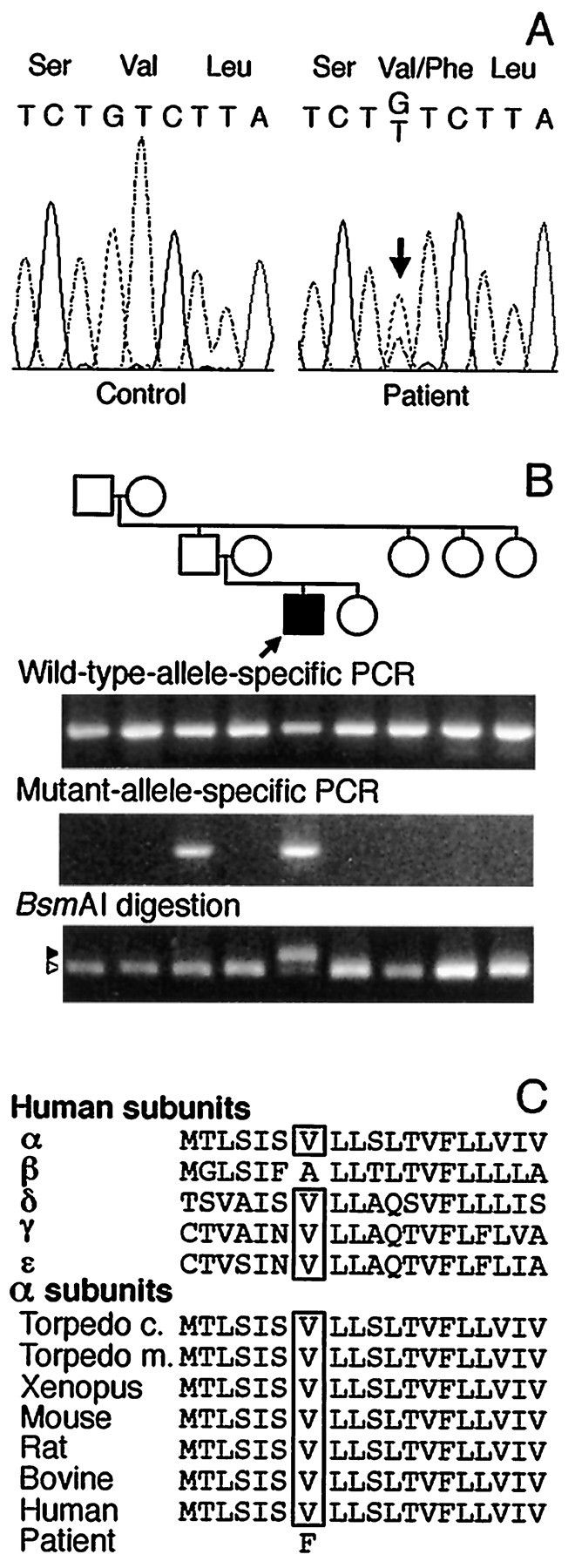
Identification and analysis of mutation in the AChR α subunit. A, Automated sequencing of α exon 7 around codon 249 in a control and the propositus. In the propositus, both G and T nucleotides are present at position 745 (arrow), indicating a heterozygous G→T transversion. This mutation changes codon 249 from a GTC for valine to a TTC for phenylalanine. B, Allele-specific PCR and mutagenic PCR plus BsmAI analysis of genomic DNA in the propositus’ family. Both wild-type and mutant-allele-specific primers amplify an expected 168 bp fragment in propositus and his father, but only the wild-type primer amplifies the expected fragment in the other family members. On BsmAI analysis after mutagenic PCR, the wild-type allele gives rise to a 134 bp fragment (open arrowhead) and a 23 bp fragment (not shown); the mutant allele yields an undigested 157 bp fragment (closed arrowhead). Both wild-type and mutant fragments appear in the propositus, but only the wild-type fragment appears in other family members. The incongruity between allele-specific PCR and restriction analysis in the father is attributable to the father being a mosaic for αV249F (see text). Thearrow indicates propositus. C, Multiple alignment of AChR M2 membrane-spanning domains. Theboxes enclose the conserved valine residues in human AChR subunits and in AChR α subunits of other species. The mutant phenylalanine is shown at the bottom.
Using genomic DNA, we searched for αV249F by allele-specific PCR in 100 normal controls, 55 other unrelated CMS patients, and in the propositus’ relatives, and detected it only in the patient’s asymptomatic father (Fig. 3B). Direct sequencing, however, did not show αV249F in the father’s genomic DNA. We then searched for αV249F in the father’s genomic DNA by mutagenic PCR and restriction analysis using BsmAI that digested only the wild-type sequence. This revealed αV249F only in the patient but not in his father or other relatives (Fig. 3B). Repeated experiments using newly drawn blood from the father gave the same results, precluding contamination of father’s DNA by that of the propositus. Detection of αV249F in the father by allele-specific PCR, but not by direct sequencing or restriction analysis, suggested paternal mosaicism for αV249F. To prove this, we cloned the PCR-amplified segment of DNA spanning αV249F from the propositus and his father. Allele-specific PCR of 20 clones from the propositus revealed that 10 harbored αV249F and 10 did not, consistent with heterozygosity. By contrast, analysis of 106 clones from the father revealed 2 clones with αV249F and 104 clones without it (Fig.4), confirming mosaicism in the father. Chimerism in the father was excluded by failure of allele-specific PCR to detect αV249F in the paternal grandparents (Fig. 3B).
Fig. 4.

Allele-specific PCR reveals mosaicism for αV247F in paternal DNA. Only 2 of 106 clones of paternal genomic DNA harbored αV247F. The figure demonstrates αV247F, represented by the 166 bp fragment, in clone number 90.
Expression studies
To evaluate pathogenicity of αV249F, we transfected 293 HEK cells with either wild-type or mutant human α plus complementary β, ε, and δ subunit cDNAs. Measurements of 125I-α-bgt binding revealed 55 ± 10% (mean ± SD for 10 determinations) surface expression of αV249F relative to wild-type AChR.
Activation of the αV249F receptor by ACh
To investigate the mechanistic consequences of αV249F, we recorded ACh-activated, single-channel currents from HEK cells expressing the αV249F AChR. We initially attempted to record currents in the presence of desensitizing concentrations of ACh (10–100 μm), because this allows identification of clusters of events attributable to a single channel; analysis of open and closed intervals within clusters would have allowed determination of rate constants for each step in a scheme for receptor activation, as described previously (Sine et al., 1990; Ohno et al., 1996). However, αV249F receptors showed no channel activity at ACh concentrations >1 μm, presumably because of enhanced desensitization. Decreasing the ACh concentration into the nanomolar range resulted in vigorous activation of αV249F receptors and the appearance of markedly prolonged channel openings (Fig. 5, left panels).
Fig. 5.
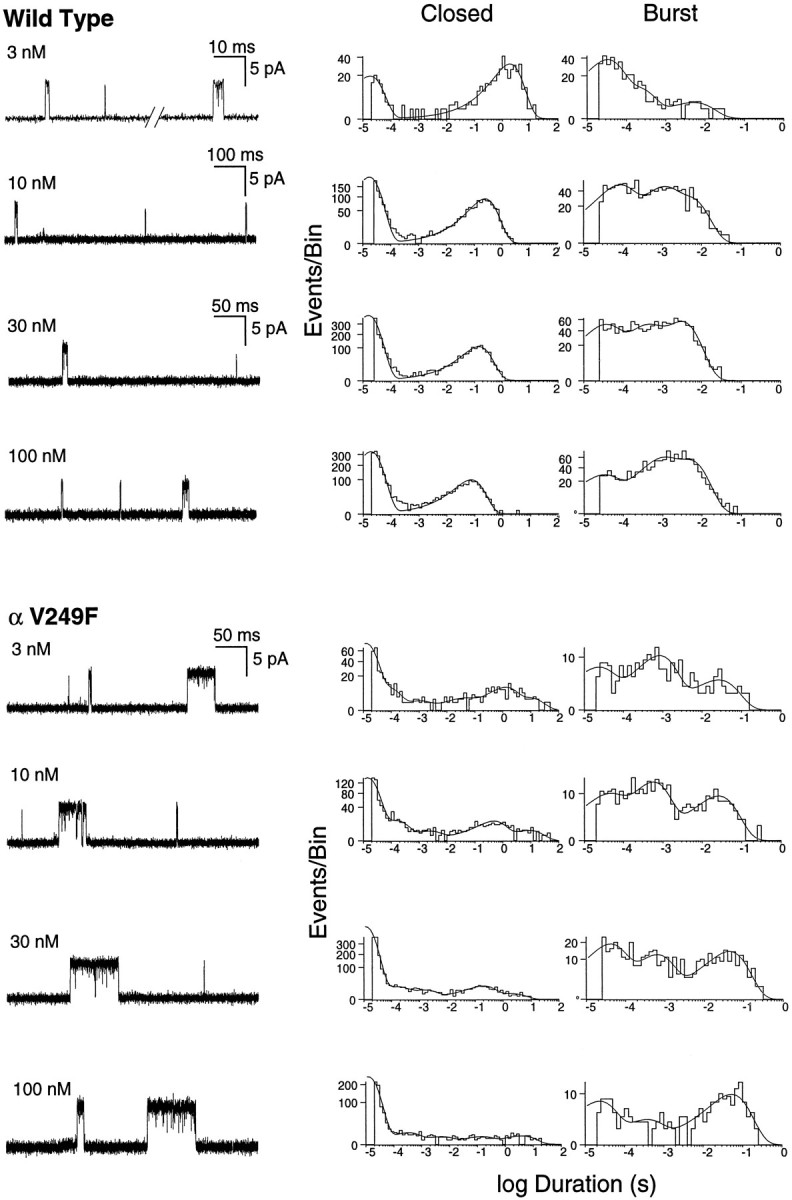
Activation of human wild-type and αV249F AChRs by nanomolar concentrations of ACh. The left panels show single-channel currents elicited by the indicated concentrations of ACh at a bandwidth of 10 kHz for wild-type and αV249F receptors. The center and right panels show corresponding closed and burst-duration histograms fitted to the sum of exponentials. Note three components of bursts for both receptor types, the increased contribution of the long-duration component with increasing ACh concentration, and the increased duration of the long component attributable to αV249F.
Current amplitudes of αV249F receptor channels were noticeably increased. The conductance of αV249F channels, 94 ± 1.0 pS (± SEM, 3 patches), was significantly greater than that for wild type, 84 ± 0.9 pS (4 patches); p < 0.001. αV249F receptors also infrequently showed subconductance states of half the full amplitude and lasting several milliseconds; these preceded, followed, or interrupted bursts of prolonged channel openings.
Effect of αV249F on burst durations
To investigate the molecular basis of the prolonged openings caused by αV249F, we recorded currents elicited by 3–100 nm ACh, constructed burst-duration histograms, and fitted the histograms by the sum of exponentials. For this analysis, bursts were defined as openings separated by closed intervals <200 μsec. For both wild-type and αV249F receptors, the distributions of burst durations show three exponential components (Fig. 5, right panels). The two brief components predominate at low ACh concentrations, whereas the long component predominates at the highest concentration. This dependence on ACh concentration suggests that long bursts correspond to receptors with two bound agonists, whereas the two types of brief bursts correspond to receptors with one bound agonist. Because the receptor contains two ACh binding sites, we hypothesize that one type of brief burst corresponds to occupancy of the αε site, whereas the other type corresponds to occupancy of the αδ site. Mean durations of singly occupied bursts are not significantly affected by αV249F, but doubly occupied bursts are prolonged ∼10-fold.
Kinetic analysis of the αV249F receptor
Determination of each rate constant underlying receptor activation relies on identifying clusters of open and closed intervals attributable to a single channel. However, because openings of αV249F receptors did not cluster, we used a novel analysis to define a subset of the activation parameters. First, we determined the time constants and relative areas of closed-duration components over the concentration range of 3–100 nm ACh. Second, we identified two types of burst events and determined the open- and closed-duration components associated with each type. Third, we determined rate and equilibrium constants for gating of the two types of bursts and, based on their dependence on ACh concentration, assigned one type to singly and the other type to doubly occupied receptors. Fourth, we determined the probability of occurrence of singly and doubly occupied bursts as a function of ACh concentration and combined the measured gating equilibrium constants to estimate the dissociation constant for ACh binding.
Analysis of closed durations
For wild-type receptors, closed-duration histograms are well described as the sum of two exponentials (Fig. 5, middle panels; 3–100 nm ACh). The long-duration component corresponds to periods between independent bursts of openings and the brief component to transient interruptions of single-channel bursts; thus, the brief component corresponds to a closed state associated with activation. By contrast, for αV249F receptors, closed-duration histograms are well described as the sum of five or six exponentials (Fig. 5, middle panels; 3–100 nm ACh). One or more of the longer components are attributed to dwells in desensitized states (Sakmann et al., 1980; Sine and Steinbach, 1987), because of profound desensitization even at nanomolar ACh concentrations, whereas one or more of the brief components are candidates for a closed state associated with activation.
Temporal association of openings and closings
To determine activation rate constants from the open- and closed-duration histograms, we first determined which component of openings is associated with which component of closings. We searched recordings for sequences of two or more closely spaced channel openings, defined as openings separated by closed intervals <1 msec; this duration was chosen to include closings belonging to the two briefest components for the αV249F receptor and could be increased to 10 msec with similar results. We then looked for bursts with different properties by examining the distribution of the mean duration of openings within bursts of closely spaced events. αV249F receptors exhibited two readily distinguishable distributions of bursts, with mean open durations of 0.49 ± 0.2 msec and 6.0 ± 1.1 msec. Wild-type receptors, by contrast, exhibited a single distribution of bursts with a mean open duration of 1.2 ± 0.3 msec. To illustrate the kinetic properties of each type of burst for αV249F, we eliminated all solitary openings from the table of detected events, eliminated one or the other type of burst based on open duration, joined the flanking long closed intervals, and constructed histograms of the resulting open and closed intervals. We used a similar procedure to select bursts for wild-type receptors, except that only solitary events were eliminated from the event table.
The histograms of selected bursts confirm that wild-type receptors give rise to only one type of coupled event, long openings interrupted by brief closings; histograms of these events reveal only one brief closed and one long burst component (Fig. 6, top panel). αV249F receptors, by contrast, give rise to two types of coupled events. The first type consists of long openings interrupted by brief closings, corresponding to a predominant brief closed and one long burst component (Fig. 6, middle panel). The second type consists of intermediate openings interrupted by intermediate closings, corresponding to one intermediate closed and one intermediate burst component (Fig. 6, bottom panel). Thus, for wild-type receptors, we assign the single type of coupled event, long openings separated by brief closings, to doubly occupied bursts because their relative frequency increases with increasing ACh concentrations. Similarly, for αV249F receptors, we assign one type of coupled event, long openings separated by brief closings, to doubly occupied bursts because their relative frequency increases with increasing ACh concentrations. We assign the other type of coupled event for αV249F, intermediate openings separated by intermediate closings, to singly occupied bursts because their relative frequency decreases with increasing ACh concentrations.
Fig. 6.

Temporal association of openings and closings elicited by wild-type and αV249F receptors. For each recording, the table of detected events was searched for consecutive openings separated by closed intervals <1 msec, and these coupled events were preserved. The remaining solitary openings were eliminated, and the flanking long closed intervals joined and retained in the table.Top panels show analysis of the only type of coupled event observed for wild-type receptors, long openings interrupted by brief closings. The center and bottom panels show analysis of the two types of coupled events observed for αV249F receptors; bursts corresponding to the first type were recognized as those with mean open durations >1 msec, whereas those of the second type had mean open durations <1 msec. To analyze each type of burst alone, the other type of burst was eliminated from the event table based on mean open duration, and the flanking closed intervals joined and retained in the table. The left panels show traces of each type of coupled event filtered at 10 kHz. Thecenter and right panels show corresponding histograms of the coupled events fitted by the sum of exponentials. Note that each type of coupled event shows one component of bursts and a major component of brief closings.
Estimation of rate and equilibrium constants for the αV249F receptor
We used the following kinetic scheme to describe activation of wild-type and αV249F receptors: where two agonists A bind to the receptor R with association ratesk+1 through k+4 and dissociate from the receptor with rates k−1through k−4. The two types of singly occupied receptors, AR and AR′, open with rates β1 and β′1, whereas open receptors AR* and AR′* close with rates α1 and α′1. Fully occupied receptors A2R open with rate β2, and open receptors A2R* close with rate α2. Scheme FS1 allows ACh to bind independently to the two sites and receptors with one or two bound agonists to open; it is a general form of the standard sequential scheme that has successfully described the kinetics of activation of Torpedo, fetal mouse, and adult human AChRs (Sine et al., 1990; Zhang et al., 1995; Ohno et al., 1996). We consider Scheme FS1 to be the simplest description of the data because of its consistency with previous work and its ability to account for three kinetically distinct types of openings.
Because brief closings and long openings are temporally associated (Fig. 6, top and middle panels) and the corresponding long bursts increase with increasing ACh concentration (Fig. 5, right panels), for wild-type and αV249F AChRs, we assign the brief closings to dwells in A2R and the long openings to dwells in A2R*. Given these assignments, the mean duration of brief closings equals (β2 +kdiss)−1, the number of brief closings per burst of long openings equals β2/kdiss, and the mean burst duration equals (1 + β2/kdiss)/α2(Colquhoun and Hawkes, 1981). The parameterkdiss is the sum of the dissociation rate constants k−1 and k−4, representing the two pathways for loosing bound ACh; if one dissociation rate constant greatly exceeds the other,kdiss equals the faster of the two rate constants. These relationships lead to estimates of α2, β2, andkdiss presented in Table 4, showing that αV249F speeds the rate of channel opening and slows the rates of channel closing and agonist dissociation. Changes in all three of these parameters contribute to the increase in burst duration observed with αV249F.
Table 4.
Kinetic parameters for activation of wild-type and αV249F receptors
| α1 | β1 | θ1 | α2 | β2 | θ2 | K1,M | kdiss | k−2 | |
|---|---|---|---|---|---|---|---|---|---|
| Wild type | 6990 ± 1320 | 59.9 ± 3.8 | 8.6E − 3 | 1520 ± 306 | 46,000 ± 6210 | 30.3 | 2.3E − 5 | 10,060 ± 2370 | 12,700 ± 597 |
| αV249F | 2260 ± 465 | 9350 ± 2850 | 4.1 | 690 ± 92 | 80,810 ± 7530 | 117 | 8.4E − 9 | 3300 ± 1060 | 8650 ± 2610 |
Rate constants are as defined in Scheme FS1 (see text), in units of sec−1. Channel opening equilibrium constants θ are ratios of corresponding opening to closing rate constants, β/α. For wild-type receptors, β1, k−2, andK1 are values determined previously from analyses of clusters (Ohno et al., 1996). For αV249F receptors, α1, β1, and k−2were determined from analyses of intermediate bursts of openings elicited by 3–10 nm ACh, as described in the text (mean values, ± SD from 4 patches). For both wild-type and αV249F receptors, α2, β2 andkdiss values were determined from analyses of bursts of long openings as described in the text; values are means from seven patches for each receptor type obtained from 3 to 100 nm ACh.
αV249F receptors give rise to a second type of burst, intermediate openings separated by intermediate closings (Fig. 6, bottom panel); because intermediate bursts are present at low ACh concentrations and diminish with increasing concentrations (Fig. 5,right panels), we assign these events to dwells in AR* and AR, respectively. At this stage of analysis, these events could be assigned equally to AR′* and AR′, but the subsequent analysis shows that the binding step to form AR is one of low affinity and the step to form A2R is one of high affinity. According to Scheme FS1, the mean duration of intermediate closings equals (β1 +k−2)−1, the number of intermediate closings per burst of intermediate openings equals β1/k−2, and the mean burst duration equals (1 + β1/k−2)/α1. These relationships assume a negligible rate for the third pathway leading away from the AR state, binding a second molecule of ACh to form A2R; this is likely because a diffusion-limited association rate constant of 108m−1 sec−1 and a concentration of 10 nm predict a rate of ∼1 sec−1 for ACh binding, whereas the apparent (β1 + k−2) is 18,000 sec−1 (Table 4).
The resulting estimates of α1, β1, and k−2 for αV249F receptors are presented in Table 4. A comparable analysis was not possible for wild-type receptors because brief openings occur in isolation rather than in bursts. However, comparison with parameters for wild-type receptors, obtained by analysis of clusters at desensitizing ACh concentrations (Ohno et al., 1996), shows that αV249F has little effect on k−2, decreases α1, and increases β1 (Table 4). Thus αV249F receptors elicit bursts of singly liganded openings and closings because β1 and k−2are of similar magnitude.
We estimated the dissociation constant for ACh binding to αV249F receptors from the dependence of the probabilities of long and intermediate bursts on ACh concentration. According to Scheme FS1, the ratio of probabilities of long to intermediate openings, P3/P2, equals [A]θ2/θ1/K1, where [A] is ACh concentration, θ2 equals β2/α2, θ1equals β1/α1, andK1 is the dissociation constant for ACh occupancy of the second binding site (Sine and Steinbach, 1984). Thus, P3/P2 should increase linearly with increasing ACh concentration with a slope of θ2/θ1/K1. Measurements of P3/P2 for αV249F show the predicted linear dependence on ACh concentration (Fig. 7A), and combined with our kinetic estimates of θ2 and θ1, yield a value of K1 of 8.4 nm. This estimate of K1 for αV249F is considerably smaller than either K1or K2 determined for wild-type receptors [23 and 116 μm, respectively (Ohno et al., 1996)] and can be understood from the observation that the probabilities of long and intermediate bursts change with nanomolar changes in ACh concentration. These findings imply that although αV249F is located in the M2 domain, the perturbation spreads to the binding site, some 30 Å away, to increase affinity for ACh.
Fig. 7.

Ratios of probabilities of long (P3) to intermediate (P2) or brief (P1) openings versus ACh concentration. For each class of opening, probability is the product of the relative area and the time constant determined by fitting dwell time histograms to the sum of exponentials. A plots P3/P2 for the αV249F receptor. The line is the least-squares fit toy = mx, with slopem = 3.38 ± 0.14 × 109 M−1. Combining the slope with θ1 and θ2 yields the dissociation constant K2 = 8.4 nm (see text). B plots P3/P1 for the αV249F receptor, with the fitted m = 2.1 ± 0.26 × 1010 M−1. C plots P3/P1 for wild-type receptor, with the fitted m = 3.8 ± 0.24 × 109M−1. D plots P3/P2 for wild-type receptor; the line is the best fit to the points from 1 to 50 nm, with m = 5.5 ± 0.83 × 108 M−1.
We checked additional predictions of Scheme FS1 by examining the dependence of the remaining types of openings on ACh concentration. For αV249F, the ratio of probabilities of long to brief openings, P3/P1, also increases linearly with increasing ACh concentration, as expected for AR′* in Scheme FS1 (Fig. 7B). However, because brief openings occur as single isolated events, the gating equilibrium constant θ′1 cannot be determined, thus preventing estimation of the dissociation constant for the second binding site. For wild-type receptors, the ratios P3/P2 and P3/P1 also increase linearly with ACh concentration (Fig.7C,D), but again both intermediate and brief openings occur as isolated events.
Simulation of channel kinetics for aV249F and wild-type receptor
To confirm our analysis method, we simulated single-channel events according to Scheme FS1 using the parameters obtained from the experimental data (Table 4). Because the microscopic rate constants underlying K1 were not determined experimentally, we chose a diffusion-limited association rate constant (108m−1sec−1), and a dissociation rate constant (1 sec−1) corresponding to K1 = 10 nm. Also, we used the experimentally determined closing rate α1′ for the second of the two classes of monoliganded openings, but assigned a value of β1′ sufficient to give enough of these events. We simulated open and closed intervals assuming Markov kinetics, constructed bursts using our experimental closed time criterion, compiled the bursts into histograms, fitted the histograms by sums of exponentials, and determined P3/P2 at different concentrations of ACh (see Materials and Methods). For the αV249F receptor, the simulated burst-duration histograms closely resemble the experimental burst-duration histograms, showing three components of bursts, with the relative area of each depending on ACh concentration (Fig.8A, left panels). Moreover, for αV249F, the ratio P3/P2 increases linearly with increasing ACh concentration with a slope identical to the expected ratio θ2/θ1/K1(Fig. 8B). The slight departure observed at 100 nm ACh is likely attributable to difficulty in accurately fitting the small component corresponding to P2 (see histogram at 100 nm ACh, Fig. 8A, left panels).
Fig. 8.

Simulated activation kinetics at nanomolar ACh concentrations for wild-type and αV249F AChRs. A shows simulated burst-duration histograms fitted by the sum of exponentials at the indicated ACh concentrations. Channel events were simulated as described in Materials and Methods using the following Scheme FS1parameters: αV249F; k+1 =k+2 = k+3 =k+4 = 108M−1sec−1,k−1 = 1 sec−1,k−2 = 8700 sec−1,k−3 = 2.6 sec−1,k−4 = 3300 sec−1, α1′ = 30,000 sec−1, β1′ = 1 sec−1, α1 = 2300 sec−1, β1 = 9400 sec−1, α2 = 690 sec−1, β2 = 80,800 sec−1: wild type: (parameters from Table 4 and Ohno et al., 1996) k+1 =k+3 = 1.0 × 108M−1 sec−1,k+2 = k+4 = 1.1 × 108 M−1sec−1, k−1 =k−3 = 2400 sec−1,k−2 = k−4 = 12,700 sec−1, α1′ = 30,000 sec−1, β1′ = 1 sec−1, α1 = 6990 sec−1, β1 = 58 sec−1, α2 = 1520 sec−1, β2 = 46,000 sec−1. B plots the ratio of probabilities of long to intermediate openings, P3/P2, against ACh concentration for each receptor type.The dashed linesindicate the theoretical slope, defined by θ2/θ1/K1.
We simulated activation kinetics for wild-type receptors using parameters in Scheme FS1 taken from our previous analysis of human wild-type receptors (Ohno et al., 1996). Because only one class of singly occupied openings was observed at the micromolar ACh concentrations used in those experiments, for the second class of singly occupied openings, we used the closing rate α1′ determined in this study, but assigned a value of β1′ sufficient to give enough of these events. The simulated burst-duration histograms again exhibit three components, with the area of each depending on ACh concentration (Fig. 8, right panel). Again, the ratio P3/P2 increases linearly with increasing ACh concentration with a slope identical to the expected ratio θ2/θ1/K1. In summary, the results from simulation of channel kinetics confirm the analysis method applied to experimental data obtained at nanomolar ACh concentrations.
Simulation of wild-type activation kinetics at nanomolar ACh concentrations allows comparison with parameters determined previously from kinetic analysis of currents activated by micromolar concentrations (Ohno et al., 1996). The ratio θ2/θ1/K1obtained at nanomolar ACh concentrations (Fig. 7C) is approximately twofold greater than obtained at micromolar concentrations. However, we consider this to be good agreement because measurements at nanomolar concentrations yield the ratio of three parameters, and measurements at micromolar concentrations yield each parameter separately. The two methods yield similar estimates of θ2 (30 at nanomolar and 24 at micromolar concentrations). The parameter θ1 was probably overestimated at micromolar ACh concentrations because the corresponding monoliganded openings were present in only a minor proportion of the data, whereas the remaining parameter K1 was consistent with data obtained over a wide range of ACh concentrations.
Activation of αV249F receptors by the competitive antagonist dimethyl-d-tubocurarine
We further examined functional consequences of αV249F by recording single-channel currents activated by the competitive antagonist dimethyl-d-tubocurarine (DMT). DMT only rarely activates wild-type receptors, whereas it frequently activates αV249F receptors (Fig. 9). The increased frequency of activation can be seen from the decrease in the mean of the major closed-duration component, from 20 sec for wild type to 200 msec for αV249F (Fig. 9). The enhancement of activation is probably >100-fold because the recording for wild type was especially active, whereas that for αV249F was typical. The recordings also show that DMT does not significantly desensitize the αV249F receptor; a major long component of closings is observed in the presence of 10 μm DMT, attributable to periods between independent activation episodes, whereas multiple components with similar areas are observed with ACh because of desensitization (compare Figs. 5 and 9, center panels). Thus, αV249F increases the ability of a competitive antagonist to activate the receptor while retaining little or no capacity to desensitize.
Fig. 9.

Activation of human wild-type and αV249F AChRs by DMT. The left panels show single-channel currents elicited by the 10 μm DMT at a bandwidth of 10 kHz. Thecenter and right panels show corresponding closed and burst-duration histograms fitted to the sum of exponentials. Note the major long-duration component of closings for both receptor types, and the 100-fold longer mean closed duration for wild type compared with αV249F.
Spontaneous opening of the αV249F receptor
In a previous study of an SCCMS, we demonstrated spontaneous openings attributable to mutations in the M2 domains of the ε and β subunits (Ohno et al., 1995; Engel et al., 1996b). Therefore, we looked for spontaneous opening of the αV249F receptor by recording from patches in the absence of ACh. Using the presence of green fluorescent protein as a marker for transfected cells and the increased conductance as a signature of the αV249F receptor, we observed spontaneous openings in every patch examined (Fig. 10), averaging 1 per second per patch over 25 patches. By contrast, spontaneous openings of the wild-type receptor were not observed, despite recording from stable patches for up to 30 min. At our standard voltage of −70 mV, αV249F receptors show a major component of spontaneous openings with a mean duration of 300 μsec and relative area of 0.85, plus minor components with means of 60 μsec and 3 msec. Spontaneous and ACh-induced openings can be distinguished readily because of differences in burst kinetics and flanking closed intervals; comparison of closed- and burst-duration histograms indicate that spontaneous openings are minimized at ACh concentrations of 3 nm and greater.
Fig. 10.

Spontaneous opening of the αV249F receptor.Current traces obtained in the absence of ACh from a transfected cell identified by presence of green fluorescent protein. Burst- and closed-duration histograms are fitted by the sums of exponentials.
Binding of agonists and antagonists at equilibrium reveal enhanced desensitization of αV249F receptors
αV249F receptors exhibit no channel activity at micromolar ACh concentrations, suggesting enhanced desensitization. Because desensitized receptors bind agonist with high affinity, we tested the desensitization hypothesis by measuring ACh binding by competition against the initial rate of 125I-α-bungarotoxin binding. Under equilibrium conditions, αV249F receptors bind ACh 300-fold more tightly than wild-type receptors (Fig.11A). The single apparent dissociation constant of 4.3 nm approaches the two dissociation constants obtained for the desensitized wild-type receptor, 4.8 and 41 nm (Ohno et al., 1996). The high affinity, together with lack of channel activity at micromolar ACh concentrations, suggests that αV249F profoundly enhances desensitization.
Fig. 11.
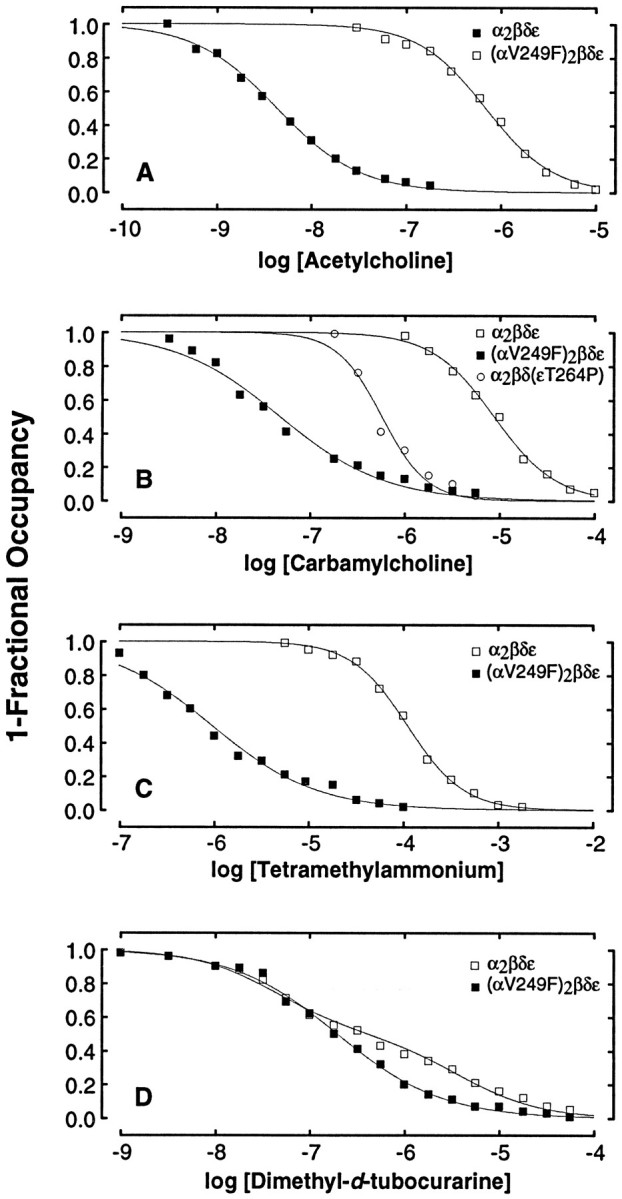
αV249F enhances agonist-binding affinity. Binding of the specified ligand was determined by competition against the initial rate of 125I-α-bgt binding. Intact HEK cells expressing the indicated receptor type were incubated in the presence of agonist for 30 min before measuring the initial rate of toxin binding. The smooth curves are fits to the Hill equation (A–C) or to an equation describing the sum of two distinct binding sites (D). Fitted parameters for each ligand are acetylcholine, wild type: Kov = 6.9 × 10−7, n = 1.2; αV249F,Kov = 4.3 × 10−9, n = 1.0; carbamylcholine, wild type: Kov = 1.6 × 10−5, n = 1.6; αV249F,Kov = 4.8 × 10−8,n = 0.8; εT264P, Kov = 9.5 × 10−7, n = 1.8. TMA, wild type: Kov = 1.1 × 10−4, n = 1.5; αV249F:Kov = 9.5 × 10−7,n = 0.8. DMT, wild type:KA = 4.4 × 10−8,KB = 3.8 × 10−6; αV249F: KA = 6.2 × 10−8, KB = 6.2 × 10−7.
To determine whether the marked enhancement of affinity depends on structure of the agonist, we examined binding of carbamylcholine (CCh), tetramethylammonium (TMA), and DMT. αV249F receptors bind CCh and TMA 300-fold more tightly than wild-type receptors (Figs.11B,C), as observed for ACh, and the relative affinities of ACh, CCh, and TMA are preserved. By contrast, αV249F receptors bind DMT only slightly more tightly than wild-type receptors, with the tighter binding restricted to the low-affinity binding site (Fig. 11D); this slight increase in affinity is likely attributable to a shift in equilibrium from the closed to the open channel state caused by αV249F. Thus, the marked increase in affinity attributable to αV249F is specific for small cholinergic agonists. Because DMT does not appreciably desensitize the αV249F receptor and binds with only slightly greater affinity, the marked increase in agonist affinity is likely attributable to enhanced desensitization.
Our previous studies ascribed slow channel myasthenic syndromes to mutations in the M2 domains of the ε and β subunits, which cause channel opening in the absence of ACh and prolonged openings in its presence (Ohno et al., 1995; Engel et al., 1996b). One of these mutations, εT264P, is three residues above the conserved leucine ring in M2, whereas αV249F is two residues below it. Thus, we compared agonist binding affinities of the two mutations to determine structural specificity. εT264P receptors bind CCh 20-fold more tightly than wild-type receptors, whereas αV249F receptors bind 300-fold more tightly (Fig. 11B). Thus, the perturbation caused by αV249F is distinct from that caused by εT264P, and the enhancement of desensitization is expected to be greater for receptors containing αV249F than εT264P.
DISCUSSION
The αV249F mutation
The slow-channel syndromes are autosomal dominant disorders (Engel, 1994b), attributable to pathological gain of function by the mutated receptor. Detection of αV249F by allele-specific PCR in the patient’s asymptomatic father at first appeared inconsistent with the dominant character as well as with the pathogenicity of the identified mutation. However, sequencing of the entire open reading frames of the α, β, δ, and ε subunit genes of AChR in the propositus revealed no other mutations, and failure to detect αV249F in paternal DNA by direct sequencing or restriction analysis suggested that only a small proportion of paternal DNA harbored the mutation. This assumption was verified by cloning of paternal DNA, which established that the father was mosaic for αV249F. For the propositus to be affected, the mutation must have been transmitted by a sperm cell carrying the mutation. Thus, αV249F must have occurred in the father’s early embryonic life to render him both a somatic and a germ-line mosaic (McGookey-Milewitz et al., 1993).
The SCCMS mutations discovered to date are in the extracellular, M1, or M2 domains of AChR subunits (Ohno et al., 1995; Sine et al., 1995;Engel et al., 1996b; Gomez et al., 1996). The M2 mutations identified previously, however, are in the outer helical region of M2 (εT264P, βV266M, and εL269F), or at the central leucine ring (βL262M) that may form the channel gate (Unwin, 1993), and the mutated residues are accessible to labeling reagents when converted to cysteine, predicting that they face the channel lumen (Akabas et al., 1994). By contrast, αV249F is two residues below the leucine ring, and accessibility studies indicate that the mutated valine does not face the channel lumen (Akabas et al., 1994). Thus, M2 mutations can cause a profound functional disturbance at a variety of locations relative to the leucine ring or the channel lumen.
Functional consequences of αV249F
The expression studies demonstrate that αV249F not only stabilizes the open and desensitized states, but it also enhances affinity of the resting state for ACh. Effects on equilibria between functional states can be explained readily by local perturbation of the M2 domain, which is a structural endpoint defining each of the three functional states. Thus, the findings show that the structure of M2 is essential for correct stabilization of each functional state. Enhancement of ACh binding affinity, on the other hand, implies spread of the perturbation from the M2 domain to the binding site, some 30 Å away. Spread of the perturbation suggests reciprocal coupling between the channel and the binding site through some linkage structure. The effects of αV249F are thus widespread and are expected to affect receptor function in several ways.
Wild-type receptors are designed to open or desensitize with low probability in the absence of ACh (Jackson, 1989). A stable closed state minimizes leak of cations and maintains the majority of receptors ready to be activated. αV249F destabilizes the resting closed state of the receptor in favor of the open channel state, as shown by the increased rate and extent of opening in both the absence and the presence of ACh. Stabilization of the open channel state is revealed by the slower rate of closing of the doubly occupied receptor. αV249F also increases equilibrium affinity for ACh, CCh, and TMA 300-fold, and the binding affinities for these small quaternary agonists are close to those for binding to desensitized wild-type receptors (Sine et al., 1994; Ohno et al., 1996). Such a large increase in apparent affinity can be explained by a change in the equilibrium between resting and desensitized states from a wild-type value of 10−4to ∼0.1 or greater (Sine et al., 1995); thus, αV249F destabilizes the resting closed state in favor of the desensitized state. Enhanced opening of the αV249F receptor overloads the sarcoplasm with cations, both tonically and during nerve impulse, whereas enhanced desensitization decreases the number of receptors that can be activated.
The expression studies also show that αV249F markedly enhances ACh affinity for at least one of the two binding sites in the resting closed state of the receptor. Evidence for enhanced affinity began with the observation of three kinetically distinct types of openings, two singly liganded and one doubly liganded. By monitoring the concentration dependence of doubly versus singly liganded openings, we followed the two possible pathways for forming the doubly occupied receptor. In the first pathway, ACh binds initially to the site with relatively low affinity, opening in bursts of events of intermediate durations and then to the site with nanomolar affinity to yield doubly occupied receptors that open in bursts of prolonged openings. Evidence for low affinity of the initial site is the fast rate of ACh dissociation (8650 sec−1) obtained from the kinetics of intermediate bursts, whereas high affinity of the subsequent site follows from changes of the probabilities of long and intermediate bursts with nanomolar changes in ACh concentration. In the second pathway, ACh initially binds to the site with nanomolar affinity and then to the site with low affinity, again leading to bursts of prolonged openings. In this pathway, the site with nanomolar affinity will be occupied until ACh can dissociate, or ∼1 sec after nerve impulse; when bound to this site, ACh elicits single openings of brief duration that persist throughout occupancy. Moreover, occupancy on the order of seconds is sufficiently long to allow transition from the resting activatable to the desensitized state, which at physiological rates of stimulation may approach the near maximal extent observed in our equilibrium binding experiments.
Comparison with other studies of M2 mutations
Previous studies revealed changes in activation, desensitization, ion permeability, and ligand specificity attributable to mutations in the M2 domain. Both naturally occurring and site-directed mutations increased channel opening in the absence of ACh and prolonged opening in its presence (Ohno et al., 1995; Auerbach et al., 1996; Engel et al., 1996b). Enhanced opening was also inferred from increased apparent affinity in dose–response studies of M2 mutations in muscle (Filatov and White, 1995,Labarca et al., 1995) and α7 neuronal (Revah et al., 1991) receptors. Heterogeneity in the positions of M2 mutated and mutant side chains suggest relatively little structural specificity in enhancing opening of the channel. Studies of mutant α7 neuronal and 5HT-3 receptors showed either increased or decreased rates of onset and extents of desensitization (Revah et al., 1991; Yakel et al., 1993). The profound desensitization we observe for αV249F contrasts with elimination of desensitization reported for mutations in α7 neuronal and GABAA receptors (Revah et al., 1991; Clements et al., 1996). Appearance of two conductance classes of channels was described for mutant α7 neuronal receptors, one of normal conductance and the other increased approximately twofold. Our findings of altered conductance properties are less striking, because we observe a single conductance, increased by only ∼10%, and infrequent subconductance states associated with it; differences may be attributable to the presence of five mutant subunits in α7 and only two mutant subunits in our studies, or to differences in the sites of the mutations and mutant side chains. Activation by competitive antagonists was reported for mutant α7 neuronal receptors (Revah et al., 1991), similar to our observation of activation of αV249F receptors by DMT. Activation by competitive antagonists is likely attributable to increase of the exceedingly low opening rate found in wild-type receptors. The novel functional consequence of αV249F is enhancement of the affinity of the resting state of the receptor for binding ACh.
Phenotypic consequences
Because of the presence of two α subunits in each AChR macromolecule, AChRs at the SCCMS EPs may harbor either wild-type or mutated α (αw or αm) subunits. If mutant and wild-type subunits assemble and are expressed as pentameric receptors with equal efficiency, AChRs containing αwαw, αwαm, and αmαm should be present in a 1:2:1 ratio. Differences in functional properties among these AChRs would be expected to introduce multiple components in the MEPC, ACh-induced current noise, and single-channel recordings, and this likely accounts for the differences among the τ values for channel bursts estimated by the different methods. For example, MEPCs decay with two detectable time constants of 2.4 msec and 43.9 msec at the SCCMS EP, but single-channel burst durations show means of 2.95 and 19.7 msec, and noise analysis shows time constants of 1.4 and 15.1 msec. Because of the continuous presence of agonist in the single-channel and noise recordings and the marked tendency of AChRs harboring two mutated α subunits to become desensitized, only AChRs with a single mutated α subunit are likely to contribute appreciably to the distribution of burst durations and the noise spectrum. The MEPC, however, which follows instantaneous application of ACh, provides a more accurate estimate of the biological activity of AChRs in vivo.
A minor proportion of channel events at the EP had a reduced conductance of 45 pS and prolonged open durations typical of γ-AChR. That a small proportion of these events had an additional longer component is explained readily by γ-AChRs harboring one or two mutated α subunits. Because we previously found a low level of γ-AChR expression in other slow-channel syndromes with mutations in the ε or β subunits and a destructive EP myopathy (Ohno et al., 1995; Engel et al., 1996b), we postulate that the γ-AChR expression is secondary to regenerative activity in the postsynaptic region. γ-AChR is also expressed in those CMS because of nonsense mutations in both alleles of the ε subunit gene, where it is the predominant species at the EP and may serve as a means of phenotypic rescue (Engel et al., 1996a).
The widespread functional consequences of αV249F are expected to compromise the safety margin of neuromuscular transmission in several ways. First, there may be reduced expression of the mutant AChR. Second, the prolonged channel activation episodes as well as tonic openings of unliganded receptors in the resting state overload the junctional sarcoplasm with cations that include calcium (Lester, 1992;Villarroel and Sakmann, 1996). This results in destruction of junctional folds with loss of AChR, widening of the synaptic space, and destruction of organelles in the junctional sarcoplasm (see Fig. 1). Third, apoptosis of a proportion of junctional nuclei (see Fig.1E), probably attributable to calcium excess in the junctional sarcoplasm (McGahon et al., 1995), likely reduces the transcription of AChR subunit genes. Fourth, the marked tendency of the mutant AChR to desensitize predicts that an appreciable fraction of the EP AChR is desensitized even in the resting state, further decreasing the number of receptors that can be activated. Moreover, acetylcholinesterase inhibitors, which prolong the life time of ACh in the synaptic space during activity, may result in desensitization of a large fraction of the EP AChR, with deleterious clinical effects. Fifth, the markedly prolonged decay of EP potentials (generally >40 msec; data not shown) predicts their temporal summation and a depolarization block of transmission during even normal physiological activity.
Footnotes
This work was supported by National Institutes of Health Grants NS6277 to A.G.E. and NS31744 to S.M.S, a Muscular Dystrophy Association (MDA) research grant to A.G.E., and an MDA postdoctoral fellowship to K.O. We thank Dr. Susan Iannaccone for the patient referral.
Correspondence should be addressed to Dr. Steven M. Sine, Receptor Biology Laboratory, Department of Physiology, Mayo Foundation, 200 First Street SW, Rochester, MN 55905.
REFERENCES
- 1.Akabas MH, Kaufmann C, Archdeacon P, Karlin A. Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the alpha subunit. Neuron. 1994;13:919–927. doi: 10.1016/0896-6273(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 2.Auerbach A, Sigurdson W, Chen J, Akk G. Voltage dependence of mouse acetylcholine receptor gating: different charge movements in di-, mono- and unliganded receptors. J Physiol (Lond) 1996;494:155–170. doi: 10.1113/jphysiol.1996.sp021482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaudet AL, Tsui L-C. A suggested nomenclature for designating mutations. Human Mut. 1993;2:245–248. doi: 10.1002/humu.1380020402. [DOI] [PubMed] [Google Scholar]
- 4.Beeson D, Brydson M, Newsom-Davis J. Nucleotide sequence of human muscle acetylcholine receptor beta subunit. Nucleic Acids Res. 1989;17:4391. doi: 10.1093/nar/17.11.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beeson D, Brydson M, Betty M, Jeremiah S, Povey S, Vincent A, Newsom-Davis J. Primary structure of the human muscle acetylcholine receptor: cDNA cloning of the gamma and epsilon subunits. Eur J Biochem. 1993;215:229–238. doi: 10.1111/j.1432-1033.1993.tb18027.x. [DOI] [PubMed] [Google Scholar]
- 6.Bouzat C, Bren N, Sine SM. Structural basis of different gating kinetics of fetal and adult acetylcholine receptors. Neuron. 1994;13:1395–1402. doi: 10.1016/0896-6273(94)90424-3. [DOI] [PubMed] [Google Scholar]
- 7.Clay J, DeFelice L. Relationship between membrane excitability and single channel open-close kinetics. Biophys J. 1983;42:151–157. doi: 10.1016/S0006-3495(83)84381-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clements JD, Tierney L, Birnir B, Pillai P, Gage P, Cox G. A point mutation of the GABAA receptor destabilizes its closed and desensitized states. Soc Neurosci Abstr. 1996;22:817. [Google Scholar]
- 9.Colquhoun D, Hawkes AG. On the stochastic properties of single ion channels. Proc R Soc Lond [Biol] 1981;211:205–235. doi: 10.1098/rspb.1981.0003. [DOI] [PubMed] [Google Scholar]
- 10.Colquhoun D, Sakmann B. Fast events in single channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol (Lond) 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engel AG. Quantitative morphological studies of muscle. In: Engel AG, Franzini-Armstrong C, editors. Myology: basic and clinical, Ed 2. McGraw-Hill; New York: 1994a. pp. 1018–1045. [Google Scholar]
- 12.Engel AG. Myasthenic syndromes. In: Engel AG, Franzini-Armstrong C, editors. Myology: basic and clinical, Ed 2. McGraw-Hill; New York: 1994b. pp. 1798–1835. [Google Scholar]
- 13.Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann Neurol. 1977a;1:315–330. doi: 10.1002/ana.410010403. [DOI] [PubMed] [Google Scholar]
- 14.Engel AG, Lindstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of the acetylcholine receptor in myasthenia gravis and in its experimental autoimmune model. Neurology. 1977b;27:307–315. doi: 10.1212/wnl.27.4.307. [DOI] [PubMed] [Google Scholar]
- 15.Engel AG, Lambert EH, Mulder DM, Torres CF, Sahashi K, Bertorini TE, Whitaker JN. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann Neurol. 1982;11:553–569. doi: 10.1002/ana.410110603. [DOI] [PubMed] [Google Scholar]
- 16.Engel AG, Nagel A, Walls TJ, Harper CM, Waisburg HA. Congenital myasthenic syndromes. I. Deficiency and short open-time of the acetylcholine receptor. Muscle Nerve. 1993;16:1284–1292. doi: 10.1002/mus.880161204. [DOI] [PubMed] [Google Scholar]
- 17.Engel AG, Ohno K, Bouzat C, Sine SM, Griggs RG. End-plate acetylcholine receptor deficiency due to nonsense mutations in the ε subunit. Ann Neurol. 1996a;40:810–817. doi: 10.1002/ana.410400521. [DOI] [PubMed] [Google Scholar]
- 18.Engel AG, Ohno K, Milone M, Wang H-L, Nakano S, Bouzat C, Pruitt JN, Hutchinson DO, Brengman JM, Bren N, Sieb JP, Sine SM. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Hum Mol Genet. 1996b;5:1217–1227. doi: 10.1093/hmg/5.9.1217. [DOI] [PubMed] [Google Scholar]
- 19.Filatov GN, White MM. The role of conserved leucines in the M2 domain of the acetylcholine receptor in channel gating. Mol Pharmacol. 1995;48:379–384. [PubMed] [Google Scholar]
- 20.Gomez CM, Maselli R, Gammack J, Lasalde J, Tamamizu S, Cornblath DR, Lehar M, McNamee M, Kuncl R. A beta-subunit mutation in the acetylcholine receptor gate causes severe slow-channel syndrome. Ann Neurol. 1996;39:712–723. doi: 10.1002/ana.410390607. [DOI] [PubMed] [Google Scholar]
- 21.Jackson MB. Perfection of a synaptic receptor: kinetics and energetics of the acetylcholine receptor. Proc Natl Acad Sci USA. 1989;86:2199–2203. doi: 10.1073/pnas.86.7.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labarca C, Nowak MW, Zhang H, Tang L, Desphpande P, Lester HA. Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature. 1995;376:514–516. doi: 10.1038/376514a0. [DOI] [PubMed] [Google Scholar]
- 23.Lee BS, Gunn RB, Kopito RR. Functional differences among nonerythroid anion exchangers expressed in a transfected human cell line. J Biol Chem. 1991;266:11448–11454. [PubMed] [Google Scholar]
- 24.Lester HA. The permeation pathway of neurotransmitter-gated ion channels. Annu Rev Biophys Biomol Struct. 1992;21:267–292. doi: 10.1146/annurev.bb.21.060192.001411. [DOI] [PubMed] [Google Scholar]
- 25.Luther MA, Schoepfer R, Whiting P, Casey B, Blatt Y, Montal MS, Montal M, Lindstrom J. A muscle acetylcholine receptor is expressed in the human cerebellar medulloblastoma cell line TE671. J Neurosci. 1989;9:1082–1096. doi: 10.1523/JNEUROSCI.09-03-01082.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGahon AJ, Martin SM, Bissonette RP, Mahboubi A, Shi Y, Mogil RJ, Nishioka WK, Green DR. The end of the (cell) line: methods for the study of apoptosis in vitro. In: Schwartz LM, Osborne BA, editors. Methods in cell biology, Vol 46, Cell death. Academic; San Diego: 1995. pp. 153–185. [DOI] [PubMed] [Google Scholar]
- 27.McGookey-Milewitz DM, Witz AM, Smith ACM, Manchaster DK, Waldstein G, Byers PH. Parental somatic and germ-line mosaicism for a multiexon deletion with unusual endpoints in a type III collagen (COL3AI) allele produces Ehlers–Danlos syndrome type IV in the heterozygous offspring. Am J Hum Genet. 1993;53:62–70. [PMC free article] [PubMed] [Google Scholar]
- 28.Milone M, Hutchinson DO, Engel AG. Patch-clamp analysis of the properties of acetylcholine receptor channels at the normal human endplate. Muscle Nerve. 1994;17:1364–1369. doi: 10.1002/mus.880171204. [DOI] [PubMed] [Google Scholar]
- 29.Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, Methfessel C, Sakmann B. Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature. 1986;321:406–411. doi: 10.1038/321406a0. [DOI] [PubMed] [Google Scholar]
- 30.Noda M, Furutani Y, Takahashi H, Toyosato M, Tanabe T, Shimizu S, Kikyotani S, Kayano T, Hirose T, Inayama S, Numa S. Cloning and sequence analysis of calf cDNA and human genomic DNA encoding alpha-subunit precursor of muscle acetylcholine receptor. Nature. 1983;305:818–823. doi: 10.1038/305818a0. [DOI] [PubMed] [Google Scholar]
- 31.Ohno K, Hutchinson DO, Milone M, Brengman JM, Bouzat C, Sine SM, Engel AG. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the ε subunit. Proc Natl Acad Sci USA. 1995;92:758–762. doi: 10.1073/pnas.92.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohno K, Wang H-L, Milone M, Bren N, Brengman JM, Nakano S, Quiram P, Pruitt JN, Sine SM, Engel AG. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor ε subunit. Neuron. 1996;17:157–170. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 33.Revah F, Bertrand D, Galzi JL, Devillers-Thiéry A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux J-P. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 34.Sakmann B, Patlak J, Neher E. Single acetylcholine-activated channels show burst kinetics in the presence of desensitizing concentrations of agonist. Nature. 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- 35.Schoepfer R, Luther M, Lindstrom J. The human medulloblastoma cell line TE671 expresses a muscle-like acetylcholine receptor. Cloning of the alpha-subunit cDNA. FEBS Lett. 1988;226:235–240. doi: 10.1016/0014-5793(88)81430-3. [DOI] [PubMed] [Google Scholar]
- 36.Sigworth FJ, Sine SM. Data transformation for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sine SM, Steinbach JH. Activation of a nicotinic acetylcholine receptor. Biophys J. 1984;45:175–185. doi: 10.1016/S0006-3495(84)84146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sine SM, Steinbach JH. Activation of acetylcholine receptors on clonal mammalian BC3H-1 cells by low concentrations of agonist. J Physiol (Lond) 1986;373:129–162. doi: 10.1113/jphysiol.1986.sp016039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sine SM, Steinbach JH. Activation of acetylcholine receptors on clonal mammalian BC3H-1 cells by high concentration of agonist. J Physiol (Lond) 1987;385:325–359. doi: 10.1113/jphysiol.1987.sp016496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sine SM, Claudio T, Sigworth FJ. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts: single-channel current kinetics reveal distinct agonist binding affinities. J Gen Physiol. 1990;96:395–437. doi: 10.1085/jgp.96.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sine SM, Quiram P, Papanikolaou F, Kreienkamp H-J, Taylor P. Conserved tyrosines in the α subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. J Biol Chem. 1994;269:8808–8816. [PubMed] [Google Scholar]
- 42.Sine SM, Ohno K, Bouzat C, Auerbach A, Milone M, Pruitt JN, Engel AG. Mutation of the acetylcholine receptor α subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 1995;15:229–239. doi: 10.1016/0896-6273(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 43.Uchitel O, Engel AG, Walls TJ, Nagel A, Atassi ZM, Bril V. Congenital myasthenic syndromes. II. A syndrome attributed to abnormal interaction of acetylcholine with its receptor. Muscle Nerve. 1993;16:1293–1301. doi: 10.1002/mus.880161205. [DOI] [PubMed] [Google Scholar]
- 44.Unwin N. Nicotinic acetylcholine receptor at 9 Å resolution. J Mol Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 45.Villarroel A, Sakmann B. Calcium permeability increase of endplate channels in rat muscle during postnatal development. J Physiol (Lond) 1996;496:331–338. doi: 10.1113/jphysiol.1996.sp021688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yakel JL, Lagrutta A, Adelman JP, North RA. Single amino acid substitution affects desensitization of the 5-hydroxytryptamine type 3 receptor expressed in Xenopus oocytes. Proc Natl Acad Sci USA. 1993;90:5030–5033. doi: 10.1073/pnas.90.11.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Chen J, Auerbach A. Activation of recombinant mouse acetylcholine receptors by acetylcholine, carbamylcholine and tetraethylammonium. J Physiol (Lond) 1995;486:189–206. doi: 10.1113/jphysiol.1995.sp020802. [DOI] [PMC free article] [PubMed] [Google Scholar]