

Abstract
Anesthetic drugs are known to interact with GABAAreceptors, both to potentiate the effects of low concentrations of GABA and to directly gate open the ion channel in the absence of GABA; however, the site(s) involved in direct gating by these drugs is not known. We have studied the ability of alphaxalone (an anesthetic steroid) and pentobarbital (an anesthetic barbiturate) to directly activate recombinant GABAA receptors containing the α1, β2, and γ2L subunits. Steroid gating was not affected when either of two mutated β2 subunits [β2(Y157S) and β2(Y205S)] are incorporated into the receptors, although these subunits greatly reduce the affinity of GABA binding. These observations indicate that steroid binding and subsequent channel gating do not require these particular residues, as already shown for barbiturates. Bicuculline or gabazine (two competitive antagonists of GABA binding) reduced the currents elicited by alphaxalone and pentobarbital from wild-type GABAA receptors; however, gabazine produced only a partial block of responses to pentobarbital or alphaxalone, and bicuculline only partially blocked responses to pentobarbital. These observations indicate that the blockers do not compete with alphaxalone or pentobarbital for a single class of sites on the GABAAreceptor. Finally, at receptors containing α1β2(Y157S)γ2L subunits, both bicuculline and gabazine showed weak agonist activity and actually potentiated responses to alphaxalone. These observations indicate that the blocking drugs can produce allosteric changes in GABAA receptors, at least those containing this mutated β2 subunit. We conclude that the sites for binding steroids and barbiturates do not overlap with the GABA-binding site. Furthermore, neither gabazine nor bicuculline competes for binding at the steroid or barbiturate sites. The data are consistent with a model in which both gabazine and bicuculline act as allosteric inhibitors of channel opening for the GABAA receptor after binding to the GABA-binding site.
Keywords: GABAA receptor, GABA, neurosteroids, bicuculline, inverse agonist, anesthetics, allosteric inhibitor
GABA activates a ligand-gated ion channel (the GABAA receptor), which underlies most rapid inhibition in the brain. Various other compounds also bind to the GABAAreceptor and can gate the channel or modulate channel function (Macdonald and Olsen, 1993). In particular, steroids and barbiturates are each able to directly gate the GABAA receptor channel (in the absence of GABA), and they can also enhance the activation produced by low concentrations of GABA. It is not known whether the same sites are involved in producing direct gating and in potentiating the effects of GABA. For the sites involved in potentiation, however, the steroid-binding site and the barbiturate-binding site are distinct from each other and are also distinct from the GABA-binding site (Macdonald and Olsen, 1993). Because the characterized sites for steroid and barbiturate binding differ from the GABA-binding site, it is puzzling that a competitive antagonist of GABA binding, bicuculline, is also a potent blocker of channel gating by steroids (Barker et al., 1987) or pentobarbital (Nicoll and Wojtowicz, 1980). We are interested in defining the sites on the GABAA receptor that are involved in direct gating by anesthetics, and we have initiated studies of channel activation by alphaxalone (an anesthetic steroid analog) and pentobarbital (an anesthetic barbiturate).
Accordingly, we have examined the ability of alphaxalone to gate mutated GABAA receptors, and we found that residues that are important in determining the binding affinity of GABA do not affect activation by steroids. We also examined the actions of blocking drugs and found that neither bicuculline nor gabazine are competitive inhibitors of currents gated by alphaxalone or pentobarbital. Finally, both gabazine and bicuculline act as weak agonists for GABAA receptors containing the β2(Y157S) mutated subunit. These data indicate that steroids and barbiturates do not bind to the GABA-binding site when they directly gate the channel of the GABAA receptor. Furthermore, the data support the idea that bicuculline and gabazine act as negative allosteric modulators of function of GABAA receptors.
MATERIALS AND METHODS
All chemicals were from Sigma (St. Louis, MO) unless specified otherwise. Gabazine (SR-95531) and alphaxalone were obtained from Research Biochemicals International (Natick, MA).
A complementary DNA construct for the rat α1 subunit of the GABA receptor was provided by Dr. A. Tobin (University of California Los Angeles). The rat γ2L and β2 subunits and the point mutants β2(Y205S) and β2(Y157S) have been described (Amin and Weiss, 1993). GABA receptor subunit cDNAs were transferred to the eucaryotic expression vector pcDNA3 (Invitrogen, San Diego, CA), for expression in QT6 cells. Direct sequencing of the mutated β2 subunits confirmed that the constructs contained the appropriate base changes (Sequenase version 2 kit; Amersham, Arlington Heights, IL).
Quail fibroblasts (QT6 cells; initially provided by Dr. J. Merlie, Washington University) were maintained in Medium 199 (Earle’s salts) containing 5% fetal bovine serum (Hyclone, Logan, UT), 10% tryptose phosphate broth (Life Technologies, Grand Island, NY), 1% dimethylsulfoxide (DMSO), and penicillin (100 U/ml) plus streptomycin (100 μg/ml) in a humidified atmosphere containing 5% CO2. Calcium phosphate precipitation was used to transfect QT6 cells (Chen and Okayama, 1987), with the additional step of an initial wash to remove tryptose phosphate broth (Phillips et al., 1991). QT6 cells were used for expression because of anomalous results obtained when subunits were expressed in HEK293 cells (Ueno et al., 1996b).
Cells that expressed a high level of protein from exogenous cDNA were identified using the bead-labeling technique described by Jurman et al. (1994). We inserted a flag epitope tag into the N-terminal region of the α1 subunit (Ueno et al., 1996b). Starting with the N terminus of the predicted mature peptide, the predicted sequence of the resulting peptide is YGQPSQDEL KDYKDDDDKLKDNTT, where the introduced residues are shown underlined. This construct was identified on the surface of intact cells using a mouse monoclonal antibody to the FLAG epitope (M2, Eastman Kodak Scientific Imaging Systems, New Haven, CT), which had been adsorbed to beads with covalently attached goat anti-mouse IgG antibody (Dynal, Great Neck, NY). Control experiments indicated that the tag had no functional effects on receptors incorporating the tagged α1 subunit (Ueno et al., 1996b).
Recordings were made using standard whole-cell methods (Hamill et al., 1981) 24–72 hr. after transfection. All experiments were performed at room temperature (21–23°C), and drugs were dissolved in external solution. In all cases, data were obtained from isolated single cells. Experiments were performed in two laboratories. In the Steinbach laboratory (Ueno et al., 1996a), drugs were applied with a polyethylene “Y tube.” The bath was perfused continuously with normal external solution from a separate perfusion line, and solution was removed from the bath with a Leiden aspirator (Medical Systems, Greenvale, NY). In the Zorumski laboratory (Hu et al., 1993), drugs were applied by pressure ejection from “puffer” pipettes positioned within 5 μm of the patch-clamped cell, using a 500 msec pulse of air pressure (10–20 psi) to the back of the pipette. The data shown in Figure1A,B,D were obtained with puffer applications, the other data with Y-tube applications. Bicuculline methiodide was dissolved in saline and used within 2 hr. Stock solutions of steroids were prepared in DMSO. The maximal concentration of DMSO in the final working solution was 0.2%, which had no effect on GABA-elicited currents (Rodgers-Neame et al., 1992). Sodium pentobarbital was dissolved in saline.
Fig. 1.

Activation of GABAA receptors containing β2 or β2(Y205S) subunits. Each panel shows concentration–response curves for an agonist: GABA (A), pentobarbital (PENT, B), alphaxalone (ALPH, C), and DHP-OH (D). In each panel, the open symbols show responses from QT6 cells transfected with α1β2γ2L subunits, whereas filled symbols show responses from cells transfected with α1β2(Y205S)γ2L subunits. GABA produced no gating of receptors containing the mutated subunit (points at 100 and 1000 μmGABA in A). For the other agonists tested, the data from receptors containing wild-type or mutated subunits were indistinguishable. The lines in A throughC show predictions derived from fitting an allosteric blocking model to data from receptors containing wild-type subunits (see Results). Symbols show mean for data from two to nine cells; error bars represent SD.
Concentration–effect curves were fit with the Hill equation using SigmaPlot (Jandel Scientific Software, San Rafael, CA). The ability of an allosteric blocking model to describe the observations was assessed by eye (see Results), using QuattroPro (Borland International, Scotts Valley, CA) to generate predicted blocking curves. Figures were produced using SigmaPlot.
RESULTS
Direct gating of GABAA receptors containing mutated β2 subunits
We initially examined GABAA receptors that contain a mutated β2 subunit, which had already been shown to have greatly reduced efficacy for gating by GABA but normal gating by pentobarbital (Amin and Weiss, 1993). Quail fibroblast cells (QT6) were transfected with cDNAs for α1 and γ2L subunits and for either wild-type β2 or β2(Y205S) mutated subunits. Responses were measured by whole-cell patch-clamp recordings. We found that direct gating by alphaxalone (Fig. 1C) or 5α-pregnan-3α-ol-20-one (DHP-OH; Fig. 1D) was indistinguishable for receptors containing the wild-type or mutated β2 subunits. We also confirmed that gating by pentobarbital was unaltered (Fig. 1B) (Amin and Weiss, 1993) and that cells expressing α1β2(Y205S)γ2L subunits showed no response to GABA (100–1000 μm; Fig.1A) (Amin and Weiss, 1993). Receptors containing a second point mutant of the β2 subunit β2(Y157S) could also be gated by 10 μm alphaxalone (Fig. 4), although the concentration–response relationship was not examined.
Fig. 4.

Responses to alphaxalone of cells expressing mutated β2 subunits. Each panel shows traces recorded from a cell exposed to 10 μm alphaxalone (dotted trace) and then to 10 μm alphaxalone plus 1 mm of an antagonist (solid trace). Actions of 1 mm gabazine are shown in the top rowand of 1 mm bicuculline in the bottom row. Cells transfected with α1β2(Y157S)γ2L subunits (left) showed potentiation between alphaxalone and either gabazine or bicuculline. Cells transfected with α1β2(Y205S)γ2L subunits showed block by either compound, but the block produced by bicuculline was reduced over that seen with wild-type receptors, whereas the block produced by gabazine was increased (see Figs. 3, 6). Calibration in each panel: 20 pA, 10 sec.
The responses to 30 mm pentobarbital showed a decrease from responses to 10 mm pentobarbital (data not shown), in agreement with previous results showing that high concentrations of pentobarbital produce “autoinhibition” (Akaike et al., 1987a). The maximal concentrations of steroids (alphaxalone and DHP-OH) were limited by the aqueous solubility, so that concentrations above 100 μm could not be used.
Inhibition by bicuculline or gabazine of currents gated by alphaxalone or pentobarbital from wild-type receptors
We then examined the ability of competitive inhibitors of GABA binding to block gating of GABAA receptors containing wild-type β2 subunits by GABA, alphaxalone, or pentobarbital. Both bicuculline and gabazine (SR 95531) have been characterized as competitive inhibitors of GABA binding to the GABAAreceptor (see Discussion). Both drugs were able to reduce currents elicited by each of the three agonists (Fig. 2). Bicuculline was approximately equally potent at blocking responses to 10 μm alphaxalone, 3 μm GABA, and 300 μm pentobarbital (Fig. 3A). When the concentration–effect curves were described by the Hill equation, the concentrations required to reduce the response by 50% (the IC50) were 0.9 μm, 0.9 μm, and 1.0 μm, whereas the Hill coefficients were 0.94, 1.01, and 0.77, respectively, for responses to alphaxalone, GABA, and pentobarbital. It is interesting that the IC50 values are so similar, because this might be expected if bicuculline blocked responses of all three agonists by binding to a single type of site. The observation that the Hill coefficients were close to 1 suggests that block can be produced after the binding of a single antagonist molecule.
Fig. 2.

Action of blocking drugs on responses elicited from cells expressing α1β2γ2L receptors. Each panel shows traces recorded from a cell exposed to an agonist (dotted trace), or the same concentration of the agonist plus 10 μm of a blocking agent (solid trace). All cells were transfected with wild-type (α1β2γ2L) subunits. Theleft column shows the action of 10 μmbicuculline, whereas the right column shows the action of gabazine. Currents were elicited with 3 μm GABA (top row), 10 μm alphaxalone (middle row), or 300 μm pentobarbital (bottom row). Calibration in each panel: 20 pA, 10 sec. These records and those shown in Figures 4 and 5 were recorded at a holding potential of 0 mV, with a reversal potential for the responses of approximately −30 mV. Hence, the evoked currents are inward. Drugs were applied with a Y tube.
Fig. 3.

Bicuculline and gabazine block responses to GABA, pentobarbital, and alphaxalone. The agonists GABA (3 μm,open circles), alphaxalone (10 μm,filled triangles), and pentobarbital (300 μm, inverted open triangles) were applied to cells transfected with α1β2γ2L subunits, in the absence of a blocking drug and then in the presence of various concentrations of bicuculline (A) or gabazine (B). The figure shows the ratio of the response in the presence of a blocker to the response in the same cell in the absence of a blocker. The lines superimposed on the data (dotted lines, 3 μm GABA; solid lines, 10 μmalphaxalone; dashed lines, 300 μmpentobarbital) show predictions derived from fitting an allosteric blocking model to data from receptors containing wild-type subunits (see Results). Symbols show mean for data from two to six cells; error bars represent SD.
We extended these observations by examining the ability of gabazine (SR 95531) to block currents gated by alphaxalone. Gabazine is more potent than bicuculline at blocking currents elicited by GABA (Fig.3B), with an IC50 for currents elicited by 3 μm GABA of ∼0.2 μm and a Hill coefficient of 1.0. Gabazine, however, could only reduce the currents elicited by 10 μm alphaxalone by ∼30%, for responses of receptors containing wild-type β2 subunits (Fig. 3B). The concentration of gabazine required to produce half the maximal block was ∼0.2 μm. Gabazine also could only produce a partial block of currents gated by 300 μm pentobarbital. The maximal reduction, again, was ∼30%, and the concentration of gabazine required to produce half the maximal block was ∼0.15 μm (Fig. 3B). Again, the IC50values are similar for currents elicited by all three agonists.
The observation that gabazine cannot produce a complete block demonstrates that it cannot compete for a single class of sites for activation by either alphaxalone or pentobarbital.
Actions of bicuculline and gabazine on receptors containing mutated β2 subunits
To explore further the interactions between alphaxalone and blocking agents, the effects of bicuculline and gabazine on GABAA receptors containing mutated β2 subunits were examined. Because the affinity of GABA is greatly reduced in receptors containing these mutated subunits (Amin and Weiss, 1993), it was expected that the blocking potency of bicuculline and gabazine would also be reduced.
Bicuculline was much less potent at blocking currents elicited by 10 μm alphaxalone from receptors containing the mutated β2(Y205S) subunit (Fig. 6A). Even at 1 mm bicuculline, the current was reduced only to 0.66 × control. The IC50 cannot be estimated, because the maximal block is not known, but it appears to be 1 mm or more.
Fig. 6.

Actions of bicuculline and gabazine on alphaxalone-elicited responses of GABAA receptors containing mutated β2 subunits. Relative responses are shown to 10 μm alphaxalone applied to cells containing α1β2γ2L subunits (open circles, dotted lines), α1β2(Y205S)γ2L subunits (filled squares, dashed lines), or α1β2(Y157S)γ2L subunits (filled triangles, solid lines). The data obtained with bicuculline are shown in A, data with gabazine are shown inB. The lines simply connect the points. Also shown are the responses of receptors containing α1β2(Y157S)γ2L subunits to blocker applied in the absence of alphaxalone (open triangles). Symbols show mean for data from two to five cells; error bars represent SD .
Surprisingly, bicuculline did not block currents activated by alphaxalone from receptors containing α1β2(Y157S)γ2L subunits. Instead, 1 mm bicuculline potentiated the response to 10 μm alphaxalone (Figs. 4,6A). The EC50 cannot be estimated, because the maximal potentiation is not known, but it must be >100 μm (Fig. 6A). Furthermore, bicuculline acted as a weak agonist at receptors containing the β2(Y205S) subunit, so that 1 mm bicuculline applied alone produced a response ∼10% the size of the response to 10 μmalphaxalone (Fig. 6A).
Gabazine was more efficacious at blocking alphaxalone-elicited responses from receptors containing β2(Y205S) subunits than for those containing wild-type β2 subunits. With wild-type β2 subunits, the current elicited by 10 μm alphaxalone in the presence of 1 mm gabazine was 0.75 × that seen in the absence of gabazine (0.75 ± 0.07, n = 3; mean ± SD), whereas with the β2(Y205S) subunit the current was reduced to 0.30 (± 0.07, n = 5; the difference is significant atp < 0.0001 by Student’s two-tailed ttest). However, the IC50 for block by gabazine was shifted to ∼100 μm (Fig. 6B).
In agreement with the observations made with bicuculline, gabazine was a weak agonist for receptors containing the β2(Y157S) subunit (Figs.5, 6B). The concentration of gabazine producing half-maximal current is likely to be between 10 and 100 μm (Fig. 5). There was strong potentiation when gabazine and alphaxalone were applied together (Figs.4, 6B). The concentration producing half-maximal potentiation appears to be between 10 and 100 μm (Fig.6B).
Fig. 5.

Partial agonist action of gabazine on cells expressing α1β2(Y157S)γ2L subunits. The responses of a single cell to applications of 10 μm alphaxalone (top trace, dotted) and 1000, 100, and 10 μm gabazine alone. Calibration in the top panel: 20 pA and 10 sec for all traces.
The concentration dependence of the actions of bicuculline and gabazine is consistent with the idea that affinities for both drugs are reduced by both point mutations, as expected. The β2(Y157S) mutation, however, also converts the blockers into weak agonists, whereas the β2(Y205S) mutation increases the efficacy of gabazine as a blocker.
Dependence of inhibition by bicuculline on the concentration of GABA, alphaxalone, or pentobarbital
Gabazine clearly does not act as a competitive inhibitor of currents elicited by steroids or barbiturates, because it produces only a partial block with wild-type receptors. To gain more insight into the mechanism of inhibition by bicuculline, we examined blocking curves at different doses of agonists using cells expressing α1β2γ2L (wild-type) GABAA receptors. The concentration–response curve for activation of these receptors by GABA can be described by the Hill equation, with an EC50 of 8 μm and a Hill coefficient of 1.7 (Fig. 1A). Activation by pentobarbital can be described by the Hill equation, with an EC50 of 590 μm and a Hill coefficient of 2.0 (Fig. 1B). Unfortunately, the low aqueous solubility of alphaxalone and other steroids limits the concentrations that can be applied, so the concentration–response curve is not well characterized (Fig. 1C) and the EC50 is not known. The data are presented as blocking curves at a given agonist concentration, rather than as concentration–response curves for agonists at given blocker concentrations. This second format is often used to demonstrate competitive inhibition, which is predicted to produce a parallel shift with no change in maximal current for the agonist. Unfortunately, the experiments could not be performed in this fashion. The maximal response for alphaxalone could not be measured under any conditions because of low aqueous solubility, whereas concentrations of pentobarbital >10 mm produce autoinhibition (see above).
GABA was tested at concentrations of 3, 10, and 30 μm, to cover the EC50 without using such high concentrations that desensitization became a major problem. As shown in Figure7A, the IC50 for bicuculline in blocking responses to GABA increased at higher GABA concentrations, from 0.9 μm with 3 μm GABA to 1.6 μm (10 μm GABA) and 5.8 μm(30 μm GABA). The block appeared to be complete at high enough bicuculline concentrations (Fig. 7A), and the Hill coefficients were close to 1 (1.01, 0.95, and 0.97, respectively).
Fig. 7.
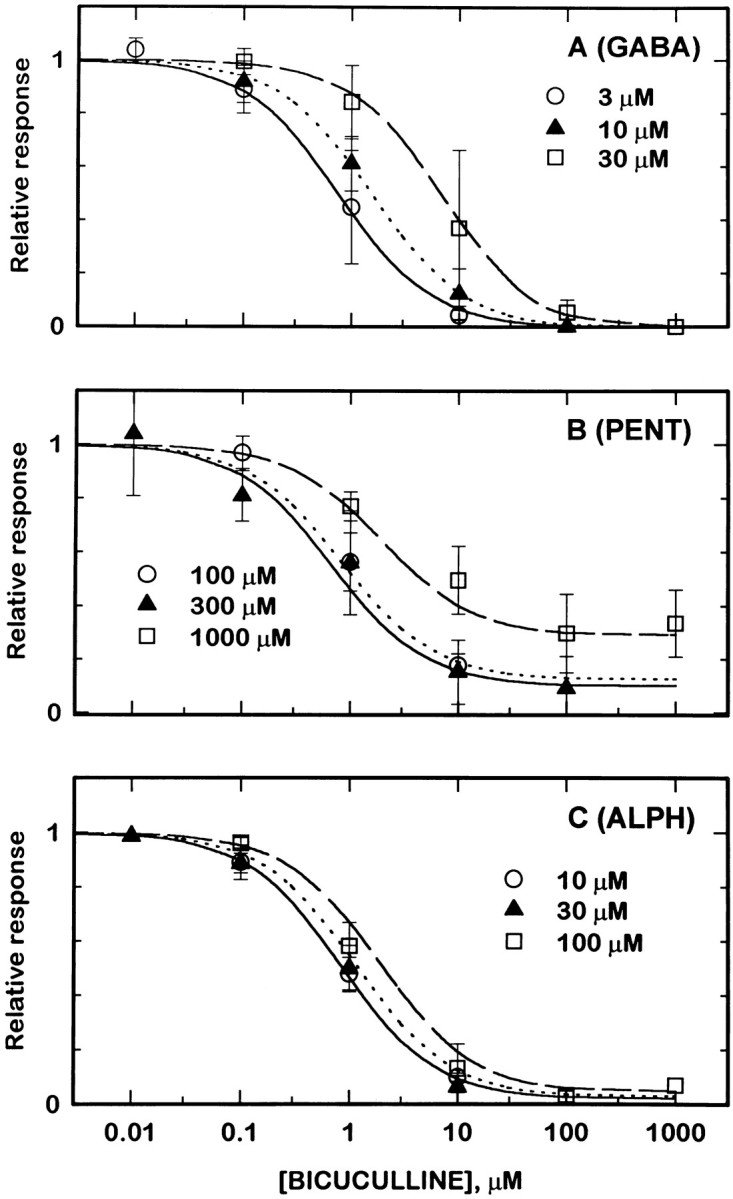
The blocking effect of bicuculline depends on the concentration of agonist used. Cells transfected with wild-type receptors (α1β2γ2L subunits). Relative responses are shown for responses to three concentrations of agonist. For each concentration or agonist, the responses in the presence of a blocker are normalized to the response of that cell to the same concentration of agonist alone.A shows data obtained with GABA as agonist: 3 μm GABA (open circles, solid line), 10 μm GABA (solid triangles, dotted line), and 30 μm GABA (open squares, dashed line). B shows data obtained with pentobarbital as agonist: 100 μm pentobarbital (open circles, solid line), 300 μm pentobarbital (solid triangles, dotted line), and 1000 μmpentobarbital (open squares, dashed line).C shows data obtained with alphaxalone as agonist: 10 μm alphaxalone (open circles, solid line), 30 μm alphaxalone (solid triangles, dotted line), and 100 μm alphaxalone (open squares, dashed line). The lines superimposed on the data show predictions derived from fitting an allosteric blocking model to data from receptors containing wild-type subunits (see Results).
The action of bicuculline on currents elicited by pentobarbital was more complex. Concentrations of 100, 300, and 1000 μmpentobarbital were used. At the highest concentration of pentobarbital, bicuculline was not able to block the response by more than ∼70%, even at 1 mm bicuculline (Fig. 7B). The IC50 values did not depend strongly on the concentration of pentobarbital, being 1.2 μm (100 μmpentobarbital), 0.9 μm (300 μm), and 2.4 μm (1000 mm). The Hill coefficients were close to 1 (0.91, 0.77, and 0.79, respectively). We were concerned that 1 mm pentobarbital might have effects not mediated by GABAA receptors, so untransfected cells were tested. One millimole pentobarbital did not produce a conductance increase in untransfected cells (six cells tested).
Alphaxalone was used at concentrations of 10, 30, and 100 μm. The IC50 for block by bicuculline increased only very slightly at higher alphaxalone concentrations (Fig.7C), from 0.9 μm to 1.0 μm and 1.3 μm. The Hill coefficients, again, were close to 1 (0.94, 0.87, and 1.2, respectively). The unblocked current at high concentrations of bicuculline did not differ significantly from zero.
The data in Figure 7 do not support the idea that bicuculline or gabazine act as open channel blockers. An open channel blocker would be expected to block more effectively as the probability of being open increased (Adams, 1976); this is not the case when block of currents elicited by different agonists or at different agonist concentrations are compared.
Bicuculline clearly does not act as a competitive antagonist for pentobarbital: the maximal block was not complete when 1 mmpentobarbital was used. The data for alphaxalone are more difficult to interpret, because the maximal concentration was limited by aqueous solubility and was likely to be less than the EC50 for gating by alphaxalone. Block of currents elicited by GABA, on the other hand, can be described by a competitive interaction between bicuculline and GABA. The calculated dissociation constant for bicuculline is ∼1.7 μm, assuming simple competition and the parameters for gating by GABA that are shown in Table 1. (In the case of gabazine interacting with GABA, the calculated dissociation constant for bicuculline is ∼0.4 μm.)
Table 1.
Values used in an allosteric blocking model
| Agonists | K1 (μm) | K2 (μm) | P1 | P2 |
|---|---|---|---|---|
| GABA | 50 | 500 | 350 | – |
| +Bicuculline | 50 | 500 | 350 | – |
| +Gabazine | 50 | 500 | 350 | – |
| Pentobarbital | 5000 | 5000 | 100 | – |
| +Bicuculline | 5000 | 5000 | 100 | 600 |
| +Gabazine | 5000 | 5000 | 100 | 100 |
| Alphaxalone | 200 | – | 6 | – |
| +Bicuculline | 200 | – | 6 | 6 |
| +Gabazine | 200 | – | 6 | 6 |
| Blockers | L1 (μm) | L2 (μm) | Q | |
| Bicuculline | 80 | 80 | 60 | |
| Gabazine | 0.6 | 0.6 | 0.6 | |
Parameters are defined in Results and Figure 8. A dash indicates that the parameter was not used in a particular fit. It was assumed that GABA bound to two sites with different microscopic dissociation constants (K1, K2). In the cases of pentobarbital, bicuculline, and gabazine, the microscopic dissociation constants are assumed to be identical. For alphaxalone, only a single site was assumed to exist. The equilibrium constants for activation are shown with only agonist bound (P1), or in the cases of pentobarbital and alphaxalone for activation with both agonist and blocker bound (P2). The blocking allosteric constant is shown as Q.
An allosteric model can describe the block by gabazine or bicuculline
The observations summarized in Figures 3 and 7 include the following points. First, the IC50 values for block by bicuculline of currents elicited by relatively low concentrations of GABA, alphaxalone, or pentobarbital are similar (Fig. 3A). Second, bicuculline cannot produce a complete block of a response to a high concentration of pentobarbital (Fig. 7B). Third, the IC50 values for block by gabazine of currents elicited by relatively low concentrations of GABA, alphaxalone, and pentobarbital are similar (Fig. 3B). Fourth, gabazine cannot produce a complete block of currents elicited by either alphaxalone or pentobarbital (Fig. 3B). Fifth, the blocking curves have Hill coefficients near 1. The similarities in IC50 values suggest that block results from the binding of antagonist to a single set of sites, no matter which agonist was used to elicit the response. The Hill coefficients suggest that binding of a single molecule of bicuculline or gabazine is sufficient to produce most of the block seen. The incomplete maximal block demonstrates that there cannot be a simple competitive interaction at a single site between the blockers and alphaxalone or pentobarbital.
One model for block that is consistent with these qualitative observations is an “allosteric model.” In this model, gabazine or bicuculline do not bind to the sites for alphaxalone or pentobarbital, but bind to distinct sites and produce inhibition by reducing the probability that the channel of the GABAA receptor will open after binding of alphaxalone or pentobarbital. In the particular version of the model that will be presented, it is assumed that bicuculline and gabazine bind to the GABA-binding site (see below). Hence, bicuculline and gabazine are assumed to act as “inverse agonists” at the GABA-binding site, as well as to prevent binding of GABA to that site.
The ability of an allosteric model to describe the data were tested by implementing simplified kinetic schemes (Fig. 8) and examining the steady-state predictions. We assumed that the receptor can adopt only three states. The first is the “resting” state (R), which has a closed channel but is activable. The resting state exists in various states of ligation by agonists and antagonists. The second state is the “active” state (R*), which is likely to be a complex of states, including the open state and some short-lived desensitized states (Maconochie et al., 1994; Jones and Westbrook, 1995). The active state, therefore, is not identical to the “open” state of the receptor. The third state is the “dead” state (r), which is a state induced after the binding of blocker. The dead state has a nonconducting channel and is mutually exclusive with the active state. The dead state was chosen as one way to model the effects of blockers. Alternative schemes would include an effect of blocker binding on agonist binding or a direct effect on the channel-opening rates for agonists. The choice of a dead state was made for two reasons. First, some evidence suggested that blockers can induce conformational changes in the GABAA receptor (see Results and Discussion). Second, the postulated dead state was treated as being determined solely by the blocking drug, rather than requiring an interaction between blocker and agonist, and so the mechanism seemed to be aesthetically preferable because it allowed us to set parameters for blocker interactions with the receptor, independent of the agonist used.
Fig. 8.

A diagram of the kinetic models used to assess the ability of a simplified allosteric model to describe the data. The models are described in Results. A shows the binding of blocker (X, bicuculline or gabazine) and the ensuing conformational change to the “dead” state (r). Note that the equilibrium constants (L1, L2, and Q) are omitted from B–D to simplify the figure. B–D show schemes for GABA, pentobarbital, and alphaxalone, respectively. Receptor states with open channels areboxed. B, GABA (G) was assumed to bind to the same two sites as blockers, so fewer heteroliganded forms of the receptor can occur. Furthermore, the channel can only activate when two GABA molecules are bound.C, Pentobarbital (B) was also assumed to bind to two sites, but in this case both pentobarbital and blockers can occupy sites on the same receptor. It was assumed that two pentobarbital molecules must be bound for a channel to activate and that dead receptors cannot activate. D, Alphaxalone (A) was assumed to bind to only a single site, but otherwise the scheme is identical to that for pentobarbital. Some binding steps (e.g., to the r state) are omitted for clarity in the figure.
We postulated the minimal number of sites required by the data as two sites for GABA, two sites for pentobarbital, and one site for alphaxalone (see below). We assumed that the blocking drugs bind to both of the GABA-binding sites and that binding of GABA and a blocker is mutually exclusive. We also constrained the parameters by requiring that there be minimal interaction between the drugs. For example, the dissociation constants for agonists are assumed to be independent of blocker binding and vice versa.
The interactions of the blocking drugs (symbolized as X; bicuculline or gabazine) with the receptor are shown in Figure 8A. Each can bind to two sites on the resting GABAA receptor (R). The number of sites was chosen to match the minimal number of GABA-binding sites, as GABA activation requires at least two sites (see below). It was assumed that the microscopic dissociation constant is the same for the two sites (L), so the microscopic dissociation constants are identical (L1 = L2 = L). After binding, the receptor can change state to the dead condition (r). For simplicity, the forward conformational change constant was assumed to be independent of the state of ligation, and Q gives the ratio Xr/XR. The observation that the Hill coefficient for block is close to 1 is consistent with this simplifying idea.
The interactions of GABA (G) are shown in Figure 8B. GABA was assumed to bind to two sites, because the Hill coefficient for gating is >1 (Fig. 1A). We show different microscopic dissociation constants for the two binding steps (K1 and K2), based on the results obtained by Maconochie et al. (1994). After two GABA molecules have bound, the resting receptor can change conformation to the active state (R*), with an activation equilibrium constant P1 = G2R*/G2R. We have neglected the contribution of open channels with only a single GABA molecule bound (Twyman et al., 1990). This was performed to simplify the model, but it seems justifiable because the Hill coefficient for activation by GABA suggests that most receptors with open channels also have two molecules of GABA bound. We assumed that GABA and the blocking drugs bind competitively to the same sites, so that the receptor cannot be activated by GABA if a blocker is bound to either site. Hence, there is only one state with an open channel, G2R*. To simplify the scheme, we assumed that parameters for blocker interaction (L, Q) are the same whether GABA is bound to one site or not.
The interactions of pentobarbital (B) are shown in Figure8C. Pentobarbital was assumed to bind to two sites, because the Hill coefficient for gating is >1 (Fig. 1B). In fitting the scheme, we assumed that the microscopic dissociation constant is identical for the two sites (because there are no data to indicate otherwise), so the microscopic dissociation constants are the same (K1 = K2 = K). We assumed that pentobarbital binding is unaffected by blocker and that channel opening could occur so long as the receptor is not dead (that is, so long as the receptor was not in the r state). Hence, there are three states with an open channel: B2R*, B2XR*, and B2X2R*. As shown in the scheme, we allowed for the possibility that channel activation could differ for receptors with only pentobarbital bound (P1) or with both pentobarbital and blocker bound (P2). Again, we assumed that parameters for blocker interaction (L, Q) are the same whether pentobarbital is bound or not.
The interactions of alphaxalone (A) are shown in Figure8D. Alphaxalone was assumed to bind to a single site, because the concentration–effect relationship had a limiting slope of only 1 (Fig. 1C). (Our data, however, would also be consistent with more than one site, depending on gating properties of various states of ligation.) The other assumptions were identical to those for pentobarbital.
The quality of the fit of the predictions to the data was assessed by eye while the parameters were adjusted. For each agonist, the concentration–response curve (Fig. 1) was fit by adjusting K and P1. In the case of GABA, we used values for K1 and K2 based on the data inMaconochie et al. (1994) and adjusted only P1. For pentobarbital and alphaxalone, the values for K and P1 were adjusted without additional constraints. As mentioned above, in the case of pentobarbital it was assumed that the microscopic dissociation constants were the same for the two sites. As shown in Figure 1A–C, the parameters used can provide an adequate description of the concentration–response curves for agonists.
The parameters for bicuculline and gabazine (L and Q) were then estimated using the data for block of currents elicited by multiple concentrations of agonists (Figs. 3, 7). Finally, the values for P2 (the activation equilibrium constant for receptors with both agonist and blocker bound) were adjusted for alphaxalone and pentobarbital (Figs. 3, 7).
Overall, it was possible to obtain an excellent match to the data (Figs. 3, 7). The parameter estimates are shown in Table 1. The overriding consideration was to have a single set of values that could be used for all conditions. Accordingly, parameters for agonists (K, P1) were fixed at the values used to describe the data in Figure 1, and the same values for L and Q were used to describe the blocking effects on currents elicited by all the agonists.
As described above, this particular model was chosen because it separated actions of agonists and antagonists. It was assumed that no interactions between agonists and antagonists occurred for binding (that is, values for K and L were assumed to be independent of occupancy). Also, the conformational change induced by antagonist (described by Q) was assumed to be independent of the binding of agonists. All other possible interactions between blocker and agonist were included in the free parameter used to describe the activation equilibrium constant for receptors with both blocker and agonist bound (P2) for alphaxalone and pentobarbital. As shown in Table 1, for most pairs of agonist and antagonist the same value was used for P1 and P2; however, it was necessary to have different values for P1 (100) and P2 (600) to describe the interaction of pentobarbital and bicuculline. If the same value were used for P1 and P2, the predicted response to 1 mm pentobarbital plus high bicuculline reached a plateau at ∼0.1 of control, rather than the 0.3 seen in the data (Fig.7B). On the face of it, this would indicate that bicuculline is a “co-agonist” for pentobarbital; however, as just discussed, the interpretation of this parameter is not obvious, because it might actually include an effect on binding (e.g., lower affinity for bicuculline when two pentobarbital molecules were bound) or conformational changes (e.g., lower value for Q when two pentobarbital molecules were bound).
The values for parameters seem to be reasonable. The intrinsic affinity of GABA for the resting receptor is likely to be rather low, based on independent results obtained using rapid applications of GABA (Maconochie et al., 1994). Pentobarbital is predicted to have even lower affinity, but direct data are not yet available for comparison. Gating by alphaxalone could only be approximated, because the maximal response could not be observed. The measured EC50 for channel gating occurs at lower agonist concentrations than the estimated affinities for the resting receptor, because the high equilibrium activation constant shifts the overall equilibrium to favor the activated state (for more discussion, see Maconochie et al., 1994).
Both GABA and pentobarbital are effective activators (P1≫1), whereas alphaxalone may be less effective. It should be emphasized that P1 is not necessarily directly related to the opening and closing rates for the receptor, because the postulated “active” state likely includes both open-channel and closed-channel states. Some independent information is available for channel activation by rapid applications of GABA (Maconochie et al., 1994). The maximal activation rate is ∼6000 sec−1. The rate for leaving the active state is in the range of 5–100 sec−1, depending on which experimental parameter is taken as the best estimate. The low concentration asymptote for the rate of current development is ∼10 sec−1, whereas the components present in the decay of current after the removal of GABA have decay rates of ∼5 sec−1 and ∼80 sec−1. Hence, P1 for GABA may lie in the range of 60–1000. The predicted maximal probability of being active is P1/(1 + P1), which is rather different from the maximal probability of being open [Po = (opening rate)/(opening rate + closing rate)]. As a consequence, the relative values for P1 do not directly address the question of the maximal responses to agonists. Depending on what fraction of time the active receptor spends in the open state, two agonists with identical values for P1 might have different maximal responses. Finally, this last point emphasizes the fact that the parameters are estimated under the assumption that activation processes have equilibrated, including some possible rapid transitions to closed states involved in the active state.
For α1β2γ2L receptors, bicuculline is much better at inducing the conformational change to the dead state (Q = 60) than gabazine (Q = 0.6). Because the measured IC50 values at low agonist concentrations are comparable for the two blockers, the actions of bicuculline are described by a lower affinity for the resting receptor than gabazine.
We conclude that it is possible to describe our data with a simplified version of an allosteric model. Hence, the overall model remains viable. Additional evidence that provides circumstantial support for an allosteric model is presented in the Discussion.
Potentiation between responses to steroids and pentobarbital
We found that steroids and pentobarbital interact essentially normally at receptors composed of α1β2(Y205S)γ2L subunits, in that the combined applications of a steroid and pentobarbital result in potentiation of the response. Responses of cells were determined when 250 μm pentobarbital was applied alone or in the presence of 10 μm DHP-OH. Responses of α1β2γ2L (wild-type) receptors were enhanced ninefold (9.3 ± 4.3-fold; mean ± SD for responses from nine cells), whereas responses from receptors containing α1β2(Y205S)γ2L subunits were enhanced sevenfold (7.2 ± 2.2; eight cells). The difference is not statistically significant. These results demonstrate that the sites involved in potentiation between steroids and barbiturates do not involve the β2(Y205) residue. The interactions between gabazine or bicuculline and alphaxalone at receptors containing the β2(Y157S) mutant also indicate that potentiation by steroids is not removed by this mutation. The data do not directly address the question of whether the binding sites involved in direct gating are the same as those involved in potentiation; however, these experiments have not demonstrated a dissociation between the two actions.
DISCUSSION
Point mutations do not affect gating by steroids
Mutation of two residues in the β2 subunit, β2(Y205S) and β2(Y157S), did not affect direct gating by steroids of GABAA receptors formed from the α1, γ2L, and mutated β2 subunits. However, these mutations reduced the affinity of bicuculline or gabazine, as assayed by effects on gating by steroids, suggesting that they form important determinants of inhibitor binding and/or a subsequent conformational change. Previous studies had shown that these mutations greatly reduced the binding affinity of GABA but did not affect gating by pentobarbital (Amin and Weiss, 1993). We conclude that these particular residues are not required for the binding of steroids or barbiturates or for the subsequent conformational changes that result in channel opening.
Mechanism for inhibition of gating produced by steroids and barbiturates
There are two basic models that are consistent with the results we obtained on the concentration dependence of block by gabazine and bicuculline. The first is the “many-sites” model. In this case, alphaxalone or pentobarbital is postulated to bind to at least two classes of sites on each GABAA receptor, occupancy of either of which can result in channel opening. Gabazine or bicuculline are postulated to act as competitive antagonists with different affinities at the two sites. The sites required for direct gating by steroids may differ from those required for gating by barbiturates. This model would include a multiplicity of parameters to specify the dissociation constants for drugs at the many sites and the many possible activation equilibrium constants. We are confident that the many-sites model could describe the data satisfactorily. One particular version of this model should be mentioned. The many sites might not be located on a single receptor, but instead might result from the expression of a heterogeneous population of receptors with different representations or arrangements of subunits. Our data cannot exclude this possibility. The concentration–response curves for pentobarbital and GABA are consistent with the idea that a single population of receptors exists, as is the observation that blocking curves have Hill coefficients near 1 (multiple populations would tend to produce low Hill coefficients in either assay).
The second model is the “allosteric” model described in Results. In this case, gabazine or bicuculline do not bind to the sites for alphaxalone or pentobarbital, but they produce inhibition by favoring an inactivated state of the receptor so that binding of alphaxalone or pentobarbital does not result in channel activation. As presented in Results, the allosteric model can accommodate qualitative observations which are more difficult to reconcile with competitive models, especially the similar IC50 values against multiple agonists and incomplete maximal block. The modeling calculations indicate that a relatively constrained allosteric model can describe major quantitative features of the data with an internally consistent set of parameters. The allosteric model also has the aesthetic appeal that the total number of postulated sites is lower than for the many-sites model and the circumstantial support that inverse agonists are known for other sites on the GABAA receptor (Macdonald and Olsen, 1993).
Whatever precise model is eventually supported, the data clearly rule out the possibility that gabazine blocks currents elicited by either steroids or barbiturates by simple competition at a single site. Results from other studies (see below) and the present observations with mutated β2 subunits support the idea that both gabazine and bicuculline bind directly to the GABA-binding site, rather than binding to yet another site on the GABAA receptor. Hence, although they may also produce conformational changes in the receptor, a primary action is to occupy the GABA-binding site. Overall, then, we conclude that at least one site for binding steroids (and one for barbiturates) is sufficiently distant from the GABA-binding site that relatively bulky blocking drugs bound to the GABA-binding site do not overlap with steroid or barbiturate sites. Furthermore, both gabazine and bicuculline can reduce activation of the GABAA receptor by an allosteric inhibition of channel activation after the binding of either alphaxalone or pentobarbital.
Point mutations alter the binding and actions of bicuculline and gabazine
Both the β2(Y157S) and β2(Y205S) mutations reduce the apparent affinity of gabazine and bicuculline in a fashion expected from the effects of the mutations on GABA binding (Amin and Weiss, 1993), as assayed from effects on alphaxalone-gated currents; however, two additional changes occur. The first is that the β2(Y157S) mutation converts both bicuculline and gabazine to weak agonists. The second is that the β2(Y205S) mutation changes the efficacy of gabazine at blocking steroid-gated currents, so that the maximal inhibition is increased over wild type. The conversion to partial agonism indicates that both drugs have the potential to cause conformational changes in GABAA receptors. Furthermore, the change in efficacy of gabazine is consistent with the idea of an allosteric mechanism for inhibition, although it is also possible that the postulated “many sites” for steroids might differ in receptors containing this mutated subunit. These observations demonstrate that both residues, β2(Y157) and β2(Y205), are involved in determining the binding affinity and subsequent conformational changes for gabazine and bicuculline.
Point mutations of a residue in the α1 subunit (F64) have also been shown to reduce the affinity of GABA, bicuculline, and gabazine (Sigel et al., 1992); however, in this case, no partial agonist activity of bicuculline or gabazine was noted. A point mutation of the glycine receptor α1 subunit has been described that changes the action of picrotoxin from inhibition to potentiation (Lynch et al., 1995).
Previous studies of bicuculline and gabazine suggest an allosteric mechanism for inhibition
Bicuculline and gabazine are viewed as classic competitive inhibitors of GABA binding to the GABAA receptor (Macdonald and Olsen, 1993), but there are indications that they can induce conformational changes in the GABAA receptor. A recent analysis of recombinant receptors expressed in HEK293 cells found that both bicuculline and gabazine reduced the binding of t-butylbicyclophosphorothionate (TBPS; Luddens and Korpi, 1995). In most cases the inhibition was only partial, and the efficacy of the two drugs depended on the subunit combination expressed. In the case of the α1β2γ2 combination (which we examined in the present studies), gabazine had no effect on TBPS binding. Bicuculline reduced the binding by ∼50%, with an IC50 of ∼1-5 μm. The greater effect of bicuculline is consistent with our observation that bicuculline is a more effective antagonist of gating by steroids or barbiturates. In terms of the allosteric model, this difference arises from a larger allosteric constant (Q) for bicuculline. There are also results from binding studies that bicuculline and gabazine interact allosterically with barbiturates or steroids. Pentobarbital has been reported to inhibit the binding of labeled bicuculline (Wong et al., 1984) and gabazine (McCabe et al., 1988) by allosteric mechanisms. Similarly, studies of TBPS binding suggest that bicuculline allosterically inhibits alphaxalone binding (Gee et al., 1987). A quantitative comparison cannot be made to our data, because the binding studies are performed on isolated receptors in an unknown physiological state. The observations, however, agree qualitatively with our interpretations, and it has already been suggested that bicuculline and gabazine may act as inverse agonists at the GABA-binding site (Luddens and Korpi, 1995).
Studies of the inhibition of GABA-elicited responses have shown that both bicuculline (Akaike et al., 1987b) and gabazine (Hamann et al., 1988) shift the concentration–response curve to higher GABA concentrations but do not depress the maximal response. These data demonstrate that the binding of GABA and the inhibitors is mutually exclusive; however, they do not demonstrate whether bicuculline or gabazine has effects on the receptor in addition to preventing GABA binding. The effects of point mutations of the β2 subunit on the apparent affinity of GABA, bicuculline, and gabazine provide support for the idea that these three drugs bind to the same site on the GABAA receptor.
It has been known for a number of years that bicuculline blocks currents elicited by pentobarbital (Nicoll and Wojtowicz, 1980) or alphaxalone (Barker et al., 1987; Peters et al., 1988). A recent study examined receptors composed of α6β3γ2S subunits expressed inXenopus oocytes (Thompson et al., 1996). Pentobarbital was an effective agonist at these receptors, but neither bicuculline nor gabazine blocked pentobarbital-gated currents. Thompson et al. (1996)concluded that pentobarbital does not bind to the GABA-binding site. They suggested that the contradiction between the lack of block observed in their work and previous observations resulted from potentiation between pentobarbital and endogenous GABA in other preparations. It seems more likely that the differences reflect properties of the particular GABAA subunit combination studied (Luddens and Korpi, 1995), and these observations are consistent with an allosteric mechanism for block of pentobarbital-gated currents.
Bicuculline can also block activation of GABAA receptors byn-octanol (Arakawa et al., 1992), isoflurane (Yang et al., 1992), or propofol (Hara et al., 1993). The general efficacy of bicuculline in blocking channel activation provides some circumstantial support for the idea that it acts allosterically to inhibit channel activation.
Implications for gating of GABAA receptors
These results support the idea that the agonists GABA, alphaxalone, and pentobarbital produce activation of the GABAA receptor after binding to different sites on the receptor. Occupation of the GABA-binding site by bicuculline or gabazine antagonizes gating by alphaxalone or pentobarbital by an allosteric mechanism. It is not known how many distinct conformational changes can occur to produce receptors with open or closed channel states. For example does bicuculline “lock the gate” or “close a second gate”? The effects of the point mutants, however, suggest that transduction of the occupancy of different agonist-binding sites into channel opening may require the presence of different specific residues in the β subunit.
Footnotes
This research was supported by National Institutes of Health (NIH) Grant PO1 GM47969 to J.H.S. and C.Z., NIH Grants AA09212 and NS35291 to D.S.W., National Institute of Mental Health Research Scientist Development Award MH00964 to C.Z., and funds from the Anesthesiology Department, Washington University School of Medicine. We thank A. Tobin for the α1 subunit cDNA.
Correspondence should be addressed to Joe Henry Steinbach, Department of Anesthesiology, Washington University School of Medicine, 660 South Euclid, St. Louis, MO 63110.
Dr. Ueno’s present address: Division of Pharmacology, National Institute of Health Sciences, 1-18-1 Kamiyoga, Tokyo 158, Japan.
REFERENCES
- 1.Adams PR. Drug blockade of open end-plate channels. J Physiol (Lond) 1976;260:531–552. doi: 10.1113/jphysiol.1976.sp011530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akaike N, Maruyama T, Tokutomi N. Kinetic properties of the pentobarbitone-gated chloride current in frog sensory neurones. J Physiol (Lond) 1987a;394:85–98. doi: 10.1113/jphysiol.1987.sp016861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akaike N, Yakushiji T, Tokutomi N, Carpenter DO. Multiple mechanisms of antagonism of γ-aminobutyric acid (GABA) responses. Cell Mol Neurobiol. 1987b;7:97–103. doi: 10.1007/BF00734993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amin J, Weiss DS. GABAA receptor needs two homologous domains of the β-subunit for activation by GABA but not by pentobarbital. Nature. 1993;366:565–569. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- 5.Arakawa O, Nakahiro M, Narahashi T. Chloride current induced by alcohols in rat dorsal root ganglion neurons. Brain Res. 1992;578:275–281. doi: 10.1016/0006-8993(92)90258-b. [DOI] [PubMed] [Google Scholar]
- 6.Barker JL, Harrison NL, Lange GD, Owen DG. Potentiation of γ-aminobutyric-acid-activated chloride conductance by a steroid anaesthetic in cultured rat spinal neurones. J Physiol (Lond) 1987;386:485–501. doi: 10.1113/jphysiol.1987.sp016547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen C, Okayama H. High-efficiency transformation of cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gee KW, Chang WC, Brinton RE, McEwen BS. GABA-dependent modulation of the Cl− ionophore by steroids in rat brain. Eur J Pharmacol. 1987;136:419–423. doi: 10.1016/0014-2999(87)90317-7. [DOI] [PubMed] [Google Scholar]
- 9.Hamann M, Desarmenien M, Desaulles E, Bader MF, Feltz P. Quantitative evaluation of the properties of a pyridazinyl GABA derivative (SR 95531) as a GABAA competitive antagonist: an electrophysiological approach. Brain Res. 1988;442:287–296. doi: 10.1016/0006-8993(88)91514-4. [DOI] [PubMed] [Google Scholar]
- 10.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 11.Hara M, Kai Y, Ikemoto Y. Propofol activates GABAA receptor-chloride ionophore complex in dissociated hippocampal pyramidal neurons of the rat. Anesthesiology. 1993;79:781–788. doi: 10.1097/00000542-199310000-00021. [DOI] [PubMed] [Google Scholar]
- 12.Hu Y, Zorumski CF, Covey DF. Neurosteroid analogues: structure-activity studies of benz[e]indene modulators of GABAA receptor function. 1. The effect of 6-methyl substitution on the electrophysiological activity of 7-substituted benz[e]indene-3-carbonitriles. J Med Chem. 1993;36:3956–3967. doi: 10.1021/jm00076a025. [DOI] [PubMed] [Google Scholar]
- 13.Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 14.Jurman ME, Boland LM, Liu Y, Yellen G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 1994;17:876–890. [PubMed] [Google Scholar]
- 15.Luddens H, Korpi ER. GABA antagonists differentiate between recombinant GABAA/benzodiazepine receptor subtypes. J Neurosci. 1995;15:6957–6962. doi: 10.1523/JNEUROSCI.15-10-06957.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lynch JW, Rajendra S, Barry PH, Schofield PR. Mutations affecting the glycine receptor agonist transduction mechanism convert the competitive antagonist, picrotoxin, into an allosteric potentiator. J Biol Chem. 1995;270:13799–13806. doi: 10.1074/jbc.270.23.13799. [DOI] [PubMed] [Google Scholar]
- 17.Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1993;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- 18.Maconochie DJ, Zempel JM, Steinbach JH. How quickly can GABAA receptors open? Neuron. 1994;12:61–71. doi: 10.1016/0896-6273(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 19.McCabe RT, Wamsley JK, Yezuita JP, Olsen RW. A novel GABAA antagonist [3H]SR 95531: microscopic analysis of binding in the rat brain and allosteric modulation by several benzodiazepine and barbiturate receptor ligands. Synapse. 1988;2:163–173. doi: 10.1002/syn.890020208. [DOI] [PubMed] [Google Scholar]
- 20.Nicoll RA, Wojtowicz JM. The effects of pentobarbital and related compounds on frog motoneurons. Brain Res. 1980;191:225–237. doi: 10.1016/0006-8993(80)90325-x. [DOI] [PubMed] [Google Scholar]
- 21.Peters JA, Kirkness EF, Callachan H, Lambert JJ, Turner AJ. Modulation of the GABAA receptor by depressant barbiturates and pregnane steroids. Br J Pharmacol. 1988;94:1257–1269. doi: 10.1111/j.1476-5381.1988.tb11646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips WD, Maimone MM, Merlie JP. Mutagenesis of the 43 kDa postsynaptic protein defines domains involved in plasma membrane targeting and AChR clustering. J Cell Biol. 1991;115:1713–1723. doi: 10.1083/jcb.115.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodgers-Neame NT, Covey DF, Hu Y, Isenberg KE, Zorumski CF. Effects of a benz[e]indene on gamma-aminobutyric acid-gated chloride currents in cultured postnatal rat hippocampal neurons. Mol Pharmacol. 1992;42:952–957. [PubMed] [Google Scholar]
- 24.Sigel E, Baur R, Kellenberger S, Malherbe P. Point mutations affecting antagonist affinity and agonist dependent gating of GABAA receptor channels. EMBO J. 1992;11:2017–2023. doi: 10.1002/j.1460-2075.1992.tb05258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson SA, Whiting PJ, Wafford KA. Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. Br J Pharmacol. 1996;117:521–527. doi: 10.1111/j.1476-5381.1996.tb15221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Twyman RE, Rogers CJ, Macdonald RL. Intraburst kinetic properties of the GABAA receptor main conductance state of mouse spinal cord neurones in culture. J Physiol (Lond) 1990;423:193–220. doi: 10.1113/jphysiol.1990.sp018018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ueno S, Zempel JM, Steinbach JH. Differences in the expression of GABAA receptors between functionally innervated and non-innervated granule neurons in neonatal rat cerebellar cultures. Brain Res. 1996a;714:49–56. doi: 10.1016/0006-8993(95)01457-8. [DOI] [PubMed] [Google Scholar]
- 28.Ueno S, Zorumski C, Bracamontes J, Steinbach JH. Endogenous subunits can cause ambiguities in the pharmacology of exogenous GABAA receptors expressed in HEK293 cells. Mol Pharmacol. 1996b;50:931–938. [PubMed] [Google Scholar]
- 29.Wong EH, Snowman AM, Leeb-Lundberg LM, Olsen RW. Barbiturates allosterically inhibit GABA antagonist and benzodiazepine inverse agonist binding. Eur J Pharmacol. 1984;102:205–212. doi: 10.1016/0014-2999(84)90252-8. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Isenberg KE, Zorumski CF. Volatile anesthetics gate a chloride current in postnatal rat hippocampal neurons. FASEB J. 1992;6:914–918. doi: 10.1096/fasebj.6.3.1740240. [DOI] [PubMed] [Google Scholar]