

Abstract
The PDAPP transgenic mouse overexpresses human amyloid precursor protein V717F (PDAPP minigene) and develops age-related cerebral amyloid-β protein (Aβ) deposits similar to senile plaques in Alzheimer’s disease. We find age-related cortical and limbic Aβ deposition that begins at 8 months and progresses to cover 20–50% of the neuropil in cingulate cortex, entorhinal cortex, and hippocampus of 18-month-old heterozygotic animals. The regional patterns of transgene expression and amyloid deposition suggest that Aβ deposits occur at the terminals of overexpressing neurons. Amyloid deposition is associated with dystrophic neurites and extensive gliosis. However, stereological analysis shows that there is no overt neuronal loss in entorhinal cortex, CA1 hippocampal subfield, or cingulate cortex through 18 months of age. In addition, there is no apparent loss of mRNA encoding neuronal synaptic, cytoskeletal, or metabolic proteins. Thus, widespread Aβ deposition in 18-month-old heterozygotic mice produces neuritic alterations and gliosis without widespread neuronal death.
Keywords: transgenic mice, Alzheimer’s disease, hippocampus, amyloid, amyloid precursor protein, cingulate cortex, entorhinal cortex, neuritic dystrophy, synaptophysin, microtubule associated protein-2, cytochrome oxidase, glial fibrillary acidic protein
Alzheimer’s disease (AD) is characterized neuropathologically by amyloid plaques, neurofibrillary tangles, and marked neuronal loss. Amyloid plaques are composed of an internal fragment of amyloid precursor protein (APP), the 4 kDa amyloid-β protein (Aβ) (Selkoe, 1994). Although APP mutations cause AD (Goate et al., 1991; Mullan et al., 1992), and Aβ is neurotoxicin vitro (Koh et al., 1990; Mattson et al., 1992; Pike et al., 1993; Cotman et al., 1994; Cotman and Su, 1996; Yankner and Lorenzo, 1996), it is uncertain whether Aβ deposition is directly linked to neurofibrillary tangle formation or neuronal loss in AD (Gomez-Isla et al., 1997). Recently, animal models of cerebral Aβ deposition have been developed using transgenic technology (Games et al., 1995; Higgins et al., 1995; Hsiao et al., 1996). The unique resource of such transgenic animal models allows us to address several questions regarding the pathophysiological role of cerebral Aβ deposition. What underlies the regional anatomic specificity of amyloid deposition? Does Aβ deposition result in disruption of the neuropil? Is Aβ neurotoxic in vivo?
The PDAPP transgenic mice (Games et al., 1995) were generated using the platelet-derived growth factor B promoter driving a human APP (hAPP) minigene (PDAPP) encoding the V717F familial AD mutation and containing modified intronic sequences allowing for alternative splicing (Rockenstein et al., 1995). These mice express the hAPP transgene at levels fourfold to sixfold higher than endogenous mouse APP (Rockenstein et al., 1995). Age-related amyloid deposition occurs in a consistent regional distribution beginning at 6–9 months and increasing with age (Johnson-Wood et al., 1997), with associated glial and neuronal morphological alterations (Masliah et al., 1996).
In this report, we examine the relationship between transgene expression, Aβ deposition, neuronal loss, and mRNA expression in 4- to 18-month-old heterozygotic PDAPP mice. In particular, we test the hypothesis that neuronal death and dysfunctional mRNA synthesis occur with aging as a consequence of hAPP V717F overproduction and Aβ deposition, using techniques of immunohistochemistry, stereology, andin situ hybridization. In AD brain, these techniques demonstrate neuritic changes within plaques (Masliah et al., 1993), neuronal loss in the hippocampal CA1 subfield, entorhinal cortex, and cingulate cortex (Vogt et al., 1992; West, 1994; Gomez-Isla et al., 1996a), and loss of neuronal synaptic, cytoskeletal, and metabolic protein mRNA with upregulation of glial fibrillary acidic protein (GFAP) mRNA associated with astrogliosis (Clark and Parhad, 1989;Simonian and Hyman, 1994; Callahan and Coleman, 1995; de la Monte et al., 1995; Hatanpaa et al., 1996). We find that the same techniques are easily adapted to the analysis of the PDAPP transgenic mice.
MATERIALS AND METHODS
Transgenic mice. The heterozygote PDAPP transgenic mice were bred from the previously established line PDAPP-109 over several generations on hybrid backgrounds representing combinations of C57BL/6, DBA, and Swiss-Webster strains (Games et al., 1995; Masliah et al., 1996; Johnson-Wood et al., 1997). Age-matched nontransgenic littermates derived from the PDAPP lines were used as controls. For amyloid burden studies and neuron counts, six transgenic and six nontransgenic mice at ages of 8, 12, and 18 months were analyzed. Forin situ hybridization studies, four transgenic and four nontransgenic mice at ages of 4, 11, and 18 months were studied.
Tissue preparation. Mice were anesthetized, and the brains were removed and snap frozen in isopentane chilled with dry ice. Forin situ hybridization, 14 μm coronal cryostat sections were thaw-mounted onto sterile Probe-On (Fisher Scientific, Houston, TX) slides coated with a sterile solution of 0.01% poly-l-lysine and fixed in ice-cold 4% paraformaldehyde in PBS for 5 min (Sirinathsinghji and Dunnett, 1993). For immunohistochemistry and stereology, brains were drop-fixed in 4% paraformaldehyde and post-fixed for 3 d before preparing 40 μm coronal vibrotome sections.
In situ hybridization. The in situ procedure has been described in detail (Sirinathsinghji and Dunnett, 1993; Page et al., 1996). Oligonucleotide probes (45 mer) were synthesized on an Applied Biosystems (Foster City, CA) DNA synthesizer (Page et al., 1996) or purchased commercially (Life Technologies, Gaithersburg, MD). The probes used were (mRNA, GenBank accession number, bases): rat synaptophysin, X06177, 1143–1187; mouse microtubule-associated protein-2 (MAP-2), M21041, 5131–5175; mouse cytochrome oxidase subunit-2 (COX-2), V00711, 7015–7059; mouse cytochrome oxidase subunit-4 (COX-4), S57870, 538–582; and mouse GFAP, K01347, 1784–1828. The probes used for hAPP isoforms are detailed by Sola et al. (1993).
Fourteen-micrometer coronal fixed cryostat sections were hybridized overnight with 35S-adenosine (DuPont NEN, Boston, MA) end-labeled 45 mer oligonucleotide probes (10,000 cpm/μl) at 42°C in sealed chambers humidified with 50% formamide/0.1% diethylpyrocarbonate water, and then washed in 1× SSC at 55°C. Slides were exposed to Amersham (Arlington Heights, IL) β-max autoradiography film for 1–18 d. Hybridization of sections with sense probes yielded no detectable signal.
Autoradiographic images of coronal sections were captured using a Bio-Rad (Hercules, CA) GS-700 imaging densitometer under maximum resolution (1200 dots per inch; pixel depth, 12) for relative optical density (ROD) measurements using Molecular Analyst software (Bio-Rad). RODs were measured directly within superimposed measurement frames across specific CNS loci. The ROD measurements were corrected for background and averaged for each region over three sections. Data were analyzed by ANOVA for genotype and region with Fisher’s exactpost hoc analysis. The power of our study was sufficient to have an 80% chance of detecting a 20% loss of neurons or mRNA signal at a confidence level of 0.05.
Immunostaining. Fixed 14 μm coronal cryostat or 40 μm vibrotome sections were treated with 0.5% Triton X-100 in Tris-buffered saline (TBS) for 20 min, blocked with 3% milk in TBS, and sequentially incubated in primary antibody [rabbit anti-GFAP (1:500), Sigma, St. Louis, MO; monoclonal anti-hAPP 8E5 (1:100), Athena Neurosciences, South San Francisco, CA; and biotinylated monoclonal anti-Aβ 3D6 (1:750), Athena Neurosciences], and secondary antibody [cy3-anti-rabbit (1:200), Jackson Immunochemicals, West Grove, PA; BODIPY-fluorescein anti-mouse (1:200), Molecular Probes, Eugene, OR; and Cy5- or horseradish peroxidase-conjugated streptavidin (1:750), Jackson, Vector Laboratories, Burlingame, CA]. 3–3′-Diaminobenzidine was the chromagen for horseradish peroxidase. Confocal images were obtained on the Bio-Rad laser confocal imaging system at an excitation wavelength of 586 nm and an emission wavelength of 605 nm for Cy3, 647 and 680 nm for Cy5, and 488 and 522 nm for BODIPY-fluorescein.
Amyloid burden quantitation. Amyloid deposition was quantified using Aβ immunostaining [monoclonal 3D6 (Johnson-Wood et al., 1997), diaminobenzidine reporter] and a Bioquant (Nashville, TN) image analysis system (Gomez-Isla et al., 1996b). Video images of each anatomic region of interest on 40 μm sections were captured, and a threshold optical density was obtained, which discriminated staining from background. Manual editing of each field eliminated artifacts. The “amyloid burden” defined as the total percentage of cortical surface area covered by amyloid deposition over three sections was calculated for CA1, cingulate, dentate gyrus molecular layer, and entorhinal cortex in each mouse.
Stereology. Neuron counts were performed using the optical disector technique (West and Gundersen, 1990) in 40 μmcresyl violet-stained coronal sections spaced 240 μmapart. The margins of the entorhinal cortex (EC) were defined in a caudal-to-rostral orientation: caudal, most caudal section containing white matter; rostral, 1 mm rostrally at the level of caudal extent of the pyriform cortex; lateral, rhinal fissure; and medial, parasubiculum (Lorente de No, 1933; Sidman et al., 1971). The entorhinal cortex was divided into layers I, II, III, and IV–VI (Lorente de No, 1933). The entire volume of the each EC layer was estimated according to the principle of Cavalieri (1966), using the Bioquant image analysis system. Neuronal counts were obtained from a systematically random-sampling scheme on sections spanning the entire EC. The number of neurons in the entire EC and each of its lamina was estimated using ∼100 optical disectors in each case. Each optical disector was a 25 × 50 μm sampling box with extended exclusion lines. Using a 100× oil immersion lens, neurons with a visible nucleolus were counted if they were not present in the initial plane of focus but came into focus as the optical plane moved through the tissue. The estimation of total neurons was calculated by multiplying the volume density of the neurons in the layers by the volume of the layers. All the counts were performed by a single examiner (M.C.I.) blinded to transgenic status.
The caudal retrosplenial/cingulate cortex was sampled in a caudal-to-rostral orientation: caudal, section containing the most caudal extent of the dentate gyrus; rostral, extending anteriorly 1.2 mm; medially, the subiculum; and laterally, the occipital cortex. This entire region was sampled in five 40-μm-thick sections taken at equally spaced intervals (240 μm), using ∼25 optical disectors in each case.
Caudal CA1 was sampled from its caudal extent anteriorly in seven 40 μm sections taken at equally spaced intervals (240 μm), using ∼25 optical disectors in each case.
The appropriateness of the sampling schemes chosen was evaluated by calculating the precision of the estimates made in each mouse, expressed as the coefficient of error (CE) (West, 1993). The CE of all cases was <0.10, suggesting that a minimal amount of variance in the counts is from the technique.
RESULTS
Regional expression of the PDAPP transgene
The PDAPP transgenic mouse expresses the three major hAPP isoforms containing 695, 751, and 770 amino acids. The isoforms containing the Kunitz protease inhibitor domains, hAPP751 and hAPP770, respectively, constitute 46.7 and 45.8% of the total hAPP mRNA (Rockenstein et al., 1995). The regional expression of the transgene was characterized byin situ hybridization with 35S-labeled oligonucleotide probes specific for each isoform in four heterozygote mice at each age of 4, 11, and 18 months. The expression pattern of each isoform was similar, with the highest expression in the hippocampus, followed by the cortex and superficial entorhinal region, then subcortical regions. White matter, amygdala, and the deep layers of the entorhinal cortex had little or no transgene expression (Fig.1). There was no change in the pattern or levels of expression with age from 4 to 18 months. Emulsion-dippedin situ sections confirmed an exclusively neuronal expression of the transgene.
Fig. 1.

Expression of hAPP isoforms in PDAPP mice.In situ hybridization showing similar hAPP695, 751, and 770 mRNA regional distribution in anterior and posterior coronal sections at 4, 11, and 18 months of age in heterozygote transgenic mice. Anterior sections demonstrate strongest message in the hippocampus, followed by the cingulate and cortical regions, and minimal signal in hippocampal white matter and amygdala; posterior sections demonstrate in addition strong message in the superficial, but not deep, layers of the entorhinal cortex. Scale bar, 1 mm.
Regional pattern of amyloid deposition
To assess the regional pattern of amyloid deposition resulting from transgene expression, we quantitated the amount of Aβ immunoreactivity in the neuropil of selected brain regions of six heterozygote mice at ages 8, 12, and 18 months (Fig.2). We focused on the hippocampus, entorhinal cortex, and cingulate cortex because these are areas of prominent amyloid deposition both in this transgenic animal and in human AD (Arnold et al., 1991). Amyloid deposition begins in the cingulate cortex of PDAPP transgenic mice by 8 months of age (amyloid burden, 0.4%). At 12 months of age, the amyloid burden of the dentate gyrus molecular layer, CA1, and cingulate and entorhinal cortex is 2–4%. Between 12 and 18 months of age, there is a profound increase in amyloid deposition in these regions, with amyloid burden on the order of 20–50%. By comparison, amyloid burden in the human neocortex in AD is generally 6–12% (Hyman et al., 1993).
Fig. 2.

Amyloid burden in PDAPP mice. Immunostaining for Aβ demonstrates age-dependent accumulation of amyloid beginning in the cingulate cortex (cing) at 8 months (A), prominent in dentate gyrus (dg), CA1, and entorhinal cortex (erc) by 12 months (B), and profound deposition in these regions by 18 months (C), quantitated by percent amyloid burden (±SD) measurements in these regions (D). Scale bar, 1 mm.
Neuron counts in CA1, cingulate, and entorhinal cortex
We next examined whether there was neuronal loss in these animals. Using unbiased stereological counting techniques, we assessed laminar neuronal counts in layers II, III, and IV–VI of the entorhinal cortex. Six transgenic and six nontransgenic mice at ages of 8, 12, and 18 months, masked for transgenic status, were examined. These ages correspond to absent, moderate, and severe cerebral Aβ deposition, respectively. The entorhinal cortex was chosen for analysis because: (1) the entorhinal cortex undergoes substantial age-related amyloid deposition in the PDAPP mouse beginning at 10–12 months of age; (2) amyloid deposits are prominent in the terminal field of its major efferent projection (the outer molecular layer of the dentate gyrus) by 12 months; and (3) the entorhinal cortex has marked neuronal depopulation early in AD (Gomez-Isla et al., 1996a). There was no difference in neuronal counts between the heterozygote transgenic mice and nontransgenic littermates at any age, whether analyzed layer by layer or as a whole (Fig. 3). Thus, there was no detectable neuronal loss in the entorhinal cortex even by 18 months of age, after 6–8 months of exposure to Aβ deposits, when amyloid burden in the entorhinal cortex is 30%, and that of the dentate gyrus molecular layer is 50%.
Fig. 3.

Neuron counts in PDAPP mice (±SD). No difference is noted in layer-by-layer neuronal counts in the entorhinal cortex of 8 (A), 12 (B), and 18 (C) month heterozygote transgenic mice compared with nontransgenic littermates. At 18 months, no significant difference is noted in the caudal CA1 or the caudal retrosplenial/cingulate region (D). Neuronal architecture is grossly preserved in wild-type mice (E) compared with nontransgenic animals (F) despite tremendous amyloid burden (G; Aβ immunostaining of section immediately adjacent to F). Counts reflect neurons in one hemisphere.cing, Cingulate cortex; erc, entorhinal cortex. Scale bar, 175 μm.
Because the majority of Aβ deposits in entorhinal cortex were morphologically “diffuse,” we considered the possibility that neuronal loss would correspond to a more “compact” morphology of amyloid deposits. We therefore examined two other areas that developed Aβ deposits with a compact morphology and that are known to have substantial neuronal loss in AD brains: caudal CA1 hippocampal subfield (West, 1994) and caudal retrosplenial/cingulate cortex (Vogt et al., 1992). Neither of these regions in the 18-month-old heterozygote transgenic animals had detectable neuronal loss (Fig. 3).
Neuronal mRNA synthesis
We also tested the hypothesis that neuronal mRNA synthesis was altered with amyloid deposition, even if the total neuronal number was unaffected. Thus, as surrogate measures of neuronal integrity, we assessed mRNA expression of neuronal synaptic and cytoskeletal proteins, synaptophysin and MAP-2, and mRNA expression of COX-2 and COX-4. These were chosen because there is a marked loss of synaptic and cytoskeletal protein mRNA and COX-2 but not COX-4 mRNA in AD brains, even in intact neurons (Clark and Parhad, 1989; Simonian and Hyman, 1994; Callahan and Coleman, 1995; de la Monte et al., 1995; Hatanpaa et al., 1996). There was no loss of regional expression of any of these mRNA’s in four transgenic and four nontransgenic mice at 4, 11, or 18 months of age (Fig. 4). This result contrasts with immunohistochemical data revealing a regional loss of both synaptophysin and MAP-2 immunoreactivity (Games et al., 1995), suggesting that neurons of origin with intact soma may have compromised axonal and dendritic processes.
Fig. 4.

mRNA expression in PDAPP mice (±SD). There is no loss of mRNA for neuronal synaptic (synaptophysin, A), cytoskeletal (MAP-2, B), or metabolic (COX-2,C; COX-4, D) proteins in 18 month PDAPP heterozygote transgenic mice compared with nontransgenic littermates.In situ hybridization demonstrates significantly increased GFAP mRNA in heterozygote transgenic mice compared with nontransgenic littermates at 18 months of age (E) (p < 0.002 by ANOVA; individual regiont tests not reaching significance), increasing in an age-related fashion in the heterozygote transgenic mice (F). dg, Dentate gyrus granule cell layer; mol, molecular layer of dentate gyrus;cing, cingulate cortex; erc, entorhinal cortex; thal, thalamus. +, Post hoc p < 0.05, 18 versus 4 and 11 months; ++, post hoc p < 0.0,5 18 versus 4 months.
Neuropil alterations
Finally, we examined whether a glial reaction in the neuropil occurred consequent to Aβ deposition. GFAP-immunoreactive astrocytes were associated with Aβ deposits and hAPP immunoreactive neurites (Fig. 5). Regional GFAP mRNA message was increased in the 18-month-old animals compared with 18-month-old nontransgenic littermates and younger transgenic animals (Fig.4E,F). GFAP mRNA was increased in parallel with cortical and hippocampal Aβ deposition and also in the thalamus, which is devoid of Aβ deposits. GFAP mRNA was not increased in 4- or 11-month-old heterozygote animals compared with nontransgenic littermates of the same age.
Fig. 5.
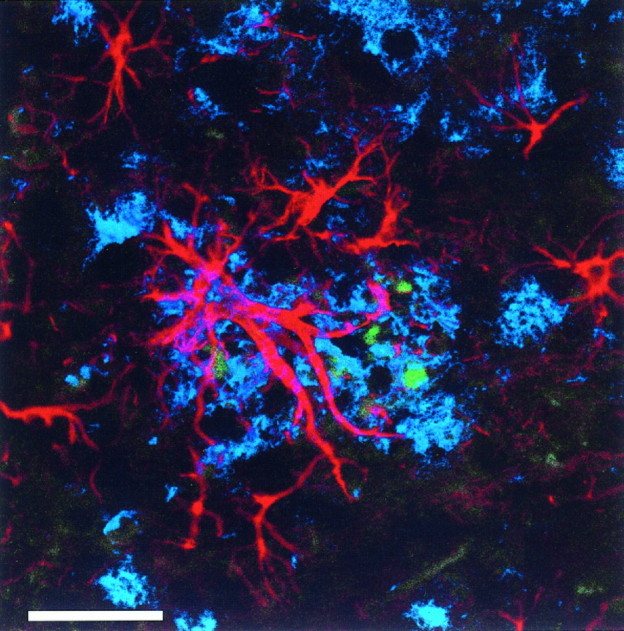
Confocal image demonstrating the association of Aβ deposition (blue) with GFAP-immunoreactive astrocytes (red) and hAPP immunoreactive structures (green) consistent with dystrophic neurites in an 18-month-old heterozygote PDAPP transgenic mouse. Scale bar, 25 μm.
DISCUSSION
These results demonstrate that the regional distribution of amyloid deposition in the PDAPP mice follows an anatomical and topographical hierarchical pattern analogous to that of AD. Cortical areas are affected more than subcortical areas, with deposits in specific laminar and cytoarchitectural zones, such as the deep layers of the entorhinal cortex, the molecular layer of CA1, and the outer molecular layer of the dentate gyrus (Hyman et al., 1986; Arnold et al., 1991). This exquisite anatomic specificity gives strong experimental support to the hypothesis that amyloid plaques develop at sites of axon terminals: (1) superficial layers of the entorhinal cortex strongly express the transgene and project as the perforant pathway to the outer molecular layer of the dentate gyrus, the precise location covered by Aβ deposition (Fig.6); (2) intrinsic hippocampal connections from the transgene expressing CA3 pyramidal neurons terminate in the molecular layer of CA1 and subiculum, which develops early amyloid deposition; (3) transgene-expressing subicular and CA1 neurons project to the deep layers of the entorhinal cortex, the latter developing amyloid plaques despite low trangene expression; and (4) the cortical afferents projecting to the amyloid laden superficial entorhinal cortex express the trangene. Thus, Aβ deposits frequently occur in terminal zones of transgene-positive neurons. However, not all areas receiving strong projections from hAPP-expressing neurons develop Aβ deposition. For example, the lateral nucleus of the amygdala, the thalamic pulvinar, and the pontine nuclei receive prominent cortical afferents without developing amyloid plaques. Additional studies have demonstrated that total protein levels of α-secretase-cleaved soluble hAPP/full-length hAPP in brain homogenates parallels hAPP expression, but that the conversion of Aβ via the amyloidogenic β-secretase pathway (as measured by APPβ) is region-specific (Johnson-Wood et al., 1997). This specificity of APP processing may account, in part, for the distribution of Aβ deposits. Therefore, the level of transgene expression is unlikely to be the sole factor determining Aβ deposition, and additional anatomic and metabolic factors play a role.
Fig. 6.

Immunostaining for Aβ in the hippocampus of an 18-month-old heterozygote transgenic mouse demonstrates the predisposition for Aβ deposition in the outer molecular layer (oml) of the dentate gyrus, which is the terminal zone for the projections of the perforant pathway. Scale bar, 100 μm.iml, Inner molecular layer; gc, granule cell layer.
The amyloid deposits in the PDAPP mouse are similar to those in AD not only in regional topography, but also in focal disruption of the neuropil eliciting a glial and neuritic response (Fig. 5). The age- and region-dependent rise in GFAP mRNA parallels the association of reactive astrocytes with extracellular Aβ deposits, providing additional evidence for Aβ-related degenerative alterations of the neuropil. Previous studies by Masliah et al. (1996) have demonstrated that the Aβ deposits in the PDAPP mouse are associated with dystrophic neurites (immunoreactive for SMI-312, hAPP, synaptophysin, and phosphorylated neurofilaments), although paired helical filaments and neurofibrillary tangles have not been identified. These results imply that Aβ deposition in the neuropil cannot be discounted as inert, nonspecific, peripheral, or a simple bystander to brain pathological changes.
Our studies also allowed us to test directly the hypothesis that Aβ deposition is neurotoxic in vivo, in light of convincingin vitro data suggesting acute neurotoxicity of Aβ (Koh et al., 1990; Mattson et al., 1992; Pike et al., 1993; Cotman et al., 1994; Cotman and Su, 1996; Yankner and Lorenzo, 1996). Aβ injection into the ventricles and brain tissue of animals has been reported to result in cellular toxicity (Kowall et al., 1991, Emre et al., 1992), although this is controversial and has not been replicated by others (Games et al., 1992; Podlisny et al., 1992; Stein-Beherns et al., 1992). Our results strongly argue against a direct acute neurotoxic mechanism of Aβ deposits in vivo in the transgenic mouse. This result is in accord with the lack of correlation between neuronal loss and amyloid deposition in AD (Wilcock and Esiri, 1982; Braak and Braak, 1991; Price et al., 1991; Terry et al., 1991; Arriagada et al., 1992; Hyman et al., 1993; Nagy et al., 1995; Gomez-Isla et al., 1996a). Nevertheless, the plaque-associated alterations of neuritic and glial morphology replicates observations in AD that specific subtypes of Aβ deposits are associated with neurodegenerative changes (Masliah et al., 1996).
Our methods would not have been sensitive to detect losses confined to a small subpopulation of neurons or to brain regions other than those examined. Indeed, degenerative changes such as vacuolization and cytoplasmic distention have been identified in some layer V neurons of the frontal cortex of PDAPP mice (Masliah et al., 1996). It remains possible that older age, or genetic, neuroanatomic, or metabolic factors absent from this mouse model, may be necessary for progression from amyloid deposition to generalized neuronal death. Evidence from Down’s syndrome brains suggest that >20 years of exposure to amyloid deposits in human brain may be required for significant AD changes (Rumble et al., 1989). Alternatively, overexpression of hAPP may be neuroprotective in mice, as has been suggested by lesion studies in hAPP transgenic mice (Mucke et al., 1996). Furthermore, intrinsic characteristics such as differences in promoter, host strain, hAPP primary structure, and levels of hAPP expression potentially influence the phenotype of transgenic mice (Hsiao et al., 1995). Nonetheless, our data suggest that human APP overexpression and Aβ deposition in the PDAPP mouse produce degenerative changes, including neuritic dystrophy and gliosis, but do not result in overt neuronal loss through 18 months of age.
Footnotes
This work was supported by National Institutes of Health Grant AG05134 and a generous gift of the Walters Family Foundation in the name of Margaret Siska. We greatly appreciate the helpful comments and suggestions by Ivan Lieberburg (Athena Neurosciences).
Correspondence should be addressed to Bradley T. Hyman, Alzheimer’s Disease Research Unit, Massachusetts General Hospital, East CNY6320, 149 13th Street, Charlestown, MA 02129.
REFERENCES
- 1.Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 2.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 3.Braak H, Braak E. Neuropathological staging of Alzheimer related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 4.Callahan LM, Coleman PD. Neurons bearing neurofibrillary tangles are responsible for selected synaptic deficits in Alzheimer’s disease. Neurobiol Aging. 1995;16:311–314. doi: 10.1016/0197-4580(95)00035-d. [DOI] [PubMed] [Google Scholar]
- 5.Cavalieri B. Geometria degli indivisibili. Unione Tipografico; Torino, Italy: 1966. [Google Scholar]
- 6.Clark AW, Parhad IM. Expression of neuronal mRNAs in Alzheimer type degeneration of the nervous system. Can J Neurol Sci. 1989;16:477–482. doi: 10.1017/s0317167100029802. [DOI] [PubMed] [Google Scholar]
- 7.Cotman CW, Su JH. Mechanisms of neuronal death in Alzheimer’s disease. Brain Pathol. 1996;6:493–506. doi: 10.1111/j.1750-3639.1996.tb00878.x. [DOI] [PubMed] [Google Scholar]
- 8.Cotman CW, Whittemore ER, Watt JA, Anderson AJ, Loo DT. Possible role of apoptosis in Alzheimer’s disease. Ann NY Acad Sci. 1994;747:36–49. doi: 10.1111/j.1749-6632.1994.tb44400.x. [DOI] [PubMed] [Google Scholar]
- 9.de la Monte SM, Ng S-C, Hsu DW. Aberrant GAP-43 gene expression in Alzheimer’s disease. Am J Pathol. 1995;147:934–946. [PMC free article] [PubMed] [Google Scholar]
- 10.Emre M, Geula C, Ransil BJ, Mesulam M-M. The acute neurotoxicity and the effects upon cholinergic axons of intracerebrally injected β amyloid in the rat brain. Neurobiol Aging. 1992;13:553–560. doi: 10.1016/0197-4580(92)90055-3. [DOI] [PubMed] [Google Scholar]
- 11.Games D, Khan KM, Soriano FG, Davis DL, Bryant K, Lieberburg I. Lack of Alzheimer pathology after β-amyloid protein injections in the brain. Neurobiol Aging. 1992;13:569–576. doi: 10.1016/0197-4580(92)90057-5. [DOI] [PubMed] [Google Scholar]
- 12.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagoplan S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 13.Goate A, Chartier-Harlin M-C, Mullan M. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–707. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 14.Gomez-Isla T, Price J, McKeel D, Morris J, Growdon J, Hyman B. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996a;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locasio JT, Perls TT, Lipsitz LA, Hyman BT. Clinical and pathological correlates of apolipoprotein E e4 in Alzheimer’s disease. Ann Neurol. 1996b;39:62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- 16.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Peterson RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 17.Hatanpaa K, Brady DR, Stoll J, Rapoport SI, Chandresekaran K. Neuronal activity and early neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1996;40:411–420. doi: 10.1002/ana.410400310. [DOI] [PubMed] [Google Scholar]
- 18.Higgins LS, Rodems JM, Catalano R, Quon D, Cordell B. Early Alzheimer disease-like histopathology increases in frequency with age in mice transgenic for beta-APP751. Proc Natl Acad Sci USA. 1995;92:4402–4406. doi: 10.1073/pnas.92.10.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- 20.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 21.Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR. Perforant pathway changes and the memory impairment of Alzheimer’s disease. Ann Neurol. 1986;20:473–482. doi: 10.1002/ana.410200406. [DOI] [PubMed] [Google Scholar]
- 22.Hyman BT, Marzloff K, Arriagada PV. The lack of accumulation of senile plaques or amyloid burden in Alzheimer’s disease suggests a dynamic balance between amyloid deposition and resolution. J Neuropathol Exp Neurol. 1993;52:594–600. doi: 10.1097/00005072-199311000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Johnson-Wood K, Lee M, Motter R, Hu R, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koh J-Y, Yang L-L, Cotman CW. β-Amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res. 1990;533:315–320. doi: 10.1016/0006-8993(90)91355-k. [DOI] [PubMed] [Google Scholar]
- 25.Kowall NW, Beal MF, Busciglio J, Duffy LK, Yankner BA. An in vivo model for the neurodegenerative effects of β amyloid and protection by substance P. Proc Natl Acad Sci USA. 1991;88:7247–7251. doi: 10.1073/pnas.88.16.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorente de No R. Studies on the structure of the cerebral cortex. I. The area entorhinalis. J Psychol Neurol. 1933;45:381–483. [Google Scholar]
- 27.Masliah E, Mallory M, Deerinck T, DeTeresa R, Lamont S, Miller A, Terry RD, Carragher B, Ellisman M. Re-evaluation of the structural organization of neuritic plaques in Alzheimer’s disease. J Neuropathol Exp Neurol. 1993;52:619–632. doi: 10.1097/00005072-199311000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer’s disease. J Neurosci. 1996;16:5795–5811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattson MP, Cheng B, Davis D. Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:379–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mucke L, Abraham CR, Masliah E. Neurotrophic and neuroprotective effects of hAPP in transgenic mice. Ann NY Acad Sci. 1996;777:82–88. doi: 10.1111/j.1749-6632.1996.tb34405.x. [DOI] [PubMed] [Google Scholar]
- 31.Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 32.Nagy Z, Esiri MM, Jobst KA. Relative roles of plaques and tangles in the dementia of Alzheimer’s disease: correlations using three sets of neuropathological criteria. Dementia. 1995;6:21–31. doi: 10.1159/000106918. [DOI] [PubMed] [Google Scholar]
- 33.Page K, Hollister R, Tanzi RE, Hyman BT. In situ hybridization analysis of presenilin 1 mRNA in Alzheimer’s disease and in lesioned rat brain. Proc Natl Acad Sci USA. 1996;93:14020–14024. doi: 10.1073/pnas.93.24.14020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Podlisny MB, Stephenson DT, Frosch MP, Lieberburg I, Clemens JA, Selkoe DJ. Synthetic β-amyloid protein fails to produce specific neurotoxicity in monkey cerebral cortex. Neurobiol Aging. 1992;13:561–568. doi: 10.1016/0197-4580(92)90056-4. [DOI] [PubMed] [Google Scholar]
- 36.Price DL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol Aging. 1991;12:295–312. doi: 10.1016/0197-4580(91)90006-6. [DOI] [PubMed] [Google Scholar]
- 37.Rockenstein E, McConlogue L, Tan H, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J Biol Chem. 1995;47:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- 38.Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, Martins R, Hockey A, Montgomery P, Beyreuther K, Masters CL. Amyloid A4 protein and its precursor in Down’s syndrome and Alzheimer’s disease. N Engl J Med. 1989;320:1446–1452. doi: 10.1056/NEJM198906013202203. [DOI] [PubMed] [Google Scholar]
- 39.Selkoe DJ. Normal and abnormal biology of the β-amyloid precursor protein. Annu Rev Neurosci. 1994;17:489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- 40.Sidman RL, Angevine JB, Pierce ET. Atlas of the mouse brain and spinal cord. Harvard UP; Cambridge, MA: 1971. [Google Scholar]
- 41.Simonian NA, Hyman BT. Functional alterations in Alzheimer’s disease: selective loss of mitochondrial-encoded cytochrome oxidase mRNA in the hippocampal formation. J Neuropathol Exp Neurol. 1994;53:508–512. doi: 10.1097/00005072-199409000-00010. [DOI] [PubMed] [Google Scholar]
- 42.Sirinathsinghji DJS, Dunnett SB. Imaging gene expression in neural grafts. In: Sharif NA, editor. Molecular imaging in neuroscience: a practical approach. Oxford UP; New York: 1993. pp. 43–70. [Google Scholar]
- 43.Sola C, Mengod G, Probst A, Palacios JM. Differential regional and cellular distribution of beta-amyloid precursor protein messenger RNAs containing and lacking the Kunitz protease inhibitor domain in the brain of human, rat and mouse. Neuroscience. 1993;53:267–295. doi: 10.1016/0306-4522(93)90304-x. [DOI] [PubMed] [Google Scholar]
- 44.Stein-Beherns B, Adams K, Yeh M, Sapolsky R. Failure of beta-amyloid protein fragment 25–35 to cause hippocampal damage in the rat. Neurobiol Aging. 1992;13:577–579. doi: 10.1016/0197-4580(92)90058-6. [DOI] [PubMed] [Google Scholar]
- 45.Terry RD, Masliah E, Salmon DP. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;41:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 46.Vogt BA, Crino PB, Vogt LJ. Reorganization of cingulate cortex in Alzheimer’s disease: neuron loss, neuritic plaques, and muscarinic receptor binding. Cereb Cortex. 1992;2:526–535. doi: 10.1093/cercor/2.6.526. [DOI] [PubMed] [Google Scholar]
- 47.West M. New stereological methods for counting neurons. Neurobiol Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- 48.West MJ. Differences in the pattern of hippocampal neuron loss in normal ageing and Alzheimer’s disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 49.West MJ, Gundersen HJG. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- 50.Wilcock GK, Esiri MM. Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 1982;56:343–356. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- 51.Yankner BA, Lorenzo A. Amyloid fibril toxicity in Alzheimer’s disease and diabetes. Ann NY Acad Sci. 1996;777:89–95. doi: 10.1111/j.1749-6632.1996.tb34406.x. [DOI] [PubMed] [Google Scholar]