

Abstract
The cellular mechanisms underlying the neuroprotective action of the immunosuppressant FK506 in experimental stroke remain uncertain, although in vitro studies have implicated an antiexcitotoxic action involving nitric oxide and calcineurin. The present in vivo study demonstrates that intraperitoneal pretreatment with 1 and 10 mg/kg FK506, doses that reduced the volume of ischemic cortical damage by 56–58%, did not decrease excitotoxic damage induced by quinolinate, NMDA, and AMPA. Similarly, intravenous FK506 did not reduce the volume of striatal quinolinate lesions at a dose (1 mg/kg) that decreased ischemic cortical damage by 63%. The temporal window for FK506 neuroprotection was defined in studies demonstrating efficacy using intravenous administration at 120 min, but not 180 min, after middle cerebral artery occlusion. The noncompetitive NMDA receptor antagonist MK801 reduced both ischemic and excitotoxic damage. Histopathological data concerning striatal quinolinate lesions were replicated in neurochemical experiments. MK801, but not FK506, attenuated the loss of glutamate decarboxylase and choline acetyltransferase activity induced by intrastriatal injection of quinolinate. The contrasting efficacy of FK506 in ischemic and excitotoxic lesion models cannot be explained by drug pharmacokinetics, because brain FK506 content rose rapidly using both treatment protocols and was sustained at a neuroprotective level for 3 d. Although these data indicate that an antiexcitotoxic mechanism is unlikely to mediate the neuroprotective action of FK506 in focal cerebral ischemia, the finding that intravenous cyclosporin A (20 mg/kg) reduced ischemic cortical damage is consistent with the proposed role of calcineurin.
Keywords: FK506, tacrolimus, stroke, neuroprotection, excitotoxicity, ischemia, MK801, dizocilpine, cyclosporin A
The immunosuppressant FK506 (tacrolimus, Prograf) recently has been introduced into clinical use for the prevention of allograft rejection. Its immunosuppressive mechanism involves inhibition of calcineurin (protein phosphatase 2B) by a complex of FK506 and the 12 kDa immunophilin FKBP12 (Liu et al., 1991, 1992; Clipstone and Crabtree, 1992; Fruman et al., 1992a), resulting in an inability to assemble the active form of the transcription factor NFAT (Bierer et al., 1990; Flanagan et al., 1991) and subsequent attenuation of T lymphocyte gene transcription (Schreiber, 1991; Liu, 1993). The immunosuppressant cyclosporin A also inhibits calcineurin in a complex with cyclophilin, another member of the immunophilin protein family (Liu et al., 1991, 1992; Clipstone and Crabtree, 1992; Fruman et al., 1992a). In contrast, the immunosuppressive mechanism of rapamycin involves blockade of interleukin-2 receptor signal transduction (Schreiber, 1991; Liu, 1993) via an interaction of a rapamycin/FKBP12 complex with a novel protein termed RAPT or FRAP (Brown et al., 1994; Chiu et al., 1994; Sabatini et al., 1994); the precise details of this pathway still have to be elucidated.
Several lines of evidence indicate a role for immunophilins and/or calcineurin in brain function and development (Lyons et al., 1994;Mulkey et al., 1994; Nichols et al., 1994; Chang et al., 1995; Liu et al., 1995; Snyder and Sabatini, 1995; Tong et al., 1995). In addition, FK506 exerts a powerful neuroprotective action in experimental models of stroke (Sharkey and Butcher, 1994), suggesting a novel therapeutic application for this drug. Although the cellular mechanism underlying this effect remains uncertain, pharmacological data confirmed the importance of immunophilin binding and suggested a role for calcineurin (Sharkey and Butcher, 1994). The presence of FKBP12 in rat brain has been demonstrated using both in situ hybridization and Western blot analysis (Steiner et al., 1992; Dawson et al., 1994;Charters et al., 1995), and colocalization with calcineurin has been reported (Steiner et al., 1992; Dawson et al., 1994). FK506 also protects cortical cell cultures against excitotoxic neuronal death, suggesting a direct drug action on brain cells that may involve nitric oxide, because FK506 prevents the dephosphorylation of nitric oxide synthase (NOS) by calcineurin in vitro (Dawson et al., 1993). However, alternative mechanisms must be considered, especially in view of the proposed role for calcium ions in neurodegeneration (Choi, 1995). FKBP12 is an integral part of the ryanodine and IP3 receptor complexes, and functional effects of FK506 on the associated intracellular Ca2+ channels have been demonstrated (Timerman et al., 1993; Zhang et al., 1993; Brillantes et al., 1994; Chen et al., 1994; Cameron et al., 1995a,b). The involvement of reactive oxygen species in the neuroprotective mechanism is also possible because FK506 inhibits superoxide production in neutrophils (Nishinaka et al., 1993), and reactive oxygen species are reported to play a role in both apoptotic neuronal death and neurodegeneration resulting from cerebral ischemia (Kinouchi et al., 1991; Greenlund et al., 1995). The present study examines the cellular mechanism underlying the neuroprotective action of FK506, with particular reference to in vivo excitotoxicity and drug pharmacokinetics.
MATERIALS AND METHODS
Materials. Quinolinate (lot Q-1375) was purchased from Sigma (Poole, UK), NMDA and AMPA from Tocris Chemicals (Bristol, UK), and MK801 (dizocilpine) from Research Biochemicals (St. Albans, UK). Excitotoxins were dissolved in sterile 50 mm PBS, pH-adjusted to 7.4 with NaOH. Endothelin-1 (Nova Biochem: lot A13210) was dissolved in sterile saline. FK506 (Fujisawa Pharmaceutical, Osaka, Japan) was dissolved in 10% ethanol in 50 mm PBS containing 1% Tween 80 for intraperitoneal studies and in 10% ethanol in saline containing 400 mg/ml polyoxyl 60 hydrogenated castor oil for intravenous studies.
Excitotoxic lesions. Male Sprague Dawley rats (280–340 gm; Charles River, Margate, UK) were anesthetized with either pentobarbitone (Sagittal; 60 mg/kg) for intraperitoneal studies or halothane (4% for induction; 1–2% for maintenance) in nitrous oxide/oxygen (80/20%; v:v) for intravenous studies. Normothermia (37 ± 1°C) was maintained by using a thermostatically controlled heating blanket connected to a rectal thermometer. Excitotoxins were injected under stereotaxic guidance over 2 min into the striatum [anteroposterior (AP) +0.5 mm; mediolateral (ML) ± 3.0 mm; dorsoventral (DV) −4.5 mm below dura], cortex (AP +0.5 mm; ML −2.5 mm; DV −1.0 mm below dura), or hippocampus (AP −4.0 mm; ML −3.5 mm; DV −3.0 mm below dura) in a volume of 1 μl (striatum) or 0.5 μl (hippocampus and cortex). The needle was left in place for a further 2 min before slowly being withdrawn. Animals were placed in an incubator to maintain normothermia until their recovery from anesthesia. Drugs were administered 30 min before excitotoxin injection in intraperitoneal studies and 1 min after excitotoxin injection in intravenous studies.
Induction of focal cerebral ischemia. Male Sprague Dawley rats (300–370 gm; Charles River) were anesthetized with either pentobarbitone (Sagittal; 60 mg/kg) for intraperitoneal studies or halothane (4% for induction; 1–2% for maintenance) in nitrous oxide/oxygen (80/20%; v:v) for intravenous studies. Normothermia (37 ± 1°C) was maintained by using a thermostatically controlled heating blanket connected to a rectal thermometer. Endothelin-1 (60 pmol in 3 μl) was injected via a 31-gauge guide cannula stereotaxically placed 0.5 mm above the middle cerebral artery (AP +0.2 mm; ML −5.9 mm; DV −7.0 mm below dura). The cannula was leftin situ for 5 min before slowly being withdrawn over 2–3 min. Animals were placed in an incubator to maintain normothermia until their recovery from anesthesia. Drugs were administered 30 min before vessel occlusion in intraperitoneal studies and 1 min after vessel occlusion in intravenous studies (except when indicated).
Histopathological assessment of brain damage. Rats were reanesthetized with pentobarbitone (Sagittal; 60 mg/kg) 3 d after injection of excitotoxins or middle cerebral artery occlusion (MCAO) and were fixed by transcardiac perfusion first with 20 ml of heparinized saline (10 U/ml), followed by 200 ml of 4% paraformaldehyde in 50 mm PBS, pH 7.4. The brain was removed intact and immersed in fixative containing 10% sucrose for at least 24 hr before cryostat sectioning. Coronal sections (20 μm thick) were cut and stained with either cresyl violet or thionine. The volume of brain damage was determined as described previously (Park et al., 1989; Sharkey and Butcher, 1994). Briefly, the area of brain damage at eight predetermined brains was assessed using light microscopy by an observer who was unaware of the treatment groups. The volume of brain damage was calculated by integration of the cross-sectional area of damage at each stereotaxic level and the distances between the various levels (Park et al., 1989; Sharkey and Butcher, 1994).
Measurement of glutamate decarboxylase (GAD) and choline acetyltransferase (ChAT) activity. Rats were killed by cervical dislocation 3 d after intrastriatal injection of quinolinate injection. The brain was removed immediately, and injected and uninjected striata were dissected and homogenized in 20 vol of ice-cold water containing 1 mm EDTA and 0.1% Triton X-100, pH 7.4. Tissue GAD and ChAT activity was determined by using minor modifications of the methods of Kanazawa et al. (1976) and Fonnum (1975), respectively. Radioactivity was determined in a Packard 2500TR liquid scintillation counter using automatic quench correction. Enzyme activity was calculated after subtraction of zero time blanks. The protein content of striatal homogenates was determined according to the method of Bradford (1976).
Measurement of mean arterial blood pressure (MABP) and rectal and brain temperature. Separate groups of animals were anesthetized with halothane (4% for induction; 1–2% for maintenance) in nitrous oxide/oxygen (80/20%; v:v) and placed in a stereotaxic frame. An intravenous catheter was inserted in the femoral artery and connected via a pressure transducer to a Kontron Supermon monitor for measurement of MABP. Rectal temperature was measured by a thermometer inserted 9 cm into the rectum, which was connected to a thermostatically controlled heating blanket. Brain temperature was measured by a miniature thin film recording probe (Ottosensor, Cleveland, OH) inserted into the striatum (AP +1.0 mm; ML −2.0 mm; DV −4 mm below dura) under stereotaxic guidance. MABP and rectal and brain temperature were recorded for 30 min before induction of focal cerebral ischemia and for 180 min after vessel occlusion.
Measurement of brain and blood FK506 content. Nonfasted rats were injected with FK506 by the intravenous (1 mg/kg) or intraperitoneal (10 mg/kg) route. Rats were anesthetized with halothane at the specified time points between 15 min and 72 hr later, and a venous blood sample was collected from the vena cava. Then the vasculature was flushed with 20 ml of heparinized saline via an intra-aortic cannula. Animals were decapitated immediately, and the whole brain (minus cerebellum and brainstem) was dissected. Blood and brain samples were stored at −70°C before determination of FK506 content. The effects of MCAO on blood and brain levels of FK506 were examined in a separate group of animals. The middle cerebral artery was occluded by intracerebral injection of endothelin-1 as described previously, and FK506 (1 mg/kg, i.v.) was injected 5 min after vessel occlusion. Animals were killed 1 and 3 hr later, and samples of ischemic and nonischemic cortex were dissected for determination of drug content.
FK506 was measured by competitive enzyme immunoassay with a mouse anti-FK506 monoclonal antibody (FKmAb) and FK506-conjugated peroxidase (FK-POD). FK506 in whole blood was extracted with methanol. Brain samples were homogenized in distilled water (10%, w:v), and FK506 was extracted with n-hexane containing 2.5% isoamyl alcohol. The extraction solvent was evaporated and the residue dissolved in FK-POD solution. The solution was added to a microtiter plate well, coated previously with goat anti-mouse IgG polyclonal antibody, and was mixed with FKmAb to determine competitive binding of FK506 and FK-POD with FKmAb. POD activity was measured usingo-phenylenediamine and hydrogen peroxide as cosubstrates. The reaction was stopped by addition of H2SO4, and optical density was measured by a microplate reader (Molecular Devices, Menlo Park, CA). FK506 content was determined by comparison with a standard curve.
RESULTS
Intraperitoneal drug administration
The effects of intraperitoneal pretreatment with FK506 and the noncompetitive NMDA receptor antagonist MK801 (dizocilpine) on the volume of brain damage associated with endothelin-induced MCAO, an experimental model of stroke, were evaluated. MK801 (5 mg/kg), administered intraperitoneally 30 min before vessel occlusion, decreased the volume of ischemic damage in the cortex by 50% (Fig.1). Similarly, FK506 reduced ischemic damage in cortex by 56 and 58% at 1 and 10 mg/kg, respectively (Fig.1). Neither drug decreased the volume of ischemic brain damage in striatum (data not shown).
Fig. 1.
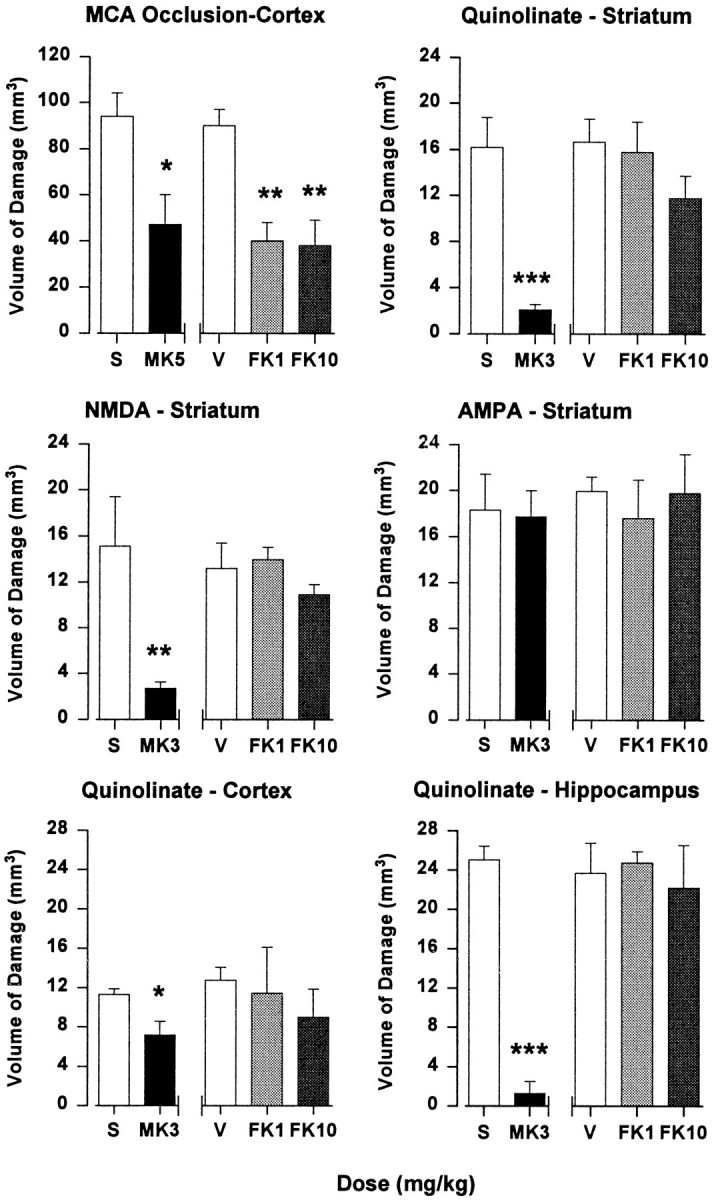
Neuroprotection studies used intraperitoneal drug administration 30 min before lesion induction. MK801 (MK3; 3 mg/kg) inhibited excitotoxic brain damage in striatum, hippocampus, and cortex induced by quinolinate (100 nmol, striatum; 50 nmol, hippocampus and cortex) and NMDA (100 nmol), and at 5 mg/kg (MK5) it reduced the volume of cortical damage induced by MCAO. FK506 (1 and 10 mg/kg; FK1 andFK10, respectively) inhibited ischemic damage, but it had no effect on excitotoxic damage. Neither drug reduced excitotoxic damage in striatum induced by AMPA (25 nmol). Data are the mean volume of brain damage (± SEM) for groups of 5–12 animals. Statistical comparisons between drug and vehicle (saline, S; FK506 vehicle, V) groups used unpaired ttests for excitotoxin data and ANOVA with post hocScheffé’s analysis for MCAO data (*p < 0.05; **p < 0.01; ***p < 0.001).
Excitotoxic striatal lesions were produced by injection of quinolinate, NMDA, or AMPA, with regional specificity evaluated by using quinolinate lesions in hippocampus and cortex. The excitotoxin doses that were used produced submaximal lesions in terms of the volume of brain damage (30–50% of maximal neuronal damage; data not shown). MK801 (3 mg/kg), administered intraperitoneally 30 min before excitotoxin injection, reduced the volume of quinolinate-induced damage in striatum, hippocampus, and cortex by 87, 95, and 37%, respectively (Fig. 1). The smaller decrease in cortex was attributable to proportionally more nonspecific damage being caused by needle penetration. The NMDA-induced striatal lesion was 82% smaller in MK801-treated rats, whereas the volume of the AMPA-induced lesion was unaffected (Fig. 1). With the use of an identical intraperitoneal administration protocol, FK506 (1 and 10 mg/kg) did not reduce the volume of excitotoxic brain damage induced by quinolinate, NMDA, or AMPA (Fig. 1).
Intravenous drug administration
MK801 (0.3 mg/kg) and FK506 (1 mg/kg) decreased the volume of ischemic brain damage in cortex after endothelin-induced MCAO by 34 and 58%, respectively (Fig. 2). In contrast to previous negative data obtained using 1 mg/kg cyclosporin A (Sharkey and Butcher, 1994), intravenous administration at 20 mg/kg decreased the volume of ischemic brain damage in cortex by 63%; in all cases, drugs were administered 1 min after excitotoxin injection (Fig. 2). Neither MK801, FK506, nor cyclosporin A reduced the volume of ischemic brain damage in striatum after MCAO (Fig. 2). With the use of an identical intravenous administration protocol, MK801 (0.3 mg/kg) reduced the volume of quinolinate-induced striatal brain damage by 45%, whereas FK506 (1 mg/kg) was ineffective.
Fig. 2.
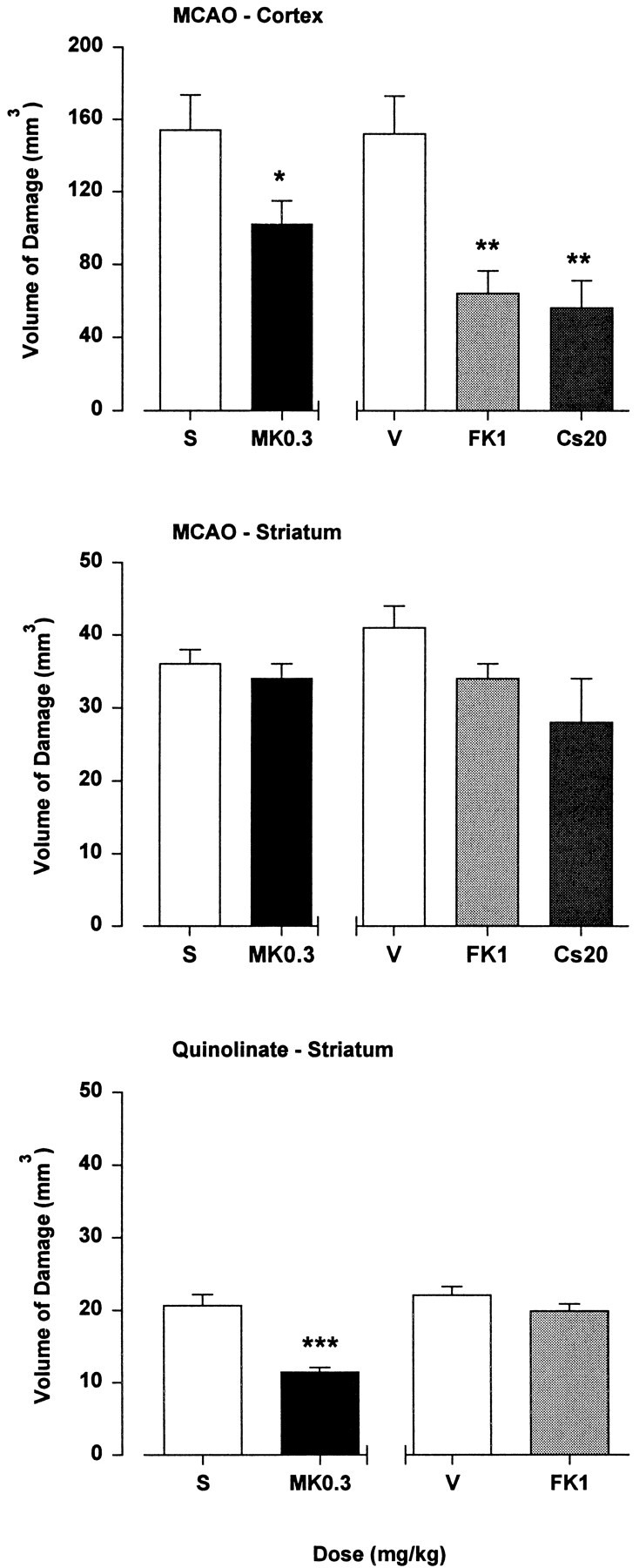
Neuroprotection studies used intravenous drug administration 1 min after lesion induction. FK506 (FK1; 1 mg/kg), MK801 (MK0.3; 0.3 mg/kg) and cyclosporin A (Cs20; 20 mg/kg) reduced the volume of ischemic brain damage in cortex, but not striatum, induced by MCAO. The volume of excitotoxic brain damage in striatum induced by quinolinate (100 nmol) was reduced by MK801 (0.3 mg/kg), whereas FK506 (1 mg/kg) was ineffective. Data are the mean volume of brain damage (± SEM) for groups of 5–12 animals. Statistical comparisons between drug and vehicle (saline, S; FK506 vehicle,V) groups used unpaired t tests for excitotoxin data and ANOVA with post hocScheffé’s analysis for MCAO data (*p < 0.05; **p < 0.01; ***p < 0.001).
GAD and ChAT activity
GAD activity, a marker for striatal GABAergic interneurons, was reduced by 44% (p < 0.05) from 4.37 ± 0.49 μmol/mg protein/hr in the contralateral striatum of vehicle-treated rats to 2.47 ± 0.27 μmol/mg protein/hr in the quinolinate-injected striatum (Fig. 3). GAD activity in the quinolinate-injected striatum of rats treated with FK506 using intraperitoneal (10 mg/kg; 30 min pretreatment) and intravenous (1 mg/kg; 1 min after excitotoxin injection) administration protocols was reduced by 44% (p < 0.05) and 46% (p < 0.05), respectively, as compared with the contralateral striatum (Fig. 3). In contrast, GAD activity was not reduced significantly in the quinolinate-injected striatum of MK801-treated rats using intraperitoneal (3 mg/kg) and intravenous (0.3 mg/mg) drug administration (Fig. 3). Similar data were obtained by using a ChAT assay to determine the survival of striatal cholinergic neurons. In this case, enzyme activity was reduced by 45% (p < 0.05) from 746 ± 94 nmol/mg protein/hr in the contralateral striatum of vehicle-treated rats to 413 ± 83 nmol/mg protein/hr in the quinolinate-injected striatum. ChAT activity in the quinolinate-injected striatum of rats treated with FK506 using intraperitoneal and intravenous administration protocols was reduced by 37% (p < 0.05) and 38% (p < 0.05), respectively, as compared with the contralateral striatum. ChAT activity was not decreased significantly in the quinolinate-injected striatum of MK801-treated rats; reductions of 9 and 25% were noted with intraperitoneal and intravenous drug administration, respectively, as compared with the contralateral striatum.
Fig. 3.

Effects of intrastriatal quinolinate injection on glutamate decarboxylase activity in striatal homogenates. When administered intraperitoneally 30 min before intrastriatal injection of 100 nmol quinolinate, MK801 [MK (i.p.); 3 mg/kg], but not FK506 [FK (i.p.); 10 mg/kg], prevented the reduction of enzyme activity noted in the quinolinate-injected hemisphere. Similarly, with intravenous administration 1 min after excitotoxin injection, MK801 [MK (i.v.); 0.3 mg/kg], but not FK506 [FK (i.v.); 1 mg/kg], prevented the reduction in enzyme activity. Data are mean enzyme activity (± SEM) in contralateral uninjected (open bars) and quinolinate-injected (filled bars) striata for groups of four animals. Statistical comparisons of enzyme activity in the two hemispheres were performed by using a paired ttest (*p < 0.05; **p < 0.01).
Delayed intravenous administration of FK506
The temporal window of therapeutic efficacy for FK506 with regard to its neuroprotective action was characterized in a separate group of animals. Although intravenous injection of FK506 (1 mg/kg) at 1, 60, and 120 min after endothelin-induced MCAO reduced the volume of cortical brain damage by 48, 46, and 57%, respectively, FK506 was ineffective when administered after 180 min (Fig.4). FK506 did not reduce the volume of striatal damage at any time point studied (data not shown). A substantial increase in the volume of ischemic brain damage also was noted in vehicle-treated rats as the duration of anesthesia was extended (Sharkey and Butcher, 1995). Although this effect was not significant when comparing anesthetic durations of 5 and 60 min, further extension to 120 and 180 min after MCAO increased the volume of cortical ischemic damage by 88 and 82%, respectively (p < 0.05).
Fig. 4.

The temporal window of neuroprotective efficacy for intravenous FK506 (1 mg/kg) administered after endothelin-induced MCAO. Data are the mean volume of ischemic brain damage (± SEM) in cortex for groups of 8–12 animals from vehicle (open bars) and FK506-treated (filled bars) rats. Statistical comparisons were performed by using ANOVA withpost hoc Scheffé’s analysis (*p < 0.05 comparison of drug and vehicle groups;†p < 0.05 comparison of vehicle groups with the 1 min vehicle group).
Physiological variables
MABP was monitored from 30 min before endothelin-induced MCAO until 180 min after vessel occlusion in animals treated intravenously with either FK506 (1 mg/kg) or FK506 vehicle; significant effects on MABP were not noted in either group (Fig.5). Rectal and brain temperature similarly were unaffected after endothelin-induced MCAO in FK506 and vehicle-treated rats (Fig. 5).
Fig. 5.

Endothelin-induced MCAO in FK506 (filled circles) and vehicle-treated (open circles) rats does not affect the mean arterial blood pressure (MABP) nor rectal or intracerebral temperatures. Rats were anesthetized continuously with halothane with measurements made from 30 min before until 180 min after vessel occlusion. Data are mean values (± SEM) from four rats per group. P is the mean preocclusion value; endothelin was injected at time 0, and FK506 (1 mg/kg) was administered intravenously 1 min after vessel occlusion. Statistical comparisons were performed by using ANOVA withpost hoc Scheffé’s analysis (p < 0.05).
Brain and blood levels of FK506
Brain and blood levels of FK506 were determined from 15 min until 72 hr after intraperitoneal (10 mg/kg) and intravenous (1 mg/kg) administration (Fig. 6). With the use of the intravenous administration route, a brain content of ∼50 ng/gm tissue was detected throughout the monitoring period (Fig. 6). In contrast, the blood level of FK506 fell rapidly in an exponential manner from 163 ng/ml at 15 min postinjection to an undetectable level at 72 hr. A slightly different pattern was noted with the intraperitoneal administration route (Fig. 6). The brain content of drug rose to a maximum of ∼400 ng/gm tissue at 12 hr after injection and thereafter fell slightly to 300 ng/gm tissue at 72 hr. It should be noted that a brain content of 135 ng/gm tissue was detected 30 min postinjection, the time at which the excitotoxic or ischemic challenge was initiated using this route of drug administration. The blood level of drug was maximal 15–30 min after injection, with FK506 levels falling rapidly thereafter to a trough level of 2 ng/ml detected at 48–72 hr.
Fig. 6.
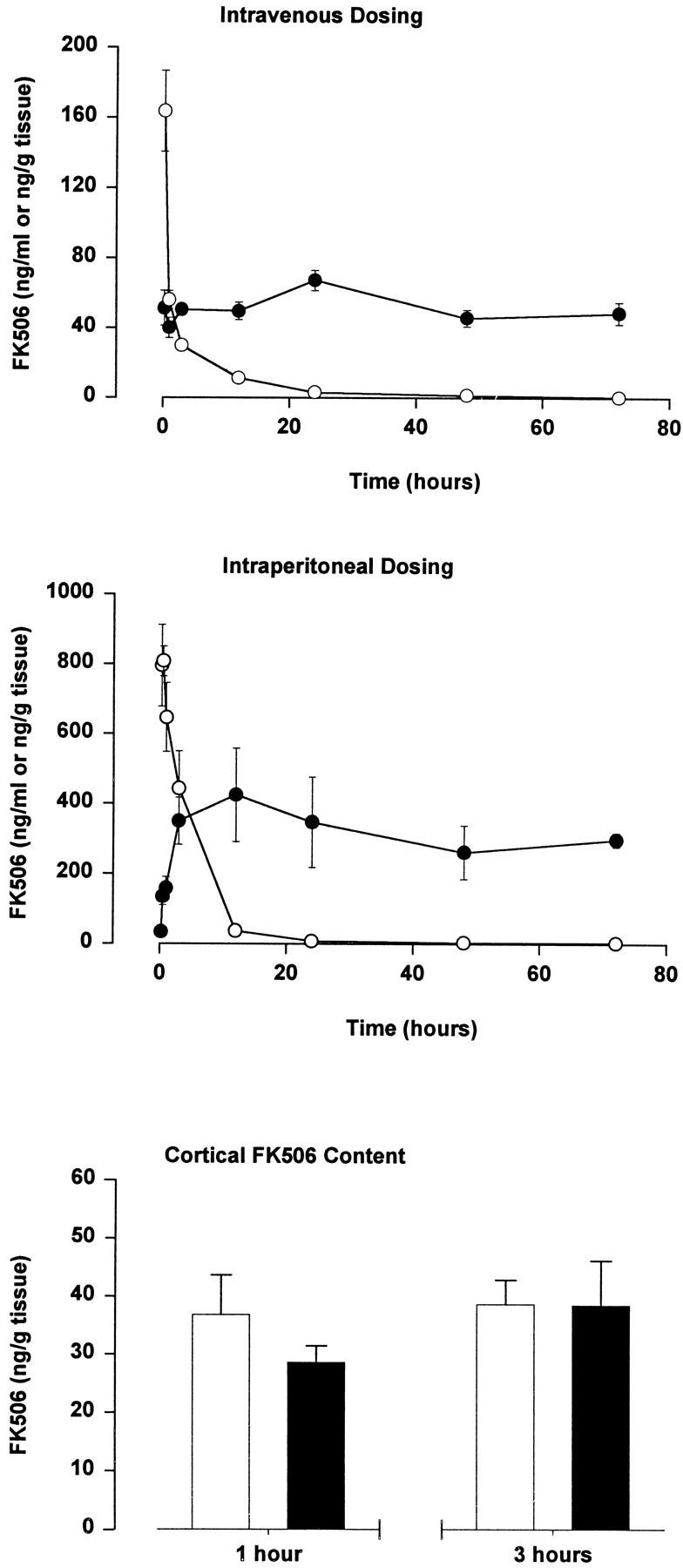
Brain and blood levels of FK506 after intravenous (1 mg/kg) and intraperitoneal (10 mg/kg) dosing. Data are mean brain (filled circles) and blood (open circles) FK506 content (± SEM) from four animals per group. Histograms show FK506 content in cortical samples obtained 1 and 3 hr after endothelin-induced middle cerebral artery occlusion. Data are mean cortical FK506 content (± SEM) in the nonischemic (open bars) and ischemic (filled bars) hemisphere from four animals per group.
Cortical FK506 content also was measured following endothelin-induced MCAO with intravenous drug (1 mg/kg) administration 5 min after vessel occlusion (Fig. 6). Drug content in the contralateral nonischemic cortex was 36.8 ± 6.83 and 37.4 ± 4.54 ng/gm tissue at 1 and 3 hr, respectively, after vessel occlusion. FK506 content in the ischemic cortex was not significantly different: 28.6 ± 2.87 ng/gm tissue and 38.5 ± 7.7 ng/gm tissue at 1 and 3 hr, respectively. Blood levels of FK506 in these animals were 47.3 ± 3.58 and 24.1 ± 3.38 ng/ml, respectively.
Further neuroprotection experiments were performed to ascertain whether the FK506 detected in brain 24–72 hr after a single intravenous injection was bioavailable. FK506 (1 mg/kg, i.v.) was administered either 24 or 72 hr before endothelin-induced MCA occlusion, and the volume of ischemic brain damage was determined 72 hr after vessel occlusion. The volume of ischemic brain damage in cortex was reduced by 64% (p < 0.05) and 39% (p < 0.05) from 132 ± 20 mm3 in vehicle-treated rats (n = 11) to 47 ± 8 mm3 (n = 10) and 82 ± 10 mm3 (n = 11) in animals pretreated with FK506 for 24 and 72 hr, respectively.
DISCUSSION
The present study confirms that FK506 exhibits a powerful neuroprotective action in an experimental model of stroke (Sharkey and Butcher, 1994). Additional intravenous studies revealed that 1 mg/kg FK506 reduces ischemic brain damage in cortex when administered 120 min, but not 180 min, after MCAO, suggesting that a critical window of opportunity exists with regard to the neuroprotective effect. The ability of intravenous FK506 to reduce cortical brain damage induced by focal cerebral ischemia was mirrored in intraperitoneal pretreatment studies. The noncompetitive NMDA receptor antagonist MK801, administered by intravenous and intraperitoneal routes as a positive control, also reduced ischemic brain damage in cortex. Neither FK506 nor MK801 prevented striatal damage after endothelin-induced MCAO in Sprague Dawley rats, presumably because of its vascular supply from the lenticulostriate artery; the lateral striatum represents end vessel territory that cannot be rescued by drug therapy (Park et al., 1989). In contrast to MK801, which reduced the volume of excitotoxic brain damage induced by NMDA receptor agonists, FK506 did not attenuate excitotoxic damage at doses that decreased ischemic brain damage. Histopathological data relevant to the striatal quinolinate lesion were replicated in separate neurochemical studies that quantified GAD and ChAT activity, markers for the viability of GABAergic and cholinergic neurons, respectively. MK801, but not FK506, attenuated the reduction in enzyme activity associated with intrastriatal injection of quinolinate. The lack of FK506 efficacy is unlikely to be attributable to regional selectivity, because negative histopathological results also were obtained using excitotoxic lesions in three structures: the hippocampus, cortex, and striatum. These data suggest that an antiexcitotoxic mechanism is unlikely to underlie the neuroprotective action of FK506 in experimental stroke.
Pharmacokinetic findings suggest that differences in the time course of excitotoxic and ischemic damage cannot explain the contrasting efficacy of FK506. The brain content of FK506 rose rapidly after intravenous dosing and was maintained at a constant level from 15 min after injection until the experimental endpoint 72 hr later. The similar degree of neuroprotection afforded by FK506 pretreatment either 24 or 72 hr before MCAO and by drug treatment immediately after vessel occlusion confirmed the bioavailability of FK506 detected in pharmacokinetic studies. Although these findings rule out the possibility that excitotoxins exert an action in the brain that outlives the half-life of the drug, an effective concentration of drug might be required immediately postinsult to observe an antiexcitotoxic effect. This is unlikely because intraperitoneal administration of FK506, at a dose that attenuated ischemic but not excitotoxic damage, resulted in a brain content of drug in excess of that required for ischemic neuroprotection using intravenous dosing, from the time of the excitotoxic challenge (30 min after intraperitoneal FK506 administration) until the experimental endpoint. The finding that FK506 content was similar in ischemic and nonischemic cortex was also of interest. These data indicate that the bioavailability of FK506 must be high because the drug penetrates readily into ischemic tissue and suggest that there is no gross perturbation of the blood–brain barrier in the endothelin model of focal cerebral ischemia.
Physiological data concerning MABP and rectal and brain temperature provided no clue to the neuroprotective mechanism of FK506 in experimental stroke. MABP was unaffected by FK506, and whereas body and brain temperature influence the severity of brain damage after focal cerebral ischemia (Morikawa et al., 1992; Xue et al., 1992), these variables were unaltered by endothelin-induced MCAO and/or FK506. These data indicate that neither a direct cardiovascular effect nor a drug-induced alteration in brain temperature mediates the neuroprotective effect of FK506. The possibility of a direct interaction between FK506 and the endothelin receptors mediating vasoconstriction in this model can be discounted because the drug, at concentrations up to 100 μm, failed to displace radiolabeled endothelin in a receptor binding assay (J. Sharkey and S. P. Butcher, unpublished data).
Further evidence to support the proposed role of calcineurin in the neuroprotective mechanism of FK506 was provided by the finding that 20 mg/kg cyclosporin A reduced ischemic brain damage. Subchronic pretreatment with equivalent doses of cyclosporin A previously had been reported to decrease brain edema after MCAO (Shiga et al., 1992). The lower potency of cyclosporin A, as compared with FK506, is presumably attributable to low blood–brain barrier permeability (Begley et al., 1990) and its lower affinity for its immunophilin binding site (Liu et al., 1992). The proposed role of calcineurin in the neuroprotective mechanism could involve a number of cellular processes. FKBP12 is associated with the ryanodine and IP3 receptor complexes (Timerman et al., 1993; Zhang et al., 1993; Brillantes et al., 1994;Chen et al., 1994; Cameron et al., 1995a) in which it may function as an anchor for calcineurin (Cameron et al., 1995b). Both FK506 and rapamycin disrupt this complex (Cameron et al., 1995b) and interfere with the associated Ca2+ channel activity (Zhang et al., 1993; Brillantes et al., 1994; Chen et al., 1994; Cameron et al., 1995a,b). In contrast, rapamycin attenuates the neuroprotective action of FK506 (Dawson et al., 1993; Sharkey and Butcher, 1994), suggesting that a drug-induced alteration in ryanodine/IP3 receptor channel activity is not involved in the neuroprotective mechanism. Alternatively, a role for NOS, an in vitro substrate for calcineurin, has been proposed on the basis of neuronal culture studies focusing on glutamate toxicity (Dawson et al., 1993). Nitric oxide-mediated toxicity is suggested to involve DNA damage with subsequent activation of poly(adenosine-5′-diphosphoribose) synthetase (PARS), ATP depletion, and cell death (Zhang et al., 1994, 1995). However, the role of nitric oxide in ischemic and excitotoxic neuronal death remains controversial, and the present findings demonstrate a clear discrepancy between in vitro and in vivodata concerning the antiexcitotoxic effect of FK506. It also should be noted that, in contrast to the situation in vitro (Dawson et al., 1991), NOS inhibitors do not block excitotoxic damage in vivo (Globus et al., 1995; Mackenzie et al., 1995).
An alternative mechanism involving peroxynitrite, a neurotoxic free radical produced from nitric oxide and superoxide (Lipton et al., 1993;Bonfoco et al., 1995), therefore is proposed. This hypothesis is based on three key experimental findings: the reduction in superoxide production by neutrophils noted in the presence of FK506 (Nishinaka et al., 1993), the reduction in ischemic brain damage in transgenic mice that overexpress superoxide dismutase (Kinouchi et al., 1991), and the inhibitory effect of superoxide dismutase and catalase on nitric oxide-mediated neurotoxicity in cortical cell cultures (Bonfoco et al., 1995). Confirmation obviously will require clarification of the effect of FK506 on superoxide and peroxynitrite production after in vivo focal cerebral ischemia and demonstration of a link between FK506-mediated inhibition of calcineurin and reduced peroxynitrite production. A final intriguing possibility is that FK506 reduces ischemic brain damage by an antiapoptotic mechanism. Activation-induced apoptosis in T and B cell lines is inhibited by FK506 (Fruman et al., 1992b; Genestier et al., 1994), and a role for calcineurin in Ca2+-triggered apoptosis in fibroblasts has been demonstrated (Shibasaki and McKeon, 1995). Peroxynitrite-induced cell death in primary neuronal cultures and a neuron-like cell line exhibits apoptotic characteristics (Bonfoco et al., 1995; Estevez et al., 1995), and evidence that apoptosis plays a key role in brain damage induced by focal cerebral ischemia has been reported (Li et al., 1995a,b; Linnik et al., 1995).
Footnotes
This work was supported by Fujisawa Pharmaceutical, Osaka, Japan, and The James A. Kennedy Bequest Fund.
Correspondence should be addressed to Dr. Steve Butcher, Fujisawa Institute of Neuroscience, Department of Pharmacology, University of Edinburgh, 1 George Square, Edinburgh EH8 9JZ, UK.
REFERENCES
- 1.Begley DJ, Squires LK, Zlokovic BV, Mitrovic DM, Hughes CCW, Revest PA, Greenwood J. Permeability of the blood–brain barrier to the immunosuppressive cyclic peptide cyclosporin A. J Neurochem. 1990;55:1222–1230. doi: 10.1111/j.1471-4159.1990.tb03128.x. [DOI] [PubMed] [Google Scholar]
- 2.Bierer BE, Mattila PS, Standaert RF, Herzenberg LA, Burakoff SJ, Crabtree G, Schreiber SL. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc Natl Acad Sci USA. 1990;87:9231–9235. doi: 10.1073/pnas.87.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonfoco E, Krainc D, Ankarcrona M, Nocitera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein using the principle of dye-protein binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 5.Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 6.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane SW, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 7.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. Calcineurin associated with the inositol 1,4,5-triphosphate receptor–FKBP12 complex modulates Ca2+ flux. Cell. 1995a;83:463–472. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 8.Cameron AM, Steiner JP, Sabatini DM, Kaplin AI, Walensky LD, Snyder SH. Immunophilin FK506 binding protein associated with inositol 1,4,5-triphosphate receptor modulates calcium flux. Proc Natl Acad Sci USA. 1995b;92:1784–1788. doi: 10.1073/pnas.92.5.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang HY, Takei K, Sydor AM, Born T, Rusnak F, Jay DG. Asymmetric retraction of growth cone filopodia following focal inactivation of calcineurin. Nature. 1995;376:686–690. doi: 10.1038/376686a0. [DOI] [PubMed] [Google Scholar]
- 10.Charters AR, Kobayashi M, Butcher SP. Developmental expression of FK506 binding protein and calcineurin in rat brain. Biochem Soc Trans. 1995;23:422S. doi: 10.1042/bst023422s. [DOI] [PubMed] [Google Scholar]
- 11.Chen SRW, Zhang L, MacLennan DH. Asymmetrical blockade of the Ca2+ release channel (ryanodine receptor) by 12 kDa FK506 binding protein. Proc Natl Acad Sci USA. 1994;91:11953–11957. doi: 10.1073/pnas.91.25.11953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast TOR, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci USA. 1994;91:12574–12578. doi: 10.1073/pnas.91.26.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 14.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signaling enzyme in T-lymphocyte activation. Nature. 1992;357:695–698. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- 15.Dawson TM, Steiner JP, Dawson VL, Dinerman JL, Uhl GR, Snyder SH. Immunosuppressant FK506 enhances phosphorylation of nitric oxide synthase and protects against glutamate neurotoxicity. Proc Natl Acad Sci USA. 1993;90:9808–9812. doi: 10.1073/pnas.90.21.9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawson TM, Steiner JP, Lyons WE, Fotuhi M, Blue M, Snyder SH. The immunophilins, FK506 binding protein and cyclophilin, are discreetly localised in the brain: relationship to calcineurin. Neuroscience. 1994;62:569–580. doi: 10.1016/0306-4522(94)90389-1. [DOI] [PubMed] [Google Scholar]
- 17.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Estevez AG, Radi R, Barbeito L, Shin JT, Thompson JA, Beckman JS. Peroxynitrite-induced cytotoxicity in PC12 cells: evidence for an apoptotic mechanism differentially modulated by neurotrophic factors. J Neurochem. 1995;65:1543–1550. doi: 10.1046/j.1471-4159.1995.65041543.x. [DOI] [PubMed] [Google Scholar]
- 19.Flanagan WM, Corthesy B, Bran RJ, Crabtree GR. Nuclear association of a T-cell transcription factor blocked by FK506 and cyclosporin A. Nature. 1991;352:803–807. doi: 10.1038/352803a0. [DOI] [PubMed] [Google Scholar]
- 20.Fonnum F. A rapid radiochemical method for the determination of choline acetyltransferase. J Neurochem. 1975;24:407–409. doi: 10.1111/j.1471-4159.1975.tb11895.x. [DOI] [PubMed] [Google Scholar]
- 21.Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK506 and cyclosporin A. Proc Natl Acad Sci USA. 1992a;89:3686–3690. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fruman DA, Mather PE, Burakoff SJ, Bierer BE. Correlation of calcineurin phosphatase activity and programmed cell death in murine T-cell hybridomas. Eur J Immunol. 1992b;22:2513–2517. doi: 10.1002/eji.1830221008. [DOI] [PubMed] [Google Scholar]
- 23.Genestier L, Dearden-Badet MT, Bonnefoy-Berard N, Lizard G, Revillard JP. Cyclosporin A and FK506 inhibit activation-induced cell death in the murine WEHI-231 B cell line. Cell Immunol. 1994;155:283–291. doi: 10.1006/cimm.1994.1122. [DOI] [PubMed] [Google Scholar]
- 24.Globus MYT, Prado R, Sanchez-Ramos J, Zhao W, Dietrich WD, Busto R, Ginsberg MD. A dual role for nitric oxide in NMDA-mediated toxicity in vivo. J Cereb Blood Flow Metab. 1995;15:904–915. doi: 10.1038/jcbfm.1995.115. [DOI] [PubMed] [Google Scholar]
- 25.Greenlund LJS, Deckwerth TL, Johnson EM. Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 26.Kanazawa I, Iverson LL, Kelly JS. Glutamate decarboxylase activity in the rat pituitary, pineal gland, dorsal root ganglion, and superior cervical ganglion. J Neurochem. 1976;27:1267–1269. doi: 10.1111/j.1471-4159.1976.tb00341.x. [DOI] [PubMed] [Google Scholar]
- 27.Kinouchi H, Epstein C, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci USA. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Chopp M, Jiang N, Zhang ZG, Zaloga C. Induction of DNA fragmentation after 10 to 120 min of focal cerebral ischemia in rats. Stroke. 1995a;26:1252–1258. doi: 10.1161/01.str.26.7.1252. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Sharov VG, Jiang N, Zaloga C, Sabbah HN, Chopp M. Ultrastructural and light microscopic evidence of apoptosis after middle cerebral artery occlusion in the rat. Am J Pathol. 1995b;146:1045–1051. [PMC free article] [PubMed] [Google Scholar]
- 30.Linnik MD, Miller JA, Sprinkle-Cavallo J, Mason PJ, Thompson FY, Montgomery LR, Schroeder KK. Apoptotic DNA fragmentation in the rat cerebral cortex induced by permanent middle cerebral artery occlusion. Mol Brain Res. 1995;32:116–124. doi: 10.1016/0169-328x(95)00069-5. [DOI] [PubMed] [Google Scholar]
- 31.Lipton SA, Choi Y, Pan Z, Lei SZ, Chen HV, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 32.Liu J. FK506 and cyclosporin: molecular probes for studying intracellular signal transduction. Trends Pharmacol Sci. 1993;14:182–188. doi: 10.1016/0165-6147(93)90206-y. [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Farmker JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin–cyclosporin A and FKBP–FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Albers MW, Wandless TJ, Luan S, Alberg DG, Belshaw PJ, Cohen P, MacKintosh C, Klee CB, Schreiber SL. Inhibition of T-cell signaling by immunophilin–ligand complexes correlates with loss of calcineurin phosphatase activity. Biochemistry. 1992;31:3896–3901. doi: 10.1021/bi00131a002. [DOI] [PubMed] [Google Scholar]
- 35.Liu J-P, Sim ATR, Robinson PJ. Calcineurin inhibition of dynamin I GTPase activity coupled to nerve terminal depolarization. Science. 1995;265:970–973. doi: 10.1126/science.8052858. [DOI] [PubMed] [Google Scholar]
- 36.Lyons WE, George EB, Dawson TM, Steiner JP, Snyder SH. Immunosuppressant FK506 promotes neurite outgrowth in cultures of PC12 cells and sensory ganglia. Proc Natl Acad Sci USA. 1994;91:3191–3195. doi: 10.1073/pnas.91.8.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mackenzie GM, Jenner P, Marsden CD. The effect of nitric oxide synthase inhibition on quinolinic acid toxicity in the rat striatum. Neuroscience. 1995;67:357–371. doi: 10.1016/0306-4522(94)00621-b. [DOI] [PubMed] [Google Scholar]
- 38.Morikawa E, Ginsberg MD, Dietrich WD, Duncan RC, Kraydieh S, Globus MYT, Busto R. The significance of brain temperature in focal cerebral ischemia—histopathological consequences of middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1992;12:380–389. doi: 10.1038/jcbfm.1992.55. [DOI] [PubMed] [Google Scholar]
- 39.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 40.Nichols RA, Suplick GR, Brown JM. Calcineurin-mediated protein dephosphorylation in brain nerve terminals regulates the release of glutamate. J Biol Chem. 1994;269:23817–23823. [PubMed] [Google Scholar]
- 41.Nishinaka Y, Sugiyama S, Yokota M, Saitoh H, Ozawa T. Protective effect of FK506 on ischemia/reperfusion-induced myocardial damage in canine heart. J Cardiovasc Pharmacol. 1993;21:448–454. doi: 10.1097/00005344-199303000-00015. [DOI] [PubMed] [Google Scholar]
- 42.Park CK, Nehl DG, Graham DI, Teasdale GI, McCulloch JM. The glutamate antagonist MK801 reduces focal ischemic brain damage in the rat. Ann Neurol. 1989;20:150–157. doi: 10.1002/ana.410240411. [DOI] [PubMed] [Google Scholar]
- 43.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT 1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 44.Schreiber SL. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- 45.Sharkey J, Butcher SP. Immunophilins mediate the neuroprotective effects of FK506 in focal cerebral ischaemia. Nature. 1994;371:336–339. doi: 10.1038/371336a0. [DOI] [PubMed] [Google Scholar]
- 46.Sharkey J, Butcher SP. Characterization of ischemic brain damage induced by stereotaxic microinjection of endothelin-1 adjacent to the rat middle cerebral artery. J Neurosci Methods. 1995;60:125–131. doi: 10.1016/0165-0270(95)00003-d. [DOI] [PubMed] [Google Scholar]
- 47.Shibasaki F, McKeon F. Calcineurin functions in Ca2+-activated cell death in mammalian cells. J Cell Biol. 1995;131:735–743. doi: 10.1083/jcb.131.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shiga Y, Onodera H, Matsuo Y, Kogure K. Cyclosporin A protects against ischemia-reperfusion injury in the brain. Brain Res. 1992;595:145–148. doi: 10.1016/0006-8993(92)91465-q. [DOI] [PubMed] [Google Scholar]
- 49.Snyder SH, Sabatini DM. Immunophilins and the nervous system. Nat Med. 1995;1:32–37. doi: 10.1038/nm0195-32. [DOI] [PubMed] [Google Scholar]
- 50.Steiner JP, Dawson TM, Fotuhi M, Glatt CE, Snowman AM, Cohen N, Snyder SH. High brain densities of the immunophilin FKBP colocalised with calcineurin. Nature. 1992;358:584–587. doi: 10.1038/358584a0. [DOI] [PubMed] [Google Scholar]
- 51.Timerman AP, Ogunbumni E, Freund E, Wiederrecht G, Marks AR, Fleischer S. The calcium release channel of sarcoplasmic reticulum is modulated by FK506 binding protein. J Biol Chem. 1993;268:22992–22999. [PubMed] [Google Scholar]
- 52.Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1511. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- 53.Xue D, Huang Z, Smith KE, Buchan AM. Immediate or delayed mild hypothermia prevents focal cerebral infarction. Brain Res. 1992;587:66–72. doi: 10.1016/0006-8993(92)91428-h. [DOI] [PubMed] [Google Scholar]
- 54.Zhang B, Zhao H, Muallem S. Ca2+-dependent kinase and phosphatase control inositol 1,4,5-triphosphate-mediated Ca2+ release. J Biol Chem. 1993;268:10997–11001. [PubMed] [Google Scholar]
- 55.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J, Pieper A, Snyder SH. Poly(ADP-ribose) synthetase activation: an early indicator of neurotoxic DNA damage. J Neurochem. 1995;65:1411–1414. doi: 10.1046/j.1471-4159.1995.65031411.x. [DOI] [PubMed] [Google Scholar]