

Abstract
Opioid receptors located on interneurons in the ventral tegmental area (VTA) inhibit GABAA-mediated synaptic transmission to dopamine projection neurons. The resulting disinhibition of dopamine cells in the VTA is thought to play a pivotal role in drug abuse; however, little is known about how this GABAA synapse is affected after chronic morphine treatment. The regulation of GABA release during acute withdrawal from morphine was studied in slices from animals treated for 6–7 d with morphine. Slices containing the VTA were prepared and maintained in morphine-free solutions, and GABAA IPSCs were recorded from dopamine cells. The amplitude of evoked IPSCs and the frequency of spontaneous miniature IPSCs measured in slices from morphine-treated guinea pigs were greater than placebo-treated controls. In addition, activation of adenylyl cyclase, with forskolin, and cAMP-dependent protein kinase, with Sp-cAMPS, caused a larger increase in IPSCs in slices from morphine-treated animals. Conversely, the kinase inhibitors staurosporine and Rp-CPT-cAMPS decreased GABA IPSCs to a greater extent after drug treatment. The results indicate that the probability of GABA release was increased during withdrawal from chronic morphine treatment and that this effect resulted from an upregulation of the cAMP-dependent cascade. Increased transmitter release from opioid-sensitive synapses during acute withdrawal may be one adaptive mechanism that results from prolonged morphine treatment.
Keywords: ventral tegmental area, dopamine, cAMP, A-kinase, GABAA, tolerance
Chronic use of opioids results in tolerance to and dependence on the drug. One of the most widely accepted mechanisms for the cellular basis of tolerance is an uncoupling of the opioid receptor from the effector so that a greater receptor occupancy is required to obtain a given response (Law et al., 1982; Chavkin and Goldstein, 1984;Puttfarcken et al., 1988). Dependence is defined by a number of abnormal responses after the abrupt withdrawal of drug (Johnson and Flemming, 1989). The expression of dependence, or withdrawal, is thought to result from the development of adaptive changes that occur in the continued presence of agonist. The first adaptive response to be recognized at the cellular level was an upregulation of adenylyl cyclase (Sharma et al., 1975). Acutely, opioids inhibit the activity of adenylyl cyclase. However, in the continued presence of agonist the inhibition declined until the activity in the presence of agonist was not different from control. Adenylyl cyclase activity measured after the removal of opioid was increased above control. This rebound increase was thought to represent a cellular expression of opioid withdrawal. Much of what is known about the cellular basis for tolerance and dependence to opioids has come from studies on the opioid regulation of adenylyl cyclase in cell lines, although the cellular basis for the upregulation is not understood (Sharma et al., 1975; Law et al., 1982; Puttfarcken et al., 1988).
The electrophysiological approach has been used successfully to demonstrate tolerance to opioids; however, the cellular expression of withdrawal has been much more difficult to identify (Christie et al., 1987; Wimpey et al., 1989; Kennedy and Henderson, 1991, 1992). In the locus coeruleus (LC), for example, tolerance to the opioid-induced inhibition in firing and the associated increase in potassium conductance has been demonstrated both in vivo and in vitro (Aghajanian, 1978; Christie et al., 1987). After withdrawal, however, the increased firing rate of LC neurons resulted primarily from augmented excitatory afferent drive (Tung et al., 1990; Akaoka and Aston-Jones, 1991).
The ventral tegmental area (VTA) is part of the endogenous reward circuit that is thought to be activated by many drugs of abuse, including opioids (Bozarth and Wise, 1981; Wise and Rompre, 1989). Within the VTA, GABAA-mediated IPSPs recorded in dopamine cells were thought to arise from excitation of local interneurons (Johnson and North, 1992a). As has been observed in many areas of the CNS, opioids directly inhibit these interneurons through activation of a potassium conductance (Nicoll et al., 1980; Madison and Nicoll, 1988;Wimpey and Chavkin, 1991; Johnson and North, 1992a). Thus, acute activation of opioid receptors within the VTA indirectly increased the activity of dopamine cells by removing GABA-mediated inhibition (Gysling and Wang, 1983). The purpose this study was to examine the GABAA IPSC during acute withdrawal from morphine and to identify potential second-messenger pathways that mediate altered function.
MATERIALS AND METHODS
Whole-cell recordings of membrane current were made from dopamine neurons in horizontal slices of guinea pig midbrain. Preparation of slices has been described previously (Cameron and Williams, 1994). Briefly, guinea pigs (300–400 gm) were anesthetized with halothane and killed. The midbrain was sliced (250 μm) in the horizontal plane using a vibratome. Slices (up to 3) containing the VTA were stored before being placed in the recording chamber and superfused (1.5 ml/min) with warmed (35°C) Krebs/bicarbonate buffer containing the following (in mm): NaCl 126, KCl 2.5, NaH2PO4 1.2, MgCl2 1.2, CaCl2 2.4, glucose 11, NaHCO3 21.4, saturated with 95% O2/5% CO2. Cells were visualized using an upright microscope with infrared illumination and recordings were made with whole-cell electrodes containing the following (mm): KCl 128, NaCl 20, MgCl2 1, EGTA 1, CaCl2 0.3, Mg-ATP 2, GTP 0.25, buffered with HEPES 10, pH 7.3. Electrode resistance was 2–4 MΩ, and acceptable access resistance was <15 MΩ, which was monitored periodically with repetitive 10 mV steps (20 msec duration) from a holding potential of −60 mV. Series resistance compensation of ∼80% was used during the entire experiment. Identification of dopamine cells was made based on the physiological properties, including a largeIh-current (previously described by Johnson and North, 1992b). A monopolar glass stimulating electrode was placed near (30–100 μm) the cell body. Neurons were voltage-clamped at a membrane potential of −60 mV. Spontaneous IPSCs were recorded in the presence of TTX (300 nm) using PClamp (60 sweeps for each condition, 1 sec/sweep), and miniature IPSC (mIPSC) amplitude was measured for each individual IPSC using Axograph 3.0. To determine accurately the IPSC amplitude, only IPSCs that were >8 pA were accepted for analysis. Results were plotted as cumulative amplitude and frequency histograms, and the effects of drugs were tested using the Komolgrov–Smirnov statistical method (Cohen et al., 1992);p < 0.05 was taken as indicating statistical significance.
Drugs were applied in known concentrations to the superfusion medium. In experiments examining the GABAA synaptic potential, the superfusion medium contained 2-amino-5-phosphonopentanoic acid (AP5; 100 μm), 6-cyano-2,3-dihydroxy-7-nitro-quinoxaline (CNQX; 10 μm), strychnine (1 μm), and eticlopride (100 nm) to block NMDA, AMPA, glycine, and dopamine D2-mediated synaptic currents, respectively. There was no effect of this solution on the holding current of the dopamine cells used in the present study. The same observation has been reported using this mixture of antagonists in guinea pig (Bonci and Williams, 1996) and rat VTA (Johnson and North, 1992b; Johnson et al., 1992). AP5 and picrotoxin were obtained from Sigma (St. Louis, MO). CNQX and eticlopride were obtained from Research Biochemicals (Natick, MA). Morphine (75 mg morphine/pellet obtained from National Institute on Drug Abuse) or placebo pellets (5 total) were implanted subcutaneously in anesthetized guinea pigs, one on day 1 and two on days 3 and 5. Experiments were carried out on days 6 and 7.
Results in the text and figures were presented as the mean ± SEM. Results between groups were compared using an unpaired ttest; p < 0.05 was taken as indicating statistical significance.
RESULTS
Whole-cell recordings were made from dopamine cells in guinea pig brain slices that were identified as cells with a prominent hyperpolarization-induced inward current (Ih) when the holding potential was stepped from −60 mV to more negative potentials (Johnson and North, 1992b). Electrically evoked GABAA IPSCs were inward at a membrane potential of −60 mV and were completely blocked by picrotoxin (100 μm). In all experiments, other synaptic currents were blocked with receptor antagonists (see Materials and Methods). Slices from both placebo- and morphine-treated animals were prepared and maintained in morphine-free solution. The opioid agonist normorphine depressed the amplitude of these GABAA-mediated IPSCs (Fig.1A). In control, the EC50of normorphine was 180 ± 83 nm and the peak inhibition was 83 ± 8% (n = 4). After chronic morphine treatment, the normorphine-induced inhibition of IPSCs had an EC50 of 380 ± 60 nm and the peak inhibition was 70 ± 3% (n = 4). These initial experiments demonstrate that this commonly used treatment protocol was sufficient to cause some tolerance to opioids in the VTA.
Fig. 1.

IPSCs from morphine-treated animals show paired-pulse depression, unlike those from controls, which show paired-pulse facilitation. A, Recording of GABAA-mediated IPSCs from a control slice using the paired-pulse protocol (left trace). Normorphine (1 μm) depressed the IPSC (middle trace), and that inhibition was reversed by the addition of naloxone to the normorphine-containing solution (right trace).B, Examples from three different cells in slices from both placebo- and morphine-treated animals. C, Cumulative results showing the distribution of paired-pulse ratio for many cells in slices from placebo (n = 41)- and morphine (n = 41)-withdrawn slices.D, The paired-pulse ratio is independent of the stimulus strength. These results are the average from four cells in each group of animals.
Morphine withdrawal augments GABA release
All experiments were carried out in morphine-free solutions at least 1 hr and as long as 8 hr after preparation of the brain slice. Characterization of GABA release after withdrawal from chronic morphine treatment was studied using two methods. The first method used a paired-pulse protocol, in which two stimuli were applied at an interval of 50 msec. In slices from the placebo-treated group, the second pulse evoked an IPSC that was generally larger than the first pulse (Fig.1B). That is, facilitation of transmitter release was observed in 78% of cells in control (s2/s1 = 1.18 ± 0.07,n = 41). In morphine-withdrawn slices, the second stimulus evoked a smaller IPSC in 80% of cells tested (s2/s1 = 0.60 ± 0.14, n = 41). Thus, during morphine withdrawal the paired-pulse protocol resulted in depression. The numbers of cells showing facilitation and depression using the paired-pulse protocol are illustrated in Figure 1C and indicate that depression was observed most often during morphine withdrawal. Facilitation or depression was not dependent on the size of the initial IPSC (Fig. 1B) and was not affected by changing the stimulus intensity (Fig. 1D). Manipulations that increase transmitter release in many sites have been found to result in a shift in the paired-pulse ratio toward depression (Mennerick and Zorumski, 1995; Salin et al., 1996). The present results suggest that the probability of GABA release was augmented during morphine withdrawal. Paired-pulse depression during morphine withdrawal was relatively persistent, because it was observed in slices as early as 1 hr and up to 8 hr after the slice was placed in morphine-free solution.
One possible explanation for the depression found during morphine withdrawal was that the GABAA receptors may be more likely to desensitize. Direct application of GABA using iontophoresis was used as a postsynaptic control to test this possibility. GABA (50–150 nA) was applied at a distance of 3–10 μm from the dopamine cell body. Two pulses of GABA (10–20 msec) were applied 100 msec apart. The amplitude of the inward current among different cells ranged from 27 pA to 4 nA, and the duration ranged from 50 to 100 msec. There was no consistent difference between the amplitude of the first and second pulses in slices from morphine-treated animals (n = 5, data not shown).
The frequency and amplitude of spontaneous mIPSCs (in 500 nm TTX) comprised the second method used to identify altered regulation of GABA release during morphine withdrawal. The rate of spontaneous mIPSCs was significantly greater in morphine-withdrawn slices [control, 2.4 ± 1.3 Hz (n = 8); morphine-withdrawn, 9.3 ± 2.8 Hz (n = 7,p < 0.05)]. There was no significant difference in the amplitude distribution of spontaneous mIPSCs in the two groups. The mean amplitude of spontaneous mIPSCs was 36.4 ± 2.8 pA in control and 35.6 ± 4.1 in morphine-withdrawn slices. Therefore, both the paired-pulse protocol and the increase in frequency of spontaneous events suggested that the probability of GABA release in the VTA was increased during withdrawal from morphine.
It was possible that the difference in GABA release between the two groups of animals resulted indirectly from activation of presynaptic receptors in response to a second transmitter. Potential candidates include GABA itself, adenosine, and 5-HT, all of which are present and have been shown to cause potent presynaptic inhibition of GABA release (Johnson et al., 1992; Wu et al., 1995). The amplitude of the IPSC (S1) was measured in the presence of both the GABAB receptor antagonist CGP35348 (100 μm) and the A1 antagonist DPCPX (1 μm). In DPCPX, the amplitude of the IPSC was increased in both groups of animals to the same extent [11 ± 7.8% in control (n = 7) and 12.3 ± 7.3 in morphine-withdrawn slices (n = 7)]. The IPSC was not affected by CGP35348 (control, n = 3; morphine-withdrawn, n = 4). Although 5-HT caused a potent presynaptic inhibition of the GABAB IPSP, it had no effect on the GABAA IPSP in rat (Johnson et al., 1992). There was no effect of the nonselective 5-HT1 agonist 5-CT (1 μm) on the GABAA IPSC in slices from control and morphine-treated guinea pigs (n = 5 for each group). Because the amplitude of the IPSC was not selectively affected by any of these manipulations in cells from morphine-withdrawn slices, the results suggest that the increase in GABAA IPSC observed in morphine-treated animals did not result from altered sensitivity to endogenous adenosine, GABA, or 5-HT.
cAMP-dependent modulation of GABA release
It has been known for many years that chronic morphine treatment augments adenylyl cyclase activity in cell lines and various parts of the CNS (Johnson and Flemming, 1989). In addition, forskolin stimulation of adenylyl cyclase has been shown to increase release of both GABA and glutamate in a variety of preparations including the VTA (Cameron and Williams, 1993; Rosenmund et al., 1994;Bonci and Williams, 1996; Salin et al., 1996). Forskolin produced a concentration-dependent increase in the amplitude of the GABAA IPSC in both control and morphine-withdrawn slices (Fig. 2). The forskolin-induced increase in morphine-withdrawn slices was significantly larger and longer lasting than that in controls (Fig. 2B,C). The sensitivity to forskolin was not changed, but the maximum effect was increased (EC50 1.8 μm in control, 1.5 μm in withdrawn slices, maximum augmentation 66% in control and 125% in withdrawn slices; Fig. 2C). The inactive analog of forskolin, dideoxyforskolin (10 μm), had no significant effect on the IPSC (percent change from control, 4.3 ± 5.1% in placebo, 5.1 ± 6.2 in morphine-withdrawn,n = 4 cells each).
Fig. 2.
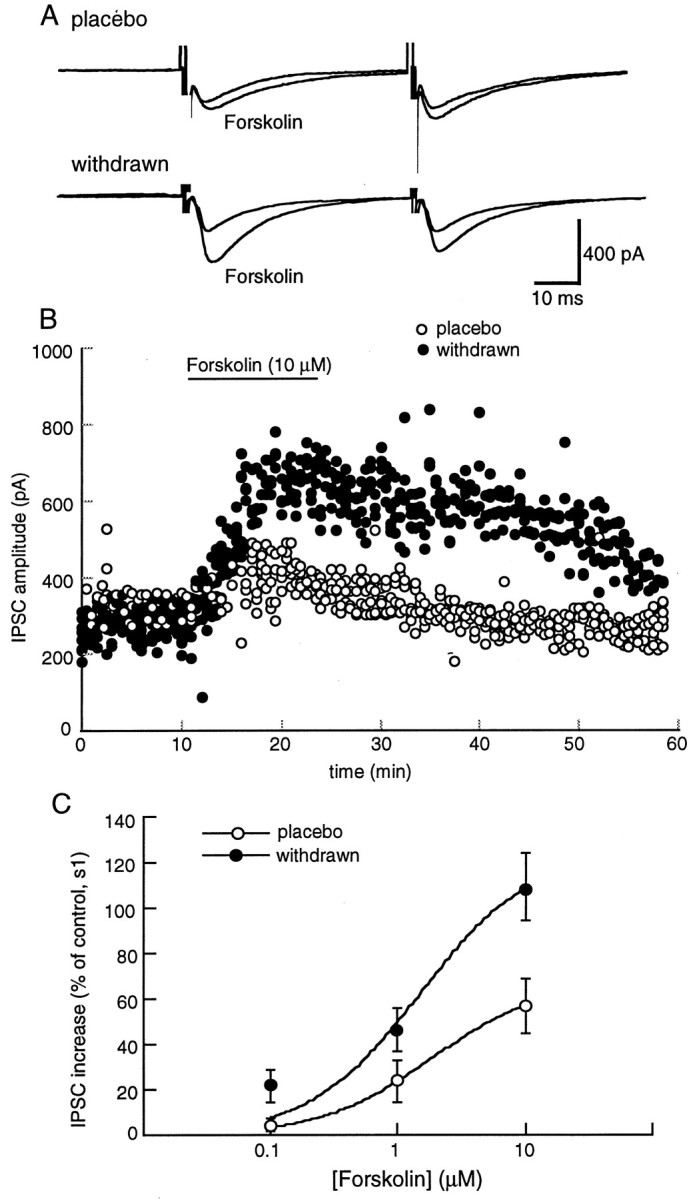
Forskolin augments the IPSC. This augmentation is significantly larger in morphine-withdrawn slices than in the placebo controls (p < 0.05). A, Examples of experiments from placebo- and morphine-treated animals.B, The amplitude of the first IPSC is plotted as a function of time in cells from placebo (open circles)- and morphine (solid circles)-withdrawn slices. Results are from four slices for each group. Forskolin (10 μm) increased the amplitude of the IPSC to a greater extent and for a more prolonged period in morphine-withdrawn slices than in controls.C, The concentration response to forskolin. The amplitude of the first IPSC in the paired-pulse protocol is plotted as a function of the concentration of forskolin. The amplitude was normalized against the mean of the first 10 IPSCs for each cell. Results were obtained from four cells from four different animals in each group. The data were fit with a least-squares regression from a logistic equation and gave estimates of the EC50 and maximum effect of forskolin of 1.8 μm and 66% increase in control and 1.5 μm and 125% increase in morphine-withdrawn slices.
The frequency of spontaneous mIPSCs was also significantly increased by forskolin (10 μm) in morphine-withdrawn slices (from 9.3 ± 2.8 to 23.6 ± 3.5 Hz, n = 7) animals as compared to controls (2.4 ± 1.3 to 3.2 ± 1.2 Hz,n = 8; Fig. 3). There was no significant difference in the amplitude of the mIPSCs in the absence and presence of forskolin in slices from either group of animals. In slices from control animals the mean amplitude was 36.4 ± 2.8 pA in control and 37.9 ± 4.3 pA in forskolin (10 μm), and in slices from morphine-withdrawn animals the mean amplitude was 35.6 ± 4.1 pA in control and 34.4 ± 3.2 pA in forskolin. An upregulation of adenylyl cyclase could account for the increase in probability of GABA release as determined by the depression of the paired-pulse ratio and the increase in spontaneous mIPSCs in morphine-withdrawn slices.
Fig. 3.

Forskolin increased the frequency of spontaneous mIPSCs to a greater extent in morphine-withdrawn slices than in placebo controls. A, Experiment from a cell in a slice from a control animal. Top traces show the occurrence of spontaneous IPSCs in control (left) and after superfusion with forskolin (10 μm, right). The three plots below the traces show an amplitude histogram (left), a cumulative probability plot of the amplitude (middle), and a cumulative probability plot of the frequency of spontaneous IPSCs from the same cell shown above. In this particular cell, forskolin had little effect on the rate and amplitude of the spontaneous IPSCs. B, Illustration of the same experiment in a cell recorded in a slice taken from a morphine-treated animal. The initial rate of activity was higher, and forskolin induced a significant increase in the rate of spontaneous IPSCs (p < 0.05). All experiments were carried out in the presence of TTX (500 nm), CNQX (10 μm), APV (100 μm), strychnine (1 μm), and eticlopride (100 nm).
Kinase-dependent modulation of GABA release
Superfusion with the cAMP analog Sp-cAMPS (100 μm, 15 min) had little or no effect on the amplitude of the IPSC (9.2 ± 3.7%, n = 3; Fig. 4); however, the same protocol caused a significant augmentation in the IPSC amplitude in withdrawn slices (46 ± 11%, n = 3,p < 0.05; Fig. 4). This experiment suggested that the forskolin-induced increase in GABAA IPSCs may involve cAMP-dependent kinase.
Fig. 4.

The cAMP analog Sp-cAMPS is more effective in increasing the IPSC in slices from morphine-withdrawn slices than placebo controls. The amplitude of the electrically evoked IPSC is plotted as a function of time. A, Top, Example of one experiment in a slice taken from a control animal. The cAMP analog Sp-cAMPS (100 μm) has no effect on the amplitude of the IPSC, whereas a low concentration of forskolin (1 μm) increased the amplitude of the IPSC.Bottom, Average of four such experiments in slices taken from four different animals. The average amplitude of IPSCs over the first 5 min was used to normalize the data. B,Top, An experiment in a single cell taken from a morphine-treated animal. In this case, cAMP analog Sp-cAMPS caused an increase in the IPSC that was about the same as that induced by forskolin. Bottom, Normalized and averaged results from four experiments.
The nonselective kinase inhibitor staurosporine (300 nm) caused a decrease of 45 ± 4.3% (n = 4) in the amplitude of the IPSC in morphine-withdrawn slices. This inhibition was larger than that found in slices from control animals (26 ± 4.3% of control, n = 4, p < 0.05; Fig.5). The ratio of IPSC amplitudes found using the paired-pulse protocol was not changed by staurosporine in control (1.41 ± 0.1 in control to 1.42 ± 0.2 in staurosporine,n = 4) but was increased in withdrawn slices (0.58 ± 0.07 in control, 1.66 ± 0.2 in staurosporine). In addition, the frequency but not the amplitude of spontaneous IPSCs was also decreased by staurosporine, an effect that was significantly greater in withdrawn slices (from 8.9 ± 1.8 Hz in control to 2.7 ± 0.8 Hz in staurosporine, n = 6) than in controls (from 2.1 ± 0.3 Hz in control to 2.0 ± 0.4 Hz in staurosporine,n = 6; Fig. 6). Finally, the forskolin-induced increase in IPSCs was blocked by pretreatment with staurosporine (Fig. 5).
Fig. 5.

The protein kinase inhibitors staurosporine (A, 300 nm) and Rp-CPT-cAMPS (B, 100 μm) decreased the IPSC in slices from both placebo- and morphine-treated animals. The inhibition is greater in morphine-withdrawn slices. In all plots, the IPSC amplitudes were normalized to the average determined over the first 5 min of the experiment. Plots on the left are from control animals (Placebo), and plots on the rightare from morphine-treated animals (Withdrawn).A, A low concentration of forskolin (1 μm) increased the amplitude of the IPSC, and staurosporine decreased the amplitude of the IPSC and blocked the forskolin-induced augmentation (n = 4). The plot labeled Withdrawnis the same experiment as shown at the left in slices taken from four morphine-treated animals. The inhibition induced by staurosporine is significantly larger than in controls (p < 0.05). B, The cAMP analog Rp-CPT-cAMPS, a cAMP-dependent kinase inhibitor, produced a greater inhibition of the amplitude of IPSCs in slices from both withdrawn slices than placebo controls (n = 4 for each group; p < 0.05). The same protocol was used as described for A.
Fig. 6.

The frequency of mIPSCs is decreased by staurosporine (300 nm). The decrease is greater in morphine-withdrawn slices than in controls. A, Experiment from a cell in a slice from a control animal. Top traces show the occurrence of spontaneous IPSCs in control (left) and after superfusion with staurosporine (300 nm, right). The three plots below the traces show an amplitude histogram (left), a cumulative probability plot of the amplitude (middle), and a cumulative probability plot of the frequency of spontaneous IPSCs from the same cell shown above. Staurosporine had little effect on the rate and amplitude of the spontaneous IPSCs.B, Illustration of the same experiment in a cell from a morphine-withdrawn slice. The initial rate of activity was higher, and staurosporine induced a significant decrease in the rate of spontaneous IPSCs (p < 0.05).
To characterize further the kinase involved in the regulation of GABA release, Rp-CPT-cAMPS, a relatively selective blocker of cAMP-dependent kinase, was used. Superfusion with Rp-CPT-cAMPS (100 μm) significantly reduced the amplitude of the IPSC (p < 0.05; Fig. 5), blocked the effect of forskolin (Fig. 5), and caused a shift of the paired-pulse ratio toward facilitation in morphine-withdrawn slices (s2/s1 from 0.6 ± 0.2 to 1.25 ± 0.2, n = 3). The same protocol had no effect in slices from control animals (s2/s1 from 1.41 ± 0.03 to 1.42 ± 0.1, n = 3). These observations suggest that the activity of cAMP-dependent kinase was higher in morphine-withdrawn slices from drug-treated animals than in controls and that the increased activity was responsible for the augmented GABA release.
The augmentation of IPSCs induced by phorbol dibutyrate (PDBU; 300 nm), a phorbol ester, was examined in slices from control and morphine-treated animals as a test for the selectivity of the cAMP-dependent augmentation of GABA release. Although PDBU increased the amplitude of the IPSC in both groups [control 38.5 ± 4.3% (n = 4), morphine 44.8 ± 6.7% (n= 3)], the augmentation was the same in each. A small depression of the paired-pulse ratio (s2/s1) was caused by PDBU in each group [control from 1.39 ± 0.1 to 0.9 ± 0.1 (n = 4), morphine-withdrawn from 0.6 ± 0.18 to 0.55 ± 0.04 (n = 3)]. Thus, it appeared that the augmentation of transmitter release during morphine withdrawal may be mediated selectively through a cAMP-dependent pathway.
Experiments aimed at further identification of sites in the cAMP cascade that were affected by morphine withdrawal were inconclusive. The adenylyl cyclase inhibitor SQ22356 (300 μm to 1 mm) was tested in an attempt to determine whether the basal level of adenylyl cyclase activity was different in the two groups of tissues. Although this compound caused an initial depression of the IPSC, continued application resulted in a large increase in the IPSC, such that the result was not interpretable. Initial experiments with the nonselective phosphatase inhibitor tautomycin (100 nm) were carried out to determine the role of phosphatases. Tautomycin caused a transient augmentation of the IPSC followed by total suppression, limiting interpretation of the results.
DISCUSSION
Morphine decreased the amplitude of the GABAA-mediated IPSC in the VTA. After chronic treatment with morphine, there was some tolerance to opioids; however, the ability of opioids to inhibit release was maintained. The results of the present study demonstrate that during the initial period after withdrawal from prolonged morphine exposure an upregulation of the adenylyl cyclase cascade resulted in an increase in the probability of GABA release from terminals that mediate GABAA IPSCs. This may result from an adaptive process at a point beyond the opioid receptor because all experiments were carried out in the absence of morphine. The persistence of the augmented release after withdrawal of morphine for periods of up to 8 hr also suggests that this adaptive mechanism was not dependent on opioid receptor activation. It is important to determine how long the augmented release persists because the release of GABA acting at GABAB receptors was depressed 1 week after termination of morphine treatment (Bonci and Williams, 1996). Our working hypothesis is that the GABAA- and GABAB-mediated synaptic inhibition results from separate sets of terminals. The augmented GABAA-mediated inhibition described here may be a measure of acute withdrawal from opioids that is relatively short-lived (1–3 d). The depression of GABAB-mediated inhibition, however, is a long-term effect of withdrawal from chronic drug treatment that is either not present or occluded by the augmented GABAA tone early in withdrawal. In fact, during acute withdrawal from morphine there may be diffusion of GABA between terminals mediating the two synaptic events, as has been reported under certain conditions in the hippocampus (Isaacson et al., 1993).
Upregulation of adenylyl cyclase/cAMP-dependent pathway
The present observations are consistent with reports of an increased activity of adenylyl cyclase and cAMP-dependent protein kinase during acute withdrawal from chronic morphine treatment in cell lines and in several brain areas (Sharma et al., 1975; Nestler and Tallman, 1988) (for review, see Johnson and Flemming, 1989; Nestler et al., 1993). Although biochemical experiments specifically in the VTA have not demonstrated a rise in cyclase activity after chronic morphine treatment, the heterogeneity of cell types in the VTA limits the interpretation of negative data (Terwilliger et al., 1991). The present study focused on a synapse known to be opioid-sensitive such that a localized upregulation of the cAMP system in a small portion of neurons and/or terminals could be detected.
There were two primary observations that indicate an upregulation of the cAMP-dependent pathway after chronic morphine treatment. The first was that forskolin caused a significantly larger increase in GABAA IPSCs in slices from morphine-treated animals than controls. This effect was mediated by adenylyl cyclase because dideoxyforskolin, an inactive analog that does not activate adenylyl cyclase, did not have any effect. The concentration response to forskolin resulted in an increase in the maximum response rather than a change in the EC50. The augmented maximum effect of forskolin could result from a number of mechanisms, including an increase in the Vmax of adenylyl cyclase, a decline in cAMP-dependent phosphodiesterase activity, or an upregulation of downstream cAMP-dependent processes. There is evidence in the literature that all three mechanisms can occur in response to chronic morphine treatment (Yu et al., 1990; Self and Nestler, 1995). The increased sensitivity to cAMP during withdrawal seems selective as suggested by the experiments with the protein kinase C activator PDBU. Although PDBU increased the amplitude of GABAA IPSCs, the increase was not different in slices from control and morphine-treated animals.
The second observation was that agents that interact directly with cAMP-dependent kinase had quantitatively different effects in morphine-withdrawn slices. The stable cAMP analog Sp-cAMPS caused only a small increase in the IPSC in control (9%) but had a significantly greater effect in slices from morphine-treated animals (46%). This experiment suggests that cAMP-dependent kinase activity was increased during acute withdrawal. In addition, the results with the kinase inhibitors Rp-CPT-cAMPS and staurosporine suggested that the basal kinase activity was elevated in slices from morphine-treated animals. Such an upregulation of basal and stimulated kinase activity after chronic morphine treatment has been reported in cell lines and various brain areas (Duman et al., 1988; Nestler and Tallman, 1988; Nestler et al., 1993).
Morphine withdrawal
There are numerous examples of an increased firing rate after withdrawal of opioids in experiments done in vivo (Fry et al., 1980; Johnson and Duggan, 1981). The identification of cellular mechanisms of morphine withdrawal has proven difficult in brain slices (Christie et al., 1987; Wimpey et al., 1989) and in cell lines (Kennedy and Henderson, 1991, 1992). That is, withdrawal from morphine has not been observed to cause a rebound effect on either potassium or calcium conductances. Recently, a subpopulation of neurons in the periaqueductal gray (PAG) from morphine-dependent animals has been observed to be strongly depolarized by the addition of naloxone to the superfusion solution (Chieng and Christie, 1996). This depolarization appeared to be a direct because it was not affected by a combination of neurotransmitter receptor antagonists or tetrodotoxin. Although acute activation of opioid receptors on neurons in the PAG has been shown to increase potassium conductance (Chieng and Christie, 1994), the depolarization induced by naloxone in tissue from morphine-dependent animals resulted from a decrease in potassium conductance (which may be a reversal of the effect of morphine contained in the superfusion solution) and another unidentified mechanism.
Reports on the excitability of neurons in the LC during morphine withdrawal were dependent on the conditions of the experiment. Inin vivo experiments, systemic injection of naloxone caused a marked increase in firing rate in morphine-treated animals (10-fold) that was largely blocked by glutamate receptor antagonists (Tung et al., 1990; Akaoka and Aston-Jones, 1991). In brain slices, the firing rate of LC neurons from morphine-treated animals has been reported to be unchanged (Andrade et al., 1983) or increased twofold over controls (Kogan et al., 1992). An increased sensitivity of LC cells to cAMP analogs after chronic morphine treatment was suggested to account for the increased firing rate (Kogan et al., 1992); however, experiments were not done after blockade of glutamate or other neurotransmitter receptors. The combination of results obtained with in vivoexperiments and the increased probability of transmitter release from opioid-sensitive terminals observed in the present study suggest that an excitatory synaptic mechanism may be important during morphine withdrawal.
Significance
Whereas the firing rate of dopamine cells in the VTA was increased acutely by morphine in vivo (Gysling and Wang, 1983), during withdrawal from morphine, activity was profoundly decreased (Diana et al., 1995). In addition, many of the signs and symptoms of morphine withdrawal were attenuated by activation of D2 dopamine receptors in the nucleus accumbens, suggesting that dopamine tone was decreased during withdrawal (Harris and Aston-Jones, 1994). We suggest that the withdrawal inhibition of dopamine cell firing and decreased dopamine tone in the nucleus accumbens results from augmented GABA tone. Results with mIPSCs suggest that the expression of this form of withdrawal occurred at the terminals of GABA interneurons. The physiological consequences resulting from the link between the acute effects of opioids and cAMP mechanisms has been difficult to demonstrate; however, the interaction among transmitter release, cAMP mechanisms, and chronic opioid treatment may prove to be a general observation. Given the number of opioid-sensitive terminals, both excitatory and inhibitory, the augmented transmitter release after chronic morphine treatment has the potential for widespread consequences.
Footnotes
This work was supported by National Institutes of Health Grant DA08163. We thank MacDonald Christie, Jeffrey Diamond, and Matthew Jones for helpful discussions and comments.
Correspondence should be addressed to Dr. John T. Williams, The Vollum Institute, L474, Oregon Health Sciences University, 3181 SW Sam Jackson Park Road, Portland, OR 97201.
REFERENCES
- 1.Aghajanian GK. Tolerance of locus coeruleus neurons to morphine and suppression of withdrawal response by clonidine. Nature. 1978;276:186–187. doi: 10.1038/276186a0. [DOI] [PubMed] [Google Scholar]
- 2.Akaoka H, Aston-Jones G. Opiate withdrawal-induced hyperactivity of locus coeruleus neurons is substantially mediated by augmented excitatory amino acid input. J Neurosci. 1991;11:3830–3839. doi: 10.1523/JNEUROSCI.11-12-03830.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrade R, Vandermaelen CP, Aghajanian GK. Morphine tolerance and dependence in the locus coeruleus: single cell studies in brain slices. Eur J Pharmacol. 1983;91:161–165. doi: 10.1016/0014-2999(83)90461-2. [DOI] [PubMed] [Google Scholar]
- 4.Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;16:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- 5.Bozarth MA, Wise RA. Intracranial self-administration of morphine into the ventral tegmental area in rats. Life Sci. 1981;28:551–555. doi: 10.1016/0024-3205(81)90148-x. [DOI] [PubMed] [Google Scholar]
- 6.Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- 7.Cameron DL, Williams JT. Cocaine inhibits GABA release in the VTA through endogenous 5-HT. J Neurosci. 1994;14:6763–6767. doi: 10.1523/JNEUROSCI.14-11-06763.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chavkin C, Goldstein A. Opioid receptor reserve in normal and morphine-tolerant guinea pig ilium myenteric plexus. Proc Natl Acad Sci USA. 1984;81:7253–7257. doi: 10.1073/pnas.81.22.7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chieng B, Christie MJ. Hyperpolarization by opioids acting on μ-receptors of a subpopulation of rat periaqueductal gray neurones in vitro. Br J Pharmacol. 1994;113:121–128. doi: 10.1111/j.1476-5381.1994.tb16183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chieng B, Christie MJ. Local opioid withdrawal in rat single periaqueductal gray neurons in vitro. J Neurosci. 1996;16:7128–7136. doi: 10.1523/JNEUROSCI.16-22-07128.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christie MJ, Williams JT, North RA. Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol. 1987;32:633–638. [PubMed] [Google Scholar]
- 12.Cohen GA, Doze VA, Madison DV. Opioid inhibition of GABA release from presynaptic terminal of rat hippocampal interneurons. Neuron. 1992;9:325–335. doi: 10.1016/0896-6273(92)90171-9. [DOI] [PubMed] [Google Scholar]
- 13.Dianan M, Pistis M, Muntoni A, Gessa G. Profound decrease of mesolimbic dopaminergic neuronal activity in morphine-withdrawn rats. J Pharmacol Exp Ther. 1995;272:781–785. [PubMed] [Google Scholar]
- 14.Duman R, Tallman J, Nestler E. Acute and chronic opiate-regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J Pharmacol Exp Ther. 1988;246:1033–1039. [PubMed] [Google Scholar]
- 15.Fry JP, Herz A, Zieglgansberger W. A demonstration of naloxone-precipitated opiate withdrawal on single neurons in the tolerance/dependent rat brain. Br J Pharmacol. 1980;68:585–592. doi: 10.1111/j.1476-5381.1980.tb14574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gysling K, Wang RY. Morphine-induced activation of A10 dopamine neurons in the rat. Brain Res. 1983;277:119–127. doi: 10.1016/0006-8993(83)90913-7. [DOI] [PubMed] [Google Scholar]
- 17.Harris GC, Aston-Jones G. Involvement of D2 dopamine receptors in the nucleus accumbens in the opiate withdrawal syndrome. Nature. 1994;371:155–157. doi: 10.1038/371155a0. [DOI] [PubMed] [Google Scholar]
- 18.Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- 19.Johnson SM, Duggan AW. Tolerance and dependence of dorsal horn neurones of the cat: the role of the opiate receptors of the substantia gelatinosa. Neuropharmacology. 1981;20:1033–1038. doi: 10.1016/0028-3908(81)90093-9. [DOI] [PubMed] [Google Scholar]
- 20.Johnson SM, Flemming W. Mechanisms of cellular adaptive sensitivity changes: application to opioid tolerance and dependence. Pharmacol Rev. 1989;41:435–488. [PubMed] [Google Scholar]
- 21.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992a;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 1992b;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson SW, Mercuri NB, North RA. 5-Hydroxytryptamine1B receptors block the GABAB synaptic potential in rat dopamine neurons. J Neurosci. 1992;12:2000–2006. doi: 10.1523/JNEUROSCI.12-05-02000.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kennedy C, Henderson G. μ-Opioid receptor inhibition of calcium current: development of homologous tolerance in single SH-SY5Y cells after chronic exposure to morphine in vitro. Mol Pharmacol. 1991;40:1000–1005. [PubMed] [Google Scholar]
- 25.Kennedy C, Henderson G. Chronic exposure to morphine does not induce dependence at the level of the calcium channel current in human SH-SY5Y cells. Neuroscience. 1992;49:937–944. doi: 10.1016/0306-4522(92)90369-d. [DOI] [PubMed] [Google Scholar]
- 26.Kogan J, Nestler E, Aghajanian G. Elevated basal firing rates and enhanced responses to 8Br-cAMP in locus coeruleus neurons in brain slices from opiate-dependent rats. Eur J Pharmacol. 1992;211:47–53. doi: 10.1016/0014-2999(92)90261-2. [DOI] [PubMed] [Google Scholar]
- 27.Law PY, Hom DS, Loh HH. Loss of opiate receptor activity in neuroblastoma × glioma NG108-15 hybrid cells after chronic opiate treatment: a multiple-step process. Mol Pharmacol. 1982;22:1–4. [PubMed] [Google Scholar]
- 28.Madison DV, Nicoll RA. Enkephalin hyperpolarizes interneurones in the rat hippocampus. J Physiol (Lond) 1988;398:123–130. doi: 10.1113/jphysiol.1988.sp017033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol (Lond) 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nestler E, Tallman J. Chronic morphine treatment increases cyclic AMP-dependent protein kinase activity in the rat locus coeruleus. Mol Pharmacol. 1988;33:127–132. [PubMed] [Google Scholar]
- 31.Nestler EJ, Hope BT, Widnell KL. Drug addiction: a model for the molecular basis of neural plasticity. Neuron. 1993;11:995–1006. doi: 10.1016/0896-6273(93)90213-b. [DOI] [PubMed] [Google Scholar]
- 32.Nicoll RA, Alger BE, Jahr CE. Enkephalin blocks inhibitory pathways in the vertebrate CNS. Nature. 1980;287:22–25. doi: 10.1038/287022a0. [DOI] [PubMed] [Google Scholar]
- 33.Puttfarcken PS, Werling LL, Cox BM. Effects of chronic morphine exposure on opioid inhibition of adenylyl cyclase in 7315c cell membranes: a useful model for the study of tolerance at μ opioid receptors. Mol Pharmacol. 1988;33:520–527. [PubMed] [Google Scholar]
- 34.Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 1994;368:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- 35.Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- 36.Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:46395. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 37.Sharma SK, Klee WA, Nirenberg M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci USA. 1975;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terwilliger RZ, Beitner-Johnson D, Sevarino KA, Crain SM, Nestler EJ. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res. 1991;548:100–110. doi: 10.1016/0006-8993(91)91111-d. [DOI] [PubMed] [Google Scholar]
- 39.Tung CS, Grenhoff J, Svensson TH. Morphine withdrawal responses of rat locus coeruleus neurons are blocked by an excitatory amino-acid antagonist. Acta Physiol Scand. 1990;138:581–582. doi: 10.1111/j.1748-1716.1990.tb08888.x. [DOI] [PubMed] [Google Scholar]
- 40.Wimpey TL, Chavkin C. Opioids activate both an inward rectifier and a novel voltage-gated potassium conductance in the hippocampal formation. Neuron. 1991;6:281–289. doi: 10.1016/0896-6273(91)90363-5. [DOI] [PubMed] [Google Scholar]
- 41.Wimpey TL, Opheim KE, Chavkin C. Effects of chronic morphine administration on the mu and delta opioid responses in the CA1 region of the rat hippocampus. J Pharmacol Exp Ther. 1989;251:405–411. [PubMed] [Google Scholar]
- 42.Wise RA, Rompre PP. Brain DA and reward. Annu Rev Psychol. 1989;40:191–225. doi: 10.1146/annurev.ps.40.020189.001203. [DOI] [PubMed] [Google Scholar]
- 43.Wu Y, Mercuri NB, Johnson SW. Presynaptic inhibition of γ-aminobutyric acid-B-mediated synaptic current by adenosine recorded in vitro in midbrain dopamine neurons. J Pharmacol Exp Ther. 1995;273:576–581. [PubMed] [Google Scholar]
- 44.Yu VC, Eiger S, Duan DS, Lameh J, Sadee W. Regulation of cyclic AMP by the μ-opioid receptor in human neuroblastoma SH-SY5Y cells. J Neurochem. 1990;55:1390–1396. doi: 10.1111/j.1471-4159.1990.tb03151.x. [DOI] [PubMed] [Google Scholar]