

Abstract
Optic nerve section in the newborn rat results in a rapid apoptotic degeneration of most axotomized retinal ganglion cells (RGCs). This massive process of neuronal death has been ascribed mainly to the interruption of a trophic factor supply from target structures rather than to the axonal damage per se. To distinguish between these two possibilities, we induced a reversible axonal transport blockade in the developing optic nerve by topical application of a local anesthetic (lidocaine). Light and electron microscopy showed no alterations in the fine structure of treated optic nerves. Retinae of treated and control rats were stained with cresyl violet and examined at different times after surgery. We found that axonal transport blockade induced only a limited number of pyknotic RGCs. Degeneration of these cells was completely prevented by inhibiting protein synthesis during lidocaine application. We conclude that the rapid degeneration of RGCs after axotomy can be ascribed only partly to the loss of retrogradely transported trophic factors.
Keywords: neuronal death, axonal transport, retinal ganglion cells, apoptosis, rat optic nerve, retrograde degeneration
Retinal ganglion cells (RGCs), as well as other mammalian CNS neurons, eventually die as a result of axonal injury (Grafstein and Ingoglia, 1982; Bregman and Reier, 1986;Hefti, 1986; Villegas-Perez et al., 1993). Transection of the optic nerve is known to induce massive RGC death by an apoptotic process in both developing (Rabacchi et al., 1994a) and adult rats (Berkelaar et al., 1994; Garcia-Valenzuela et al., 1994). The molecular nature of the signal triggering the loss of axotomized neurons is largely unknown. According to the trophic theory, the effects of axotomy can be ascribed to the disconnection of neurons from their targets, resulting in the sudden loss of retrogradely transported trophic molecules (Oppenheim, 1991; Snider et al., 1992; Johnson and Deckwerth, 1993). An alternative hypothesis is that axonal injury per se can induce cell death. In particular, acute alterations in cellular homeostasis and ionic imbalances subsequent to plasmalemmal leakage, such as Ca2+ deregulation, have been implicated (Choi, 1992;Snider et al., 1992).
Especially in the neonate, a lack of target-derived trophic signals is proposed as the main cause of death for axotomized RGCs. It is known that trophic factors, such as NGF and BDNF, are synthesized in the major retinal projection fields, the optic tectum (Hofer et al., 1990;Maisonpierre et al., 1990; Friedman et al., 1991) and the dorsal lateral geniculate nucleus (dLGN) (Schoups et al., 1995). Exogenous administration of NGF and BDNF, as well as other growth factors, can partly prevent RGC death after optic nerve lesion (Carmignoto et al., 1989; Mey and Thanos, 1993; Cohen et al., 1994; Mansour-Robaey et al., 1994; Rabacchi et al., 1994b). Moreover, the survival of highly purified RGCs in culture can be enhanced by both tectal cells (McCaffery et al., 1982; Armson and Bennett, 1983) and soluble tectal factors (Meyer-Franke et al., 1995).
This evidence, however, is indirect and does not exclude the possibility that axonal injury per se contributes to the cell death process. To determine whether cell death after axotomy is attributable to trophic factor deprivation or to axonal damage, we produced a reversible functional disconnection between RGCs and central targets in neonatal animals. Accordingly, we blocked axonal transport in the optic nerve by topical application of a local anesthetic (lidocaine) (Byers et al., 1973; Bisby, 1975; Fink and Kish, 1976). This drug was chosen because of its relatively low neurotoxicity, as compared with other more conventional transport blockers (Byers et al., 1973; Kalichman, 1993; Selander, 1993). Application of lidocaine at suitable doses indeed blocked axonal transport without damaging optic nerve structures. This blockade induced a significant increase in dying RGCs, consistent with a loss of trophic support. Cell death was dramatically higher, however, after direct nerve injury. We therefore conclude that the rapid and massive death of RGCs after axotomy can be ascribed only partly to the loss of retrogradely transported trophic factors.
MATERIALS AND METHODS
Single lidocaine application. A total of 96 Long–Evans rats at P0 (day of birth) were used in this study. Rat pups were anesthetized by hypothermia, and a small pellet of gelfoam soaked in lidocaine (Bayer, 0.2–0.3% solution), or in saline as control, was applied intracranially onto the exposed surface of the optic nerve on one side. After 1 or 3 hr the pellet was removed and the exposed nerve was rinsed with saline. Rats were then returned to their mother until they were killed (24–36 hr after treatment). In 16 animals, 250 nl of 1% cycloheximide (Sigma, St. Louis, MO) or saline was injected intraocularly 18 hr after surgery. The injections were performed under hypothermia anesthesia, with a pulled glass micropipette inserted at the ora serrata.
Double lidocaine application. In 13 rats, a second 3 hr lidocaine application was administered 10 hr after the first. The same animals received two intraocular injections of 1% cycloheximide or saline (250 nl) at 17 and 20 hr after the first application of the anesthetic. Rats were killed 36–72 hr after the first lidocaine administration.
Optic nerve section. Rat pups were anesthetized by hypothermia, and suction was applied to remove the medial portion of the cerebral hemisphere overlying the optic nerve. The transection was performed at ∼2 mm from the posterior pole of the eye. In four rats, a pellet of gelfoam soaked in 0.2% lidocaine was applied onto the transected stump of the nerve and left in place for 3 hr. All animals were killed 24 hr after lesion.
Analysis of axonal transport. The effects of lidocaine on axonal flow in the optic nerve were monitored by following the retrograde transport of horseradish peroxidase (HRP type VI, Sigma), injected bilaterally into the superior colliculi at different time intervals. Rat pups were anesthetized with ether, and two injections (1 μl each) of 30% HRP (in saline containing 2% DMSO) were made slowly on both sides. Two hours later, rats were perfused with a solution of 1.25% glutaraldehyde and 1% paraformaldehyde in 0.1 mphosphate buffer, pH 7.4. Optic nerves were dissected and cryoprotected in 30% sucrose. Serial sections were cut with a cryostat at 20–35 μm in the longitudinal plane and mounted onto gelatinized slides. Sections were then reacted for HRP according to the tetramethylbenzidine procedure (Mesulam, 1982) and examined with dark-field microscopy.
Histology and counting procedures. At the indicated times, rats were perfused through the heart with fixative containing 2.5% glutaraldehyde, 1% paraformaldehyde, and 0.5 mmCaCl2 in 0.1 m cacodylate buffer, pH 7.2. Retinal whole mounts were prepared and stained with cresyl violet (0.1%). The number of pyknotic as well as surviving cells was counted in 20–50 fields (100 × 100 μm) in the ganglion cell layer of each whole-mounted retina. In the newborn rat, virtually all neurons found in this layer are indeed ganglion cells (Perry et al., 1983;Rabacchi et al., 1994a,b). Retinal areas were determined with a computer-aided image analyzer (MCID 4; Imaging Research, St. Catharines, Ontario, Canada). The total number of cells per retina was determined by multiplying the average number of cells per unit area times the total area of each retina. All counts were performed by following a blind procedure.
Morphology of the optic nerves. The integrity of the optic nerve was assessed by light and electron microscopy in both lidocaine-treated and control animals. Optic nerves were dissected from perfused animals, post-fixed in osmium tetroxide, stained en bloc with uranyl acetate, dehydrated in increasing ethanol concentrations, and then embedded in Epon-Araldite resin. Sections were obtained by ultramicrotomy at 100 μm intervals along the entire length of each nerve. Semithin, 1-μm-thick sections were stained with toluidine blue and examined under a light microscope. Adjacent ultrathin sections were collected on grids, stained with uranyl acetate and lead citrate, and photographed at the electron microscope. We evaluated RGC death only in the animals in which optic nerves were morphologically analyzed and were found to be unaltered by the lidocaine treatment. Nerves and corresponding retinae were otherwise discarded.
RESULTS
Reversible inhibition of axonal transport
Previous work in vitro (Fink et al., 1972; Anderson and Edstrom, 1973; Byers et al., 1973) and in vivo (Bisby, 1975;Fink and Kish, 1976) has shown that lidocaine application produces a reversible blockade of rapid axonal transport. The delay in recovery is dependent on both the exposure time and the concentration of lidocaine used. To test the blockade efficacy in our system, lidocaine was applied onto the optic nerve, and concurrently HRP was injected into both superior colliculi. Two hours later, HRP distribution was evaluated in longitudinal sections of the optic nerve (Caleo, 1996). In lidocaine-treated nerves, the progression of HRP was clearly arrested, starting at the point of anesthetic application (Fig.1A). We made sure that blockade occurred shortly after treatment onset. Indeed, retrograde transport was completely impaired within 1 hr after application of lidocaine. Control nerves treated with physiological solution showed no such effect, and the tracer covered the entire length of the nerve (Fig. 1B).
Fig. 1.

Blockade of HRP axonal transport by lidocaine. Dark-field photomicrographs of longitudinal sections from two optic nerves treated with lidocaine (A) or saline as control (B). Migration of label is clearly inhibited in lidocaine-treated nerve; administration of vehicle alone gives no effect. Optic chiasm is visible on the left part of each section. Scale bar, 300 μm.
The persistence of fast transport blockade was then analyzed. The functionality of fast transport was checked at 3, 8, and 13 hr after 1 hr lidocaine application. We found that axonal transport was still blocked 3 hr (Fig. 2A) and 8 hr (Fig. 2B) after application but had completely recovered after 13 hr (Fig. 2C). Three hour anesthetic applications arrested axonal transport for <15 hr, when complete recovery was observed (data not shown).
Fig. 2.

Dark-field photomicrographs showing time course of fast transport inhibition in the optic nerve. Retrograde progression of HRP is still impaired at 3 hr (A) and 8 hr (B), but not at 13 hr (C) after 1 hr lidocaine application. Optic chiasm is on theleft. Scale bar, 350 μm.
Morphological analysis of optic nerves
It is reported in the literature that lidocaine, as well as other transport blockers, can induce nonspecific neurotoxic effects, especially at high doses and long periods of exposure (Byers et al., 1973; Kalichman, 1993; Selander, 1993). To exclude the presence of such effects in our preparation, and therefore rule out the possibility of injury-induced cell death, we analyzed the fine morphology of treated optic nerves at the light and electron microscopic level. Nerves were prepared for analysis both acutely and 24 hr after the lidocaine treatment. Here we report only the results of preparations in which the morphological analysis showed no structural alterations in any portion of the lidocaine-treated nerves, which were therefore indistinguishable from control and normal nerves (Fig.3).
Fig. 3.
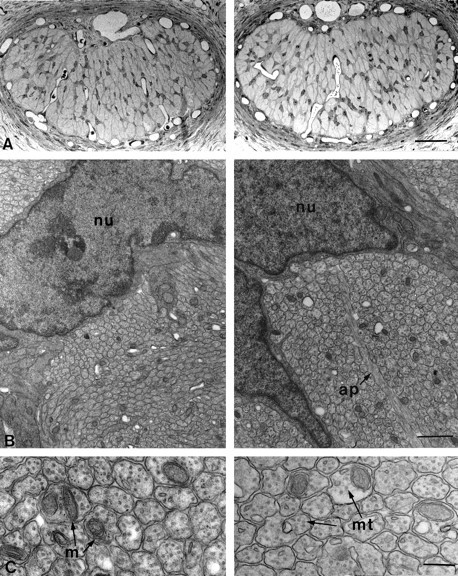
Morphology of lidocaine-treated (left) and vehicle-treated (right) optic nerves 24 hr after surgery. A, Light micrographs of semithin transverse sections of treated and control nerves in the proximity of the site of gelfoam application. There are no obvious differences between the two sections. Scale bar, 50 μm.B, Low-power electron micrographs of the specimens shown in A. Once again no qualitative differences can be seen between treated and control animals. Axons are clearly grouped in bundles separated by astrocyte processes (ap).nu, Astrocytic nuclei. Scale bar, 1 μm.C, High magnification electron micrographs showing intact, unmyelinated axons with their complement of mitochondria (m) and microtubules (mt). Scale bar, 200 nm.
At the light microscopic level, the general morphology appeared well preserved, with numerous astrocytic processes forming a regular plexus across the nerve surface (Fig. 3A). Electron microscopy showed normal axon profiles, with no signs of pathological alterations and organization into regular bundles delimited by astrocytes (Fig.3B). The intra-axonal compartment again exhibited a normal appearance and the characteristic presence of vesicular profiles, mitochondria, microtubules, and neurofilaments (Fig. 3C). Thus, our treatment was able to induce a transient block of axonal transport without interfering with the ultrastructure of the axons.
Induction of RGC death
Previous studies have shown that optic nerve section in newborn rats leads to virtually complete death of RGCs within 2–3 d, with a peak in pyknosis between 20 and 36 hr after lesion (Perry et al., 1983;Beazley et al., 1987; Rabacchi et al., 1994a). We therefore compared the effects of injury and axonal transport blockade in retinal whole mounts prepared 24 hr after surgery. After lidocaine application, pyknotic profiles appeared in the ganglion cell layer (Fig.4A). The number of ganglion cells displaying apoptotic morphology was enhanced approximately fourfold (Fig. 5) in treated versus control animals [3530 ± 290 (SE) vs 910 ± 280 (SE); p < 0.001], but was still 10–15 times lower than that obtained in retinae that underwent axotomy (44,120 ± 4340). Saline application had no effect, and only rare dying cells could be observed (Fig. 4B). Pyknosis in vehicle-treated animals was perfectly comparable to the level of natural cell death present at this age (Perry et al., 1983; Rabacchi et al., 1994a). Effects produced by 1 or 3 hr lidocaine application were not significantly different, and therefore the data from the two experimental groups were pooled (Fig. 5).
Fig. 4.
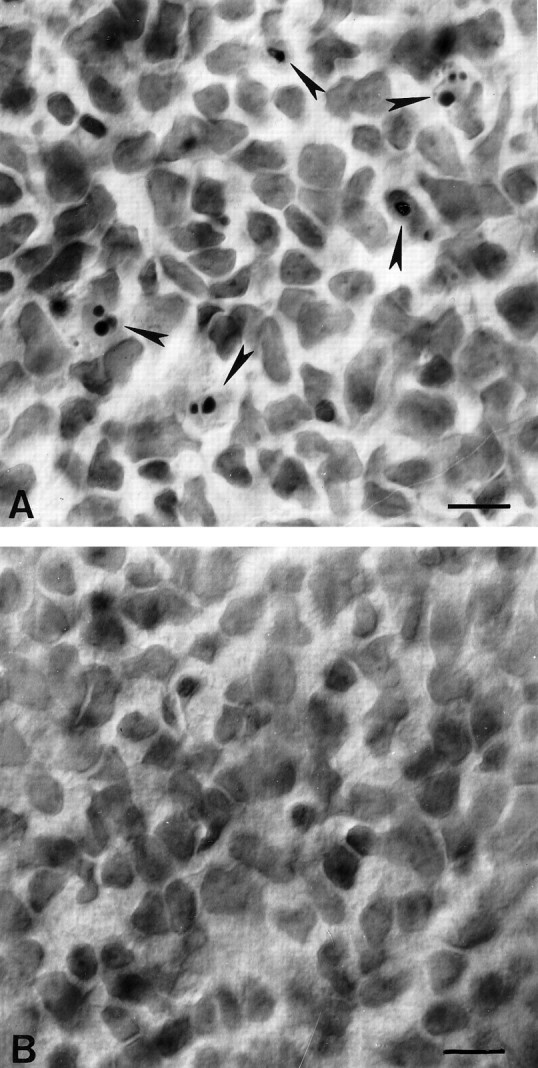
Photomicrographs of representative regions of the ganglion cell layer in whole-mounted retinae stained with cresyl violet. A, Twenty-four hours after lidocaine application onto the optic nerve, pyknotic nuclei (arrows) appear in the ganglion cell layer. B, Control retina, 24 hr after saline application. No pyknotic profiles are visible in this field. Scale bar, 10 μm.
Fig. 5.

Effects of 1 or 3 hr lidocaine application on pyknosis in the RGC layer. The number of pyknotic ganglion cells is significantly increased in treated animals 24 hr after surgery. Error bars indicate SE; n = 16 for lidocaine;n = 11 for saline treatment.
To check for any effect of the anesthetic on the apoptotic process, lidocaine was applied onto the transected stump of the optic nerve immediately after the lesion. No significant difference in the number of dying RGCs could be found at 24 hr in the lidocaine-treated group (39,360 ± 5130) with respect to lesion alone (44,120 ± 4340) (Fig. 6). This rules out any substantial anti-apoptotic activity of the stump-applied lidocaine that might counteract the effects of trophic deprivation.
Fig. 6.

Mean number of pyknotic RGCs 24 hr after optic nerve section. No significant difference can be detected between axotomy (n = 4) and axotomy + lidocaine (n = 4). Error bars indicate SE.
Effects of cycloheximide on RGC survival
The apoptotic process after optic nerve section can be delayed by protein synthesis inhibitors (Garcia-Valenzuela et al., 1994; Rabacchi et al., 1994a). Administration of cycloheximide 18 hr after lesion prevents the appearance of pyknotic cells at 24 hr. At 36 hr after lesion, however, protein synthesis resumes, and the number of pyknotic profiles increases dramatically (Rabacchi et al., 1994a). The interpretation of these data is that RGCs saved by cycloheximide 24 hr after lesion are eventually destined to die, because communication between RGCs and central targets is irreversibly lost.
Our data show that a transient axonal transport blockade is sufficient to induce a low level of pyknotic RGCs in neonatal animals, probably by depleting ganglion cells of target-derived trophic molecules. Because of the reversibility of the treatment, death of these cells should be completely avoided if protein synthesis blockers are administered in an appropriate time interval, until axonal transport is reestablished. Accordingly, we injected cycloheximide intraocularly 18 hr after lidocaine treatment. We found that cycloheximide maintained pyknotic cells at control levels not only at 24 hr but also 36 hr after lidocaine application (Fig. 7). This result suggests that our treatment produced a reversible separation between RGCs and central targets and that cycloheximide inhibited the death program until proper survival conditions were restored.
Fig. 7.

Effects of intraocular cycloheximide on RGC death after lidocaine application onto the optic nerve. Pyknosis is low at both 24 and 36 hr after surgery. At 24 hr, n = 3 for saline, n = 4 for cycloheximide; at 36 hr,n = 3 for saline, n = 6 for cycloheximide. Error bars indicate SE.
Prolonged blockade of axonal transport
It could be argued that the duration of axonal transport blockade produced by 1 or 3 hr lidocaine was not long enough to induce a level of pyknosis as high as that after optic nerve section. Therefore, we prolonged the blockade by giving a second 3 hr lidocaine application 10 hr after the first. We indeed found that the number of pyknotic cells significantly increased 36–42 hr after surgery with respect to the single application protocol (8190 ± 3000 vs 1190 ± 200;p < 0.05).
Pyknotic cells are estimated to last only 1–4 hr in the tissue (Horsburgh and Sefton, 1987; Harvey and Robertson, 1992; Galli-Resta and Ensini, 1996), because they are rapidly cleared by microglial cells. To determine the total number of ganglion cells affected by the blockade, counts of surviving cells have to be performed. Our quantitative analysis showed that the vast majority of RGCs survived the lidocaine treatment. Approximately 147,000 (146,980 ± 16,680) living cells were counted at 36–42 hr (Fig.8). In three additional animals examined 72 hr after surgery, a total of 135,900 ± 13,400 (SE) ganglion cells were found. Because an average of approximately 160,000 RGCs is reported in the literature for normal, age-matched rats (Perry et al., 1983), these results show that only a minority of RGCs are sensitive to axonal transport blockade in our experimental conditions.
Fig. 8.

Effects of double lidocaine application and cycloheximide administration 36–42 hr after treatment. Open bars represent the number of surviving cells, whereassolid bars represent the number of pyknotic cells. Error bars indicate SE.
This small proportion of transport-sensitive RGCs could not be detected by counting living cells after cycloheximide treatment. The number of living RGCs at 36–42 hr was greater, but not significantly different, between cycloheximide- and saline-injected rats (Fig. 8). The low level of induced death and the high inter-individual variability probably accounted for this result.
DISCUSSION
Transection of the optic nerve in newborn rats leads to the rapid apoptotic degeneration of the vast majority of RGCs. Within 24 hr, a dramatic increase in pyknosis and a corresponding reduction in surviving cells occurs (Beazley et al., 1987; Rabacchi et al., 1994a,b). Here we asked whether cell death after axotomy can be attributed entirely to the interruption of trophic factor supply from target structures. A transient blockade of axonal transport was produced in the neonatal rat optic nerve by topical application of lidocaine. Our results show that a significant increase in pyknotic cells occurs after blockade of axonal transport, consistent with a loss of trophic supply; however, cell death reaches dramatically high levels only after injury, suggesting that other factors may be involved in lesion-induced apoptosis.
Effects of injury and trophic deprivation on RGC survival
Application of lidocaine rapidly induced a blockade of axonal transport, as monitored by HRP transfer from the optic tectum to the eye. The effects of lidocaine and related molecules on axonal transport are well documented in the literature (Fink et al., 1972; Byers et al., 1973; Bisby, 1975; Fink and Kish, 1976; Lavoie et al., 1989). Lidocaine is believed to produce its inhibitory effect by an uncoupling of oxidative phosphorylation (Haschke and Fink, 1975), with a consequent interruption of ATP supply to the transport mechanism. This mechanism of block suggests that along with retrograde transport of HRP, retrograde transport of endogenous target-derived trophic factors would be affected as well.
At concentrations lower than required for interference with transport, lidocaine prevents the propagation of action potentials by blocking Na+ channels. Blockade of membrane excitability, however, cannot be responsible for the observed increase in the level of RGC death, because it has been shown that silencing electrical activity produces no change in the number of dying ganglion cells (Fawcett et al., 1984; O’Leary et al., 1986a,b; Friedman and Shatz, 1990; Galli-Resta et al., 1993).
In the first 10 postnatal days, more than half of the entire rat RGC population disappears because of natural cell death, which is currently thought to reflect a competition for limiting amounts of survival signals secreted by the targets (Oppenheim, 1991; Johnson and Deckwerth, 1993; Korsching, 1993; Silos-Santiago et al., 1995). Our results are consistent with a role for axonal transport in RGC survival. Indeed, a transient disconnection of RGCs from their central targets significantly increases the amount of cell death. The duration of blockade consistently correlates with the extent of cell loss under experimental conditions in which morphological analysis rules out any direct injury or pathological alteration of the nerve.
Our results are in agreement with previous studies (Hughes and McLoon, 1979; Pearson et al., 1981; Tong et al., 1982; Carpenter et al., 1986;Vanselow et al., 1990) showing that removal of target support in the absence of axonal injury is sufficient to increase the level of RGC death. Early ablation of the tectum (before RGC innervation) leads to a dramatic increase in the number of degenerating ganglion cells in the chick (Hughes and McLoon, 1979; Vanselow et al., 1990). Excitotoxic lesion of the neonatal rat superior colliculus (sparing retinocollicular axons) results in a substantial reduction of the number of RGCs that survive the period of natural cell death (Carpenter et al., 1986). Finally, surgical ablation of the occipital cortex in the neonatal cat leads to retrograde degeneration of the dLGN and consequent transneuronal loss of RGCs that project to the dLGN (Pearson et al., 1981; Tong et al., 1982). In all of these paradigms, cell loss is simply attributable to deprivation of contact with the target population.
Transection of the optic nerve also dramatically increases the rate of natural RGC death. Within 24 hr, the number of RGCs is substantially reduced (Rabacchi et al., 1994a,b). We have now shown, however, that this rapid and massive death of RGCs cannot be accounted for by trophic deprivation alone. Double lidocaine applications, which extended the blockade period up to 20–24 hr, produced only a limited amount of cell death. The high number of surviving ganglion cells at 36–42 hr after treatment confirmed their resistance to disconnection from target support. Thus, injury and trophic deprivation differentially affect the process of RGC death.
A similar conclusion can be drawn by a detailed comparison of the kinetics of RGC death after surgical ablation or excitotoxic lesion of the neonatal rat superior colliculus. At 24 hr after lesion, a 10-fold increase in pyknotic cells is observed with surgical removal of the tectum and its afferent fibers (Harvey and Robertson, 1992; Cui and Harvey, 1995). Merely a twofold increase in cell death occurs after selective postsynaptic ablation by kainic acid injection (Horsburgh and Sefton, 1987). It seems likely, therefore, that after axotomy some “injury-related” factors may come into play and promote neuronal degeneration.
In particular, many authors point to the perturbation of Ca2+ homeostasis as playing a critical role in triggering the process of lesion-induced apoptosis (Nicotera et al., 1992; Trump and Berezesky, 1992; Choi, 1996). Massive Ca2+ influx, for example, is known to mediate excitotoxic effects, which probably actively take part in neuronal loss induced by acute insults (Choi, 1992; Schreiber and Baudry, 1995). Altered Ca2+ levels interfere with mitochondrial function, promote the formation of damaging free radicals, and enhance protease activity, thus leading to derepression of the central components of the cell death machinery (Nicotera et al., 1992; Trump and Berezesky, 1992; Martin and Green, 1995; Yuan, 1995).
An unresolved issue is whether a more prolonged blockade of axonal transport can result in a substantial decrease of surviving RGCs. It seems likely that a longer perturbation would affect a larger proportion of RGCs, consistent with previous experiments (Hughes and McLoon, 1979; Carpenter et al., 1986; Vanselow et al., 1990) designed to induce a protracted loss of target support. The kinetics of this process have never been analyzed in detail; however, the present results together with the available literature (Carpenter et al., 1986;Cui and Harvey, 1995) suggest that the massive and almost synchronous loss of RGCs after a lesion is a rapid process quite unlike deprivation-induced death.
Cycloheximide-mediated rescue of RGCs
We have revealed a subpopulation of RGCs that is sensitive to axonal transport blockade. These cells undergo an apoptotic type of cell death, as defined by the combination of morphological (pyknosis) and interventional (inhibition by cycloheximide) criteria. A requirement for protein synthesis likely reflects the need to produce a set of molecules that activate a constitutively existing cell death machinery (Raff et al., 1993; Jacobson et al., 1994; Steller, 1995;Weil et al., 1996). Proteases of the interleukin-1β converting enzyme family constitute the main components of this machinery (Martin and Green, 1995; Martinou and Sadoul, 1996). If the apoptotic degeneration of RGCs in our system is actually attributable to the temporary lack of trophic factors, these cells should be rescued by blocking protein synthesis until axonal transport is reestablished.
Indeed, cycloheximide experiments showed that RGC degeneration can be completely avoided by blocking the death program during the period of trophic deprivation. A plausible explanation for this result is that cycloheximide administration rescues treated cells during the period of susceptibility to apoptosis. When protein synthesis resumes, execution of the death program is no longer triggered, because the appropriate influx of trophic signals is restored. Commitment to death is therefore tightly controlled by a set of antagonizing proteins that can reaffirm or forestall the death sentence.
In conclusion, our results show that the rapid degeneration of RGCs after axotomy can be ascribed only partly to the loss of retrograde transport of target-derived trophic factors. The role of trophic deprivation after brain insults should therefore be reconsidered in the literature. Our findings also underscore the complex and timed balance between survival and death signals in determining cellular fate. We have directly demonstrated that neurons can be protected from transient death stimuli by means of a suitable exogenous intervention. This result has obvious implications for the clinical treatment of specific kinds of pathologies.
Footnotes
This work was partially supported by European Economic Community through Biotech contract BIO4-CT96 0774 and by Telethon Project 934. We are grateful to Professor V. H. Perry and Dr. T. Hensch for critical reading of this manuscript. We also thank Mr. A. Tacchi for histological help and Mrs. B. Margheritti and Mr. A. Bertini for photographic work.
M.F. and M.C. contributed equally to this study.
Correspondence should be addressed to Enrica Strettoi, Istituto di Neurofisiologia del Consiglio Nazionale delle Ricerche, Via S. Zeno 51, 56127 Pisa, Italy.
Dr. Fagiolini’s present address: Department of Physiology, University of California at San Francisco, San Francisco, CA 94143-0444.
REFERENCES
- 1.Anderson K-E, Edstrom A. Effects of nerve blocking agents on fast axonal transport of proteins in frog sciatic nerves in vitro. Brain Res. 1973;50:125–134. doi: 10.1016/0006-8993(73)90599-4. [DOI] [PubMed] [Google Scholar]
- 2.Armson PF, Bennett MR. Neonatal retinal ganglion cell cultures of high purity: effect of superior colliculus on their survival. Neurosci Lett. 1983;38:181–186. doi: 10.1016/0304-3940(83)90037-x. [DOI] [PubMed] [Google Scholar]
- 3.Beazley LD, Perry VH, Baker B, Darby EJ. An investigation into the role of ganglion cells in the regulation of division and death of other retinal cells. Dev Brain Res. 1987;33:169–184. doi: 10.1016/0165-3806(87)90151-9. [DOI] [PubMed] [Google Scholar]
- 4.Berkelaar M, Clarke DB, Wang Y-C, Bray GM, Aguayo AJ. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J Neurosci. 1994;14:4368–4374. doi: 10.1523/JNEUROSCI.14-07-04368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bisby MA. Inhibition of axonal transport in nerves chronically treated with local anesthetics. Exp Neurol. 1975;47:481–489. doi: 10.1016/0014-4886(75)90080-1. [DOI] [PubMed] [Google Scholar]
- 6.Bregman BS, Reier PJ. Neural tissue transplants rescue axotomized rubrospinal cells from retrograde death. J Comp Neurol. 1986;244:86–95. doi: 10.1002/cne.902440107. [DOI] [PubMed] [Google Scholar]
- 7.Byers MR, Fink BR, Kennedy RD, Middaugh ME, Hendrickson AE. Effects of lidocaine on axonal morphology, microtubules, and rapid transport in rabbit vagus nerve in vitro. J Neurobiol. 1973;4:125–143. doi: 10.1002/neu.480040205. [DOI] [PubMed] [Google Scholar]
- 8.Caleo M. Different rates of horseradish peroxidase transport in the optic nerve of neonatal and adult rats. Neuroscience. 1996;72:725–730. doi: 10.1016/0306-4522(95)00574-9. [DOI] [PubMed] [Google Scholar]
- 9.Carmignoto G, Maffei L, Candeo P, Canella R, Comelli C. Effect of NGF on the survival of retinal ganglion cells following optic nerve section. J Neurosci. 1989;9:1263–1272. doi: 10.1523/JNEUROSCI.09-04-01263.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carpenter P, Sefton AJ, Dreher B, Lim W-L. Role of target tissue in regulating the development of retinal ganglion cells in the albino rat: effects of kainate lesions in the superior colliculus. J Comp Neurol. 1986;251:240–259. doi: 10.1002/cne.902510208. [DOI] [PubMed] [Google Scholar]
- 11.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 12.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 13.Cohen A, Bray GM, Aguayo AJ. Neurotrophin-4/5 (NT-4/5) increases adult rat retinal ganglion cells survival and neurite outgrowth in vitro. J Neurobiol. 1994;25:953–959. doi: 10.1002/neu.480250805. [DOI] [PubMed] [Google Scholar]
- 14.Cui Q, Harvey AR. At least two mechanisms are involved in the death of retinal ganglion cells following target ablation in neonatal rats. J Neurosci. 1995;15:8143–8155. doi: 10.1523/JNEUROSCI.15-12-08143.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fawcett JW, O’Leary DDM, Cowan WM. Activity and the control of ganglion cell death in the rat retina. Proc Natl Acad Sci USA. 1984;81:5589–5593. doi: 10.1073/pnas.81.17.5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fink BR, Kish SJ. Reversible inhibition of rapid axonal transport in vivo by lidocaine hydrochloride. Anesthesiology. 1976;44:139–146. doi: 10.1097/00000542-197602000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Fink BR, Kennedy RD, Hendrickson AE, Middaugh ME. Lidocaine inhibition of rapid axonal transport. Anesthesiology. 1972;36:422–432. doi: 10.1097/00000542-197205000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Friedman S, Shatz CJ. The effects of prenatal intracranial infusion of tetrodotoxin on naturally occurring retinal ganglion cell death and optic nerve ultrastructure. Eur J Neurosci. 1990;2:243–253. doi: 10.1111/j.1460-9568.1990.tb00416.x. [DOI] [PubMed] [Google Scholar]
- 19.Friedman WJ, Olson L, Persson H. Cells that express brain-derived neurotrophic factor mRNA in the developing postnatal rat brain. Eur J Neurosci. 1991;3:688–697. doi: 10.1111/j.1460-9568.1991.tb00854.x. [DOI] [PubMed] [Google Scholar]
- 20.Galli-Resta L, Ensini M. An intrinsic time limit between genesis and death of individual neurons in the developing retinal ganglion cell layer. J Neurosci. 1996;16:2318–2324. doi: 10.1523/JNEUROSCI.16-07-02318.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galli-Resta L, Ensini M, Fusco E, Gravina A, Margheritti B. Afferent spontaneous electrical activity promotes the survival of target cells in the developing retinotectal system of the rat. J Neurosci. 1993;13:243–250. doi: 10.1523/JNEUROSCI.13-01-00243.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Valenzuela E, Gorczyca W, Darzynkiewicz Z, Sharma SC. Apoptosis in adult retinal ganglion cells after axotomy. J Neurobiol. 1994;25:431–438. doi: 10.1002/neu.480250408. [DOI] [PubMed] [Google Scholar]
- 23.Grafstein B, Ingoglia NA. Intracranial transection of the optic nerve in adult mice: preliminary observations. Exp Neurol. 1982;76:318–330. doi: 10.1016/0014-4886(82)90212-6. [DOI] [PubMed] [Google Scholar]
- 24.Harvey AR, Robertson D. Time-course and extent of retinal ganglion cell death following ablation of the superior colliculus in neonatal rats. J Comp Neurol. 1992;325:83–94. doi: 10.1002/cne.903250108. [DOI] [PubMed] [Google Scholar]
- 25.Haschke RH, Fink BR. Lidocaine effects on brain mitochondrial metabolism in vitro. Anesthesiology. 1975;42:737–740. doi: 10.1097/00000542-197506000-00018. [DOI] [PubMed] [Google Scholar]
- 26.Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J Neurosci. 1986;6:2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde Y-A. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 1990;9:2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horsburgh GM, Sefton AJ. Cellular degeneration and synaptogenesis in the developing retina of the rat. J Comp Neurol. 1987;263:553–566. doi: 10.1002/cne.902630407. [DOI] [PubMed] [Google Scholar]
- 29.Hughes WF, McLoon SC. Ganglion cell death during normal retinal development in the chick: comparison with cell death induced by early target field destruction. Exp Neurol. 1979;66:587–601. doi: 10.1016/0014-4886(79)90204-8. [DOI] [PubMed] [Google Scholar]
- 30.Jacobson MD, Burne JF, Raff MC. Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO J. 1994;13:1899–1910. doi: 10.1002/j.1460-2075.1994.tb06459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson EM, Deckwerth TL. Molecular mechanisms of developmental neuronal death. Annu Rev Neurosci. 1993;16:31–46. doi: 10.1146/annurev.ne.16.030193.000335. [DOI] [PubMed] [Google Scholar]
- 32.Kalichman MW. Physiologic mechanisms by which local anesthetics may cause injury to nerve and spinal cord. Reg Anaesth. 1993;18:448–452. [PubMed] [Google Scholar]
- 33.Korsching S. The neurotrophic factor concept: a re-examination. J Neurosci. 1993;13:2739–2748. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lavoie P-A, Khazen T, Fition PR. Mechanisms of the inhibition of fast axonal transport by local anesthetics. Neuropharmacology. 1989;2:175–181. doi: 10.1016/0028-3908(89)90054-3. [DOI] [PubMed] [Google Scholar]
- 35.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 36.Mansour-Robaey S, Clarke DB, Wang Y-C, Bray GM, Aguayo AJ. Effects of ocular injury and the administration of brain-derived neurotrophic factor (BDNF) on the survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci USA. 1994;91:1632–1636. doi: 10.1073/pnas.91.5.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 38.Martinou J-C, Sadoul R. ICE-like proteases execute the neuronal death program. Curr Opin Neurobiol. 1996;6:609–614. doi: 10.1016/s0959-4388(96)80092-4. [DOI] [PubMed] [Google Scholar]
- 39.McCaffery CA, Bennett MR, Dreher MR. The survival of neonatal rat retinal ganglion cells in vitro is enhanced in the presence of appropriate parts of the brain. Exp Brain Res. 1982;48:377–386. doi: 10.1007/BF00238614. [DOI] [PubMed] [Google Scholar]
- 40.Mesulam M-M. Principles of horseradish peroxidase neurohistochemistry and their applications for tracing neural pathways-axonal transport, enzyme histochemistry and light microscopic analysis. In: Mesulam M-M, editor. Tracing neural connections with horseradish peroxidase. Wiley; Chichester, UK: 1982. pp. 1–151. [Google Scholar]
- 41.Mey J, Thanos S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993;602:304–317. doi: 10.1016/0006-8993(93)90695-j. [DOI] [PubMed] [Google Scholar]
- 42.Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 43.Nicotera P, Bellomo G, Orrenius S. Calcium-mediated mechanisms in chemically induced cell death. Annu Rev Pharmacol Toxicol. 1992;32:449–470. doi: 10.1146/annurev.pa.32.040192.002313. [DOI] [PubMed] [Google Scholar]
- 44.O’Leary DDM, Crespo D, Fawcett JW, Cowan WM. The effect of intraocular tetrodotoxin on the postnatal reduction in the numbers of optic nerve axons in the rat. Dev Brain Res. 1986a;30:96–103. doi: 10.1016/s0006-8993(86)80014-2. [DOI] [PubMed] [Google Scholar]
- 45.O’Leary DDM, Fawcett JW, Cowan WM. Topographic targeting errors in the retinocollicular projection and their elimination by selective ganglion cell death. J Neurosci. 1986b;6:3692–3705. doi: 10.1523/JNEUROSCI.06-12-03692.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 47.Pearson HE, Labar DR, Payne BR, Cornwell P, Aggarwal N. Transneuronal retrograde degeneration in the cat retina following neonatal ablation of visual cortex. Brain Res. 1981;212:470–475. doi: 10.1016/0006-8993(81)90480-7. [DOI] [PubMed] [Google Scholar]
- 48.Perry VH, Henderson Z, Linden R. Postnatal changes in retinal ganglion cell and optic axon populations in the pigmented rat. J Comp Neurol. 1983;219:356–368. doi: 10.1002/cne.902190309. [DOI] [PubMed] [Google Scholar]
- 49.Rabacchi SA, Bonfanti L, Liu X-H, Maffei L. Apoptotic cell death induced by optic nerve lesion in the neonatal rat. J Neurosci. 1994a;14:5292–5301. doi: 10.1523/JNEUROSCI.14-09-05292.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rabacchi SA, Ensini M, Bonfanti L, Gravina A, Maffei L. Nerve growth factor reduces apoptosis of axotomized retinal ganglion cells in the neonatal rat. Neuroscience. 1994b;63:969–973. doi: 10.1016/0306-4522(94)90565-7. [DOI] [PubMed] [Google Scholar]
- 51.Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 52.Schoups AA, Elliott RC, Friedman WJ, Black IB. NGF and BDNF are differentially modulated by visual experience in the developing geniculocortical pathway. Dev Brain Res. 1995;86:326–334. doi: 10.1016/0165-3806(95)00043-d. [DOI] [PubMed] [Google Scholar]
- 53.Schreiber SS, Baudry M. Selective neuronal vulnerability in the hippocampus: a role for gene expression? Trends Neurosci. 1995;18:446–451. doi: 10.1016/0166-2236(95)94495-q. [DOI] [PubMed] [Google Scholar]
- 54.Selander D. Neurotoxicity of local anesthetics: animal data. Reg Anaesth. 1993;18:461–468. [PubMed] [Google Scholar]
- 55.Silos-Santiago I, Greenlund LJS, Johnson EM, Snider WD. Molecular genetics of neuronal survival. Curr Opin Neurobiol. 1995;5:42–49. doi: 10.1016/0959-4388(95)80085-9. [DOI] [PubMed] [Google Scholar]
- 56.Snider WD, Elliott JL, Yan Q. Axotomy-induced neuronal death during development. J Neurobiol. 1992;23:1231–1246. doi: 10.1002/neu.480230913. [DOI] [PubMed] [Google Scholar]
- 57.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 58.Tong L, Spear PD, Kalil RE, Callahan EC. Loss of retinal X-cells in cats with neonatal or adult visual cortex damage. Science. 1982;217:72–75. doi: 10.1126/science.7089543. [DOI] [PubMed] [Google Scholar]
- 59.Trump BF, Berezesky IK. The role of cytosolic Ca++ in cell injury, necrosis and apoptosis. Curr Opin Cell Biol. 1992;4:227–232. doi: 10.1016/0955-0674(92)90037-d. [DOI] [PubMed] [Google Scholar]
- 60.Vanselow J, Dütting D, Thanos S. Target dependence of chick retinal ganglion cells during embryogenesis: cell survival and dendritic development. J Comp Neurol. 1990;295:235–247. doi: 10.1002/cne.902950207. [DOI] [PubMed] [Google Scholar]
- 61.Villegas-Perez MP, Vidal Sanz M, Rasminsky M, Bray GM, Aguayo AJ. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J Neurobiol. 1993;24:23–36. doi: 10.1002/neu.480240103. [DOI] [PubMed] [Google Scholar]
- 62.Weil M, Jacobson MD, Coles HSR, Davies TJ, Gardner RL, Raff KD, Raff MC. Constitutive expression of the machinery for programmed cell death. J Cell Biol. 1996;133:1053–1059. doi: 10.1083/jcb.133.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan J. Molecular control of life and death. Curr Opin Cell Biol. 1995;7:211–214. doi: 10.1016/0955-0674(95)80030-1. [DOI] [PubMed] [Google Scholar]