

Abstract
We examined the interactions among three classes of peripherally-acting antinociceptive agents (μ-opioid, α2-adrenergic, and A1-adenosine) in the development of tolerance and dependence to their antinociceptive effects. Antinociception was determined by assessing the degree of inhibition of prostaglandin E2 (PGE2)-induced mechanical hyperalgesia, using the Randall-Selitto paw-withdrawal test.
Tolerance developed within 4 hr to the antinociceptive effect of the α2-adrenergic agonist clonidine; dependence also occurred at that time, demonstrated as a withdrawal hyperalgesia that was precipitated by the α2-receptor antagonist yohimbine. These findings are similar to those reported previously for tolerance and dependence to μ and A1 peripheral antinociception (Aley et al., 1995).
Furthermore, cross-tolerance and cross-withdrawal between μ, A1, and α2 agonists occurred. The observations of cross-tolerance and cross-withdrawal suggest that all three receptors are located on the same primary afferent nociceptors. In addition, the observations suggest that the mechanisms of tolerance and dependence to the antinociceptive effects of μ, A1, and α2 are mediated by a common mechanism.
Although any of the agonists administered alone produce antinociception, we found that μ, A1, and α2 receptors may not act independently to produce antinociception, but rather may require the physical presence of the other receptors to produce antinociception by any one agonist. This was suggested by the finding that clonidine (α2-agonist) antinociception was blocked not only by yohimbine (α2-antagonist) but also by PACPX (A1-antagonist) and by naloxone (μ-antagonist), and that DAMGO (μ-agonist) antinociception and CPA (A1-agonist) antinociception were blocked not only by naloxone (μ-antagonist) and PACPX (A1-antagonist), respectively, but also by yohimbine (α2-antagonist). This cross-antagonism of antinociception occurred at the ID80 dose for each antagonist at its homologous receptor. To test the hypothesis that the physical presence of μ-opioid receptor is required not only for μ antinociception but also for α2 antinociception, antisense oligodeoxynucleotides (ODNs) for the μ-opioid and α2C-adrenergic receptors were administered intrathecally to reduce the expression of these receptors on primary afferent neurons. These studies demonstrated that μ-opioid ODN administration decreased not only μ-opioid but also α2-adrenergic antinociception; A1 antinociception was unaffected. In contrast, α2C-adrenergic ODN decreased antinociception induced by all three classes of antinociceptive agents.
In conclusion, these data suggest that peripheral antinociception induced by μ, α2, and A1 agonists requires the physical presence of multiple receptors. We propose that there is a μ, A1, α2 receptor complex mediating antinociception in the periphery. In addition, there is cross-tolerance and cross-dependence between μ, A1, and α2antinociception, suggesting that their underlying mechanisms are related.
Keywords: pain, analgesia, dorsal root ganglion, opioid, antisense oligodeoxynucleotide, receptor cross-talk
Both μ-opioid and A1-adenosine agonists have been shown to produce a potent antinociception when administered in the periphery (Taiwo and Levine, 1990; Aley et al., 1995). This antinociception is detected in the presence of hyperalgesia produced by numerous inflammatory mediators including PGE2(Taiwo and Levine, 1990). Many of the effects produced by μ and A1 agonists are mediated through a common second messenger, specifically, activation of an inhibitory guanine nucleotide binding (Gi) protein (Sharma et al., 1975; Law et al., 1981;Childers and LaRiviere, 1984; Mankman et al., 1988).
We have found that tolerance and dependence develop to both μ and A1 peripheral antinociception. In addition, a symmetric cross-tolerance and cross-dependence exists between the μ and A1 antinociceptive mechanisms (Aley et al., 1995).
α2-adrenergic agonists have also been shown to produce antinociception when administered peripherally (Khasar et al., 1995). Because many of the effects of α2 agonists, like those produced by μ and A1 agonists, involve Giprotein signaling (Sharma et al., 1975; Law et al., 1981; Childers and LaRiviere, 1984; Mankman et al., 1988), we hypothesized that they all produce antinociception in the periphery through common cellular mechanisms. In addition, we hypothesized that there would be symmetric cross-tolerance and cross-dependence between the peripheral antinociceptive actions of these three agonists. Because α2-adrenergic, μ-opioid, and A1-adenosine agonists are widely used clinically, interactions between their peripheral antinociceptive effects is of significant interest.
MATERIALS AND METHODS
Animals
Experiments were performed on male Sprague Dawley rats (250–300 gm, Bantin and Kingman, Fremont, CA). Animals were housed in groups of two under a 12 hr light/dark cycle (light on 6.0 hr). Food and water were available ad libitum. All testing was done between 10.0 and 16.0 hr. Experiments were carried out under approval of the Institutional Animal Care Committee of the University of California, San Francisco.
Behavioral testing
The nociceptive flexion reflex was quantified with a Basile Analgesymeter (Stoelting, Chicago, IL), which applies a linearly increasing mechanical force to the dorsum of the hindpaw of the rat. Before the experiments, rats were exposed to the procedure for 3 d (1 hr daily at 5 min intervals, i.e., 12 exposures), a procedure that produces a stable baseline threshold measurement and enhances the ability to detect the action of hyperalgesic agents (Taiwo et al., 1989; Aley et al., 1995). On the day of the experiment, rats were exposed to the same procedure, and the mean of the last 6 of the 12 readings was considered to be the baseline mechanical nociceptive threshold. The mean baseline threshold in these experiments was 110.9 ± 0.4 gm (n = 466; mean ± SEM). Mechanical threshold was again determined at different time points (15, 20, and 25 min) after various treatments. The mean of these three readings was defined as the paw-withdrawal threshold post-treatment for that paw, and this value was used to calculate the percentage change from the baseline threshold [% change in threshold = (pretreatment threshold - post-treatment threshold)/(pretreatment threshold)] × 100.
Drug administration
The drugs used in this study were PGE2 [100 ng; direct-acting hyperalgesic inflammatory mediator (Pitchford and Levine, 1991, Gold et al., 1994)], DAMGO (μ-opioid receptor agonist), clonidine (α2-adrenergic receptor agonist), CPA (A1-adenosine receptor agonist), naloxone methyl iodide (opioid receptor antagonist), yohimbine HCl (α2-adrenergic receptor antagonist), and PACPX (A1-adenosine receptor antagonist), all from Research Biochemicals International (Natick, MA). The selection of the drug doses used in this study was based on the dose–response curves determined during this study or from previous work done in this laboratory (Aley et al., 1995). The stock solution of PGE2(1 μg/2.5 μl) was prepared in 10% ethanol and further dilutions were made in saline; the final concentration of ethanol was ≤1%. DAMGO, clonidine, CPA, naloxone, yohimbine, and PACPX were dissolved in saline. When drug combinations were used, they were administered from the same syringe so that the drug mentioned first reached the intradermal site first; the two drugs were separated in the syringe by a small air bubble to avoid the problem of diffusion. When an antagonist was included to antagonize the effect of an agonist, it was always injected first. The ID80 dose of each antagonist (i.e., naloxone 200 ng, yohimbine 100 ng, and PACPX 100 ng), calculated from its dose–response curve for reversal of the effect of its homologous agonist, was used throughout the study. All the drugs except the oligodeoxynucleotides were administered intradermally in a volume of 2.5 μl/paw.
Intrathecal cannulation
To administer drugs intrathecally, a catheter (PE-10 polyethylene tubing) was inserted caudally 8.5 cm into the subdural space through a midline incision made in the atlanto-occipital membrane of rats anesthetized with pentobarbital (50 mg/kg, i.p.); the external end of the catheter was secured to the skull with screws and dental acrylic (Yaksh and Rudy, 1976). The skin incision was sutured closed, and the animals were allowed to recover. Two days after surgery, rats that showed no motor deficits were used for experimental studies involving intrathecal administration of ODNs.
Antisense oligodeoxynucleotides
μ-Opioid receptor oligodeoxynucleotides. The μ-opioid receptor antisense and sense ODNs used in this study were synthesized using a Nucleic Acid Synthesizer model 391 (PCR Mate; Applied Biosystems, Foster City, CA). The μ-opioid receptor antisense ODN, 5′-CGCCCCAGCCTCTTCCTCT-3′, is directed against the 5′-untranslated region of μ-opioid receptor-1 (MOR-1) clone, located between bases 87 and 69 upstream from the initiating ATG. The sense ODN, 5′-AGAGGAAGAGGCTGGGGCG-3′ (Rossi et al., 1994), is complementary to the antisense sequence. Concentrations of ODN stocks were determined by spectrophotometry. Before their use, ODNs were lyophilized and resuspended in 0.9% NaCl to a concentration of 1 μg/10 μl.
Rats were divided into three groups: one group was untreated (without cannulae), a second group was treated with sense ODN (1 μg), and the third group was treated with antisense ODN (1 μg/rat). Using a microsyringe (Hamilton, Nevada City, UT), a dose of 1 μg ODN was administered to each rat intrathecally, in a volume of 10 μl, followed by 10 μl of saline (the dead space of the intrathecal catheter), on alternate days (days 1, 3, and 5). Behavioral tests were done 24 hr after the last dose of ODN. We have found that antisense ODN against MOR-1 attenuates μ-opioid receptor-like immunoreactivity in the dorsal horn of the spinal cord and peripheral nerve, DAMGO-induced inhibition of calcium current in cultured dorsal root ganglion neurons and DAMGO-induced inhibition of PGE2-induced hyperalgesia (Khasar et al., 1996).
α2C-opioid receptor oligodeoxynucleotides. We have demonstrated previously that the α2-adrenergic receptor mediating peripheral antinociception has the pharmacological characteristics of the α2C subtype (Khasar et al., 1995). Therefore, we also synthesized the antisense and sense ODNs for the α2C receptor subtype, using Nucleic Acid Synthesizer model 391 PCR Mate. The α2C receptor antisense ODN, 5′-ACCTGCGGAGTACTG-3′, was developed by Lingen and Ordway (1995). The sense ODN sequence 5′-CAGTACTCCGCAGGT-3′ is complementary to the antisense sequence.
Rats were divided into three groups: one group was untreated (without cannulae), a second group was treated with sense ODN (1 μg/rat), and the third group was treated with antisense ODN (1 μg). Treatment and behavioral testing were as described for μ-opioid receptor ODN experiments.
Abbreviations for the drugs used in this study and their actions are shown in Table 1; experimental protocols are shown in Table 2.
Table 1.
Abbreviations of agents used
| Abbreviation(s) | Agent | Action |
|---|---|---|
| PGE2or E2 | Prostaglandin E2 | Hyperalgesic inflammatory mediator |
| DAMGO or D | [d-Ala2,N-Me-Phe4,gly5-ol] enkephalin | μ-opioid receptor agonist |
| N | Naloxone | Opioid receptor antagonist |
| Cl | Clonidine | α2-adrenergic agonist |
| Yo | Yohimbine | α2-adrenergic antagonist |
| CPA | N6-cyclopentyl-adenosine | A1-adenosine receptor agonist |
| PACPX | 1,3-dipropyl-8-(2-amino-4-chlorophenyl)-xanthine | A1-adenosine receptor antagonist |
| μ-antisense | μ-opioid receptor oligodeoxynucleotide (ODN) | Downregulation of μ-opioid receptors |
| μ-sense | No effect | |
| α2-antisense | α2C-adrenergic receptor oligodeoxynucleotide (ODN) | Downregulation of α2C-adrenergic receptors |
| α2-sense | No effect |
Table 2.
Experimental protocols
| Group | N | Treatment | Dose(s) |
|---|---|---|---|
| I–A | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 16 | Clonidine + PGE2 | 100 ng + 100 ng |
| 3 | 6 | Clonidine hourly × 3 | 100 ng × 3 |
| 4 | 12 | Clonidine hourly × 3, fourth hour clonidine + PGE2 | 100 ng × 3, 100 ng + 100 ng |
| I–B | |||
| 1 | 12 | DAMGO hourly × 3, fourth hour clonidine + PGE2 | 1 μg × 3, 100 ng + 100 ng |
| 2 | 12 | CPA hourly × 3, fourth hour clonidine + PGE2 | 1 μg × 3, 100 ng + 100 ng |
| 3 | 16 | Clonidine + PGE2 | 100 ng + 100 ng |
| 4 | 12 | Clonidine hourly × 3, fourth hour DAMGO + PGE2 | 100 ng × 3, 1 μg + 100 ng |
| 5 | 6 | DAMGO + PGE2 | 1 μg + 100 ng |
| 6 | 6 | DAMGO hourly × 3 | 1 μg × 3 |
| 7 | 6 | CPA hourly × 3 | 1 μg × 3 |
| 8 | 12 | Clonidine hourly × 3, fourth hour CPA + PGE2 | 100 ng × 3, 1 μg + 100 ng |
| 9 | 6 | CPA + PGE2 | 1 μg + 100 ng |
| II-A | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 16 | Clonidine + PGE2 | 100 ng + 100 ng |
| 3 | 6 | Clonidine × 3 | 100 ng × 3 |
| 4 | 8 | Clonidine hourly × 3, fourth hour yohimbine | 100 ng × 3, 100 ng |
| II-B | |||
| 1 | 10 | Clonidine hourly × 3, fourth hour naloxone | 100 ng × 3, 200 ng |
| 2 | 10 | Clonidine hourly × 3, fourth hour PACPX | 100 ng × 3, 100 ng |
| 3 | 6 | Clonidine × 3 | 100 ng × 3 |
| 4 | 10 | DAMGO hourly × 3, fourth hour yohimbine | 1 μg × 3, 100 ng |
| 5 | 6 | DAMGO × 3 | 1 μg × 3 |
| 6 | 10 | CPA hourly × 3, fourth hour yohimbine | 1 μg × 3, 100 ng |
| 7 | 6 | CPA hourly × 3 | 1 μg × 3 |
| III-A | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 6 | DAMGO + PGE2 | 1 μg + 100 ng |
| 3 | 6 | Naloxone + DAMGO + PGE2 | 1 ng + 1 μg + 100 ng |
| 4 | 6 | Naloxone + DAMGO + PGE2 | 10 ng + 1 μg + 100 ng |
| 5 | 6 | Naloxone + DAMGO + PGE2 | 100 ng + 1 μg + 100 ng |
| 6 | 6 | Naloxone + DAMGO + PGE2 | 1 μg + 1 μg + 100 ng |
| III-B | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 16 | Clonidine + PGE2 | 100 ng + 100 ng |
| 3 | 6 | Yohimbine + clonidine + PGE2 | 1 ng + 100 ng + 100 ng |
| 4 | 6 | Yohimbine + clonidine + PGE2 | 10 ng + 100 ng + 100 ng |
| 5 | 6 | Yohimbine + clonidine + PGE2 | 100 ng + 100 ng + 100 ng |
| 6 | 6 | Yohimbine + clonidine + PGE2 | 1 μg + 100 ng + 100 ng |
| III-C | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 6 | CPA + PGE2 | 1 μg + 100 ng |
| 3 | 6 | PACPX + CPA + PGE2 | 1 ng + 1 μg + 100 ng |
| 4 | 6 | PACPX + CPA + PGE2 | 10 ng + 1 μg + 100 ng |
| 5 | 6 | PACPX + CPA + PGE2 | 100 ng + 1 μg + 100 ng |
| 6 | 6 | PACPX + CPA + PGE2 | 1 μg + 1 μg + 100 ng |
| IV-A | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 16 | Clonidine + PGE2 | 100 ng + 100 ng |
| 3 | 6 | Yohimbine + clonidine + PGE2 | 100 ng + 100 ng + 100 ng |
| 4 | 10 | Naloxone + clonidine + PGE2 | 200 ng + 100 ng + 100 ng |
| 5 | 10 | PACPX + clonidine + PGE2 | 100 ng + 100 ng + 100 ng |
| IV-B | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 6 | DAMGO + PGE2 | 1 μg + 100 ng |
| 3 | 6 | Naloxone + DAMGO + PGE2 | 200 ng + 1 μg + 100 ng |
| 4 | 8 | Yohimbine + DAMGO + PGE2 | 100 ng + 1 μg + 100 ng |
| 5 | 6 | PACPX + DAMGO + PGE2 | 100 ng + 1 μg + 100 ng |
| IV-C | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 6 | CPA + PGE2 | 1 μg + 100 ng |
| Group | N | Treatment | Dose(s) |
| IV-C | |||
| 3 | 6 | PACPX + CPA + PGE2 | 100 ng + 1 μg + 100 ng |
| 4 | 8 | Naloxone + CPA + PGE2 | 200 ng + 1 μg + 100 ng |
| 5 | 6 | Yohimbine + CPA + PGE2 | 100 ng + 1 μg + 100 ng |
| V-A | |||
| 1 | 8 | Clonidine hourly × 3, fourth hour yohimbine | 100 ng × 3, 100 ng |
| 2 | 6 | Clonidine hourly × 3, fourth hour clonidine + yohimbine | 100 ng × 3, 100 ng + 100 ng |
| 3 | 8 | Clonidine hourly × 3, fourth hour DAMGO + yohimbine | 100 ng × 3, 1 μg + 100 ng |
| 4 | 8 | Clonidine hourly × 3, fourth hour CPA + yohimbine | 100 ng × 3, 1 μg + 100 ng |
| V-B | |||
| 1 | 6 | DAMGO hourly × 3, fourth hour naloxone | 1 μg × 3, 200 ng |
| 2 | 6 | DAMGO hourly × 3, fourth hour DAMGO + naloxone | 1 μg + 1 μg + 200 ng |
| 3 | 8 | DAMGO hourly × 3, fourth hour clonidine + naloxone | 1 μg + 100 ng + 200 ng |
| 4 | 8 | DAMGO hourly × 3, fourth hour CPA + naloxone | 1 μg + 1 μg + 200 ng |
| V-C | |||
| 1 | 8 | CPA hourly × 3, fourth hour PACPX | 1 μg × 3, 100 ng |
| 2 | 6 | CPA hourly × 3, fourth hour CPA + PACPX | 1 μg + 1 μg + 100 ng |
| 3 | 8 | CPA hourly × 3, fourth hour clonidine + PACPX | 1 μg + 100 ng + 100 ng |
| 4 | 8 | CPA hourly × 3, fourth hour DAMGO + PACPX | 1 μg + 1 μg + 100 ng |
| VI-A | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 6 | DAMGO + PGE2 | 1 μg + 100 ng |
| 3 | 6 | μ-antisense intrathecally alternate days × 3, 24 hr after DAMGO + PGE2 | 1 μg × 3, 1 μg + 100 ng |
| 4 | 6 | μ-sense intrathecally alternate days × 3, 24 hr after DAMGO + PGE2 | 1 μg × 3, 1 μg + 100 ng |
| VI-B | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 16 | Clonidine + PGE2 | 100 ng + 100 ng |
| 3 | 6 | μ-antisense intrathecally alternate days × 3, 24 hr after clonidine + PGE2 | 1 μg × 3, 100 ng + 100 ng |
| 4 | 6 | μ-sense intrathecally alternate days × 3, 24 hr after clonidine + PGE2 | 1 μg × 3, 100 ng + 100 ng |
| VI-C | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 6 | CPA + PGE2 | 1 μg + 100 ng |
| 3 | 6 | μ-antisense intrathecally alternate days × 3, 24 hr after CPA + PGE2 | 1 μg × 3, 1 μg + 100 ng |
| 4 | 6 | μ-sense intrathecally alternate days × 3, 24 hr after CPA + PGE2 | 1 μg × 3, 1 μg + 100 ng |
| VII-A | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 6 | DAMGO + PGE2 | 1 μg + 100 ng |
| 3 | 6 | α2-antisense intrathecally alternate days × 3, 24 hr after DAMGO + PGE2 | 1 μg × 3, 1 μg + 100 ng |
| 4 | 6 | α2-sense intrathecally alternate days × 3, 24 hr after DAMGO + PGE2 | 1 μg × 3, 1 μg + 100 ng |
| VII-B | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 16 | Clonidine + PGE2 | 100 ng + 100 ng |
| 3 | 6 | α2-antisense intrathecally alternate days × 3, 24 hr after clonidine + PGE2 | 1 μg × 3, 100 ng + 100 ng |
| 4 | 6 | α2-sense intrathecally alternate days × 3, 24 hr after clonidine + PGE2 | 1 μg × 3, 100 ng + 100 ng |
| VII-C | |||
| 1 | 24 | PGE2 | 100 ng |
| 2 | 6 | CPA + PGE2 | 1 μg + 100 ng |
| 3 | 6 | α2-antisense intrathecally days × 3, 24 hr after CPA + PGE2 | 1 μg × 3, 1 μg + 100 ng |
| 4 | 6 | α2-sense intrathecally alternate days × 3, 24 hr after CPA + PGE2 | 1 μg × 3, 1 μg + 100 ng |
Abbreviations: PGE2, Prostaglandin E2 (EP receptor agonist); DAMGO, [d-Ala2, N-Me-Phe4, gly5-ol] (μ-opioid receptor agonist); Cl, clonidine (α2 agonist); Yo, yohimbine (α2 antagonist); CPA, N6-cyclopentyl adenosine (A1-adenosine agonist); PACPX, 1,3-dipropyl-8-(2-amino-4-chlorophenyl)-xanthine (A1-adenosine antagonist). There are repetitions for the sake of comparison.
Statistical analysis
Data are presented as mean ± SEM of six or more observations in each of the experimental groups. Statistical significance was determined by ANOVA, followed by Scheffe’s post hoc test; p < 0.05 was considered statistically significant. Some data are repeated for comparison (see figure legends).
RESULTS
Tolerance to clonidine antinociception
In the present study, we found that after repeated administration, clonidine (100 ng) produces tolerance for its inhibition of PGE2 (100 ng)-induced hyperalgesia (Fig.1A), similar to that seen for μ and A1 agonists (Aley et al., 1995).
Fig. 1.

A, Repeated administration of clonidine produces tolerance to antinociception. Effect of PGE2 (E2), clonidine plus PGE2(Cl+E2), clonidine once hourly for 3 hr (Clx3), clonidine once hourly for 3 hr, and at the fourth hour clonidine plus PGE2 (Clx3, Cl+E2) on mechanical paw withdrawal threshold in the rat.B, Bidirectional cross-tolerance develops among A1, α2, and μ antinociception. Effect of clonidine plus PGE2 (Cl+E2), DAMGO once hourly for 3 hr (Dx3), DAMGO once hourly for 3 hr and at the fourth hour clonidine plus PGE2(Dx3,Cl+E2), CPA once hourly for 3 hr and at the fourth hour clonidine plus PGE2 (CPAx3, Cl+E2), CPA once hourly for 3 hr (CPAx3), clonidine once hourly for 3 hr and at the fourth hour DAMGO plus PGE2 (Clx3, D+E2), and clonidine once hourly for 3 hr and at the fourth hour CPA plus PGE2 (Clx3, CPA+E2) on mechanical paw withdrawal threshold in the rat.
Bidirectional cross-tolerance for μ, A1, and α2 peripheral antinociception
In paws made tolerant to DAMGO or CPA, clonidine failed to produce a significant antinociceptive effect when injected at the fourth hour (Fig. 1B). Also, in paws made tolerant to clonidine, DAMGO and CPA were no longer antinociceptive (Fig. 1B). These observations suggest that there is bidirectional cross-tolerance between α2, μ, and A1 to their peripheral antinociceptive effects.
Yohimbine precipitated withdrawal hyperalgesia in clonidine-tolerant paws
In the present study, we found that after induction of tolerance with three hourly injections of clonidine (100 ng), administration of its receptor antagonist yohimbine (100 ng) precipitated a withdrawal hyperalgesia, revealing the development of dependence (Fig.2A).
Fig. 2.

A, Yohimbine precipitates withdrawal hyperalgesia in clonidine tolerant paws. Effect of PGE2 (E2), clonidine plus PGE2(Cl+E2), clonidine once hourly for 3 hr (Clx3), clonidine once hourly for 3 hr (Clx3), clonidine once hourly for 3 hr, and at the fourth hour yohimbine (Clx3, Yo) on mechanical paw-withdrawal threshold in the rat. B, Bidirectional cross-withdrawal develops among A1, α2, and μ antinociception. Effect of clonidine once hourly for 3 hr and at the fourth hour naloxone (Clx3, N), clonidine once hourly for 3 hr and at the fourth hour PACPX (Clx3, PACPX), clonidine once hourly for 3 hr (Clx3), DAMGO once hourly for 3 hr and at the fourth hour yohimbine (Dx3, Yo), DAMGO once hourly for 3 hr (Dx3), CPA once hourly for 3 hr (CPAx3), CPA once hourly for 3 hr and at the fourth hour yohimbine (CPAx3, Yo), and CPA once hourly for 3 hr (CPAx3) on mechanical paw withdrawal threshold in the rat.
Bidirectional cross-withdrawal for μ, A1, and α2 tolerance/antinociception
In paws made tolerant to clonidine, administration of the μ- and A1-antagonists, naloxone and PACPX, respectively, precipitated withdrawal hyperalgesia. In paws made tolerant to DAMGO and CPA, yohimbine precipitated withdrawal hyperalgesia (Fig.2B). These observations suggest that there is bidirectional cross-withdrawal between μ, A1, and α2 after the development of peripheral tolerance to their peripheral antinociceptive effects.
Multiple receptors involved in μ, A1, and α2 antinociception
Naloxone, yohimbine, and PACPX dose-dependently blocked the antinociceptive effects of their homologous agonists DAMGO, clonidine, and CPA, respectively (Fig. 3A–C). However, in addition, naloxone (200 ng) also blocked clonidine antinociception but not CPA antinociception (Fig.4A,C), and PACPX (100 ng) blocked clonidine antinociception but not DAMGO antinociception (Fig.4A,B). Yohimbine (100 ng) blocked clonidine, DAMGO, and CPA antinociception (Fig. 4A–C). These data suggest that the α2 receptor is involved not only in α2 antinociception but also in μ and A1antinociception. In addition, the data suggest that the μ and A1 receptors are involved in α2antinociception.
Fig. 3.

μ, α2, and A1antagonists dose-dependently block μ, α2, and A1 antinociception, respectively. A, Naloxone dose-dependently blocks DAMGO antinociception. Effect of PGE2 (E2), DAMGO plus PGE2(D+E2) and various doses of naloxone (1 ng to 1 μg), and DAMGO plus PGE2 (N+D+E2), on mechanical paw withdrawal threshold in the rat. B, Yohimbine dose-dependently blocks clonidine antinociception. Effect of PGE2 (E2), clonidine plus PGE2(Cl+E2), and various doses of yohimbine (1 ng to 1 μg) and clonidine plus PGE2 (Yo+Cl+E2) on mechanical paw withdrawal threshold in the rat. C, PACPX dose-dependently blocks CPA antinociception. Effect of PGE2(E2), DAMGO plus PGE2(CPA+E2), and various doses of PACPX (1 ng to 1 μg) and CPA plus PGE2 (PACPX+CPA+E2) on mechanical paw withdrawal threshold in the rat.
Fig. 4.

Multiple receptors are involved in μ, α2, and A1 antinociception. A, Clonidine α2 antinociception is blocked not only by yohimbine but also by naloxone and PACPX. Effect of PGE2(E2), clonidine plus PGE2(Cl+E2), yohimbine plus clonidine plus PGE2(Yo+Cl+E2), naloxone plus clonidine plus PGE2 (N+Cl+E2), and PACPX plus clonidine plus PGE2 (PACPX+Cl+E2) on mechanical paw withdrawal threshold in the rat. B, DAMGO μ antinociception is blocked not only by naloxone but also by yohimbine. Effect of PGE2 (E2), DAMGO plus PGE2 (D+E2), naloxone plus DAMGO plus PGE2 (N+D+E2), yohimbine plus DAMGO plus PGE2 (Yo+D+E2), and PACPX plus DAMGO plus PGE2 (PACPX+D+E2) on mechanical paw withdrawal threshold in the rat. C, CPA A1antinociception is blocked not only by PACPX but also by yohimbine. Effect of PGE2 (E2), CPA plus PGE2 (CPA+E2), PACPX plus CPA plus PGE2 (PACPX+CPA+E2), naloxone plus CPA plus PGE2 (N+CPA+E2), and yohimbine plus CPA plus PGE2 (Yo+CPA+E2) on mechanical paw withdrawal threshold in the rat.
Multiple receptors involved in μ, A1, and α2 tolerance and withdrawal
A similar profile of receptor interactions was seen for tolerance and withdrawal to μ, A1, and α2antinociception. This was demonstrated by examining which receptor agonists (μ, A1, and α2) could block antagonist-induced withdrawal hyperalgesia. Yohimbine-induced withdrawal, in clonidine-tolerant paws, was blocked by coadministration of clonidine with yohimbine, as well as by coadministration of DAMGO and CPA with yohimbine (Fig. 5A). Naloxone-induced withdrawal, in DAMGO-tolerant paws, was blocked by coinjection of DAMGO with naloxone or clonidine with naloxone, but was not blocked by coadministration of CPA with naloxone (Fig.5B). Similarly, PACPX-induced withdrawal in CPA-tolerant paws was blocked by coinjection of CPA with PACPX or clonidine with PACPX, but was not blocked by coadministration of DAMGO with PACPX (Fig. 5C). These data suggest, as in previous experiments, that μ, α2, and A1 receptors are involved in α2 antinociception, tolerance, and withdrawal; that μ and α2 receptors are involved in μ antinociception, tolerance, and withdrawal; and that A1 and α2are involved in A1 antinociception, tolerance, and withdrawal.
Fig. 5.

Multiple receptors are involved in α2, μ, and A1 tolerance and withdrawal.A, Yohimbine withdrawal is blocked not only by clonidine but also by DAMGO and CPA. Effect of clonidine once hourly for 3 hr and at the fourth hour yohimbine (Clx3,Yo), clonidine once hourly for 3 hr and at the fourth hour clonidine plus yohimbine (Clx3,Cl+Yo), clonidine once hourly for 3 hr and at the fourth hour DAMGO plus yohimbine (Clx3,D+Yo), and clonidine once hourly for 3 hr and at the fourth hour CPA plus yohimbine (Clx3,CPA+Yo) on mechanical paw withdrawal threshold in the rat. B, Naloxone withdrawal is blocked not only by DAMGO but also by clonidine. Effect of DAMGO once hourly for 3 hr and at the fourth hour naloxone (Dx3,N), DAMGO once hourly for 3 hr and at the fourth hour DAMGO plus naloxone (Dx3,D+N), DAMGO once hourly for 3 hr and at the fourth hour clonidine plus naloxone (Dx3,Cl+N), and DAMGO once hourly for 3 hr and at the fourth hour CPA plus naloxone (Dx3,CPA+N) on mechanical paw withdrawal threshold in the rat. C, PACPX withdrawal is blocked not only by CPA but also by clonidine. Effect of CPA once hourly for 3 hr and at the fourth hour PACPX (CPAx3,PACPX), CPA once hourly for 3 hr and at the fourth hour CPA plus PACPX (CPAx3,CPA+PACPX), CPA once hourly for 3 hr and at the fourth hour clonidine plus PACPX (CPAx3,Cl+PACPX), and CPA once hourly for 3 hr and at the fourth hour DAMGO plus naloxone (CPAx3,D+PACPX) on mechanical paw withdrawal threshold in the rat.
Antisense ODN treatment supports the hypothesis that multiple receptors are involved in μ, α2, and A1antinociception
Antisense μ-opioid receptor ODN significantly attenuated not only the μ antinociception but also α2 antinociception 24 hr after the last injection of the antisense ODN. In contrast, A1 antinociception was unaffected by μ ODN treatment (Fig. 6A–C). Sense μ-opioid receptor ODN was without effect on PGE2hyperalgesia, μ antinociception, or α2 antinociception (Fig. 6A–C).
Fig. 6.

Antisense μ ODN treatment blocks not only μ antinociception but also α2 antinociception. A1 antinociception is unaffected. A, Effect of PGE2 (E2), DAMGO plus PGE2(D+E2), μ-antisense (AS) ODN 1 μg intrathecally on alternate days × 3 and DAMGO plus PGE2 [μ-(AS)x3,D+E2], μ-sense (S) ODN 1 μg intrathecally on alternate days × 3, and DAMGO plus PGE2 [μ-(S)x3,D+E2] on mechanical paw-withdrawal threshold. B, Effect of PGE2 (E2), clonidine plus PGE2(Cl+E2), μ-(AS) ODN 1 μg intrathecally on alternate days × 3, and clonidine plus PGE2[μ-(AS)x3,Cl+E2], μ-(S) ODN 1 μg intrathecally on alternate days × 3, and clonidine plus PGE2[μ-(S)x3,Cl+E2] on mechanical paw-withdrawal threshold. C, Effect of PGE2(E2), CPA plus PGE2 (CPA+E2), μ-(AS) ODN 1 μg intrathecally on alternate days × 3, CPA plus PGE2 [μ-(AS)x3,CPA+E2], μ-(S) ODN 1 μg intrathecally on alternate days × 3, and CPA plus PGE2 [μ-(S)x3,CPA+E2] on mechanical paw withdrawal threshold in the rat.
Antisense ODN for the α2C-adrenergic receptor significantly attenuated not only α2 antinociception but also μ and A1 antinociception (Fig.7A–C). Sense α2C-adrenergic receptor ODN was without effect on PGE2 hyperalgesia, μ antinociception, or α2 antinociception.
Fig. 7.

Antisense α2C ODN treatment blocks not only α2 antinociception but also μ and A1 antinociception. A, Effect of PGE2 (E2), clonidine plus PGE2(Cl+E2), α2-(AS) ODN 1 μg intrathecally on alternate days × 3, and clonidine plus PGE2[α2-(AS)x3,CCl+E2], α2-(S) ODN 1 μg intrathecally on alternate days × 3, and clonidine plus PGE2 [α2-(S)x3,Cl+E2] on mechanical paw withdrawal threshold in the rat. B, Effect of PGE2 (E2), DAMGO plus PGE2 (D+E2), α2-(AS) ODN 1 μg intrathecally on alternate days × 3, and DAMGO plus PGE2 [α2-(AS)x3,D+E2], α2-(S) ODN 1 μg intrathecally on alternate days × 3, and DAMGO plus PGE2[α2-(S)x3,D+E2] on mechanical paw withdrawal threshold in the rat. C, Effect of PGE2(E2), CPA plus PGE2 (CPA + E2), α2-(AS) ODN 1 μg intrathecally on alternate days × 3, and CPA plus PGE2 [α2-(AS)x3,CPA+E2], α2-(S) ODN 1 μg intrathecally on alternate days × 3, and CPA plus PGE2[α2-(S)x3,CPA+E2] on mechanical paw withdrawal threshold in the rat.
The data from these antisense ODN experiments suggest that α2-adrenergic receptors are required for μ and A1 antinociception. In addition, they suggest that the μ-opioid receptor is required for α2 antinociception. Thus, multiple receptors are involved in the production of antinociception by μ, α2, and A1agonists.
DISCUSSION
In this study, we tested the hypothesis that peripheral antinociception produced by μ, α2, and A1agonists exhibit cross-tolerance after repeated exposure to these agents. This hypothesis arose from previous data demonstrating that μ, α2, and A1 receptors can signal via a common second messenger, namely activation of an inhibitory G-protein (Sharma et al., 1975; Law et al., 1981; Childers and Rivere, 1989; Mankmann et al., 1988). In all experiments evaluating cross-tolerance and cross-dependence, complete symmetry was found for μ, α2, and A1. These findings support the hypothesis that there is a common signaling pathway for these three receptors. Although the mechanisms of tolerance and dependence in primary afferent nociceptors is unknown, in other systems the protein kinase C second messenger system has been implicated (Mao et al., 1995;Mayer et al., 1995). The role of this second messenger system in the tolerance and dependence to peripheral antinociception is currently being investigated. These data also suggest that clinically these pharmacologies may not be cross-substituted in dependent individuals.
As a control for the cross-withdrawal experiments, we tested whether there was blockade of antinociception by heterologous antagonists in naive animals. Unexpectedly, we found that the α2antagonist blocked not only α2 antinociception but also A1 and μ antinociception, that both μ and A1 antagonists blocked α2 antinociception, and that there was no such heterologous antagonism between μ and A1 ligands in antinociception. The absence of an interaction between μ and A1 antinociception is unlikely to be a result of an inadequate dose of antagonist because even at a very high dose (1 μg) no cross-antagonism was observed (unpublished observations).
There are known mechanisms by which antagonists can heterologously antagonize the actions of an agonist at another receptor class. First, when a receptor ligand is not highly selective, at sufficiently high doses it will bind to another receptor to displace a heterologous ligand (Cicero et al., 1974; Spiehler et al., 1978; Blank et al., 1983). However, clonidine binding is not displaced from neuronal membranes by morphine or naloxone (Golombiowska-Nikitkin et al., 1980). Second, there may be physical interactions between the receptors in the cell membrane. Such interactions have been suggested to explain effects of agonist combinations that are greater than additive (synergistic) or less than additive (antagonistic) than the effects seen at the different receptors. For example, an α2 agonist attenuates both A1 and μ mediated inhibition of norepinephrine release from sympathetic nerve endings; this interaction has been suggested to occur at the level of the receptor (Bucher et al., 1992). Furthermore, Bentley et al. (1983) have hypothesized that α-adrenoceptor and opioid receptors may be linked, either via second messenger systems or physically in the membrane.
We hypothesize from the data in our experiments that the α2 receptor is arranged topologically between the μ and A1 receptors to form a receptor complex (Fig.8). This hypothesis is supported by the observation that antisense oligodeoxynucleotides against μ-opioid receptor reduced not only μ antinociception, but also α2 antinociception, while preserving A1 antinociception, whereas the α2C antisense oligodeoxynucleotide reduced the antinociceptive effects of all three receptor systems, α2, A1, and μ. The preservation of A1 antinociception after μ antisense treatment but not after α2C antisense treatment suggests that the effect of receptor attenuation in the terminal is of short-range topologically. The same profile of interactions between α2, μ, and A1 were found in assessing the ability of a heterologous agonist to block withdrawal induced by the homologous antagonist in a tolerant paw.
Fig. 8.
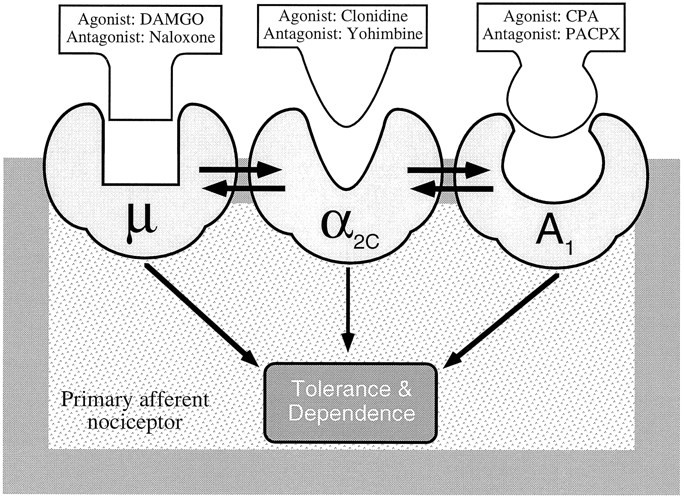
Schematic diagram of hypothesized topological/physical arrangement of the three receptors for peripheral antinociception in the cell membrane. μ (DAMGO), α2C (Clonidine), and A1(CPA) agonism all result in peripheral antinociception mediated through a common second messenger pathway, leading to complete symmetrical cross-tolerance and cross-dependence. However, the asymmetrical interactions are proposed to be a result of the central position of the α2C receptor leading to bidirectional interactions between this receptor and the two other receptors but no interaction between the μ and A1 receptors.
In summary, we have demonstrated that there is a symmetry for the three agonists for production of tolerance and dependence. There was also an unexpected interaction, which seems to occur at the level of the receptor, between α2 and μ receptors and α2 and A1 receptors, but not between μ and A1 receptors. The results suggest the hypothesis that these three receptors may be coupled physically in the plasma membrane.
Footnotes
Correspondence should be addressed to Dr. Jon D. Levine, Department of Anatomy, University of California-San Francisco, 521 Parnassus Avenue, San Francisco, CA 94143-0452.
REFERENCES
- 1.Aley KO, Green PG, Levine JD. Opioid and adenosine peripheral antinociception are subject to tolerance and withdrawal. J Neurosci. 1995;15:8031–8038. doi: 10.1523/JNEUROSCI.15-12-08031.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bentley GA, Newton SH, Starr J. Studies on the antinociceptive action of α-agonist drugs and their interactions with opioid mechanisms. Br J Pharmacol. 1983;79:125–134. doi: 10.1111/j.1476-5381.1983.tb10504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blank MS, Diez JA, Roberts DL. Monoaminergic antagonists which block naloxone-induced release of luteinizing hormone bind selectively to hypothalamic opiate receptors. Brain Res. 1983;279:153–158. doi: 10.1016/0006-8993(83)90173-7. [DOI] [PubMed] [Google Scholar]
- 4.Bucher B, Corriu C, Stoclet JC. Prejunctional opioid μ-receptors and adenosine A1-receptors on the sympathetic nerve endings of the rat tail artery interact with the α2-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol. 1992;345:37–43. doi: 10.1007/BF00175467. [DOI] [PubMed] [Google Scholar]
- 5.Childers SR, LaRiviere G. Modification of guanine nucleotide-regulatory components in brain membranes. I. Relationship of guanosine 5′-triphosphate effects on opiate receptor binding and coupling receptors with adenylate cyclase. J Neurosci. 1984;4:2764–2771. doi: 10.1523/JNEUROSCI.04-11-02764.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cicero TJ, Wilcox CE, Meyer ER. Effect of α-adrenergic blockers on naloxone-binding in brain. Biochem Pharmacol. 1974;23:2349–2352. doi: 10.1016/0006-2952(74)90570-x. [DOI] [PubMed] [Google Scholar]
- 7.Gold MS, White DM, Ahlgren SC, Guo M, Levine JD. Catecholamine-induced mechanical sensitization of cutaneous nociceptors in the rat. Neurosci Lett. 1994;175:166–170. doi: 10.1016/0304-3940(94)91105-3. [DOI] [PubMed] [Google Scholar]
- 8.Golombiowska-Nikitkin K, Pilc A, Ventulani J. Opiate and specific receptor binding of [3H]-clonidine. J Pharm Pharmacol. 1980;32:70–71. doi: 10.1111/j.2042-7158.1980.tb12852.x. [DOI] [PubMed] [Google Scholar]
- 9.Khasar SG, Green PG, Chou B, Levine JD. Peripheral nociceptive effects of α2-adrenergic receptor agonists in the rat. Neuroscience. 1995;66:427–432. doi: 10.1016/0306-4522(94)00562-j. [DOI] [PubMed] [Google Scholar]
- 10.Khasar SG, Gold MS, Dastmalchi S, Levine JD. Selective attenuation of μ-opioid receptor-mediated effects in rat sensory neurons by intrathecal administration of antisense oligodeoxynucleotides. Neurosci Lett. 1996;218:17–20. doi: 10.1016/0304-3940(96)13111-6. [DOI] [PubMed] [Google Scholar]
- 11.Law PY, Wu J, Koehler E, Loh HH. Demonstration and characterization of opiate inhibition of the striatal adenylate cyclase activity. J Neurochem. 1981;36:1834–1846. doi: 10.1111/j.1471-4159.1981.tb00438.x. [DOI] [PubMed] [Google Scholar]
- 12.Lingen LU, Ordway G. Antisense knock-down of α2C-adrenoceptors in rat striatum. Neurosci Abstr. 1995;21:1612. [Google Scholar]
- 13.Mankman MH, Dvorkin B, Crain SM. Modulation of adenylate cyclase activity of mouse spinal cord-ganglion explants by opioids, serotonin and pertussis toxin. Brain Res. 1988;445:303–313. doi: 10.1016/0006-8993(88)91193-6. [DOI] [PubMed] [Google Scholar]
- 14.Mao J, Price DD, Phillips L, Lu J, Mayer D. Increase in protein kinase C γ immunoreactivity in the spinal cord of rats associated with tolerance to analgesic effects of morphine. Brain Res. 1995;677:257–267. doi: 10.1016/0006-8993(95)00161-i. [DOI] [PubMed] [Google Scholar]
- 15.Mayer DJ, Mao J, Price DD. The development of morphine tolerance and dependence is associated with translocation of protein kinase C. Pain. 1995;61:365–374. doi: 10.1016/0304-3959(95)00023-L. [DOI] [PubMed] [Google Scholar]
- 16.Pitchford S, Levine JD. Prostaglandins sensitize nociceptors in cell culture. Neurosci Lett. 1991;132:105–108. doi: 10.1016/0304-3940(91)90444-x. [DOI] [PubMed] [Google Scholar]
- 17.Rossi G, Pan YX, Cheng J, Pasternak GW. Blockade of morphine analgesia by an antisense oligodeoxynucleotide against the μ receptor. Life Sci. 1994;54:375–379. doi: 10.1016/0024-3205(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 18.Sharma SK, Nirenberg M, Klee WA. Morphine receptors as regulators of adenylate cyclase activity. Proc Natl Acad Sci USA. 1975;72:590–594. doi: 10.1073/pnas.72.2.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spiehler V, Fairhurst AS, Randall LO. The interaction of phenoxybenzamine with the mouse brain opiate receptor. Mol Pharmacol. 1978;14:587–595. [PubMed] [Google Scholar]
- 20.Taiwo YO, Coderre TJ, Levine JD. The contribution of training to sensitivity in the nociceptive paw-withdrawal test. Brain Res. 1989;487:148–151. doi: 10.1016/0006-8993(89)90950-5. [DOI] [PubMed] [Google Scholar]
- 21.Taiwo YO, Levine JD. Direct cutaneous hyperalgesia induced by adenosine. Neuroscience. 1990;38:757–762. doi: 10.1016/0306-4522(90)90068-f. [DOI] [PubMed] [Google Scholar]
- 22.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]