

Abstract
Acetylcholine has long been implicated in nocturnal phase adjustment of circadian rhythms, yet the subject remains controversial. Although the suprachiasmatic nucleus (SCN), site of the circadian clock, contains no intrinsic cholinergic somata, it receives choline acetyltransferase-immunopositive projections from basal forebrain and mesopontine tegmental nuclei that contribute to sleep and wakefulness. We have demonstrated that the SCN of inbred rats in a hypothalamic brain slice is sensitive to cholinergic phase adjustment via muscarinic receptors (mAChRs) only at night. We used this paradigm to probe the muscarinic signal transduction mechanism and the site(s) gating nocturnal responsiveness. The cholinergic agonist carbachol altered the circadian rhythm of SCN neuronal activity in a pattern closely resembling that for analogs of cGMP; nocturnal gating of clock sensitivity to each is preserved in vitro. Specific inhibitors of guanylyl cyclase (GC) and cGMP-dependent protein kinase (PKG), key elements in the cGMP signal transduction cascade, blocked phase shifts induced by carbachol. Further, carbachol administration to the SCN at night increased cGMP production and PKG activity. The carbachol-induced increase in cGMP was blocked both by atropine, an mAChR antagonist, and by LY83583, a GC inhibitor. We conclude that (1) mAChR regulation of the SCN is mediated via GC→cGMP→PKG, (2) nocturnal gating of this pathway is controlled by the circadian clock, and (3) a gating site is positioned downstream from cGMP. This study is among the first to identify a functional context for mAChR–cGMP coupling in the CNS.
Keywords: suprachiasmatic nucleus, acetylcholine, carbachol, circadian rhythm, guanylyl cyclase, muscarinic receptor, cyclic GMP, cGMP-dependent protein kinase, KT5823, LY83583, enzyme activity assay, scintillation proximity assay, sleep
The near 24 hr oscillations of physiological and behavioral functions in mammals are controlled by a central pacemaker, or clock, within the suprachiasmatic nucleus (SCN) of the hypothalamus. The SCN receives diverse neural and hormonal inputs by which signals communicating environmental and organismic states are transmitted to the SCN and can modify its phase (for review, see Inouye and Shibata, 1994; Moore, 1996). Clock properties are endogenous to the SCN; spontaneous neuronal activity continues to oscillate in a circadian rhythm under constant conditions in a hypothalamic slice of SCN from rat (Green and Gillette, 1982; Prosser and Gillette, 1989), and the phasing of this rhythm can be reset by treating the SCN slice with the transmitters or peptides contained in afferent fibers (for review, seeGillette et al., 1995). Although the circadian clock is sensitive to resetting by diverse neurotransmitters, the sensitivity to each is restricted, or gated, to discrete phases of the clock’s 24 hr cycle (Gillette, 1996).
One class of afferent projections to the SCN contains acetylcholine (ACh), as revealed by choline acetyltransferase (ChAT, biosynthetic for ACh) immunostaining (van den Pol and Tsujimoto, 1985; Ichikawa and Hirata, 1986; Rao et al., 1987; Tago et al., 1987). Bidirectional tract-tracing combined with immunocytochemistry for ChAT localized cholinergic neurons that project to the rat SCN to the major cholinergic regions of the CNS: the basal forebrain as well as the peduncular pontine tegmental and the lateral dorsal tegmental nuclei of the brainstem (Bina et al., 1993). These cholinergic nuclei contribute to state changes in multiple brain sites, including those underlying sleep and wakefulness (Semba, 1991; Steriade and McCarley, 1990).
The functional significance of the cholinergic projections to the SCN has been unclear. Attempts to address this issue in vivohave involved administration of carbachol, a general cholinergic agonist. Although these studies present variability in both design and data, there is concordance that phasing of the circadian system can be modulated by cholinergic stimulation at night (Zatz and Brownstein, 1979; Zatz and Herkenham, 1981; Ernest and Turek, 1983, 1985; Meijer et al., 1988; Wee et al., 1992). Within and near the rat SCN, some neurons are cholinoceptive (Nishino and Koizumi, 1977; Shibata et al., 1983;Kow and Pfaff, 1984; Miller et al., 1987; Liu, 1995), and modest evidence exists for nicotinic or muscarinic ACh receptors (mAChRs) (Fuchs and Hoppens, 1978; van der Zee et al., 1991). Direct and specific cholinergic stimulation of the SCN in a brain slice preparation induces large phase advances in the SCN neuronal activity rhythm; this response is mediated by an mAChR, possibly an M1 subtype (Liu and Gillette, 1996).
ACh has been linked to cGMP production in brain and cultured neurons through a muscarinic mechanism (Tonnaer, 1991; Castoldi et al., 1993;Hu and El-Fakahany, 1993; Mathes and Thompson, 1996). However, functional contexts for this coupling in the CNS have not been reported. We hypothesized that cholinergic activation of a cGMP pathway within the SCN mediates nocturnal phase resetting. Following up on our preliminary report (Liu and Gillette, 1994), we tested this hypothesis by attempting to block the phase shifts induced by carbachol through inhibiting cGMP-dependent protein kinase (PKG) and guanylyl cyclase (GC). We then measured directly the activity of PKG and the production of cGMP in the SCN in response to carbachol. We also tested the ability of the muscarinic antagonist atropine and of a GC inhibitor to block the effect of carbachol on cGMP production. Our results demonstrate that carbachol advances the phase of the SCN clock at night via mAChR-mediated activation of a GC/cGMP/PKG pathway.
MATERIALS AND METHODS
Animals and brain slice preparation. Eight-week-old Long–Evans rats from our inbred colony were used in this study. This colony has been inbred in our Illinois facility for more than 32 generations and, thus, exceeds the level of inbreeding required for genetic homogeneity. Animals were maintained and tissue prepared in full accord with institutional and federal guidelines for the humane treatment of animals. The rats were kept on a schedule of 12:12 hr light/dark cycle, with access to food and water ad libitum. They were killed between circadian time 7–10 (CT 7–10, where CT 0 is defined as the beginning of the light period of the animal colony), and the brain was quickly dissected from the skull. The brain was then manually sectioned to form a block of tissue containing the hypothalamic region. This block of tissue was transferred to a mechanical tissue chopper, where 500 μm coronal slices were made. The hypothalamic slices containing the SCN were then transferred to the brain slice dish, where they were maintained for up to 3 d. Under these conditions, the SCN generates near 24 hr oscillations in neuronal activity (Green and Gillette, 1982) that are stable over 3 d (Prosser and Gillette, 1989). The unperturbed sinusoidal pattern of ensemble activity is stable, running very close to 24 hr. It is predictably high in the day (peaking mid-day near CT 7) and is low at night (Gillette and Prosser, 1988), so that measure of the time-of-peak can be used as an accurate assessment of the phase of the circadian oscillation (Gillette et al., 1995).
The brain slice dish, consisting of an inner and an outer chamber, was modeled after Hatton et al. (1980). The outer chamber of the dish was filled with dH2O warmed to 37°C and continuously bubbled with 95% O2/5% CO2. Brain slices were maintained at the gas/liquid interface within the inner chamber and perifused at 35 ml/hr with warmed, oxygenated Earle’s balanced salt solution (EBBS, Life Technologies, Gaithersburg, MD) supplemented to 24.6 mm glucose, 26.3 mm NaHCO3, and 0.005% gentamicin, pH 7.3–7.4.
In the experiments measuring cGMP levels and PKG activity, a reduced SCN slice was made using iridectomy scissors under a dissecting microscope and then transferred into the brain slice chamber. At the appropriate CT (>2 hr later), the reduced slice was either (1) frozen at −80°C until assay for PKG activity, or (2) transferred to equilibrated EBSS at 37°C in an Eppendorf tube gassed with 95% O2/5% CO2 and maintained stably for 60 min in IBMX (100 μm) for assay of cGMP accumulation. For the cGMP measurements, inhibitors, such as atropine and LY83583, were added after 45 min and for a total of 15 min; carbachol was added to the tube during the last 3–4 min, after which cGMP was extracted by EtOH (100%). The reduced SCN slice contains the paired SCN, a thin rim of hypothalamic tissue around the SCN, and the underlying optic chiasm. The circadian oscillation in neuronal firing is maintained in reduced SCN slices (Gillette and Reppert, 1987).
Extracellular electrical recording and data analysis. The method for recording the circadian rhythm of the ensemble of SCN neurons has been detailed and thoroughly validated (Prosser and Gillette, 1989); it is only briefly summarized here. Extracellular signals of single neurons were recorded using glass microelectrodes filled with 5 m NaCl. An electrode was lowered into the SCN by a hydraulic microdrive until the signal from a single cell was encountered. Electrical signals exceeding twice the level of the background noise were isolated using a window discriminator, observed for stability over at least 2 min, and counted by computer. The firing rate of each cell was monitored over two consecutive 120 sec periods, using 10 sec bins. The electrode was then repositioned within the SCN so as to sample throughout the nucleus and advanced until another cell was encountered.
The firing rates of individual SCN neurons recorded during each experiment were grouped into 2 hr running averages using 15 min lags. The time-of-peak was determined by visual inspection of a plot of these values for the symmetrically highest point. Phase shifts were determined by comparing the time-of-peak electrical activity in treated slices with that of vehicle-treated slices. Differences among groups were evaluated by ANOVA, with Duncan’s test for post hoccomparisons.
Experimental treatments. The protocol for drug administration to the SCN used in this study has been described (Liu and Gillette, 1996). Briefly, brain slices were equilibrated in the recording chamber for ≥ 2 hr before administration of the reagent (dissolved in EBSS then prepared to match the physical characteristics of normal medium in the chamber). Agonists were administered in a microdrop (1 μl) applied to the surface of each SCN; this covered only the SCN tissue of the slice. After 5 min, the surface of the slice was rinsed toward the optic chiasm with warmed, gassed EBSS, and perifusion with normal medium was resumed. Antagonists were applied as a static bath 10 min before, 5 min during, and 15 min after the treatment. Results were compared with controls treated in the same way with microdrops of EBSS only.
Assay of PKG activity. Endogenous PKG activity was measured according to Glass and Krebs (1982) using the PKG-specific heptapeptide substrate for PKG RKRSRAE (Peninsula Labs). Reduced SCN slices were exposed to carbachol for from 0 to 10 min, then frozen on dry ice. Single reduced slices were sonicated cold for 10 sec in 50 μl buffer containing glycerophosphate 10 mm and a cocktail of protease inhibitors [phenylmethylsulfonyl fluoride (100 μm), phenanthroline (10 mm), leupeptin (2 μm), pepstatin (0.2 μm), and aprotinin (1%) (Sigma, St. Louis, MO)], BSA (0.1 mg/ml), KT5720 (protein kinase A inhibitor, 0.1 μm), Tris (20 mm), pH 7.4, Mg2+ acetate (20 mm), and isobutylmethylxanthine (IBMX, 100 μm), and then centrifuged at 2000 × g for 3 min at 4°C. A 30 μl aliquot of supernatant was taken and mixed with 15 μl of buffer containing the heptapeptide substrate RKRSRAE (400 μg/ml), [32P]ATP (1 μCi/tube), and unlabeled ATP (14 μm), and then incubated for 2 min at 37°C. The reaction was stopped by cooling on ice and adding 10 μl of 1N HCl. A 35 μl aliquot of the reaction product was placed on filter paper (Whatman P81 disks) and air-dried. The filter paper was washed with 0.5% orthophosphoric acid until no more radioactivity appeared in the effluent. The dried filter paper was placed in a scintillation vial with 5 ml scintillation fluid and counted for 2 min. Activity is expressed as a percentage of the average activity immediately before carbachol administration (100%).
Scintillation proximity assay (SPA) of cGMP levels. The SPA assay for cGMP was performed according to the manufacturer’s directions (Amersham, Arlington Heights, IL). The acetylation procedure was followed both for the standard curve and for the samples. Briefly, single frozen reduced slices, ∼30 μg of protein (Bradford, 1976), in 100 μl of EBSS without phenol red, were treated with 150 μl of 100% ethanol and the supernatant pipetted off to a fresh tube. The slices were resuspended and washed 1 time with 65% ethanol, and the supernatants of each slice pooled. These pooled supernatants were lyophilyzed to dryness and resuspended in 100 μl of 0.05m acetate buffer containing 0.01% sodium azide. Equal amounts of cGMP standard, radioactive tracer, primary antibody, and secondary antibody conjugated to beads were added. The mixtures were shaken overnight and counted in a Beckman scintillation counter. Effects of treatment conditions on cGMP levels were compared by ANOVA, with Fisher’s post hoc test.
RESULTS
Comparison of the phase–response relationships of carbachol and 8-Br-cGMP
Cholinergic effects on phasing of SCN circadian rhythms have been studied after a 5 min exposure to carbachol and other cholinergic agonists in a 1 μl microdrop (Liu and Gillette, 1996), whereas the effects of 8-Br-cGMP have been assessed after 1 hr bath administration (Prosser et al., 1989). Therefore, we evaluated the phase shifts induced by microdrop versus bath treatments. We applied carbachol (100 μm) or 8-Br-cGMP (500 μm) as a 1 μl microdrop to the surface of the SCN in vitro for 5 min at CT 18 and compared the effects on the subsequent time-of-peak activity in the circadian rhythm SCN neuronal firing. The pattern of this endogenous circadian rhythm is normally a near 24 hr oscillation that peaks midsubjective day near CT 7 (7 hr into the light portion of the entrained light/dark cycle of the rat) (Fig.1A). Mean time-of-peak for media-treated controls in this study was CT 6.8 ± 0.1 (n = 5). The 5 min exposure to carbachol at CT 18 (midnight) induced a 6.0 hr phase advance (Fig. 1B) in the SCN neuronal activity rhythm (which is in agreement with +6.3 ± 0.2 hr, n = 3, Liu and Gillette, 1996). 8-Br-cGMP administered in this way induced a 6.0 ± 0.3 hr phase advance (n = 3) (Fig. 1C). These advances caused by brief, localized application of cGMP analog are not significantly different (p > 0.05) from phase resetting induced by bathing the SCN for 1 hr in 500 μm8-Br-cGMP (+6.5 ± 0.2 hr, n = 3, Prosser et al., 1989). Thus, a 5 min exposure of the SCN to either carbachol or 8-Br-cGMP in a microdrop is sufficient to initiate the same phase shift as that observed after bathing the whole hypothalamic slice in the cGMP analog for 1 hr at CT 18. Together, these data suggest that SCN sensitivity to carbachol may be functionally related to its sensitivity to cGMP analogs.
Fig. 1.

Carbachol and 8-Br-cGMP applied by microdrop at CT 18 induce similar phase advances of SCN circadian rhythms in brain slices from inbred rats. A, In control brain slices, the SCN spontaneously generates a circadian rhythm of neuronal activity that peaks near CT 7 on days 2 and 3 in vitro (replotted from Ding et al., 1994). B, When a microdrop of carbachol (Carb, 100 μm in 1 μl) was applied to the SCN at CT 18 for 5 min, peak activity was advanced by 6.0 hr on subsequent days. C, When 8-Br-cGMP (500 μm in 1 μl) was applied to the SCN using this microdrop protocol, peak activity was advanced by 6.5 hr. The 2 hr running averages ± SEMs of ensemble SCN neuronal firing rates are plotted against the CT of recording. Horizontal bars represent subjective night (the time of lights-off in the donor colony).Thin dashed line indicates the mean time of peak activity in media-treated slices (CT 6.8 ± 0.1;n = 5).
When we compared the phase–response relationships between the CT of treatment and responses of the SCN clock to carbachol (100 μm for 5 min in a 1 μl microdrop, Liu and Gillette, 1996) with those to 8-Br-cGMP (500 μm for 60 min in the bath, Prosser et al., 1989), the patterns were strikingly similar (Fig.2). This similarity included the circadian timing of SCN sensitivity to both stimuli as well as the amplitude and direction of the phase shifts in the SCN rhythm. The SCN was generally unresponsive to carbachol and 8-Br-cGMP treatments when either was administered during subjective day but responded with phase advances to treatments administered throughout subjective night. The largest phase advances to either carbachol or 8-Br-cGMP (+6.3 and +6.5 hr, respectively) were induced when the treatment was administered at CT 18. We have demonstrated that this nocturnal effect of carbachol on SCN phasing is mediated via an mAChR (Liu and Gillette, 1996); thus, it follows that the signal may activate a cGMP-dependent pathway to initiate phase resetting.
Fig. 2.

Comparison of the phase–response relationships for carbachol and 8-Br-cGMP in resetting the SCN circadian clock. The phase shifts induced by carbachol (100 μm in 1 μl microdrops, solid circles, Liu and Gillette, 1996) and by 8-Br-cGMP (500 μm in the bathing medium, open circles, Prosser et al., 1989) are plotted against the CT at which the treatment was administered. Each point is the result of a single experiment in which the time-of-peak neuronal activity on the day after treatment (as in Fig. 1C) was derived from recording 4–9 neurons/hr over 8–14 hr to define the peak in neuronal activity. Symbols as in Figure 1.
PKG and GC inhibitors block the response to carbachol
To explore the possibility that carbachol acts via a cGMP-dependent pathway, we tested selective inhibitors of steps in cGMP signaling. First, the SCN slice was preincubated with an inhibitor specific for cGMP-dependent PKG, KT5823, to determine whether this could block the phase advance induced by carbachol. In addition to activating PKG, 8-Br-cGMP can act directly on cGMP-dependent phosphodiesterases or ion channels to exert its effect (for review, seeCorbin et al., 1990). To evaluate the potential contribution of these non-PKG-mediated mechanisms, we also tested whether KT5823 could block the phase advance induced by 8-Br-cGMP. Microdrop application of carbachol or 8-Br-cGMP for 5 min at CT 18 induced robust phase advances of >6 hr (Fig.3A,B). KT5823 (0.1 μm), when applied in the bath during the period surrounding agonist treatment, blocked the phase-advancing effects of both carbachol (100 μm) and 8-Br-cGMP (500 μm) (+1.1 ± 0.3 hr, n = 3 and +1.2 ± 0.3 hr, n = 3, respectively) (Fig.3C,D); the timing of the peak of the neuronal rhythm was not significantly different from the effect of KT5823 alone (+1.1 ± 0.2 hr, n = 3;p > 0.05) (Fig. 3E). These results suggest that both carbachol and 8-Br-cGMP reset the SCN clock through activation of PKG and, further, that carbachol may increase endogenous cGMP by stimulating GC.
Fig. 3.
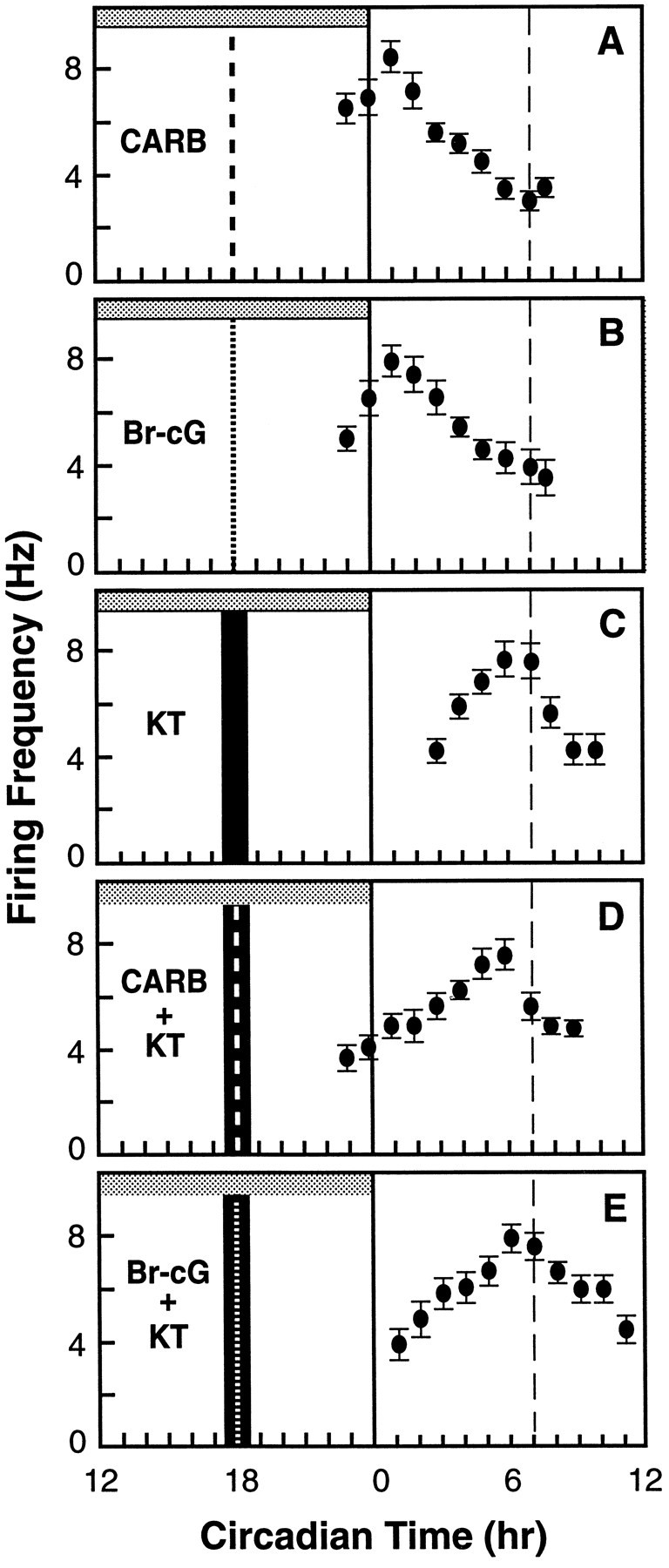
The PKG-specific inhibitor KT5823 blocks the phase-advancing effects of both carbachol and 8-Br-cGMP. A microdrop of carbachol (heavy vertical dashed line) or 8-BR-cGMP (vertical dotted line) applied for 5 min at CT 18 caused 6.4 and 6.0 hr phase advances, respectively (A,B), in the time of the next peak. A 30 min bath application of KT5823 (0.1 μm) (vertical solid bar) induced a small phase advance of ∼1 hr (C). The effects of carbachol and 8-Br-cGMP were blocked by the KT5823 pulse (D, E). Symbols are as in Figure 1.
To determine whether the phase shift induced by carbachol requires activation of GC, the biosynthetic enzyme for cGMP, we evaluated the effect of LY83583, a specific inhibitor of GC. A 30 min exposure of the SCN slice to LY83583 (3 μm) at CT 18 had no effect on the time-of-peak in neuronal activity in the following circadian cycle (Fig. 4A). The same LY83583 treatment fully blocked the phase-advancing effect of carbachol (Fig.4B) (mean phase advance = 0.3 ± 0.2 hr,n = 3) (Fig. 5). However, it did not block the effect of 8-Br-cGMP (Fig. 4C) (+6.3 ± 0.3 hr, n = 3) (Fig. 5). The fact that the same phase-advancing effect was induced by the cGMP analog in the presence of the GC inhibitor is consistent with cGMP acting downstream from GC in the signaling pathway. These experiments, together with the similarity of phase–response relationships between carbachol and 8-Br-cGMP, support the hypothesis that the cholinergic agonist carbachol regulates the SCN through a GC/cGMP/PKG-dependent mechanism.
Fig. 4.
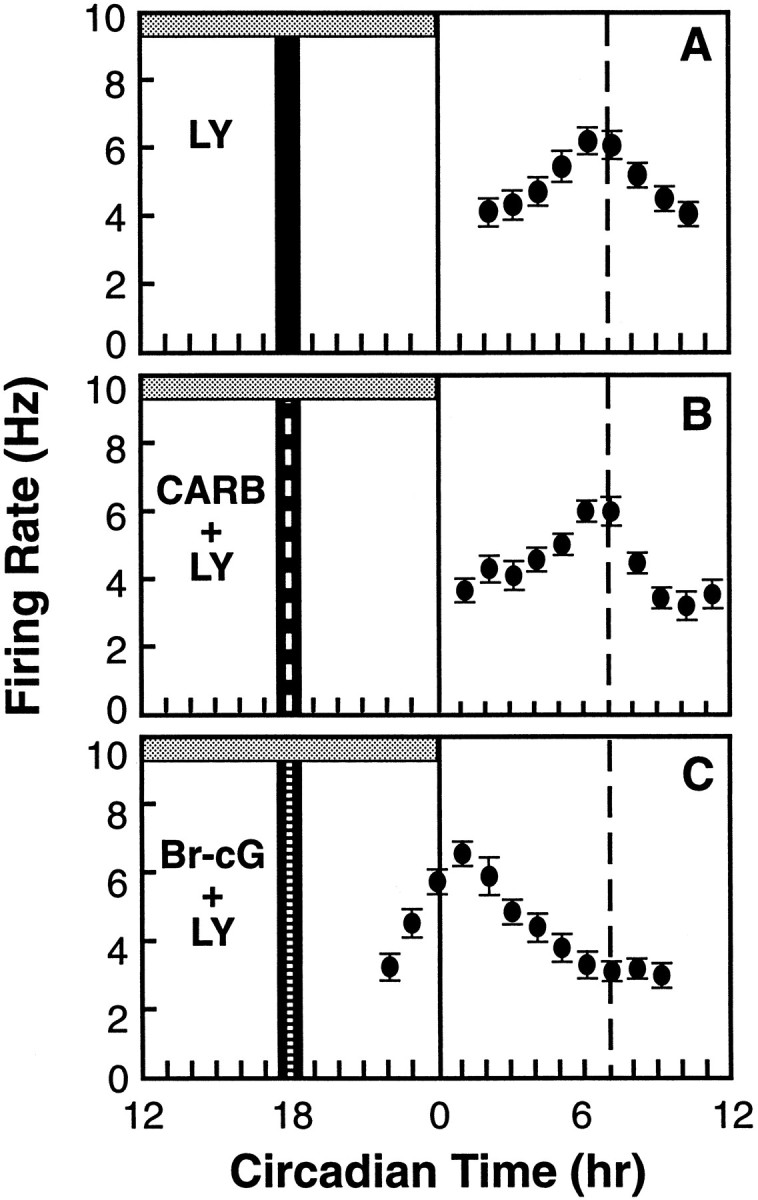
The GC inhibitor LY83583 blocks the effect of carbachol but not that of 8-Br-cGMP. A 30 min bath exposure to LY83583 (3 μm, vertical solid bar) did not affect the time-of-peak of the SCN firing rate (A). LY83583 blocked the phase-advancing effects of carbachol but not of 8-Br-cGMP (B, C; compare with Fig.3A, B). Symbols are as in Figure 1.
Fig. 5.

Effects of KT5823 (KT, 0.1 μm) and LY83583 (LY, 3 μm) on the phase advances induced by a microdrop of carbachol (CARB, 100 μm) or 8-Br-cGMP (500 μm). The phase advances induced by CARB, 8-Br-cGMP, and 8-Br-cGMP + LY were highly significantly different from controls (**p < 0.01), whereas the other treatments were not (p > 0.05). Average phase advances induced by each treatment are represented by bars, with treatments labeled on top of each bar. Plotted are the mean ± SEM (n = 3 for each).
Carbachol rapidly stimulates cGMP production and PKG activity in the SCN
These experiments with inhibitors indicate that increased GC synthesis of cGMP and subsequent PKG activation are necessary for carbachol to induce resetting of the circadian clock. Therefore, we directly evaluated the effect of carbachol on each of these effectors. First, to assess the rate of the activation process, we examined the time course of PKG phosphotransferase activity toward a PKG-selective substrate in response to carbachol treatment. To ensure that we measured responses localized within the SCN, the brain slice was surgically reduced to include only the 500 μm slice of SCN centered along the rostrocaudal axis, a thin rim of surrounding hypothalamus, and the adherent optic chiasm. The phosphodiesterase inhibitor IBMX (200 μm, 1 hr preincubation) was included to preserve cyclic nucleotides generated by the treatment; this increased basal PKG activity by ∼30%, to 6500 cpm, on average. This value was then used as the normalized basal level before carbachol treatment. Carbachol application rapidly and significantly increased PKG activity;32P incorporation rose by up to 40% between 2 and 5 min of exposure to carbachol, with statistical significance at 5 min compared with 0 (p < 0.005), 1 and 10 min (bothp < 0.05) (Fig. 6). The control procedure, which involved the same manipulation as with carbachol but using only medium, did not increase the PKG activity after 3 min (100 ± 15%, n = 4; p > 0.05). Therefore, carbachol administration induces a rapid and transient activation of PKG in the SCN at CT 18, the time when it induces phase resetting.
Fig. 6.

Carbachol administration activates PKG within the SCN. PKG activity in extracts of reduced SCN slices was measured by monitoring 32P incorporation into the preferred artificial PKG substrate peptide RKRSRAE. The general phosphodiesterase inhibitor IBMX (200 μm) and the specific PKA inhibitor KT 5720 (0.1 μm) were included in the reaction. The average activity of PKG in each reduced slice immediately before carbachol treatment (0 min) was taken as 100%. The PKG activity in the SCN slice exposed to carbachol (100 μm) for 1, 2, 3, 5, and 10 min was measured and normalized to a percentage of the basal activity. Transient increase in the PKG activity was observed, with 5 min showing significant elevation (asterisk) over 0 (p < 0.005), 1 and 10 min (bothp < 0.05). The number of samples in each group is noted.
To confirm that carbachol treatment increased cGMP levels, we measured total cGMP in the tissue and assay medium. Based on the time course of the increase in PKG activity (Fig. 6), we examined the cGMP level at 3 min after carbachol or control treatments. After treatment at CT 18, cGMP levels increased dramatically (fivefold, on average) in SCN treated with carbachol (Fig. 7). The cGMP level in vehicle-treated controls was 63.6 ± 4.6 fmol/reduced slice, whereas that in carbachol-treated samples was 291.8 ± 78.0 fmol/reduced slice (n = 7 and 8, respectively;p < 0.02).
Fig. 7.

Carbachol increases cGMP production in the reduced SCN slice via an mAChR. Levels of cGMP in the SCN slices and assay medium were measured in the presence of IBMX by SPA (Amersham). When reduced SCN slices were treated with carbachol (100 μm) for 3 min at CT 18, the level of cGMP increased significantly, from 63.6 ± 4.6 fmol/reduced slice in vehicle-treated samples to 291.8 ± 78.0 fmol/reduced slice (n = 7 and 8;p < 0.002). This carbachol-induced increase in the cGMP was completely blocked by the muscarinic antagonist atropine (1 μm) as well as by the GC inhibitor LY83583 (3 μm), which themselves had no effect on the cGMP level in the SCN (n = 4 and 3, respectively;p > 0.05). Results of individual experiments (open circles) overlay the bars representing the mean ± SEM.
Our previous results demonstrated that phase shifts of SCN rhythms by cholinergic agonists are mediated by an M1-like mAChR mechanism (Liu and Gillette, 1996). To evaluate the pathway by which carbachol increases cGMP production in the SCN slice, we examined the effects of the muscarinic antagonist atropine and of the GC inhibitor LY83583 at CT 18. Atropine (1 μm) itself had no effect on the phase of the SCN circadian rhythm but blocked the ability of carbachol to induce phase advances (Liu and Gillette, 1996). Pretreatment of the reduced SCN slice with atropine in the presence of IBMX completely inhibited the carbachol-stimulated increase in cGMP; thus, cGMP levels were equivalent to when atropine alone was added (65.9 ± 6.3 fmol/reduced slice and 58.5 ± 5.0 fmol/reduced slice,n = 4 and 3, respectively; p < 0.05). Likewise, a 15 min exposure to LY83583 was without effect and also fully blocked the carbachol-induced stimulation of cGMP (65.2 ± 1.2 fmol/reduced slice and 58.2 ± 7.1 fmol/reduced slice, respectively; n = 3 for each; p < 0.05). These results demonstrate that administration of carbachol to the SCN at CT 18 initiates an mAChR-mediated signaling cascade that increases cGMP production and, in turn, activates PKG.
DISCUSSION
In the present study, we compared the effect of carbachol on the circadian rhythm of SCN neuronal activity with the effect of a cGMP analog, 8-Br-cGMP, studied previously by Prosser et al. (1989). We found that the phase advance initiated by a 5 min application of carbachol was of the same amplitude as that induced by a 5- or 60 min exposure to 8-Br-cGMP. The phase–response relationships between timing of SCN sensitivity to carbachol and 8-Br-cGMP and their phase-resetting effects are fully overlapping; sensitivity to each is restricted to the subjective night. The specific PKG inhibitor KT5823 blocked the phase advances induced at midsubjective night (CT 18) by microdrop application of carbachol or 8-Br-cGMP to the SCN, whereas the GC inhibitor LY83583 blocked the phase advances induced by carbachol but not 8-Br-cGMP. PKG activity in the reduced SCN slice was transiently increased by 5 min after carbachol administration. Exposure of the SCN to carbachol rapidly increased cGMP levels; this increase was blocked by inhibiting either mAChRs or GC. We conclude that muscarinic cholinergic regulation of the SCN circadian clock at night is mediated via activation of a GC/cGMP/PKG signaling pathway.
Evidence that ACh activates a nocturnally gated GC/cGMP/PKG pathway
Our conclusions are supported by three lines of evidence. First, both the coincidence in the circadian timing of SCN sensitivities and the similarity of responses to carbachol and 8-Br-cGMP suggest that they may use a common pathway to induce phase shifts. The circadian clock in the SCN can be reset by a number of intercellular messengers, including serotonin (Prosser et al., 1990; Medanic and Gillette, 1992), neuropeptide Y (Medanic and Gillette, 1993), glutamate (Ding et al., 1994), and melatonin (McArthur et al., 1997). Yet, the ability of the SCN to respond to each of these stimuli is restricted, or gated, to discrete domains of the circadian cycle. Further, these changes in gating occur in SCN under constant conditions in vitro; therefore, differential gating must be a clock-controlled process (Gillette, 1996). Responses of the SCN to several candidate intracellular messengers are also differentially gated (Prosser and Gillette, 1989; Prosser et al., 1989; Ding et al., 1994, 1997). This implies that a critical level of clock-controlled gating liesdownstream from the second messenger(s) activated by various neurotransmitter systems (Gillette, 1996). It follows that the apparent similarity in the responses of the SCN clock to carbachol and 8-Br-cGMP suggests that they act via a common signaling pathway that can access the clock mechanism only at night and that gating of this response can be controlled at a site downstream from cGMP.
Second, both the PKG inhibitor KT5823 and the GC inhibitor LY83583 blocked the effect of carbachol on clock phasing. KT5823 is a highly selective inhibitor of PKG (Ki = 0.2 μm for PKG, Ki > 10 μm for PKA, and Ki = 4 μm for PKC) (Glass and Krebs, 1982). Thus, it is unlikely that KT5823, used at 0.1 μm in these experiments, acted through nonspecific inhibition of other kinases. LY83583, a widely used GC inhibitor that also inhibited cGMP production in the SCN (Fig. 7), did not block the effect of 8-Br-cGMP. This is consistent with cGMP lying downstream from GC in the signaling pathway by which the clock mechanism is accessed. The observation that two inhibitors acting at different levels of the same cGMP signal transduction pathway each inhibited the effects of carbachol in the SCN indicates that the carbachol-induced nocturnal phase shift is caused by specific activation of GC and PKG.
Third, carbachol significantly increased both PKG activity and cGMP production in the SCN. We monitored 32P incorporation into the preferred substrate of PKG (Glass and Krebs, 1982; Weber, 1995) in the presence of a selective PKA inhibitor, KT 5720, conditions unlikely to measure phosphorylation caused by a kinase other than PKG. Furthermore, KT5823 is an effective inhibitor of this kinase in this tissue; it blocks 32P incorporation into natural substrate proteins of SCN tissue by endogenous PKG (L. Faiman, unpublished observations). The time course of the increase in PKG activity in the SCN by carbachol is similar to the time course of the cGMP increase in rat superior cervical ganglia (Ando et al., 1994). To further test our hypothesis, a highly specific cGMP immunoassay was used to measure cGMP production in response to carbachol; a significant increase in cGMP was observed within 3 min. The relatively smaller amplitude of increase in the PKG activity stimulated by carbachol compared with enhancement of actual cGMP levels could reflect a limited amount of PKG available for activation by cGMP. Nevertheless, the increases in both the cGMP level and PKG activity in SCN stimulated by carbachol strongly support the conclusion that carbachol acts through a cGMP/PKG pathway to induce phase shifts.
Coupling of mAChR-activation to a cGMP pathway
The increase in cGMP induced by carbachol was blocked by 1 μm atropine, indicating involvement of an mAChR. Our study of the pharmacology of cholinergic regulation of the SCN circadian rhythm concluded that the nocturnal response to cholinergics is mediated by an M1-like mAChR subtype (Liu and Gillette, 1996). The relative potencies of muscarinics inducing phase advance at CT 18 are ACh > McN-A-343 > carbachol ∼ muscarine, and relative efficacies of antagonists in blocking carbachol are atropine > pirenzepine > 4-DAMP. Molecular cloning has revealed five subtypes of mAChRs, m1–m5 (Bonner et al., 1987, 1988), the first four of which correspond to the M1–M4 receptors identified pharmacologically (Waelbroeck et al., 1986, 1990; Ehlert and Tran, 1990; Watson and Abbot, 1992). These muscarinic subtypes can impinge on a range of signal transduction pathways, including activating phospholipase C through m1, m3, and m5 receptors, inhibiting adenylyl cyclase through m2 and m4 receptors, as well as activating G-protein-mediated ion channels (Hulme et al., 1990).
Coupling of mAChR activation and cGMP production has been demonstrated in both peripheral and central nervous tissue and in cell culture. For example, stimulation of mAChRs causes cGMP accumulation in ganglionic tissues of various species (Wamsley et al., 1979; Frey and McIssac, 1981). More recently, ACh agonists have been reported to increase cGMP synthesis via an mAChR in brain tissue and in neuroblastoma cells (Tonnaer, 1991; Castoldi et al., 1993; Hu and El-Fakahany, 1993; Mathes and Thompson, 1996). The link between the mAChR and cGMP synthesis is indirect, because no G-protein-activated GCs have been found. Instead, signaling through mAChRs appears to involve an intermediary, such as Ca2+/nitric oxide (NO) (Schultz et al., 1973; Castoldi et al., 1993; Hu and El-Fakahany, 1993; Ando et al., 1994; Mathes and Thompson, 1996), CO (Ingi et al., 1996), and/or a metabolite of arachidonic acid (Snider et al., 1984; McKinney and Richelson, 1986). It remains to be tested whether NO, CO, arachidonic acid, or multiple pathways are involved in the muscarinic regulation of the SCN circadian rhythm.
cGMP and circadian clock regulation by cholinergic signals
ACh is among the longest-studied regulators of circadian timing. Cholinergics have been administered at a range of sites in the periphery and CNS, and their effects on circadian rhythms have been measured at various endpoints (behavior, hormone secretion, neuronal activity). Because the results have not produced a congruent profile of timing of sensitivity, site of efficacy, or receptor–effector coupling, a question still remains as to the role of cholinergic signaling in the regulation of the circadian system. Various cholinergic sensitivities at multiple brain and peripheral levels subserving circadian rhythmic processes are likely to underlie the lack of coherence in these observations.
Yet despite the diversity in these experimental designs, the resulting windows of circadian sensitivity to cholinergics have strong correlations with the window of circadian sensitivity to light. Light is the most powerful nocturnal regulator of circadian rhythms. Retinal fibers project directly both to the SCN, site of the circadian clock, and to the basal forebrain cholinergic neurons, which function in sleep and arousal (Semba, 1991; Steriade and McCarley, 1990). Thus, light could activate neurons in the basal forebrain complex to release ACh onto SCN neurons during glutamatergic neurotransmission from projections of the retinohypothalamic tract. Cholinergic–glutamatergic interactions are well documented in brain sites modified by learning (Aigner, 1995). It follows that the phase-resetting signal at the SCN may represent the integration of ongoing activities in brain regions subserving sleep and wakefulness with those from other brain sites regulating the biological clock. Indeed, the GC/cGMP/PKG pathway, which we here demonstrate mediates coupling of SCN mAChRs and circadian clock processes, contributes to light-stimulated nocturnal phase resetting in the phylogenetically distant clocks of both hamsters (Weber et al., 1995; Mathur et al., 1996) and sea hares (Eskin et al., 1984).
We have approached the contentious issue of whether and in what way cholinergic stimulation affects the circadian clock by studying a brain slice preparation and selectively manipulating the neurochemical environment of this isolated SCN. Although we do not exclude a nicotinic response at other times of day (see Traschel et al., 1995), our evaluation of cholinergic mechanisms generates remarkably coherent support for our initial hypothesis: nocturnal cholinergic stimulation of mAChRs within the SCN can reset the circadian clock by stimulating GC, generating cGMP, and activating PKG. Important support for this pathway would be contributed by demonstration that M1 mAChRs are localized in proximity to cholinergic terminals within the SCN.
Although mechanisms that restrict the cholinergic response to the clock’s nocturnal domain are unknown, access is likely regulated along signaling pathways. Because cGMP analogs reset the SCN at night, but not daytime, at least one gating site lies downstream from cGMP. Precisely which steps downstream from cGMP are gated by the clock, what molecular mechanisms regulate their open state, and how they facilitate clock access during state-dependent changes in other brain regions, such as those contributing to sleep and wakefulness, will be important to establish. Nevertheless, this demonstration of mediation of nocturnal clock resetting is among the first studies to identify a specific functional consequence of the coupling of mAChRs and the cGMP signaling pathway in the CNS.
Footnotes
This work was supported by Public Health Service Grants NS22155 and NS33240 from the National Institute of Neurological Disorders and Stroke. We thank Liana Kuriashkina for excellent technical assistance, E. Todd Weber and Shelley Tischkau for technical advice, and Dong Chen and Tom Tcheng for thoughtful comments.
Correspondence should be addressed to Prof. Martha U. Gillette, Department of Cell and Structural Biology, University of Illinois, B107 Chemical and Life Sciences Laboratory, MC-123, 601 South Goodwin Avenue, Urbana, IL 61801.
Dr. Liu’s present address: Laboratory of Developmental Chronobiology, Children’s Service, GRJ 1226, Massachusetts General Hospital, Boston, MA 02114.
REFERENCES
- 1.Aigner TG. Pharmacology of memory: cholinergic-glutamatergic interactions. Curr Biol. 1995;5:155–160. doi: 10.1016/0959-4388(95)80021-2. [DOI] [PubMed] [Google Scholar]
- 2.Ando M, Tatematsu T, Kunii S, Nagata Y. The intercellular communication via nitric oxide and its regulation in coupling of cyclic GMP synthesis upon stimulation of muscarinic cholinergic receptors in rat superior cervical sympathetic ganglia. Brain Res. 1994;650:283–288. doi: 10.1016/0006-8993(94)91793-0. [DOI] [PubMed] [Google Scholar]
- 3.Bina KG, Rusak B, Semba K. Localization of cholinergic neurons in the forebrain and brainstem that project to the suprachiasmatic nucleus of the hypothalamus in rats. J Comp Neurol. 1993;335:295–307. doi: 10.1002/cne.903350212. [DOI] [PubMed] [Google Scholar]
- 4.Bonner TI, Buckley NJ, Young AC, Brann MR. Identification of a family of muscarinic acetylcholine receptor genes. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- 5.Bonner TI, Young AC, Brann MR, Buckley NJ. Cloning and expression of the human and rat m5 muscarinic acetylcholine receptor genes. Neuron. 1988;1:403–410. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]
- 6.Bradford MM. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 7.Castoldi AF, Manzo L, Costa LG. Cyclic GMP formation induced by muscarinic receptors is mediated by nitric oxide synthesis in rat cortical primary cultures. Brain Res. 1993;610:57–61. doi: 10.1016/0006-8993(93)91216-f. [DOI] [PubMed] [Google Scholar]
- 8.Corbin JD, Thomas MK, Wolfe L, Shabb JB, Woodford TA, Francis SH. New insights into cGMP action. In: Nishizuka Y, Endo M, Tanaka C, editors. The biology and medicine of signal transduction. Raven; New York: 1990. pp. 411–417. [PubMed] [Google Scholar]
- 9.Ding JM, Chen D, Weber ET, Faiman LE, Rea MA, Gillette MU. Resetting the SCN biological clock: mediation of nocturnal circadian shifts by glutamate and NO. Science. 1994;266:1713–1717. doi: 10.1126/science.7527589. [DOI] [PubMed] [Google Scholar]
- 10.Ding JM, Faiman LE, Hurst WJ, Kuriashkina LR, Gillette MU. Resetting the biological clock: mediation of nocturnal CREB phosphorylation through light, glutamate and NO. J Neurosci. 1997;17:748–756. doi: 10.1523/JNEUROSCI.17-02-00667.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Earnest DJ, Turek FW. Role for acetylcholine in mediating effects of light on reproduction. Science. 1983;219:77–79. doi: 10.1126/science.6849121. [DOI] [PubMed] [Google Scholar]
- 12.Earnest DJ, Turek FW. Neurochemical basis for the photic control of circadian rhythms and seasonal reproductive cycles: role for acetylcholine. Proc Natl Acad Sci USA. 1985;82:4277–4281. doi: 10.1073/pnas.82.12.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehlert F, Tran LLP. Regional distribution of M1, M2 and non M1, non M2 subtypes of muscarinic binding sites in rat brain. J Pharmacol Exp Ther. 1990;255:1148–1157. [PubMed] [Google Scholar]
- 14.Eskin A, Takahashi JS, Zatz M, Block GD. Cyclic guanosine 3′:5′-monophosphate mimics the effects of light on a circadian pacemaker in the eye of Aplysia. J Neurosci. 1984;4:2466–2471. doi: 10.1523/JNEUROSCI.04-10-02466.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frey EA, McIssac RJ. A comparison of cyclic guanosine 3′, 5′-monophosphate and muscarinic excitatory responses in the superior cervical ganglion of the rat. J Pharmacol Exp Ther. 1981;218:115–121. [PubMed] [Google Scholar]
- 16.Fuchs JL, Hoppens KS. Alpha-bungarotoxin binding in relation to functional organization of the rat suprachiasmatic nucleus. Brain Res. 1987;407:9–16. doi: 10.1016/0006-8993(87)91214-5. [DOI] [PubMed] [Google Scholar]
- 17.Gillette MU (1996) Regulation of entrainment pathways by the suprachiasmatic circadian clock: sensitivities to second messengers. In: Hypothalamic integration of circadian rhythms (Buijs R, Romijn H, Pennartz C, Mirmiran M, eds), pp 119–130 Elsevier: Amsterdam. [DOI] [PubMed]
- 18.Gillette MU, Prosser RA. Circadian rhythm of the rat suprachiasmatic brain slice is rapidly reset by daytime application of cAMP analogs. Brain Res. 1988;474:348–352. doi: 10.1016/0006-8993(88)90449-0. [DOI] [PubMed] [Google Scholar]
- 19.Gillette MU, Reppert SM. The hypothalamic suprachiasmatic nuclei: circadian patterns of vasopressin secretion and neuronal activity in vitro. Brain Res Bull. 1987;19:135–139. doi: 10.1016/0361-9230(87)90176-6. [DOI] [PubMed] [Google Scholar]
- 20.Gillette MU, Medanic M, McArthur AJ, Liu C, Ding JM, Faiman LE, Weber ET, Tcheng TK, Gallman EA. Circadian clocks and their adjustment (Ciba Foundation Symposium 183), pp 134-153. Wiley; Chichester, UK: 1995. Intrinsic neuronal rhythms in the suprachiasmatic nuclei and their adjustment. [DOI] [PubMed] [Google Scholar]
- 21.Glass DB, Krebs EG. Phosphorylation by guanosine 3′:5′-monophosphate-dependent protein kinase of synthetic peptide analogs of a site phosphorylated in histone H2B. J Biol Chem. 1982;257:1196–1200. [PubMed] [Google Scholar]
- 22.Green DJ, Gillette R. Circadian rhythm of firing rate recorded from single cells in the rat suprachiasmatic slice. Brain Res. 1982;245:198–200. doi: 10.1016/0006-8993(82)90361-4. [DOI] [PubMed] [Google Scholar]
- 23.Hatton GI, Doran AD, Salm AK, Tweedle CD. Brain slice preparation: hypothalamus. Brain Res Bull. 1980;5:405–414. doi: 10.1016/s0361-9230(80)80010-4. [DOI] [PubMed] [Google Scholar]
- 24.Hu J, El-Fakahany EE. Role of intercellular and intracellular communication by nitric oxide in coupling of muscarinic receptors to activation of guanylate cyclase in neuronal cells. J Neurochem. 1993;61:578–585. doi: 10.1111/j.1471-4159.1993.tb02161.x. [DOI] [PubMed] [Google Scholar]
- 25.Hulme E, Birdsall N, Buckley J. Muscarinic receptor subtypes. Annu Rev Pharmacol Toxicol. 1990;30:633–673. doi: 10.1146/annurev.pa.30.040190.003221. [DOI] [PubMed] [Google Scholar]
- 26.Ichikawa T, Hirata Y. Organization of choline acetyltransferase-containing structures in the forebrain of the rat. J Neurosci. 1986;6:281–292. doi: 10.1523/JNEUROSCI.06-01-00281.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ingi T, Cheng J, Ronnett GV. Carbon monoxide: an endogenous modulator of the nitric oxide-cyclic GMP signaling system. Neuron. 1996;16:835–842. doi: 10.1016/s0896-6273(00)80103-8. [DOI] [PubMed] [Google Scholar]
- 28.Inouye ST, Shibata S. Neurochemical organization of circadian rhythm in the suprachiasmatic nucleus. Neurosci Res. 1994;20:109–130. doi: 10.1016/0168-0102(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 29.Kow L, Pfaff DW. Suprachiasmatic neurons in tissue slices from ovariectomized rats: electrophysiological and neuropharmacological characterization and the effects of estrogen treatment. Brain Res. 1984;297:275–286. doi: 10.1016/0006-8993(84)90568-7. [DOI] [PubMed] [Google Scholar]
- 30.Liu C (1995) Cholinergic regulation of a mammalian circadian clock. PhD thesis, University of Illinois at Urbana-Champaign, pp 72–87.
- 31.Liu C, Gillette MU. Carbachol directly resets the circadian rhythm of SCN neuronal activity in vitro during subjective night through a cGMP/PKG-dependent mechanism. Soc Neurosci Abstr. 1994;20:159. [Google Scholar]
- 32.Liu C, Gillette MU. Cholinergic regulation of the SCN circadian rhythm via a muscarinic mechanism at night. J Neurosci. 1996;16:744–751. doi: 10.1523/JNEUROSCI.16-02-00744.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathes C, Thompson SH. The nitric oxide/cGMP pathway couples muscarinic receptors to the activation of Ca+2 influx. J Neurosci. 1996;16:1702–1709. doi: 10.1523/JNEUROSCI.16-05-01702.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathur A, Golobek DA, Ralph MR. cGMP-dependent protein kinase inhibitors block light-induced phase advances of circadian rhythms in vivo. Am J Physiol. 1996;270:R1031–R1036. doi: 10.1152/ajpregu.1996.270.5.R1031. [DOI] [PubMed] [Google Scholar]
- 35.McArthur AJ, Hunt AE, Gillette MU. Melatonin action and signal transduction in the rat suprachiasmatic circadian clock: activation of protein kinase C at dusk and dawn. Endocrinology. 1997;138:627–634. doi: 10.1210/endo.138.2.4925. [DOI] [PubMed] [Google Scholar]
- 36.McKinney M, Richelson E. Blockade of N1E-115 murine neuroblastoma muscarinic receptor function by agents that affect the metabolism of arachidonic acid. Biochem Pharmacol. 1986;35:2389–2397. doi: 10.1016/0006-2952(86)90466-1. [DOI] [PubMed] [Google Scholar]
- 37.Medanic M, Gillette MU. Serotonin regulates the phase of the rat suprachiasmatic circadian pacemaker in vitro only during the subjective day. J Physiol (Lond) 1992;450:629–642. doi: 10.1113/jphysiol.1992.sp019147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medanic M, Gillette MU. Suprachiasmatic circadian pacemaker of rat shows two windows of sensitivity to neuropeptide Y in vitro. Brain Res. 1993;620:281–286. doi: 10.1016/0006-8993(93)90166-k. [DOI] [PubMed] [Google Scholar]
- 39.Meijer JH, van der Zee EA, Dietz M. The effects of intraventricular carbachol injection on the free-running activity rhythm of the hamster. J Biol Rhythms. 1988;3:333–348. doi: 10.1177/074873048800300403. [DOI] [PubMed] [Google Scholar]
- 40.Miller JD, Murakami DM, Fuller CA. The response of suprachiasmatic neurons of the rat hypothalamus to photic and nicotinic stimuli. J Neurosci. 1987;7:978–986. doi: 10.1523/JNEUROSCI.07-04-00978.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore RY. Organization of the circadian timing system. In: Buijs R, Romijn H, Pennartz C, Mirmiran M, editors. Hypothalamic integration of circadian rhythms. Elsevier; Amsterdam: 1996. pp. 101–117. [Google Scholar]
- 42.Nishino H, Koizumi K. Responses of neurons in the suprachiasmatic nuclei of the hypothalamus to putative transmitters. Brain Res. 1977;120:167–172. doi: 10.1016/0006-8993(77)90509-1. [DOI] [PubMed] [Google Scholar]
- 43.Prosser RA, Gillette MU. The mammalian circadian clock in the suprachiasmatic nuclei is reset in vitro by cAMP. J Neurosci. 1989;9:1073–1081. doi: 10.1523/JNEUROSCI.09-03-01073.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prosser RA, McArthur AJ, Gillette MU. cGMP induces phase shifts of a mammalian circadian pacemaker at night, in antiphase to cAMP effects. Proc Natl Acad Sci USA. 1989;86:6812–6815. doi: 10.1073/pnas.86.17.6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prosser RA, Miller JD, Heller HC. A serotonin agonist phase-shifts the circadian clock in the suprachiasmatic nuclei in vitro. Brain Res. 1990;534:336–339. doi: 10.1016/0006-8993(90)90153-3. [DOI] [PubMed] [Google Scholar]
- 46.Rao ZR, Yamano M, Wanaka A, Tatehata T, Shiosaka S, Tohyama M. Distribution of cholinergic neurons and fibers in the hypothalamus of the rat using choline acetyltransferase as a marker. Neuroscience. 1987;20:923–934. doi: 10.1016/0306-4522(87)90253-3. [DOI] [PubMed] [Google Scholar]
- 47.Schultz G, Hardman JG, Schultz K, Baird CE, Sutherland EW. The importance of calcium ions for the regulation of guanosine 3′:5′-monophosphate levels. Proc Natl Acad Sci USA. 1973;70:3889–3893. doi: 10.1073/pnas.70.12.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Semba K. The cholinergic basal forebrain: a critical role in cortical arousal. In: Napier TC, Kalivas PW, Hanin I, editors. The basal forebrain: anatomy to function. Plenum; New York: 1991. pp. 197–218. [DOI] [PubMed] [Google Scholar]
- 49.Shibata S, Liou SY, Ueki S. Different effects of amino acids, acetylcholine and monoamines on neuronal activity of suprachiasmatic nucleus in rat pups and adults. Neurosci Lett. 1983;39:187–192. doi: 10.1016/0304-3940(83)90075-7. [DOI] [PubMed] [Google Scholar]
- 50.Snider RM, McKinney M, Forray C, Richelson E. Neurotransmitters mediate cyclic GMP formation by involvement of arachidonic acid and lipoxygenase. Proc Natl Acad Sci USA. 1984;81:3905–3909. doi: 10.1073/pnas.81.12.3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steriade M, McCarley RW. Brainstem control of wakefulness and sleep. Plenum; New York: 1990. [Google Scholar]
- 52.Tago H, McGeer PL, Bruce G, Hersh LB. Distribution of choline acetyltransferase-containing neurons of the hypothalamus. Brain Res. 1987;415:49–62. doi: 10.1016/0006-8993(87)90268-x. [DOI] [PubMed] [Google Scholar]
- 53.Tonnaer JADM, Cheung CL, Boer TD. cGMP formation and phosphoinositide turnover in rat brain slices are mediated by pharmacologically distinct muscarinic acetylcholine receptors. Eur J Pharmacol. 1991;207:183–188. doi: 10.1016/0922-4106(91)90029-h. [DOI] [PubMed] [Google Scholar]
- 54.Trachsel L, Heller HC, Miller JD. Nicotine phase-advances the circadian neuronal activity rhythm in rat suprachiasmatic nuclei explants. Neuroscience. 1995;65:797–803. doi: 10.1016/0306-4522(94)00506-z. [DOI] [PubMed] [Google Scholar]
- 55.van den Pol AN, Tsujimoto KL. Neurotransmitters of the hypothalamic suprachiasmatic nucleus: immunocytochemical analysis of 25 neuronal antigens. Neuroscience. 1985;15:1049–1086. doi: 10.1016/0306-4522(85)90254-4. [DOI] [PubMed] [Google Scholar]
- 56.van der Zee EA, Streefland C, Strosberg AD, Schroder H, Luiten PGM. Colocalization of muscarinic and nicotinic receptors in cholinoceptive neurons of the suprachiasmatic region in young and aged rats. Brain Res. 1991;542:348–352. doi: 10.1016/0006-8993(91)91590-w. [DOI] [PubMed] [Google Scholar]
- 57.Waelbroeck M, Gillard M, Robberecht P, Christophe J. Kinetic studies of [3H]N-methylscopolamine binding to muscarinic receptors in the rat central nervous system: evidence for the existence of three classes of binding sites. Mol Pharmacol. 1986;30:305–314. [PubMed] [Google Scholar]
- 58.Waelbroeck M, Tastenoy M, Camus J, Christophe J. Binding of selective antagonists to four muscarinic receptors (M1 to M4) in rat forebrain. Mol Pharmacol. 1990;38:267–273. [PubMed] [Google Scholar]
- 59.Wamsley JK, West JR, Black AC, Jr, Williams TH. Muscarinic cholinergic and preganglionic physiological stimulation increase cyclic GMP levels in guinea pig superior cervical ganglia. J Neurochem. 1979;32:1033–1035. doi: 10.1111/j.1471-4159.1979.tb04590.x. [DOI] [PubMed] [Google Scholar]
- 60.Watson S, Abbot A (1992) TIPS receptor nomenclature supplement. Trends Pharmacol Sci [Suppl]19. [PubMed]
- 61.Weber ET (1995) The roles of nitric oxide, cGMP and cGMP-dependent protein kinase in generation and modulation of circadian rhythms by the suprachiasmatic nucleus. PhD thesis, University of Illinois at Urbana-Champaign, pp 33–38; 67–69.
- 62.Weber ET, Gannon RL, Rea MA. cGMP-dependent protein kinase blocks light-induced phase advances of circadian rhythms in vivo. Neurosci Lett. 1995;197:227–230. doi: 10.1016/0304-3940(95)11961-u. [DOI] [PubMed] [Google Scholar]
- 63.Wee BEF, Anderson KD, Kouchis NS, Turek FW. Administration of carbachol into the lateral ventricle and suprachiasmatic nucleus (SCN) produces dose-dependent phase shifts in the circadian rhythm of locomotor activity. Neurosci Lett. 1992;137:211–215. doi: 10.1016/0304-3940(92)90406-w. [DOI] [PubMed] [Google Scholar]
- 64.Zatz M, Brownstein MJ. Intraventricular carbachol mimics the effects of light on the circadian rhythm in the rat pineal gland. Science. 1979;203:358–361. doi: 10.1126/science.32619. [DOI] [PubMed] [Google Scholar]
- 65.Zatz M, Herkenham MA. Intraventricular carbachol mimics the phase-shifting effect of light on the circadian rhythm of wheel-running activity. Brain Res. 1981;212:234–238. doi: 10.1016/0006-8993(81)90059-7. [DOI] [PubMed] [Google Scholar]