

Abstract
A relationship between the apolipoprotein E (apoE) genotype and the risk to develop Alzheimer’s disease has been established recently. Apolipoprotein synthesis is implicated in developmental processes and in neuronal repair of the adult nervous system.
In the present study, we investigated the influence of the apolipoprotein polymorphism on the severity of neuronal degeneration and the extent of plastic dendritic remodeling in Alzheimer’s disease. Changes in length and arborization of dendrites of Golgi-impregnated neurons in the basal nucleus of Meynert, locus coeruleus, raphe magnus nucleus, medial amygdaloid nucleus, pedunculopontine tegmental nucleus, and substantia nigra were analyzed after three-dimensional reconstruction. Patients with either one or two apoE ε4 alleles not only showed a more severe degeneration in all areas investigated than in patients lacking the apoE 4 allele but also revealed significantly less plastic dendritic changes. ApoE ε4 allele copy number, furthermore, had a significant effect on the pattern of dendritic arborization. Moreover, the relationship between the intensity of dendritic growth and both the extent of neuronal degeneration and the stage of the disease seen in patients lacking the apoE ε4 allele was very weak in the presence of one ε4 allele and completely lost in patients homozygous for the ε4 allele.
The results provide direct evidence that neuronal reorganization is affected severely in patients with Alzheimer’s disease carrying the apoE ε4 allele. This impairment of neuronal repair might lead to a more rapid functional decompensation, thereby contributing to an earlier onset and more rapid progression of the disease.
Keywords: Alzheimer’s disease, apolipoprotein E, degeneration, dendritic sprouting, plasticity, regeneration
In the adult CNS affected by age- or disease-related degeneration, mechanisms of compensation and repair are activated in an attempt to counteract functional sequelae of neuronal loss (Scheff et al., 1990; Lapchak et al., 1991; Fritschy and Grzanna, 1992; Lippa et al., 1992; Ramirez and Ulfhake, 1992; Arendt et al., 1995d,e). As a consequence, degenerative events become manifest as a disorder only after exceeding a critical threshold, thereby exhausting the capacity of compensation (Marsden, 1981; Arendt and Bigl, 1987). Under certain conditions, however, these compensational processes might be maladaptive and eventually even contribute to the development and progression of the disease. This seems to be the situation in Alzheimer’s disease (AD) (Geddes et al., 1985; Arendt et al., 1986;Butcher and Woolf, 1989), where an aberrant dendritic growth is observed (Scheibel and Tomiyasu, 1978; Ferrer et al., 1983; Arendt et al., 1986, 1991, 1995b,c; Ihara et al., 1988; McKee et al., 1989) instead of the regular dendritic growth seen during aging and in a variety of related degenerative disorders (Buell and Coleman, 1979;Graveland et al., 1985; Coleman and Flood, 1987; Arendt et al., 1995b,c).
The activation of processes involved in the reorganization of membrane components is a major requirement for restructuring the dendritic organization. In the brain, apolipoprotein E (apoE) is the major lipoprotein involved in lipid transport and metabolism (Elshourbagy et al., 1985; Mahley, 1988; Kinoshita et al., 1993; Poirier et al., 1993a). ApoE synthesis is implicated in neuronal growth and in repair after injury of both peripheral and central neurons (Ignatius et al., 1986; Snipes et al., 1986; Poirier et al., 1991a,b).
There are three major isoforms of apoE (E2, E3, and E4), which are products of the three alleles ε2, ε3, and ε4 located at a single gene locus (Mahley, 1988). It has been shown recently that the ε4 allele is a major risk factor for late-onset familial AD and probably also for sporadic AD (Corder et al., 1993; Mayeux et al., 1993; Poirier et al., 1993b; Rebeck et al., 1993; Saunders et al., 1993; Strittmatter et al., 1993a). Moreover, AD patients with ε4 alleles usually show an earlier age of onset (Corder et al., 1993; Basun et al., 1995; Locke et al., 1995) and a more rapid progression of the disease (Bennett et al., 1995), suggesting a causal link between apoE polymorphism and the development of AD (for review, see Roses et al., 1994).
ApoE is detectable immunohistochemically in senile plaques, neurofibrillary tangles (NFT), and cerebrovascular amyloid in AD (Namba et al., 1991; Wisniewski and Frangione, 1992; Rebeck et al., 1993;Strittmatter et al., 1993a,b; Kida et al., 1994; Benzing and Mufson, 1995). An enhanced burden of both β/A4-amyloid (Schmechel et al., 1993; Strittmatter et al., 1993a,b; Czech et al., 1994; Gearing et al., 1995; Nagy et al., 1995; Ohm et al., 1995; Oyama et al., 1995) and NFT (Nagy et al., 1995; Ohm et al., 1995) was gene dose dependently seen in apoE ε4-carriers. Although isoform-specific interactions of apoE with both β/A4-amyloid (Strittmatter et al., 1993a,b; Wisniewski et al., 1993) and tau (Cotton et al., 1994; Strittmatter et al., 1994a,b;Whitson et al., 1994) have been reported, the mechanism behind the pathological effects of the apoE polymorphism remains unknown.
In the present study, we provide direct evidence that dendritic remodeling, which occurs in AD on subcortical neurons and which is likely to represent a response to neurodegenerative changes, is impaired severely in patients carrying the apoE ε4 allele. This deficiency might be regarded as a failure of plastic neuronal response that could contribute to a more rapid decompensation, resulting in an earlier onset and a more rapid progression of the disease.
MATERIALS AND METHODS
Cases. Brains were obtained from 64 patients with AD and from 20 age-matched control patients. The profile of cases is summarized in Table 1. The clinical diagnosis of AD was based on the occurrence of significant intellectual dysfunction, i.e., the presence of deficits in at least four aspects of cognitive and social behavior. Other causes of dementia were excluded by medical, psychiatric, and paraclinical examination (American Psychiatric Association, 1987). Each case met the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and Alzheimer’s Disease and Related Disorders Association (ADRDA) criteria for definite diagnosis of Alzheimer’s disease (McKhann et al., 1984), based on the presence of NFTs and neuritic plaques observed in the hippocampal formation and neocortical areas, as recommended (Khatchaturian, 1985). Clinical severity of the disease was assessed according to the Functional Assessment Stages of Alzheimer’s disease (FAST) according to Reisberg et al. (1982). Rating of major stages (5, 6, 7) and its substages (a, b, c, etc.) was based on a regular clinical evaluation during the last month of life with the final examination performed within <2 weeks before death. Because the distinctions between FAST substages are less marked than between major stages, two scales, one including substages and the other consisting only of major stages, were used to analyze the influence of the clinical stage of AD on both neuronal degeneration and dendritic plasticity. No major differences, however, were obtained using either one of the scales. Therefore, only data obtained with the full scale, including substages, are reported. To minimize the likelihood of an artificial influence by premortem hypoxia and hypovolemia, we matched cases of AD and controls with respect to the Premortem Severity Index (PMSI) of Monfort et al. (1985). Brains used as normal controls were obtained at routine autopsy from patients dying without a history of neuropsychiatric disorder or mental impairment. There had to be clear evidence that the patient was alert, well oriented, and capable of functioning relatively independently shortly before death. No pathological signs were detected by neuropathological examination. None of the control cases carried an apoE ε4 allele. The entire procedure of case recruitment, acquisition of the patients’ personal data, performing the autopsy, and handling the autoptic material has been approved by the Ethical Committee of Leipzig University.
Table 1.
Profile of cases
| Controls | Alzheimer’s disease/apolipoprotein E genotype | |||
|---|---|---|---|---|
| ε3/ε3 | ε3/ε4 | ε4/ε4 | ||
| Number of cases | 20 | 34 | 19 | 11 |
| Sex (male/female) | 9/11 | 15/19 | 8/11 | 5/6 |
| Age in years (±SD) | 81 ± 8.1 | 85 ± 6.6 | 76 ± 7.3 | 77 ± 7.9 |
| FAST | ||||
| 5 | 6 | 4 | 2 | |
| 6b | 5 | 2 | 1 | |
| 6d | 6 | 4 | 2 | |
| 6e | 5 | 3 | 2 | |
| 7a | 5 | 4 | 2 | |
| 7b | 7 | 2 | 2 | |
| PMSI (±SD) | 2.2 ± 0.4 | 2.6 ± 0.7 | 2.7 ± 0.6 | 2.7 ± 0.5 |
| Cause of death | ||||
| Bronchopneumonia | 5 | 30 | 17 | 9 |
| Myocardial infarction | 11 | — | — | 2 |
| Embolism of lung | 2 | 1 | — | — |
| Sepsis | 2 | 3 | 2 | — |
| Postmortem delay in hr (±SD) | 2.8 ± 0.3 | 3.3 ± 0.5 | 3.1 ± 0.4 | 3.0 ± 0.7 |
| Fresh brain weight in gm (±SD) | 1282 ± 74 | 1210 ± 53 | 1208 ± 64 | 1164 ± 86 |
Tissue preparation. Samples of the cerebral cortex were dissected bilaterally from Brodmann areas 8 and 22. White matter was removed carefully, and specimens were snap-frozen and stored at −80°C. To prevent artifacts by postmortem delay (Williams et al., 1978; Buell, 1982; De Ruiter, 1983), we immersed the remaining parts of the brains in 4% formaldehyde/0.1 m sodium phosphate buffer adjusted to pH 7.2 within <3.5 hr postmortem. After fixation for ∼1 month, cerebellum and brainstem were removed, and brains were cut in the coronal plane into slabs of ∼5 mm thickness. One hemisphere was processed further for Golgi impregnation, the other hemisphere for immunohistochemical techniques and Nissl staining.
ApoE isotyping. Genomic DNA was isolated by treating 15–25 mg of frozen brain tissue with proteinase K (Sambrook et al., 1989). The digest was deproteinized by phenol/chloroform/isoamyl alcohol extractions, recovered by ethanol precipitation, dried, and resuspended in buffer. A 227 bp sequence of the apoE gene was amplified by PCR using primers and reaction conditions as described (Wenham et al., 1991). Genotyping was performed after digestion with the restriction enzyme CfoI (Wenham et al., 1991) and separation of fragments on a 10% polyacrylamide gel (Hixson and Vernier, 1990).
Golgi impregnation. Tissue slabs 5 mm thick were trimmed to blocks varying in size between 3 and 15 cm2. Blocks were incubated at room temperature for 8–10 d in a solution of 5% formaldehyde, 3% potassium dichromate, and 12.5% sucrose. Solution was freshly prepared every day. Then tissue was rinsed and kept for a further 3–6 d in 0.75% silver nitrate at room temperature in the dark. After dehydration and celloidin embedding, serial sections 240 μm thick were cut in the coronal plane. Blocks were oriented carefully before cutting to ensure that sectioning was parallel to the frontal reference plane. Sections were mounted between two coverslips to be observable from both sides.
Histochemistry. Tissue blocks containing the basal nucleus of Meynert of one hemisphere were immersed in 30% sucrose for cryoprotection and cut in the coronal plane on a freezing microtome at a thickness of 30 μm. Sections were treated for 15 min with 0.3% H2O2 in methanol to destroy endogenous peroxidase and preincubated in 0.3% nonfat dried milk and 0.1% gelatin in 0.1 m PBS for blocking. Free-floating sections were incubated at room temperature overnight in one of the following primary antibodies in 0.1 m PBS also containing 0.01% Triton X-100: rat monoclonal anti-choline acetyltransferase (ChAT; Boehringer Mannheim, Mannheim, Germany; 1:200) or mouse monoclonal anti-NGF-receptor p75 (p75NGFR; clone ME 20.4, 1:200). Immunoreaction was detected with peroxidase-labeled anti-rat Ig (Boehringer Mannheim; 1:500) or biotinylated sheep anti-mouse Ig (Amersham, Buckinghamshire, UK; 1:300), the ExtrAvidin–peroxidase complex (Sigma-Aldrich GmbH, Deisenhofen, Germany; 1:300) and 0.04% 3,3′-diaminobenzidine/0.015% H2O2 in 0.1m PBS. For intensification, selected sections were, instead, treated with 0.02% 3,3′-diaminobenzidine, 0.015% H2O2, and 0.4% nickel ammonium sulfate in 0.5m Tris-HCl, pH 8. Sections were rinsed, mounted on chrome-alum-coated slides, dehydrated, and coverslipped with Entellan (Merck, Darmstadt, Germany).
Quantitative analysis of Golgi-impregnated neurons. A three-dimensional morphometric analysis of Golgi-impregnated reticular neurons in the basal nucleus of Meynert, locus coeruleus, raphe magnus nucleus, medial amygdaloid nucleus, pedunculopontine tegmental nucleus, and substantia nigra was performed as described (Arendt et al., 1995a,c). The morphological delineation of the basal nucleus was based on a detailed cytological and histochemical study (Fischer and Fischer, 1987). Reconstruction of neurons and quantitative measurements were made in three dimensions with reference to a defined Cartesian system of axis by digitizing the neuron directly from serial sections with help of a semiautomatic image analyzing system (Orthoplan-3D, Leitz Wetzlar and analySIS, Münster, Germany). From the digitized image, the total dendritic length per neuron and the number of dendritic segments of each order using the centrifugal ordering system were calculated (Arendt et al., 1986, 1995a). No correction for tissue shrinkage was made. Data acquisition was performed blind to diagnosis.
Neuronal cell counts. Numerical neuronal density, defined as the number of nucleolated neurons per tissue volume, was determined after Nissl staining in every tenth section (30 μm thickness) throughout the brain areas listed above with help of an automatic image analyzing system (analySIS). To allow a direct comparison of figures obtained for different areas and different subgroups of patients, we expressed data as percentages of control values. In the Ch4 part (Mesulam et al., 1983) of the basal nucleus of Meynert, cell counts were performed additionally on every 20th section processed for ChAT or p75NGFR immunohistochemistry. Total neuronal number was determined by interpolating over the entire structure.
Activity of ChAT. Determination of activity of ChAT was performed on homogenates of tissue samples in 0.25 msucrose containing 0.2% Triton X-100 according to the method of Fonnum (1969) in the modification of Bigl (1975), using [3H]acetyl CoA (specific activity 1.5 MBq/μmol, Amersham) as substrate. Protein content was determined according toPeterson (1977).
Statistics. For each case, 300 Golgi-impregnated neurons were analyzed, and data were averaged to determine a case mean. These case means were averaged to obtain group means, the differences of which were analyzed with parametric statistical tests (program SPSS/PC+, SPCC, Chicago, IL). One way ANOVA and orthogonalt tests were used to compare individual subgroups of patients with each other and with the control group. Analysis of linear regression was used to analyze the relationship between dendritic growth and neuronal loss, between dendritic growth and clinical stage, and between regenerative capacity and clinical stage of the disease.
RESULTS
Plastic dendritic changes of subcortical neurons
Golgi-impregnated reticular neurons were sampled in the basal nucleus of Meynert (Fig. 1), locus coeruleus, raphe magnus nucleus, medial amygdaloid nucleus, pedunculopontine tegmental nucleus, and substantia nigra. The three-dimensional analysis of the dendritic tree of reticular neurons revealed an increase in dendritic length in patients with AD, as compared with controls in all areas investigated (Table 2). This increase in length of dendrites varied for the different areas. It ranged from ∼8% in the substantia nigra to ∼26% in the basal nucleus of Meynert when all AD patients were averaged. The intensity of changes in dendritic length within a given area, however, markedly differed for subgroups of patients grouped according to their apoE genotypes. Dendritic changes were most pronounced in patients carrying apoE ε3/3. They were clearly marked less strongly in patients with the genotypes apoE ε3/4 or ε4/4 (Table 2). This influence of the apoE ε4 allele on the intensity of plastic dendritic changes was present most clearly in the basal nucleus of Meynert and the locus coeruleus but also could be detected in all other areas investigated.
Fig. 1.
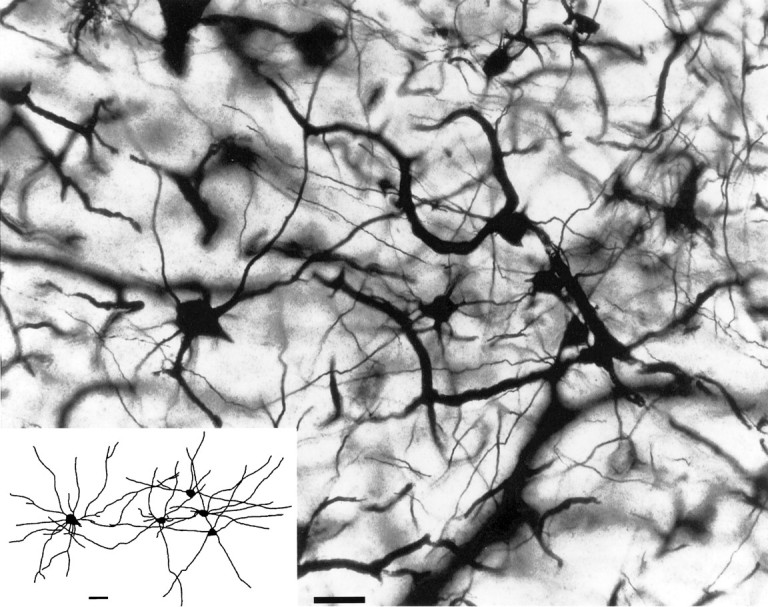
Cluster of Golgi-impregnated reticular neurons in the basal nucleus of Meynert in a patient with AD.Inset, Two-dimensional projection of the three-dimensional image reconstructed from serial sections. Scale bar, 50 μm.
Table 2.
Effects of the ApoE genotype on the loss of subcortical neurons and on dendritic growth in AD
| Control | Alzheimer’s disease | |||
|---|---|---|---|---|
| ApoE 3/3 | ApoE 3/4 | ApoE 4/4 | ||
| Basal nucleus of Meynert | ||||
| Dendritic length (mm) | 2.612 ± 0.049 | 3.600 ± 0.0472-165 | 3.353 ± 0.0452-165## | 3.275 ± 0.0362-165### |
| Neuronal density (%) | 100 | 36.3 ± 3.82-165 | 26.6 ± 3.92-165 | 13.4 ± 2.92-165## |
| Locus coeruleus | ||||
| Dendritic length (mm) | 2.230 ± 0.041 | 3.021 ± 0.0312-165 | 2.838 ± 0.0272-165### | 2.732 ± 0.0192-165### |
| Neuronal density (%) | 100 | 49.9 ± 3.52-165 | 33.1 ± 5.32-165## | 14.2 ± 5.02-165### |
| Raphe magnus nucleus | ||||
| Dendritic length (mm) | 2.520 ± 0.026 | 2.982 ± 0.0212-165 | 2.922 ± 0.0272-165 | 2.855 ± 0.0292-165### |
| Neuronal density (%) | 100 | 70.3 ± 2.22-165 | 62.5 ± 8.52-165 | 49.1 ± 3.72-165### |
| Medial amygdaloid nucleus | ||||
| Dendritic length (mm) | 2.342 ± 0.026 | 2.776 ± 0.0222-165 | 2.730 ± 0.0282-165 | 2.654 ± 0.0302-165## |
| Neuronal density (%) | 100 | 75.1 ± 2.12-165 | 66.3 ± 2.82-165# | 44.1 ± 3.72-165### |
| Pedunculopontine tegmental nucleus | ||||
| Dendritic length (mm) | 2.410 ± 0.024 | 2.867 ± 0.0202-165 | 2.805 ± 0.0322-165 | 2.733 ± 0.0252-165## |
| Neuronal density (%) | 100 | 77.7 ± 3.82-165 | 58.8 ± 3.42-165## | 35.4 ± 5.12-165### |
| Substantia nigra | ||||
| Dendritic length (mm) | 2.155 ± 0.037 | 2.389 ± 0.0302-165 | 2.299 ± 0.0272-160 | 2.256 ± 0.008# |
| Neuronal density (%) | 100 | 87.8 ± 1.32-165 | 77.3 ± 2.72-165### | 62.6 ± 4.82-165### |
Data are mean ± SEM. Significantly different from control:
F2-165: p < 0.001;
F2-160: p < 0.01 (Student’st test). Significantly different from AD subgroup ApoE 3/3: ###p < 0.001; ##p < 0.01; #p < 0.05 (Student’s t test). For group size, see Table 1.
To characterize changes in the pattern of arborization that might accompany the observed process of dendritic growth and to study the distribution of newly formed dendritic segments within the dendritic tree, we analyzed the frequency distribution of all dendritic segments of different orders (Fig. 2). The centrifugal system of ordering segments was used, i.e., primary dendritic segments correspond to segment order 1; the highest order of segments represents most distal branches. Although no subgroup-specific changes were noted for the total number of segments (all p > 0.1), subgroups of patients were significantly different with respect to the distribution of newly formed dendritic segments (Table3). In patients with apoE ε3/3, newly formed segments were mostly of higher order, i.e., more distally localized. Growth processes were approximately equally present in distal and proximal parts of the dendritic tree in patients with apoE ε3/4 and were most frequently localized in proximity to the soma in patients with apoE ε4/4.
Fig. 2.


Changes in the total number of dendritic segments and in the frequency distribution of segments obtained for different orders on Golgi-impregnated reticular neurons in different subgroups of patients with AD, as compared with controls (centrifugal system of ordering, i.e., primary segments correspond to order 1; most distal branches correspond to highest segment order). Open columns, Control values; open pluscross-hatched columns, AD patients. Data are mean values. For each case, 300 neurons were analyzed. For group size, see Table 1; for summary of statistical analysis, compare Table 3.
Table 3.
Statistical summary for the analysis of frequency distribution of dendritic segments in AD compared with controls
| Effect | ApoE 3/3 | ApoE 3/4 | ApoE 4/4 | ||||
|---|---|---|---|---|---|---|---|
| df | F | df | F | df | F | ||
| Basal nucleus of Meynert | |||||||
| Control | AD | 1, 52 | 72.123-165 | 1, 37 | 143.343-165 | 1, 29 | 226.823-165 |
| AD × Lin segm.or. | 1, 52 | 32.343-165 | 1, 37 | 63.703-165 | 1, 29 | 108.193-165 | |
| AD: ApoE 3/4 | AD × Lin segm.or. | 1, 51 | 8.293-160 | 1, 28 | 9.163-160 | ||
| AD: ApoE 4/4 | AD × Lin segm.or. | 1, 43 | 21.223-165 | ||||
| Locus coeruleus | |||||||
| Control | AD | 1, 52 | 43.783-165 | 1, 37 | 82.103-165 | 1, 29 | 136.163-165 |
| AD × Lin segm.or. | 1, 52 | 33.103-165 | 1, 37 | 71.903-165 | 1, 29 | 112.343-165 | |
| AD: ApoE 3/4 | AD × Lin segm.or. | 1, 51 | 7.563-160 | 1, 28 | 8.723-160 | ||
| AD: ApoE 4/4 | AD × Lin segm.or. | 1, 43 | 18.323-165 | ||||
| Raphe magnus nucleus | |||||||
| Control | AD | 1, 52 | 45.283-165 | 1, 37 | 93.103-165 | 1, 29 | 129.453-165 |
| AD × Lin segm.or. | 1, 52 | 29.343-165 | 1, 37 | 58.223-165 | 1, 29 | 89.453-165 | |
| AD: ApoE 3/4 | AD × Lin segm.or. | 1, 51 | 5.163-150 | 1, 28 | 4.823-150 | ||
| AD: ApoE 4/4 | AD × Lin segm.or. | 1, 43 | 8.293-160 | ||||
| Medial amygdaloid nucleus | |||||||
| Control | AD | 1, 52 | 41.183-165 | 1, 37 | 83.193-165 | 1, 29 | 124.123-165 |
| AD × Lin segm.or. | 1, 52 | 23.183-165 | 1, 37 | 48.223-165 | 1, 29 | 75.603-165 | |
| AD: ApoE 3/4 | AD × Lin segm.or. | 1, 51 | 4.563-150 | 1, 28 | 5.283-150 | ||
| AD: ApoE 4/4 | AD × Lin segm.or. | 1, 43 | 7.783-160 | ||||
| Pedunculopontine tegmental nucleus | |||||||
| Control | AD | 1, 52 | 28.923-165 | 1, 37 | 62.103-165 | 1, 29 | 89.523-165 |
| AD × Lin segm.or. | 1, 52 | 22.123-165 | 1, 37 | 43.293-165 | 1, 29 | 63.173-165 | |
| AD: ApoE 3/4 | AD × Lin segm.or. | 1, 51 | 5.103-150 | 1, 28 | 6.233-150 | ||
| AD: ApoE 4/4 | AD × Lin segm.or. | 1, 43 | 7.563-160 | ||||
| Substantia nigra | |||||||
| Control | AD | 1, 52 | 21.123-165 | 1, 37 | 31.193-165 | 1, 29 | 67.323-165 |
| AD × Lin segm.or. | 1, 52 | 14.203-165 | 1, 37 | 19.123-165 | 1, 29 | 38.173-165 | |
| AD: ApoE 3/4 | AD × Lin segm.or. | 1, 51 | 4.783-150 | 1, 28 | 4.313-150 | ||
| AD: ApoE 4/4 | AD × Lin segm.or. | 1, 43 | 7.303-160 | ||||
Data are F values for the comparison of subgroups of patients with AD with the control group and for comparison between AD subgroups obtained by one-way ANOVA.
F3-150: p < 0.05;
F3-160: p < 0.01;
F3-165: p < 0.001; for group size, see Table 1. Lin segm.or., Linear trend of segment order.
Degeneration of neurons
Neuronal degeneration, characterized by formation of NFT and neuronal loss, was observed in all areas investigated. Neuronal loss was most severe in the basal nucleus of Meynert, followed in decreasing order of severity by the locus coeruleus, raphe magnus nucleus, medial amygdaloid nucleus, pedunculopontine tegmental nucleus, and substantia nigra. Again, the extent of changes differed for different subgroups of patients according to their apoE genotype (Table 2). Loss of neurons was more severe in patients carrying one apoE ε4 allele, as compared with patients with apoE ε3/3, and was even higher in patients with apoE ε4/4. These differences applied to all areas analyzed in the present study.
Relationship between degeneration and dendritic reorganization
To investigate whether the process of dendritic reorganization in AD might be related to the extent of neuronal loss, we analyzed the dependency of these two parameters by statistical methods (Fig. 3). A highly significant relationship between dendritic growth and neuronal loss was obtained for patients with apoE ε3/3 in all areas investigated. The relationship of these two parameters, however, was only marginally significant in patients with the apoE ε3/4 genotype. The small increase in dendritic length observed in patients with apoE ε4/4 was unrelated to the extent of neuronal loss.
Fig. 3.

Relationship between the loss of neurons and the extent of dendritic growth in subcortical brain areas separately analyzed for different subgroups of patients with AD. Analyses of linear regression were performed, and correlation coefficients according to Bravais–Pearson were calculated. For group size, see Table 1.
To further characterize subgroups of AD patients concerning differences in the dependency of dendritic growth on the extent of neuronal loss, we calculated the individual ratios of the two parameters. This ratio of the relative increase in dendritic length per relative neuronal loss gives an indication of the overall extent of regeneration/repair in the different structures that might occur in response to degeneration of a given extent. This parameter was, therefore, designated as “reparative capacity.” For example, a reparative capacity with a value of “1” would indicate that the newly formed dendritic elements are quantitatively equivalent to those that have been lost by neuronal degeneration. It should be borne in mind, however, that this parameter of reparative capacity solely represents a measure of the mathematical dependency of the two processes, i.e., cell loss and dendritic growth. A strong statistical relationship obtained by this method does not necessarily imply a causal relationship between the two processes. The reparative capacity emerged as a particularly useful parameter with a high potential to discriminate different subgroups of AD patients (Fig. 4). In comparison to patients with AD lacking the apoE ε4 allele, the value of this parameter was reduced by up to 68 and 85% in patients carrying one or two apoE ε4 alleles, respectively.
Fig. 4.

Differences in the reparative capacity of patients with AD grouped according to their apoE genotypes. Student’st tests were used to compare individual group means ± SEM. For group size, see Table 1.
Stage-dependent differences of dendritic plasticity
The intensity of dendritic growth observed in patients with AD not only might depend on the extent of neuronal degeneration but also might be influenced by the progression of the disease and might, thus, vary for different disease stages. The influence of the stage of the disease on both the intensity of dendritic growth and the reparative capacity was, therefore, evaluated for each subgroup of AD patients separately (Fig. 5 and Table 4). During early stages of the disease, the extent of dendritic growth was similar for all subgroups of AD patients, independent of its apoE genotype (Fig. 5,left panels). The progression of the disease, however, had a significantly different influence on further dendritic changes (Table4). Whereas in patients with apoE ε3/3 dendritic growth continuously increased over the progression of the disease, this increase was only marginally significant in the medial amygdaloid nucleus and insignificantly small in all other areas in patients with apoE ε3/4. Disease progression had no influence on dendritic growth in patients with apoE ε4/4. Contrary to changes in dendritic length, reparative capacity tended to decline over the progression of the disease (Fig. 5,right panels). Again, changes were significant for the apoE ε3/3 genotype but only marginally significant or insignificantly small in the presence of one or two apoE ε4 alleles (Table 4). Although at early stages of the disease subgroups of patients clearly differed with respect to their reparative capacity, these differences declined considerably during the progression of the disease (Fig. 5).
Fig. 5.

Relationship between the dendritic growth and reparative capacity of reticular neurons in subcortical brain areas and the clinical stage of AD, assessed according to FAST. Different subgroups of patients classified according to their apoE genotypes were compared by ANOVA (effect, subgroup × linear trend of disease progression). df, apoE 3/3 versus apoE 3/4, 1, 51; apoE 3/3 versus apoE 4/4, 1, 43; apoE 3/4 versus apoE 4/4, 1, 28; for both dendritic growth and reparative capacity, all p < 0.01. For group size, see Table 1; for analysis of linear regression, compare Table4.
Table 4.
Analysis of stage dependency of dendritic growth and reparative capacity of different subgroups of AD patientsa
| ApoE 3/3 dendr.gr. | ApoE 3/4 regen.cap. | ApoE 4/4 dendr.gr. | regen.cap. | dendr.gr. | regen.cap. | |
|---|---|---|---|---|---|---|
| Basal nucleus of Meynert | 0.984-165 | 0.924-160 | 0.74 | 0.884-150 | 0.41 | 0.80 |
| Locus coeruleus | 0.984-165 | 0.894-150 | 0.80 | 0.934-160 | 0.68 | 0.13 |
| Raphe magnus nucleus | 0.934-160 | 0.964-160 | 0.57 | 0.984-165 | 0.44 | 0.12 |
| Medial amygdaloid nucleus | 0.944-160 | 0.914-150 | 0.884-150 | 0.81 | 0.72 | 0.23 |
| Pedunculopontine tegmental nucleus | 0.884-150 | 0.924-160 | 0.59 | 0.46 | 0.01 | 0.38 |
| Substantia nigra | 0.994-165 | 0.964-160 | 0.66 | 0.65 | 0.09 | 0.18 |
Analysis of linear regression of dendritic growth (dendr.gr.) and reparative capacity (regen.cap.) on the clinical stage of the disease assessed according to FAST (Reisberg et al., 1982).
The correlation coefficient is significantly different from zero for
F4-165: p < 0.001;
F4-160: p < 0.01;
F4-150: p < 0.05. For group size, see Table 1.
Degeneration of cortical cholinergic axon terminals
Activity of ChAT in the frontal cortex (Brodmann area 8) and temporal cortex (Brodmann area 20) was reduced in all cases with AD. Differences of these changes related to subgroups of patients with different apoE genotypes were pronounced even more clearly than the loss of cholinergic neurons in the basal nucleus of Meynert identified by anti-ChAT and anti-p75NGFR immunoreactivity (Table5). Whereas ChAT activity was reduced by 35–40% in cases with apoE ε3/3, reductions of ∼70% and even >90% were observed in cases with apoE ε3/4 and 4/4, respectively.
Table 5.
Changes in the number of cholinergic neurons in the basal nucleus of Meynert and the activity of ChAT in the cerebral cortex in different subgroups of patients with AD
| Control | Alzheimer’s disease | |||
|---|---|---|---|---|
| ApoE 3/3 | ApoE 3/4 | ApoE 4/4 | ||
| Neuronal number | ||||
| ChAT-immunoreactive | 178,467 ± 4230 | 64,834 ± 48525-165 | 47,519 ± 51155-165# | 23,850 ± 46355-165### |
| p75NGFR-immunoreactive | 182,389 ± 4820 | 66,972 ± 52105-165 | 49,608 ± 48645-165# | 25,160 ± 46455-165### |
| ChAT activity (nmol/mg protein × h) | ||||
| Brodmann area 8 | 11.9 ± 0.70 | 7.14 ± 0.525-165 | 3.33 ± 0.345-165### | 0.83 ± 0.225-165### |
| Brodmann area 20 | 8.3 ± 0.40 | 5.47 ± 0.255-165 | 2.40 ± 0.255-165### | 0.55 ± 0.055-165### |
Comparing the loss of cholinergic neurons in the basal nucleus giving rise to the cortical cholinergic innervation with changes in ChAT activity at cortical target sites might give some indication on adaptive changes of cholinergic neurotransmission related to plastic phenomena occurring in remaining neurons. These plastic changes at the sites of cholinergic axon terminals varied among different subgroups of patients, as shown in Figure 6. In patients with apoE ε3/4, activity of ChAT at cortical sites was reduced to a similar extent as the number of cholinergic basal forebrain neurons (ratio of ChAT activity to cell number close to 1). Cortical ChAT activity was affected less severely than cell number in the basal nucleus in patients with apoE ε3/3 (ratio of ChAT activity to cell number >1), whereas cortical ChAT activity was reduced even more dramatically than neuronal number in the basal forebrain in patients with apoE ε4/4 (ratio of ChAT activity to cell number <1).
Fig. 6.
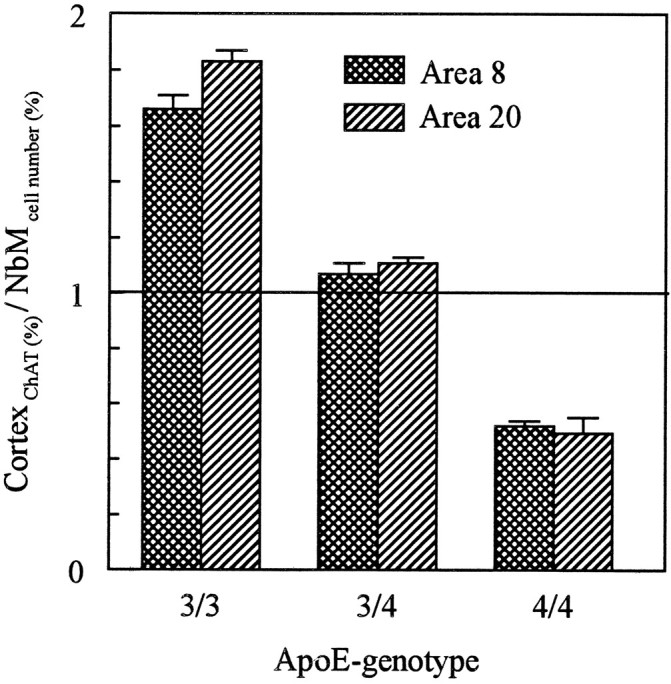
Ratio between ChAT activity in the cerebral cortex and number of cholinergic neurons in the basal nucleus of Meynert in patients with AD carrying different apoE genotypes. Values of both ChAT activity and neuronal number (determined on every 20th section processed for anti-ChAT immunocytochemistry) are expressed as percentages of control values. Student’s t tests were used to compare mean values of individual groups; allp < 0.001. For group size, see Table 1.
DISCUSSION
Chronic neuronal degeneration in a number of disorders related to quite different etiologies, such as AD, postalcoholic Korsakoff’s disease, Parkinson’s disease, or Huntington’s chorea, is accompanied by growth and reorganization of the dendritic tree of certain types of neurons (Graveland et al., 1985; Arendt et al., 1986, 1995b,c; Scott, 1993). These changes have been regarded as an attempt of the nervous system to counteract the functional impairments resulting from degenerative events.
In AD, plastic changes that occur in response to degeneration in cortical and subcortical areas show severe aberrations with respect to their localization, morphological appearance (Scheibel and Tomiyasu, 1978; Scheibel, 1979; Ferrer et al., 1983, 1990; Probst et al., 1983;Arendt et al., 1986, 1995a–c; Arendt and Zvegintseva, 1987; Ihara et al., 1988; Arendt and Brückner, 1991, 1992), and composition of cytoskeletal elements (McKee et al., 1989). The reasons for these aberrancies are unknown.
The results of the present study demonstrate that, in a number of subcortical brain areas, the extent of neuronal degeneration as well as the intensity and pattern of plastic dendritic changes in AD is related to the apoE genotype (For synopsis of the major findings, see Table6). Because none of the control cases carried an apoE ε4 allele, the present study does not allow any conclusion on whether apoE polymorphism might affect neuronal number and branching in patients without AD.
Table 6.
Synopsis of the influence of apoE polymorphism on degeneration and dendritic reorganization on subcortical neurons in AD
| ApoE 3/3 | ApoE 3/4 | ApoE 4/4 | |
|---|---|---|---|
| Degeneration | |||
| Neuronal density (depending on brain region)6-a | 30–90% | 20–80% | 10–70% |
| Dendritic reorganization | |||
| Dendritic length (depending on brain region)6-a | 110–140% | 100–130% | 100–130% |
| Localization of newly formed dendritic branches | Preferentially distally localized | Intermediate | Preferentially proxi mally localized |
| Relationship between degeneration and dendritic reorganization | |||
| Relationship between neuronal loss and dendritic growth | Significantly positively correlated | Marginally significant | Insignificantly small |
| Reparative capacity | 0.6–0.9 | 0.3–0.6 | 0.1–0.3 |
| Ratio: dendritic growth/neuronal loss (depending on brain region)6-a | |||
| Stage dependency of dendritic reorganization | |||
| Relationship between disease stage and dendritic growth | Significantly positively correlated | Marginally significant | Insignificantly small |
| Relationship between disease stage and reparative capacity | Significantly negatively correlated | Marginally significant | Insignificantly small |
| Degeneration and reorganization of the cortical cholinergic afferentation | |||
| Cortical activity of ChAT | 60–70% | 20–40% | <10% |
| Number of ChAT-positive neurons in the basal nucleus of Meynert | 30–40% | 20–40% | 10–20% |
| Ratio of cortical ChAT (%) versus number (%) of ChAT-positive neurons in the basal nucleus | >1 | ∼1 | <1 |
The regional intensity of both degenerative and plastic dendritic changes varies in the following order: basal nucleus > locus coeruleus > raphe magnus nucleus > medial amygdaloid nucleus > pedunculopontine tegmental nucleus > substantia nigra.
ApoE gene dose and degeneration of subcortical neurons
A severe neuronal degeneration was observed in the basal nucleus of Meynert, locus coeruleus, the median raphe nucleus, medial amygdaloid nucleus, pedunculopontine tegmental nucleus, and substantia nigra, thereby confirming a number of earlier studies (Herzog and Kemper, 1980; Arendt et al., 1983; Mann et al., 1983; Shortridge et al., 1985; Yamamoto and Hirano, 1985; German et al., 1987; Jellinger, 1988; Woolf et al., 1989; Esiri et al., 1990; Goto et al., 1990;Aletrino et al., 1992; Förstl et al., 1992; Halliday et al., 1992; Scott et al., 1992). The overall extent of degeneration and the gradual variation of cell loss between different areas are also in agreement with previous reports (Zweig et al., 1988; Chan-Palay et al., 1992; Arendt et al., 1995c).
In the present study, the severity of degeneration in these areas was found to vary among different patients, depending on their apoE genotype. A gene dosage effect of the apoE ε4 allele on the severity of degeneration of cholinergic basal forebrain neurons, as revealed in the present study, had been reported previously by Poirier et al. (Poirier, 1994; Poirier et al., 1995) and others (Soininen et al., 1995). The concept has, therefore, been put forward that the integrity of cholinergic neurons may be compromised selectively in apoE ε4 carriers (Poirier, 1994). The present study demonstrates that effects mediated by the apoE ε4 allele are not confined to the cholinergic basal forebrain neurons. Effects related to the apoE-polymorphism might, therefore, be of more global relevance to the process of neuronal degeneration in AD.
ApoE polymorphism and dendritic plasticity in AD
ApoE ε4 allele copy number in patients with AD showed an inverse relationship to the extent of plastic neuronal remodeling. In AD patients lacking the apoE ε4 allele, reactive dendritic growth parallels both the extent of neuronal degeneration and the progression of the disease. Although conclusions of dynamic events from static images need to be drawn with caution, it is tempting to regard this correlative relationship as an indication of a “coupling” between the process of dendritic reorganization and the functional demands of compensating degenerative events. In these patients, newly formed dendritic branches were localized mainly on segments of higher order, resulting in an extensive pattern of growth. A similar pattern of dendritic growth has been established previously for normal aging (Arendt et al., 1995b).
In patients carrying one apoE ε4 allele, the increase in dendritic length was much less pronounced. Plastic dendritic changes were reduced further in patients homozygous for the ε4 allele. These effects of the apoE polymorphism on reactive dendritic growth in AD also could be the reason for previous discrepancies between different studies on the extent of dendritic growth in AD (Buell and Coleman, 1979, 1981; Arendt et al., 1986, 1995b,c; Flood et al., 1991).
In ApoE ε4-carriers, newly formed dendritic elements were gene dose dependently shifted from distal segments to more proximal parts of the dendritic tree. This different distribution of growth processes resulted in an intensive pattern of growth that already was observed previously in AD (Arendt et al., 1995b). The intensity of growth, furthermore, was related only weakly to both the extent of neuronal loss and the progression of the disease in patients carrying one apoE ε4 allele. It was completely independent of these two parameters in patients homozygous for the ε4 allele. These findings might indicate an “uncoupling” of dendritic growth processes from their functional requirements under these conditions.
The gene dosage effects of the apoE ε4 allele on plastic neuronal changes observed in the present study were not restricted to dendrites and similarly could be detected on axons. In patients lacking apoE ε4, ChAT activity, determined in the frontal or parietal cortex, was reduced less drastically than the number of cholinergic neurons in the basal forebrain, giving rise to the cholinergic cortical innervation. In patients homozygous for ε4, on the contrary, loss of ChAT activity was more pronounced than loss of cholinergic neurons, whereas the situation was intermediate for patients carryingone ε4 allele. These findings imply either an upregulation of ChAT activity in surviving neurons in the absence of the apoE ε4 allele or compensatory axonal sprouting of surviving neurons. The present data do not allow us to distinguish between these two possibilities but clearly show the impairment of these plastic axonal processes in patients carrying ε4 allele.
ApoE genotype and stage dependency of reparative capacity
In the present study, the mean age of patients with apoE ε4 alleles was ∼8 years lower than for patients without ε4. This earlier age of onset is a consistent finding associated with apoE ε4/4 (Corder et al., 1993; Soininen, 1995). Other studies, furthermore, have reported on a more rapid progression of the disease in these patients (Bennett et al., 1995). These findings implicate that patients with apoE ε4 alleles might reach more advanced stages of the disease at a younger age than patients without ε4. Previous studies have indicated that younger AD patients show a more severe degeneration (Bird et al., 1983; Rossor et al., 1984; Perry et al., 1992; Arendt et al., 1995b; Soininen et al., 1995) and a more intense plastic response of subcortical neurons (Arendt et al., 1995c) than older patients. Comparative studies on carriers and noncarriers of the ε4 allele might, therefore, be biased by age-related effects. In the present study, two measures were taken to minimize the likelihood of such influence. First, effects related to different apoE genotypes were matched according to the clinical stage of the disease. Second, a parameter, the reparative capacity, was defined, which allowed us to distinguish effects of the ε4 allele on qualitative, rather than quantitative, grounds. Differences in the reparative capacity related to the presence of the apoE ε4 allele were independent of the clinical stage of the disease. These results clearly show that the gene dose of the apoE ε4 allele has an effect on the intensity of reactive dendritic remodeling.
Although dendritic elements continue to grow during the progression of AD in patients lacking the apoE ε4 allele, reparative capacity continuously declines in these patients. For the most advanced stages, they converge with the low levels of reparative capacity seen in the presence of the apoE ε4 allele. This observation indicates that, even in patients lacking the apoE ε4 allele, the presumptive reparative capacity is progressively exhausted. Differences in the mechanism attributed to the apoE-polymorphism might, therefore, be particularly relevant in early stages of the disease.
The present results support the assumption of an involvement of apoE in the repair of central neurons, as suggested by Poirier et al. (1991 a,b, 1993a) and Roses (1994). This proposed mechanism for apoE is supported further by recent observations on apoE knockout mice that show a severe impairment of age and lesion-related plastic synaptic changes (Masliah et al., 1995a,b).
An impairment of the expression of the ε4 gene and/or the functional properties of the E 4 protein might be related causally to the aberrancies of neuronal repair in AD and might, thus, explain the effect of apoE-polymorphism on the onset and development of AD at the cellular level.
It remains to be determined whether the molecular mechanisms behind these effects are related to genotype-specific alterations of apoE levels in the brain (Blennow et al., 1994; Bertrand et al., 1995), to isoform-specific differences in interactions with cellular proteins that mediate neurotrophic (Nathan et al., 1994; Bellosta et al., 1995;Holtzman et al., 1995) or cytotoxic (Crutcher et al., 1994; Clay et al., 1995) effects, or to molecular interactions with the β/A4-amyloid (Wisniewski and Frangione, 1992; Strittmatter et al., 1993b; Wisniewski et al., 1993; Gallo et al., 1994; Castano et al., 1995; Evans et al., 1995) or microtubule-associated proteins (Cotton et al., 1994; Huang et al., 1994, 1995; Strittmatter et al., 1994a,b;Whitson et al., 1994).
Despite these clear-cut effects of the ε4 allele on neuronal plasticity in AD, the plastic neuronal response was not completely absent, even in patients homozygous for the ε4 allele. These observations indicate that plastic reorganization under conditions of neurodegeneration is not disturbed completely but appears to be uncoupled from its functional demands. It might, therefore, be hypothesized that effects mediated by the presence of the apoE ε4 allele are not the direct cause of the disease. These effects, instead, might lead to a more rapid functional decompensation of a neuronal system under the conditions of a slowly progressing degeneration, which results in a more early onset and more rapid progression of the disease.
Footnotes
This work was supported by the Bundesministerium für Bildung, Forschung und Technologie (BMBF), Interdisciplinary Centre for Clinical Research at the University of Leipzig (01KS9504, Project C1).
Correspondence should be addressed to Dr. Thomas Arendt, Paul Flechsig Institute, Jahnallee 59, 04109 Leipzig, Germany.
REFERENCES
- 1.Aletrino MA, Vogels OJM, Vandomburg PHMF. Cell loss in the nucleus-raphe-dorsalis in Alzheimer’s disease. Neurobiol Aging. 1992;13:461–468. doi: 10.1016/0197-4580(92)90073-7. [DOI] [PubMed] [Google Scholar]
- 2.American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM-IIIR), 3rd Ed, rev. American Psychiatric Association; Washington, DC: 1987. [Google Scholar]
- 3.Arendt T, Bigl V. Alzheimer’s disease as a presumptive threshold phenomenon. Neurobiol Aging. 1987;8:552–554. doi: 10.1016/0197-4580(87)90131-x. [DOI] [PubMed] [Google Scholar]
- 4.Arendt T, Brückner MK. Preserved neuronal plasticity in the reticular core during aging and in Alzheimer’s disease. In: Wurtman RJ, Corkin SH, Growdon JH, editors. Alzheimer’s disease: advances in basic research and therapies. Center for Brain Sciences and Metabolism Charitable Trust; Cambridge, MA: 1991. pp. 250–256. [Google Scholar]
- 5.Arendt T, Brückner MK. Perisomatic sprouts immunoreactive for nerve growth factor receptor and neurofibrillary degeneration affect different neuronal populations in the basal nucleus in patients with Alzheimer’s disease. Neurosci Lett. 1992;148:63–66. doi: 10.1016/0304-3940(92)90805-h. [DOI] [PubMed] [Google Scholar]
- 6.Arendt T, Zvegintseva H. Alzheimer’s disease: increase in dendritic branching of reticular neurons in the basal nucleus—a sign of regeneration? In: Dowdell MJ, Hawthorne JN, editors. Cellular and molecular basis of cholinergic function. VCH Verlagsgesellschaft; Weinheim, Germany: 1987. pp. 869–873. [Google Scholar]
- 7.Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans, and Korsakoff’s disease. Acta Neuropathol (Berl) 1983;61:101–108. doi: 10.1007/BF00697388. [DOI] [PubMed] [Google Scholar]
- 8.Arendt T, Zvegintseva HG, Leontovich TA. Dendritic changes in the basal nucleus of Meynert and in the diagonal band nucleus in Alzheimer’s disease—a quantitative Golgi investigation. Neuroscience. 1986;19:1265–1278. doi: 10.1016/0306-4522(86)90141-7. [DOI] [PubMed] [Google Scholar]
- 9.Arendt T, Brückner MK, Bigl V. Maintenance of neuronal plasticity in the reticular core and changes in the trophic activity in Alzheimer’s disease. Ann NY Acad Sci. 1991;640:210–214. doi: 10.1111/j.1749-6632.1991.tb00219.x. [DOI] [PubMed] [Google Scholar]
- 10.Arendt T, Marcova L, Bigl V, Brückner MK. Dendritic reorganisation in the basal forebrain under degenerative conditions and its defects in Alzheimer’s disease. I. Dendritic organisation of the normal human basal forebrain. J Comp Neurol. 1995a;151:169–188. doi: 10.1002/cne.903510202. [DOI] [PubMed] [Google Scholar]
- 11.Arendt T, Brückner MK, Bigl V, Marcova L. Dendritic reorganisation in the basal forebrain under degenerative conditions and its defects in Alzheimer’s disease. II. Aging, Korsakoff’s disease, Parkinson’s disease, and Alzheimer’s disease. J Comp Neurol. 1995b;151:189–222. doi: 10.1002/cne.903510203. [DOI] [PubMed] [Google Scholar]
- 12.Arendt T, Brückner MK, Bigl V, Marcova L. Dendritic reorganisation in the basal forebrain under degenerative conditions and its defects in Alzheimer’s disease. III. The basal forebrain compared with other subcortical areas. J Comp Neurol. 1995c;151:223–246. doi: 10.1002/cne.903510204. [DOI] [PubMed] [Google Scholar]
- 13.Arendt T, Brückner MK, Pagliusi S, Krell T. Degeneration of rat cholinergic basal forebrain neurons and reactive changes in nerve growth factor expression after chronic neurotoxic injury. I. Degeneration and plastic response of basal forebrain neurons. Neuroscience. 1995d;65:633–645. doi: 10.1016/0306-4522(94)00526-b. [DOI] [PubMed] [Google Scholar]
- 14.Arendt T, Brückner MK, Krell T, Pagliusi S, Kruska L, Heumann R. Degeneration of rat cholinergic basal forebrain neurons and reactive changes in nerve growth factor expression after chronic neurotoxic injury. II. Reactive expression of the nerve growth factor gene in astrocytes. Neuroscience. 1995e;65:647–659. doi: 10.1016/0306-4522(94)00523-8. [DOI] [PubMed] [Google Scholar]
- 15.Basun H, Grut M, Winblad B, Lannfelt L. Apolipoprotein epsilon4 allele and disease progression in patients with late-onset Alzheimer’s disease. Neurosci Lett. 1995;183:32–34. doi: 10.1016/0304-3940(94)11107-t. [DOI] [PubMed] [Google Scholar]
- 16.Bellosta S, Nathan BP, Orth M, Dong LM, Mahley RW, Pitas RE. Stable expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells produce differential effects on neurite outgrowth. J Biol Chem. 1995;270:27063–27071. doi: 10.1074/jbc.270.45.27063. [DOI] [PubMed] [Google Scholar]
- 17.Bennett C, Crawford F, Osborne A, Diaz P, Hoyne J, Lopez R, Roques P, Duara R, Rossor M, Mullan M. Evidence that the apoE locus influences rate of disease progression in late-onset familial Alzheimer’s disease but is not causative. Am J Med Genet. 1995;60:1–6. doi: 10.1002/ajmg.1320600102. [DOI] [PubMed] [Google Scholar]
- 18.Benzing WC, Mufson EJ. Apolipoprotein E immunoreactivity within neurofibrillary tangles: relationship to tau and PHF in Alzheimer’s disease. Exp Neurol. 1995;132:162–171. doi: 10.1016/0014-4886(95)90021-7. [DOI] [PubMed] [Google Scholar]
- 19.Bertrand P, Poirier J, Oda T, Finch CE, Pasinetti GM. Association of apolipoprotein-E genotype with brain levels of apolipoprotein-E and apolipoprotein J (clusterin) in Alzheimer’s disease. Mol Brain Res. 1995;33:174–178. doi: 10.1016/0169-328x(95)00097-c. [DOI] [PubMed] [Google Scholar]
- 20.Bigl V. Modified microassay for the rapid and sensitive determination of choline acetyltransferase activity using 3H-acetyl CoA and Fonnum’s extraction procedure. Acta Biol Med Germ. 1975;34:1437–1440. [PubMed] [Google Scholar]
- 21.Bird TD, Stranahan S, Sumi SM, Raskind M. Alzheimer’s disease: choline acetyltransferase activity in brain tissue from clinical and pathological subgroups. Ann Neurol. 1983;14:284–293. doi: 10.1002/ana.410140306. [DOI] [PubMed] [Google Scholar]
- 22.Blennow K, Hesse C, Fredman P. Cerebrospinal fluid apolipoprotein E is reduced in Alzheimer’s disease. NeuroReport. 1994;5:2534–2536. doi: 10.1097/00001756-199412000-00032. [DOI] [PubMed] [Google Scholar]
- 23.Buell SJ. Golgi-Cox and rapid Golgi method as applied to autopsied human brain tissue: widely disparate results. J Neuropathol Exp Neurol. 1982;41:500–507. doi: 10.1097/00005072-198209000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Buell SJ, Coleman PD. Dendritic growth in the aged human brain and failure of growth in senile dementia. Science. 1979;206:854–856. doi: 10.1126/science.493989. [DOI] [PubMed] [Google Scholar]
- 25.Buell SJ, Coleman PD. Quantitative evidence for selective dendritic growth in normal human aging but not in senile dementia. Brain Res. 1981;214:23–41. doi: 10.1016/0006-8993(81)90436-4. [DOI] [PubMed] [Google Scholar]
- 26.Butcher LL, Woolf NJ. Neurotrophic agents may exacerbate the pathological cascade of Alzheimer’s disease. Neurobiol Aging. 1989;10:557–570. doi: 10.1016/0197-4580(89)90130-9. [DOI] [PubMed] [Google Scholar]
- 27.Castano EM, Prelli F, Pras M, Frangione B. Apolipoprotein E carboxyl-terminal fragments are complexed to amyloid-A and amyloid-L—implications for amyloidogenesis and Alzheimer’s disease. J Biol Chem. 1995;270:17610–17615. doi: 10.1074/jbc.270.29.17610. [DOI] [PubMed] [Google Scholar]
- 28.Chan-Palay V, Hochli M, Jentsch B, Leonard B, Zetzsche T. Raphe serotonin neurons in the human brain stem in normal controls and patients with senile dementia of the Alzheimer type and Parkinson’s disease—relationship to the monoamine oxidase enzyme localization. Dementia. 1992;3:253–269. [Google Scholar]
- 29.Clay MA, Anantharamaiah GM, Mistry MJ, Balasubramaniam A, Harmony JAK. Localization of a domain in apolipoprotein E with both cytostatic and cytotoxic activity. Biochemistry. 1995;34:11142–11151. doi: 10.1021/bi00035a020. [DOI] [PubMed] [Google Scholar]
- 30.Coleman PD, Flood DG. Neuron numbers and dendritic extent in normal aging and Alzheimer’s disease. Neurobiol Aging. 1987;8:521–545. doi: 10.1016/0197-4580(87)90127-8. [DOI] [PubMed] [Google Scholar]
- 31.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 32.Cotton P. Constellation of risks and processes seen in search for Alzheimer’s clues. JAMA. 1994;271:89–91. [PubMed] [Google Scholar]
- 33.Crutcher KA, Clay A, Scott SA, Tian X, Tolar M, Harmony JAK. Neurite degeneration elicited by apolipoprotein E peptides. Exp Neurol. 1994;130:120–126. doi: 10.1006/exnr.1994.1191. [DOI] [PubMed] [Google Scholar]
- 34.Czech C, Förstl H, Hentschel F, Monning U, Besthorn C, Geigerkabisch C, Sattel H, Masters C, Beyreuther K. Apolipoprotein E4 gene dose in clinically diagnosed Alzheimer’s disease—prevalence, plasma cholesterol levels, and cerebrovascular change. Eur Arch Psychiatry Clin Neurosci. 1994;243:291–292. doi: 10.1007/BF02191587. [DOI] [PubMed] [Google Scholar]
- 35.De Ruiter JP. The influence of postmortem fixation delay on the reliability of the Golgi silver impregnation. Brain Res. 1983;266:143–147. doi: 10.1016/0006-8993(83)91317-3. [DOI] [PubMed] [Google Scholar]
- 36.Elshourbagy NA, Laio WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc Natl Acad Sci USA. 1985;82:203–207. doi: 10.1073/pnas.82.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esiri MM, Pearson RCA, Steele JE, Bowen DM, Powell TPS. A quantitative study of the neurofibrillary tangles and the choline acetyltransferase activity in the cerebral cortex and the amygdala in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1990;53:161–165. doi: 10.1136/jnnp.53.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans KC, Berger EP, Cho CG, Weisgraber KH, Lansbury PT. Apolipoprotein-E is a kinetic but not a thermodynamic inhibitor of amyloid formation—implications for the pathogenesis and treatment of Alzheimer’s disease. Proc Natl Acad Sci USA. 1995;92:763–767. doi: 10.1073/pnas.92.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrer I, Aymami A, Rovira A, Grau-Veciana JM. Growth of abnormal neurites in atypical Alzheimer’s disease: a study with the Golgi method. Acta Neuropathol (Berl) 1983;59:167–170. doi: 10.1007/BF00703200. [DOI] [PubMed] [Google Scholar]
- 40.Ferrer I, Guionnet N, Cruz-Sanchez F, Tunon T. Neuronal alterations in patients with dementia: a Golgi study on biopsy samples. Neurosci Lett. 1990;114:11–16. doi: 10.1016/0304-3940(90)90420-e. [DOI] [PubMed] [Google Scholar]
- 41.Fischer S, Fischer S (1987) Der Nucleus basalis Meynert im Alternsgang beim Menschen—Quantitativ morphologische Untersuchung. PhD thesis, University of Leipzig.
- 42.Flood DG, Stechna SB, Hanks SD. Stability of dendritic extent in neurons of the vertical limb of the diagonal band of Broca in Alzheimer’s disease. In: Iqbal K, McLachlan DRC, Winblad B, Wisniewski HM, editors. Alzheimer’s disease: basic mechanisms, diagnosis, and therapeutic strategies. Wiley; New York: 1991. pp. 137–143. [Google Scholar]
- 43.Fonnum F. Radiochemical microassay for the determination of choline acetyltransferase and acetylcholinesterase activities. Biochem J. 1969;115:465–472. doi: 10.1042/bj1150465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Förstl H, Burns A, Luthert P, Cairns N, Lantos P, Levy R. Clinical and neuropathological correlates of depression in Alzheimer’s disease. Psychol Med. 1992;22:877–884. doi: 10.1017/s0033291700038459. [DOI] [PubMed] [Google Scholar]
- 45.Fritschy JM, Grzanna R. Restoration of ascending noradrenergic projections by residual locus coeruleus neurons—compensatory response to neurotoxin-induced cell death in the adult rat brain. J Comp Neurol. 1992;321:421–441. doi: 10.1002/cne.903210309. [DOI] [PubMed] [Google Scholar]
- 46.Gallo G, Wisniewski T, Choimiura NH, Ghiso J, Frangione B. Potential role of apolipoprotein-E in fibrillogenesis. Am J Pathol. 1994;145:526–530. [PMC free article] [PubMed] [Google Scholar]
- 47.Gearing M, Schneider JA, Robins RS, Hollister RD, Mori H, Games D, Hyman BT, Mirra SS. Regional variation in the distribution of apolipoprotein-E and Aβ in Alzheimer’s disease. J Neuropathol Exp Neurol. 1995;54:833–841. doi: 10.1097/00005072-199511000-00010. [DOI] [PubMed] [Google Scholar]
- 48.Geddes JW, Monaghan DT, Cotman CW, Lott IT, Kim RC, Chui HC. Plasticity of hippocampal circuitry in Alzheimer’s disease. Science. 1985;230:1179–1181. doi: 10.1126/science.4071042. [DOI] [PubMed] [Google Scholar]
- 49.German DC, White CL, Sparkman DR. Alzheimer’s disease: neurofibrillary tangles in nuclei that project to cerebral cortex. Neuroscience. 1987;21:305–312. doi: 10.1016/0306-4522(87)90123-0. [DOI] [PubMed] [Google Scholar]
- 50.Goto S, Hirano A, Matsumoto S. Immunohistochemical study of the striatal efferents and nigral dopaminergic neurons in parkinsonism-dementia complex on Guam in comparison with those in Parkinson’s disease and Alzheimer’s disease. Ann Neurol. 1990;27:520–527. doi: 10.1002/ana.410270511. [DOI] [PubMed] [Google Scholar]
- 51.Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- 52.Halliday GM, McCann HL, Pamphlett R, Brooks WS, Creasey H, McCusker E, Cotton RGH, Broe GA, Harper CG. Brainstem serotonin synthesizing neurons in Alzheimer’s disease—a clinicopathological correlation. Acta Neuropathol (Berl) 1992;84:638–650. doi: 10.1007/BF00227741. [DOI] [PubMed] [Google Scholar]
- 53.Herzog AG, Kemper TL. Amygdaloid changes in aging and dementia. Arch Neurol. 1980;37:625–629. doi: 10.1001/archneur.1980.00500590049006. [DOI] [PubMed] [Google Scholar]
- 54.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- 55.Holtzman DM, Pitas RE, Kilbridge J, Nathan B, Mahley RW, Bu G, Schwartz AL. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc Natl Acad Sci USA. 1995;92:9480–9484. doi: 10.1073/pnas.92.21.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang DY, Goedert M, Jakes R, Weisgraber KH, Garner CC, Saunders AM, Pericak-Vance MA, Schmechel DE, Roses AD, Strittmatter WJ. Isoform-specific interactions of apolipoprotein E with the microtubule-associated protein MAP2c: implications for Alzheimer’s disease. Neurosci Lett. 1994;182:55–58. doi: 10.1016/0304-3940(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 57.Huang DY, Weisgraber KH, Goedert M, Saunders AM, Roses AD, Strittmatter WJ. ApoE3 binding to tau tandem repeat I is abolished by tau serine262 phosphorylation. Neurosci Lett. 1995;192:209–212. doi: 10.1016/0304-3940(95)11649-h. [DOI] [PubMed] [Google Scholar]
- 58.Ignatius MJ, Gebicke-Harter PJ, Skene JH, Schilling JW, Weisgraber KH, Mahley RW, Shooter EM. Expression of apolipoprotein E during nerve degeneration and regeneration. Proc Natl Acad Sci USA. 1986;83:1125–1129. doi: 10.1073/pnas.83.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ihara Y. Massive somatodendritic sprouting of cortical neurons in Alzheimer’s disease. Brain Res. 1988;459:138–144. doi: 10.1016/0006-8993(88)90293-4. [DOI] [PubMed] [Google Scholar]
- 60.Jellinger K. The pedunculopontine nucleus in Parkinson’s disease, progressive supranuclear palsy, and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1988;51:540–543. doi: 10.1136/jnnp.51.4.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khatchaturian ZS. Diagnosis of Alzheimer’s disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 62.Kida E, Golabek AA, Wisniewski T, Wisniewski KE. Regional differences in apolipoprotein E immunoreactivity in diffuse plaques in Alzheimer’s disease brain. Neurosci Lett. 1994;167:73–76. doi: 10.1016/0304-3940(94)91030-8. [DOI] [PubMed] [Google Scholar]
- 63.Kinoshita M, Arai H, Fukasawa M, Watanabe T, Tsukamoto K, Hashimoto Y, Inoue K, Kurokawa K, Teramoto T. Apolipoprotein E enhances lipid exchange between lipoproteins mediated by cholesteryl ester transfer protein. J Lipid Res. 1993;34:261–268. [PubMed] [Google Scholar]
- 64.Lapchak PA, Jenden DJ, Hefti F. Compensatory elevation of acetylcholine synthesis in vivo by cholinergic neurons surviving partial lesions of the septohippocampal pathway. J Neurosci. 1991;11:2821–2828. doi: 10.1523/JNEUROSCI.11-09-02821.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lippa CF, Hamos JE, Pulaskisalo D, Degennaro LJ, Drachmann DA. Alzheimer’s disease and aging—effects of perforant pathway perikarya and synapse. Neurobiol Aging. 1992;13:405–411. doi: 10.1016/0197-4580(92)90115-e. [DOI] [PubMed] [Google Scholar]
- 66.Locke PA, Conneally PM, Tanzi RE, Gusella JF, Haines- JL. Apolipoprotein-E4 allele and Alzheimer’s disease—examination of allelic association and effect on age at onset in both early-onset and late-onset cases. Genet Epidemiol. 1995;12:83–92. doi: 10.1002/gepi.1370120108. [DOI] [PubMed] [Google Scholar]
- 67.Mahley RW. Apolipoprotein E: cholesterol transprotein with expanding role in cell biology. Science. 1988;240:622–629. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 68.Mann DMA, Yates PO, Hawkes J. The pathology of the human locus coeruleus. Clin Neuropathol. 1983;2:1–7. [PubMed] [Google Scholar]
- 69.Marsden CD. Extrapyramidal diseases. In: Davison AN, Thompson RHS, editors. The molecular basis of neuropathology. Arnold; London: 1981. pp. 345–383. [Google Scholar]
- 70.Masliah E, Mallory M, Alford M, Ge N, Mucke L. Abnormal synaptic regeneration in happ(695), transgenic, and apoE knockout mice. In: Iqbal K, editor. Research advances in Alzheimer’s disease and related disorders. Wiley; Chichester, UK: 1995a. pp. 405–414. [Google Scholar]
- 71.Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, Roses AD. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp Neurol. 1995b;136:107–122. doi: 10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- 72.Mayeux R, Sterm Y, Ottman R, Tatemichi TK, Tang MX, Maestre G, Ngai C, Tycko B, Ginsberg H. The apolipoprotein E4 allele in patients with Alzheimer’s disease. Ann Neurol. 1993;34:752–754. doi: 10.1002/ana.410340527. [DOI] [PubMed] [Google Scholar]
- 73.McKee AC, Kowall NW, Kosik KS. Microtubular reorganization and dendritic growth response in Alzheimer’s disease. Ann Neurol. 1989;26:652–659. doi: 10.1002/ana.410260511. [DOI] [PubMed] [Google Scholar]
- 74.McKhann G, Drachmann D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 75.Mesulam M-M, Mufson EJ, Levey AI, Wainer BH. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- 76.Monfort JC, Javoy-Agid F, Hauw JJ, Dubois B, Agid Y. Brain glutamate decarboxylase in Parkinson’s disease with particular reference to premortem severity index. Brain. 1985;108:301–313. doi: 10.1093/brain/108.2.301. [DOI] [PubMed] [Google Scholar]
- 77.Nagy ZS, Esiri MM, Jobst KA, Johnston C, Litchfield S, Sim E, Smith AD. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience. 1995;69:757–761. doi: 10.1016/0306-4522(95)00331-c. [DOI] [PubMed] [Google Scholar]
- 78.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 79.Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–852. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- 80.Ohm TG, Kirca M, Bohl J, Scharnagl H, Gross W, März W. Apolipoprotein E polymorphism influences not only cerebral senile plaque load but also Alzheimer type neurofibrillary tangle formation. Neuroscience. 1995;66:583–587. doi: 10.1016/0306-4522(94)00596-w. [DOI] [PubMed] [Google Scholar]
- 81.Oyama F, Shimada H, Oyama R, Ihara Y. Apolipoprotein-E genotype, Alzheimer’s pathologies, and related gene expression in the aged population. Mol Brain Res. 1995;29:92–98. doi: 10.1016/0169-328x(94)00233-5. [DOI] [PubMed] [Google Scholar]
- 82.Perry EK, Johnson M, Kerwin JM, Piggott MA, Court JA, Shaw PJ, Ince PG, Brown A, Perry RH. Convergent cholinergic activities in aging and Alzheimer’s disease. Neurobiol Aging. 1992;13:393–400. doi: 10.1016/0197-4580(92)90113-c. [DOI] [PubMed] [Google Scholar]
- 83.Peterson GL. A simplification of the protein assay method of Lowry et al., which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 84.Poirier J. Apolipoprotein E in animal models of CNS injury and in Alzheimer’s disease. Trends Neurosci. 1994;17:525–530. doi: 10.1016/0166-2236(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 85.Poirier J, Hess M, May PC, Finch CE. Cloning of hippocampal poly(A) RNA sequences that increase after entorhinal cortex lesion in adult rat. Mol Brain Res. 1991a;9:191–195. doi: 10.1016/0169-328x(91)90002-f. [DOI] [PubMed] [Google Scholar]
- 86.Poirier J, Hess M, May PC, Finch CE. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Mol Brain Res. 1991b;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- 87.Poirier J, Baccichet A, Dea D, Gauthier S. Cholesterol synthesis and lipoprotein reuptake during synaptic remodelling in hippocampus in adult rats. Neuroscience. 1993a;55:81–90. doi: 10.1016/0306-4522(93)90456-p. [DOI] [PubMed] [Google Scholar]
- 88.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993b;342:697–699. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 89.Poirier J, Delisle MC, Quirion R, Aubert I, Farlow M, Lahiri D, Hui S, Bertrand P, Nalbantoglu J, Gilfix BM, Gauthier S. Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci USA. 1995;92:12260–12264. doi: 10.1073/pnas.92.26.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Probst A, Basler V, Bron B, Ulrich J. Neuritic plaques in senile dementia of Alzheimer type: a Golgi analysis in the hippocampal region. Brain Res. 1983;268:249–254. doi: 10.1016/0006-8993(83)90490-0. [DOI] [PubMed] [Google Scholar]
- 91.Ramirez V, Ulfhake B. Anatomy of dendrites in motoneurons supplying the intrinsic muscles of the foot sole in the aged cat—evidence for dendritic growth and neo-synaptogenesis. J Comp Neurol. 1992;316:1–16. doi: 10.1002/cne.903160102. [DOI] [PubMed] [Google Scholar]
- 92.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 93.Reisberg B, Ferris SH, deLeon MJ, Crook T. The global deterioration scale for assessment of primary degenerative dementia. Am J Psychiatry. 1982;139:1136–1139. doi: 10.1176/ajp.139.9.1136. [DOI] [PubMed] [Google Scholar]
- 94.Roses AD (1994) Apolipoprotein E affects the rate of Alzheimer disease expression: β-amyloid burden is a secondary consequence dependent on apoE genotype and duration of disease. J Neuropathol Exp Neurol 429–437. [DOI] [PubMed]
- 95.Rossor MN, Iversen LL, Reynolds GP, Mountjoy CQ, Roth M. Neurochemical characteristics of early and late onset types of Alzheimer’s disease. Br Med J. 1984;288:961–964. doi: 10.1136/bmj.288.6422.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual, 2nd Ed. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 97.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-Maclachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Association of apolipoprotein E allele 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 98.Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- 99.Scheibel AB. Dendritic changes in senile and presenile dementia. In: Katzman R, editor. Congenital and acquired cognitive disorders. Raven; New York: 1979. pp. 107–122. [PubMed] [Google Scholar]
- 100.Scheibel AB, Tomiyasu U. Dendritic sprouting in Alzheimer’s presenile dementia. Exp Neurol. 1978;60:1–8. doi: 10.1016/0014-4886(78)90164-4. [DOI] [PubMed] [Google Scholar]
- 101.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein-E genotype in late- onset Alzheimer’s disease. Proc Natl Acad Sci USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Scott SA. Dendritic atrophy and remodeling of amygdaloid neurons in Alzheimer’s disease. Dementia. 1993;4:264–272. doi: 10.1159/000107332. [DOI] [PubMed] [Google Scholar]
- 103.Scott SA, DeKosky ST, Sparks DL, Knox CA, Scheff SW. Amygdala cell loss and atrophy in Alzheimer’s disease. Ann Neurol. 1992;32:555–563. doi: 10.1002/ana.410320412. [DOI] [PubMed] [Google Scholar]
- 104.Shortridge BA, Vogel FS, Burger PC. Topographic relationship between neurofibrillary change and acetylcholinesterase-rich neurons in the upper brainstem of patients with senile dementia of the Alzheimer’s type and Down’s syndrome. Clin Neuropathol. 1985;4:227–237. [PubMed] [Google Scholar]
- 105.Snipes GJ, McGuire CB, Norder JJ, Freeman JA. Nerve injury stimulates the secretion of apolipoprotein E by nonneuronal cells. Proc Natl Acad Sci USA. 1986;83:1130–1134. doi: 10.1073/pnas.83.4.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Soininen H, Kosunen O, Helisalmi S, Mannermaa A, Paljärvi L, Talasniemi S, Ryynänen M, Riekkinen P. A severe loss of choline acetyltransferase in the frontal cortex of Alzheimer patients carrying apolipoprotein epsilon4 allele. Neurosci Lett. 1995;187:79–82. doi: 10.1016/0304-3940(95)11343-6. [DOI] [PubMed] [Google Scholar]
- 107.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer’s disease. Proc Natl Acad Sci USA. 1993a;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein-E to synthetic amyloid beta-peptide isoform-specific effects and implications for late-onset Alzheimer’s disease. Proc Natl Acad Sci USA. 1993b;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R, Huang DY, Pericakvance M, Schmechel D, Roses AD. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer’s disease. Proc Natl Acad Sci USA. 1994a;91:11138–11186. doi: 10.1073/pnas.91.23.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Strittmatter WJ, Weisgraber KH, Goedert M, Saunders AM, Huang D, Corder EH, Dong LM, Jakes R, Alberts MJ, Gilbert JR, Han S-H, Hulette C, Einstein G, Schmechel DE, Pericak-Vance MA, Roses AD. Hypothesis: microtubule instability and paired helical filament formation in the Alzheimer disease brain are related to apolipoprotein E genotype. Exp Neurol. 1994b;125:163–171. doi: 10.1006/exnr.1994.1019. [DOI] [PubMed] [Google Scholar]
- 111.Wenham PR, Price WH, Blandell G. Apolipoprotein E genotyping by one-stage PCR. Lancet. 1991;337:1158–1159. doi: 10.1016/0140-6736(91)92823-k. [DOI] [PubMed] [Google Scholar]
- 112.Whitson JS, Mims MP, Strittmatter WJ, Yamaki T, Morrisett JD, Appel SH. Attenuation of the neurotoxic effects of Aβ amyloid peptide by apolipoprotein E. Biochem Biophys Res Commun. 1994;199:163–170. doi: 10.1006/bbrc.1994.1209. [DOI] [PubMed] [Google Scholar]
- 113.Williams RS, Ferrante RJ, Caviness VS., Jr The Golgi rapid method in clinical neuropathology: the morphological consequences of suboptimal fixation. J Neuropathol Exp Neurol. 1978;37:13–33. doi: 10.1097/00005072-197801000-00002. [DOI] [PubMed] [Google Scholar]
- 114.Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 115.Wisniewski T, Golabek A, Matsubara E, Frangione GJ. Apolipoprotein E: binding to soluble Alzheimer’s β-amyloid. Biochem Biophys Res Commun. 1993;192:359–365. doi: 10.1006/bbrc.1993.1423. [DOI] [PubMed] [Google Scholar]
- 116.Woolf NJ, Jacobs RW, Butcher LL. The pontomesencephalotegmental cholinergic system does not degenerate in Alzheimer’s disease. Neurosci Lett. 1989;96:277–282. doi: 10.1016/0304-3940(89)90391-1. [DOI] [PubMed] [Google Scholar]
- 117.Yamamoto T, Hirano A. Nucleus raphe dorsalis in Alzheimer’s disease: neurofibrillary tangles and loss of large neurons. Ann Neurol. 1985;17:573–577. doi: 10.1002/ana.410170608. [DOI] [PubMed] [Google Scholar]
- 118.Zweig RM, Ross CA, Hedreen JC, Steele C, Cardillo JE, Whitehouse P, Folstein MF, Price DL. The neuropathology of aminergic nuclei in Alzheimer’s disease. Ann Neurol. 1988;24:233–242. doi: 10.1002/ana.410240210. [DOI] [PubMed] [Google Scholar]