

Abstract
Oxidative stress is believed to play important roles in neuronal cell death associated with many different neurodegenerative conditions (e.g., Alzheimer’s disease, Parkinson’s disease, and cerebral ischemia), and it is believed also that apoptosis is an important mode of cell death in these disorders. Membrane lipid peroxidation has been documented in the brain regions affected in these disorders as well as in cell culture and in vivo models. We now provide evidence that 4-hydroxynonenal (HNE), an aldehydic product of membrane lipid peroxidation, is a key mediator of neuronal apoptosis induced by oxidative stress. HNE induced apoptosis in PC12 cells and primary rat hippocampal neurons. Oxidative insults (FeSO4 and amyloid β-peptide) induced lipid peroxidation, cellular accumulation of HNE, and apoptosis. Bcl-2 prevented apoptosis of PC12 cells induced by oxidative stress and HNE. Antioxidants that suppress lipid peroxidation protected against apoptosis induced by oxidative insults, but not that induced by HNE. Glutathione, which binds HNE, protected neurons against apoptosis induced by oxidative stress and HNE. PC12 cells expressing Bcl-2 exhibited higher levels of glutathione and lower levels of HNE after oxidative stress. Collectively, the data identify that HNE is a novel nonprotein mediator of oxidative stress-induced neuronal apoptosis and suggest that the antiapoptotic action of glutathione may involve detoxification of HNE.
Keywords: Alzheimer’s disease, amyloid β-peptide, Bcl-2, glutathione, hippocampal neurons, iron, lipid peroxidation, mitochondria, programmed cell death, reactive oxygen species, vitamin E
Oxidative stress may contribute to neuronal dysfunction and death in an array of disorders, including stroke, Alzheimer’s disease (AD), and Parkinson’s disease (for review, seeJesberger and Richardson, 1991; Mattson et al., 1996; Simonian and Coyle, 1996). Reactive oxygen species (ROS) are generated in several metabolic pathways, a major source being mitochondrially derived superoxide anion radical, which gives rise to hydrogen peroxide, which is converted further to hydroxyl radical, which induces membrane lipid peroxidation (Evans, 1993). Systems that detoxify ROS include the enzymes superoxide dismutase, catalase, and glutathione peroxidase and the thiol tripeptide glutathione (GSH). Oxidative stress occurs in neurons exposed to excitotoxins (Lafon-Cazal et al., 1993; Dugan et al., 1995; Mattson et al., 1995a), metabolic poisons (Beal et al., 1995), ischemia (Chan, 1996), and amyloid β-peptide (Aβ) (Behl et al., 1994; Goodman and Mattson, 1994). By damaging ion transport proteins and other regulatory systems in membranes, lipid peroxidation may be particularly detrimental to neurons (Mark et al., 1995,1997).
Apoptotic cell death is characterized by cell shrinkage, cell surface blebbing, chromatin condensation, and DNA fragmentation with maintenance of membrane and organellar integrity; macromolecular synthesis inhibitors can prevent apoptosis, suggesting an active or “programmed” mechanism of cell death (for review, see Bredesen, 1995; Johnson et al., 1995; Steller, 1995; Thompson, 1995). Necrosis is another form of cell death in which cells swell, mitochondria and other organelles are damaged, and the plasma membrane ruptures. Neuronal apoptosis may occur in stroke (Linnik et al., 1993; MacManus et al., 1993; Nitatori et al., 1995), AD (Loo et al., 1993; Su et al., 1994;Anderson et al., 1995), and Huntington’s disease (Portera-Cailliau et al., 1995). The involvement of ROS in apoptosis is suggested by studies showing that apoptotic stimuli induce accumulation of ROS in neurons and that antioxidants can prevent apoptosis (Kane et al., 1993;Whittemore et al., 1994; Ferrari et al., 1995; Greenlund et al., 1995;Mark et al., 1995).
Proapoptotic gene products such as interleukin-1β-converting enzyme (ICE) (Troy et al., 1996a) and antiapoptotic gene products such as Bcl-2 (Hockenbery et al., 1993; Kane et al., 1993; Mah et al., 1993;Reed, 1994; Wiedau-Pazos et al., 1996) may play roles in oxidative stress-induced apoptosis. On the other hand, nonprotein mediators of apoptosis have not been identified. 4-Hydroxynonenal (HNE), an aldehydic product of lipid peroxidation, is cytotoxic to non-neuronal cells at concentrations reached when cells are exposed to various oxidative insults (Esterbauer et al., 1991). HNE forms covalent cross-links with proteins via Michael addition to lysine, cysteine, and histidine residues (Uchida and Stadtman, 1993; Uchida et al., 1994); HNE normally is detoxified by conjugation with GSH (Spitz et al., 1990;Grune et al., 1994; Ullrich et al., 1994). It is not known whether HNE induces apoptosis nor whether HNE is involved mechanistically in oxidative stress-induced apoptosis. We now report that HNE is generated in response to apoptotic oxidative insults and can induce neuronal apoptosis at pathophysiologically relevant concentrations.
MATERIALS AND METHODS
Materials. HNE, propanal, pentanal, heptanal, and nonenal were purchased from Cayman Chemical (Ann Arbor, MI). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), hexanal, malondialdehyde, vitamin E (α-tocopherol), propyl gallate, glutathione (GSH)-ethyl ester, cycloheximide, actinomycin-D, ATA, and buthionine sulfoximine (BSO) were obtained from Sigma (St. Louis, MO). Nonaldehyde was from Aldrich (Milwaukee, WI), andtrans-2-nonenal was from Wako Pure Chemicals (Osaka, Japan). Aldehydes were prepared as 200–500× stocks in ethanol; ethanol vehicle (0.2–0.5% final concentration) was added to control cultures. Aβ25–35 was purchased from Bachem (Torrence, CA) and dissolved in water at a concentration of 2 mm 2–4 hr before experiments. Hoescht 33342, propidium iodide, rhodamine 123, and monochlorobimane were obtained from Molecular Probes (Eugene, OR). The antibody against HNE-conjugated proteins was generated and characterized as described in our previous study (Waeg et al., 1996). FITC–Annexin-V was obtained from Nexins Research.
PC12 and rat hippocampal cell cultures and experimental treatments. Control vector-transfected PC12 cells (PC12-V) and PC12 cells expressing high levels of human Bcl-2 (PC12-Bcl2) were established by methods described in our previous studies (Kane et al., 1993; Zhong et al., 1993a). Cultures were maintained at 37°C (5% CO2 atmosphere) in RPMI-1640 medium supplemented 10% with heat-inactivated horse serum and 5% with heat-inactivated fetal bovine serum; immediately before experimental treatment the medium was replaced with RPMI-1640 containing 1% fetal calf serum. Rat hippocampal cell cultures were established from 18 d embryos, as described previously (Mattson et al., 1995b); experiments were performed in cells that had been in culture for 6–8 d. Immediately before experimental treatment the culture medium was replaced with Locke’s solution containing (in mm): NaCl 154, KCl 5.6, CaCl2 2.3, MgCl2 1.0, NaHCO3 3.6, glucose 5, and HEPES 5, pH 7.2.
Analyses of necrosis and apoptosis. Numbers of trypan blue-positive (necrotic) and -negative cells were counted in four 20× fields per culture (typically 70–100 cells/20× field), and the percentage of trypan blue-positive cells in each culture was calculated. Under control conditions <5% of the cells were trypan blue-positive. Methods used to establish apoptotic cell death included Hoescht and propidium iodide staining of DNA, annexin-V binding, and suppression of cell death by macromolecular synthesis inhibitors. For Hoescht and propidium iodide staining, cells were fixed in 4% paraformaldehyde, membranes were permeabilized with 0.2% Triton X-100, and cells were stained with the fluorescent DNA-binding dyes Hoescht 33342 or propidium iodide, as described previously (Mark et al., 1995). Hoescht-stained cells were visualized and photographed under epifluorescence illumination (340 nm excitation and 510 nm barrier filter) with a 40× oil immersion objective (200 cells per culture were counted, and counts were made in at least four separate cultures per treatment condition; analyses were performed without knowledge of the treatment history of the cultures). The percentage of apoptotic cells (cells with condensed and fragmented DNA) in each culture was determined. Images of propidium iodide-stained cells were acquired with a confocal laser scanning microscope (488 nm excitation and 510 nm barrier filter; Molecular Dynamics, Sunnyvale, CA ) with a 60× oil immersion objective. Alterations in plasma membrane phospholipids linked to apoptosis were detected by fluorescently labeled annexin-V, as described previously (Vermes et al., 1995; Van Engeland et al., 1996). After experimental treatment, cells were exposed for 10 min to FITC–annexin-V, using conditions recommended by the manufacturer (Nexins Research), and confocal images of annexin-V fluorescence were acquired.
Quantification of lipid peroxidation and HNE levels. The thiobarbituric acid reactive substances (TBARS) method was used to assess membrane lipid peroxidation, as described previously (Goodman et al., 1996); values were normalized to protein content. Levels of HNE were quantified by dot-blot analysis. After experimental treatment, culture medium was removed and stored at −80°C. Cells were washed three times with PBS, scraped in a lysis buffer containing 0.1% SDS, and sonicated. Samples (50 μl of medium; or 106cells, which corresponds to ∼50 μl cell volume) were blotted onto a nitrocellulose sheet with a dot-blot apparatus (Bio-Rad, Hercules, CA); a standard curve was generated by medium or cell homogenates containing HNE at concentrations ranging from 0.1–5 μm. Then the membrane was washed with TTBS, blocked by incubation in the presence of 5% milk, exposed to HRP-conjugated anti-mouse secondary antibody, and detected by a chemiluminescence kit (Amersham, Arlington Heights, IL). Densitometric analysis of dots in images of the blots was used to calculate levels of HNE. It should be noted that this dot-blot method is not truly quantitative and that the values reported are only an estimate of the actual HNE levels.
Immunocytochemistry and Western blot analyses. Methods for Western blot and immunocytochemical analyses of HNE–protein conjugates were similar to those described previously (Waeg et al., 1996; Mark et al., 1997). For immunostaining, cultured cells were fixed for 30 min in 4% paraformaldehyde/PBS, and membranes were permeabilized by incubation in 0.2% Triton X-100 in PBS. Cells were incubated for 1 hr in blocking serum (1% normal horse serum in PBS), for 3 hr in the presence of anti-HNE mouse monoclonal antibody (1:100 in PBS), for 1 hr in PBS containing biotinylated horse anti-mouse secondary antibody, and for 30 min in ABC reagent (Vector Laboratories, Burlingame, CA). Levels of cellular immunoreactivity were assessed by using a semiquantitative method described previously (Mattson, 1992); staining intensity of cell bodies was scored on a scale from 0 to 3 (0, no staining; 1, weak staining; 2, moderate staining; 3, intense staining). A total of 400 cells in four separate cultures per condition (100 cells/culture) were scored in a blinded manner without knowledge of their treatment history. For Western blot analysis, solubilized cell proteins were separated by electrophoresis in a 10% polyacrylamide gel, transferred to a nitrocellulose sheet, and immunoreacted with an antibody against HNE-conjugated proteins (clone 1 g4; Waeg et al., 1996); The nitrocellulose sheet was processed further with HRP-conjugated anti-mouse secondary antibody and a chemiluminescence detection method (Amersham).
Assessments of mitochondrial function. The conversion of the dye MTT to formazan crystals in cells has been shown to be related to mitochondrial respiratory chain activity (Musser and Oseroff, 1994) and mitochondrial redox state (Shearman et al., 1995). Levels of cellular MTT reduction were quantified as described previously (Mattson et al., 1995b). The dye rhodamine 123 was used as a measure of mitochondrial transmembrane potential by methods described previously (Mattson et al., 1993).
Quantification of GSH levels. Two methods were used to quantify cellular GSH levels. The first method used monochlorobimane, a fluorescent probe for GSH (Barhoumi et al., 1995), and procedures described in our previous study (Kane et al., 1993). The second method was based on an enzymatic recycling procedure in which GSH is oxidized sequentially by 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) and reduced by NADPH in the presence of glutathione reductase and followed a protocol similar to that reported by Tietze (1969). A GSH standard curve was generated by analysis of serial dilutions of pure GSH. GSH levels were adjusted to sample protein content (determined with a Pierce BCA kit, Rockford, IL), and values were expressed as U/mg protein.
RESULTS
Oxidative insults and HNE induce delayed apoptosis in PC12 cells
Cultured control (PC12-V) and Bcl-2-expressing (PC12-Bcl2) PC12 cells were exposed to increasing concentrations of FeSO4, Aβ, and HNE, and mitochondrial function and cell survival were quantified by MTT and trypan blue exclusion assays, respectively. FeSO4 and Aβ are known to induce lipid peroxidation and to kill cultured neurons (Poli et al., 1985; Zhang et al., 1993; Behl et al., 1994; Goodman and Mattson, 1994; Muller and Krieglstein, 1995; Goodman et al., 1996). Exposure of PC12-V or PC12-Bcl2 cells to high concentrations of FeSO4, Aβ, and HNE resulted in rapid decreases in levels of MTT reduction to <20% of basal levels within 6 hr of exposure (Fig.1A,B). Exposure of PC12-V cells to lower concentrations of each insult (1 mmFeSO4, 50 μm Aβ, or 10 μm HNE) resulted in a moderate (20–30%) decrease in levels of MTT reduction during the first 12–24 hr; values stayed at this level through 48 hr of exposure and then decreased further (to 10–30% of basal levels) during a subsequent 24 hr period. In contrast, levels of MTT reduction were maintained at 80–90% of basal levels in PC12-Bcl2 cells exposed to the lower concentrations of FeSO4, Aβ, and HNE throughout the entire 72 hr exposure period (Fig. 1A,B). Using rhodamine 123 fluorescence as an indicator of mitochondrial transmembrane potential (Mattson et al., 1993), we found that 10 μm HNE caused a significant (>50%) decrease in rhodamine 123 fluorescence in PC12-V cells, but not in PC12-Bcl2 cells, during a 24 hr exposure period (data not shown). High concentrations of FeSO4, Aβ25–35, and HNE induced relatively rapid cell death in both PC12-V and PC12-Bcl2 cells that occurred within 12–24 hr of exposure (Fig.1C,D). Lower concentrations of FeSO4, Aβ, and HNE induced delayed uptake of trypan blue in PC12-V cells that occurred 48–72 hr post-treatment; the delayed uptake of trypan blue likely was attributable to secondary necrosis (Slater et al., 1995). In contrast to PC12-V cells, PC12-Bcl2 cells were not killed by the lower concentrations of FeSO4, Aβ, or HNE (Fig. 1C,D), consistent with an apoptotic mechanism of cell death.
Fig. 1.

Oxidative insults and HNE induce rapid and delayed cell death in PC12 cells: Bcl-2 prevents delayed cell death.A, B, PC12-V (A) and PC12-Bcl2 (B) cells were exposed to the indicated concentrations of HNE, Aβ, or FeSO4 for the indicated time periods, and levels of MTT reduction were quantified (mean and SEM of determinations made in four culture wells/condition).C, D, PC12-V cells (C) and PC12-Bcl2 cells (D) were exposed to vehicle (Control) or the indicated concentrations of HNE, Aβ, orFeSO4, and trypan blue-positive and nonstaining cells were counted. Values are the mean percentage of trypan blue-positive cells in four separate cultures.
To establish whether the delayed cell death induced by oxidative insults and HNE in PC12-V cells was apoptotic, we examined nuclear condensation and fragmentation by using fluorescent DNA-binding dyes Hoescht 33342 and propidium iodide (Fig. 2). Exposure of PC12-V cells to concentrations of FeSO4, Aβ, and HNE that caused delayed death resulted in the appearance of many cells exhibiting nuclear condensation and fragmentation, which occurred progressively beginning at ∼30 hr post-treatment (Fig.2A,D). In contrast, no PC12-Bcl2 cells exhibited nuclear signs of apoptosis during the 72 hr exposure period to FeSO4, Aβ, or HNE. An early event in apoptosis is a loss of plasma membrane asymmetry, resulting in the exposure of phosphatidylserine on the cell surface, which binds annexin-V (Martin et al., 1995). In control PC12-V cultures annexin-V-positive cells were rare (<5% of the cells). When PC12-V cells were exposed to 10 μm HNE, there was a relatively rapid and progressive appearance of annexin-V-positive cells such that ∼25 and 45% of the cells were annexin-V-positive by 4 and 16 hr post-treatment, respectively (Fig. 2B). Confocal images suggested that the annexin-V was associated with the cell surface (data not shown). Because several aldehydes are liberated when membrane lipids are peroxidized, we determined whether other aldehydes also induce apoptosis. Among nine different aldehydes examined (HNE, propanal, pentanal, hexanal, heptanal, nonenal, malondialdehyde, nonaldehyde, andtrans-2-nonenal), only HNE induced nuclear condensation and DNA fragmentation (Fig. 2C) and annexin-V binding (data not shown).
Fig. 2.

Oxidative insults and HNE induce apoptotic nuclear and plasma membrane alterations in PC12 cells: prevention by Bcl-2.A, Cultures of PC12-V cells and PC12-Bcl2 cells were exposed to vehicle (Control) or the indicated concentrations of HNE, Aβ, orFeSO4. At the indicated time points cells were stained with Hoescht dye, and the percentage of cells with condensed and fragmented (apoptotic) nuclei was determined (mean and SEM of determinations made in four separate cultures). No cells expressing Bcl-2 exhibited condensed and fragmented nuclei (PC12-Bcl2line represents combined data from cells exposed to each treatment condition). B, Annexin-V-positive cells were counted in PC12-V and PC12-Bcl2 cultures exposed for 4 hr to 10 μm HNE or for 16 hr to vehicle (Control) or 10 μm HNE. Values are the mean and SEM of determinations made in four separate cultures. *p < 0.001 compared with values for control cultures and PC12-Bcl2 cultures (ANOVA with Scheffé’spost hoc tests). C, PC12-V cell cultures were exposed for 48 hr to vehicle (Control) or the indicated aldehydes (10 μm). Cells were stained with Hoescht dye, and the percentages of cells with condensed and fragmented nuclei were determined. Values are the mean and SEM of determinations made in four separate cultures. *p < 0.001 compared with each of the other values (ANOVA with Scheffé’spost hoc tests). D, Confocal laser scanning microscope images of propidium iodide fluorescence in untreated control PC12-V cells (left), PC12-V cells exposed for 48 hr to 10 μm HNE (middle), and PC12-Bcl-2 cells exposed for 48 hr to 10 μm HNE (right). Note that many PC12-V cells treated with HNE exhibit DNA condensation and fragmentation (arrowheads), whereas cells expressing Bcl-2 did not.
Macromolecular synthesis inhibitors and endonuclease inhibitors can prevent apoptosis (Bastitatou and Greene, 1991; Rukenstein et al., 1991). Nuclear condensation and fragmentation induced by HNE, FeSO4, and Aβ was prevented mainly in PC12-V cultures cotreated with either the protein synthesis inhibitor cycloheximide, the RNA synthesis inhibitor actinomycin-D, or the Ca2+–Mg2+-dependent endonuclease inhibitor aurintricarboxylic acid (ATA; Table 1).
Table 1.
Effects of macromolecular synthesis inhibitors, an endonuclease inhibitor, and antioxidants on apoptotic cell death induced by oxidative insults and HNE in PC12 cells
| Insult | Vehicle | Cells with apoptotic nuclei (%) | ||||
|---|---|---|---|---|---|---|
| Cyclohex | Act-D | ATA | VitE | PG | ||
| Cont (2 hr) | 3 ± 1.2 | 1 ± 0.3 | 2 ± 0.6 | 1 ± 0.3 | 0 ± 0.3 | 1 ± 0.3 |
| Cont (72 hr) | 24 ± 4.7 | 11 ± 2.5 | 10 ± 3.3 | 9 ± 2.4 | 10 ± 2.1 | 13 ± 3.1 |
| FeSO4 | 75 ± 3.2* | 16 ± 2.71-160 | 22 ± 3.31-160 | 13 ± 3.31-160 | 13 ± 2.81-160 | 21 ± 2.11-160 |
| Aβ | 72 ± 3.3* | 21 ± 3.41-160 | 23 ± 4.31-160 | 12 ± 2.21-160 | 11 ± 3.51-160 | 14 ± 3.21-160 |
| HNE | 71 ± 6.7* | 29 ± 4.31-160 | 29 ± 4.01-160 | 26 ± 3.21-160 | 59 ± 3.3 | 65 ± 3.8 |
Cultures were pretreated for 2 hr with the indicated agents: 0.2% ethanol (Vehicle), 10 μm cycloheximide (Cyclohex), 5 μm actinomycin-D (Act-D), 100 μmaurintricarboxylic acid (ATA), 50 μg/ml vitamin E (VitE), 10 μm propyl gallate (PG), or 1 mmglutathione-ethyl ester (GSH). Some cultures were fixed at that point (Cont, 2 hr), while parallel cultures were exposed for 72 hr to 0.2% ethanol (Cont 72 hr), 1 mm FeSO4, 50 μm Aβ, or 10 μm HNE. Cells then were fixed and stained with Hoescht dye, and percentages of cells exhibiting nuclear condensation and fragmentation were determined. Values are the mean and SEM of determinations made in three or four separate cultures.
p < 0.001 compared with value for control vehicle-treated (72 hr) cultures;
F1-160: p < 0.01 compared with value for vehicle-treated cultures exposed to the same insult. ANOVA with Scheffé’s post hoc tests. Preliminary studies showed that 10 μm cycloheximide reduced levels of protein synthesis by >90% during a 24 hr exposure period (data not shown).
Oxidative insults and HNE induce apoptosis in hippocampal neurons: protection by GSH
Although studies of PC12 cells have provided valuable insight into mechanisms of neural cell apoptosis (Bastitatou and Greene, 1991;Rukenstein et al., 1991; Ferrari et al., 1995; Troy et al., 1996a), PC12 cells do not exhibit several important features of primary neurons, including expression of glutamate receptors and synapse formation. We therefore turned to mature primary hippocampal cell cultures. Whereas <5% of hippocampal neurons exhibited apoptotic nuclei in vehicle-treated control cultures, 70–80% of the neurons exhibited nuclear condensation and fragmentation in cultures exposed to 2 μm HNE (Fig. 3). Lower concentrations of HNE caused progressively less apoptotic neuronal death (0.5 μm HNE, 18 ± 3.0%; 1 μm HNE, 49 ± 4.1%; n = 4 cultures), whereas higher levels (5–10 μm) induced rapid necrosis (data not shown). Eight other aldehydes (2 μm) did not induce apoptosis (Fig.3A) or necrosis (data not shown). Aβ and FeSO4also induced apoptosis in the cultured hippocampal neurons (Fig.3B). Previous studies showed that GSH can conjugate with, and thereby detoxify, HNE in non-neuronal cells (Spitz et al., 1990). Pretreatment of hippocampal cultures with a membrane-permeant form of GSH (GSH-diethyl ester) afforded significant protection against apoptosis induced by each oxidative insult and HNE (Fig.3B).
Fig. 3.
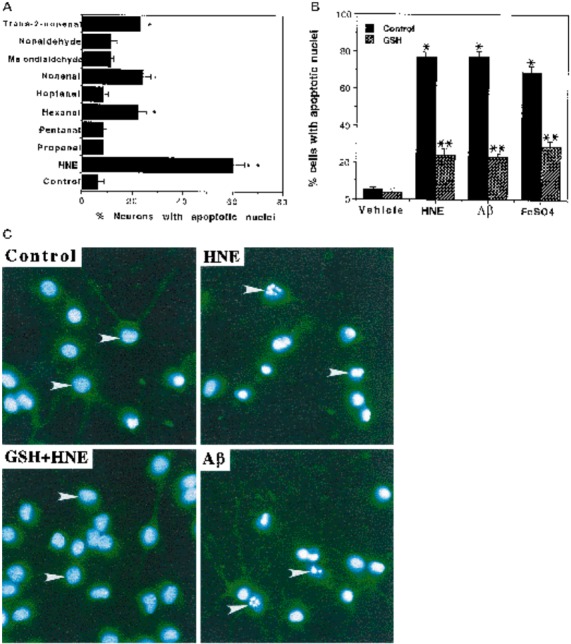
Oxidative insults and HNE induce apoptosis in primary hippocampal neurons: prevention by GSH. A, Cultures were exposed to 2 μm of each aldehyde (see Fig. 2C for aldehyde structures) or 0.2% ethanol (Control) for 16 hr, and percentages of neurons with condensed and fragmented nuclei were quantified. Values are the mean and SEM of determinations made in four separate cultures. *p < 0.05, **p < 0.001 compared with control value (ANOVA with Scheffé’s post hoc tests). B, Cultures were exposed for 16 hr to 0.2% ethanol (Control) or the indicated treatments, at which time cells were stained with Hoescht dye and the percentages of neurons with condensed and fragmented nuclei were quantified. Treatment concentrations were HNE, 10 μm; Aβ, 10 μm;FeSO4, 2 μm;GSH, 1 mm. Values are the mean and SEM of determinations made in four separate cultures. *p< 0.01 compared with control value; **p < 0.01, ***p < 0.05 compared with corresponding values for cultures not cotreated with GSH (ANOVA with Scheffé’spost hoc tests). C, Images of Hoescht dye fluorescence in hippocampal neurons from cultures exposed for 16 hr to 0.2% ethanol (Control), 2 μmHNE, 1 mm GSH-ethyl-ester plus 2 μm HNE (GSH+HNE), or 10 μmAβ. HNE and Aβ induced nuclear condensation and fragmentation in most neurons (arrowheads), whereas fluorescence remained uniformly distributed throughout the nucleus in neurons in control cultures and neurons in cultures cotreated with GSH and HNE (arrowheads).
Oxidative insults induce lipid peroxidation and formation of HNE–protein conjugates in neural cells: suppression by GSH and Bcl-2
A TBARS fluorescence assay, which measures malondialdehyde levels (Kovachich and Mishra, 1980), was used to quantify membrane lipid peroxidation. Exposure of PC12-V cells or hippocampal neurons to apoptotic concentrations of FeSO4 and Aβ resulted in marked increases in TBARS fluorescence (Fig.4A). HNE did not affect TBARS levels in either cell type, consistent with its being a downstream effector of oxidative stress-induced apoptosis rather than an initiator of oxidative stress. If HNE mediates oxidative stress-induced apoptosis, then the oxidative insults should induce HNE accumulation in neurons to levels capable of inducing apoptosis. HNE levels were measured with an anti-HNE antibody (Waeg et al., 1996) in dot-blot, immunocytochemical, and Western blot assays. Hippocampal cells were exposed to 2 μm FeSO4 or 1 μm HNE, and levels of HNE in the culture medium and cells were quantified at increasing time points after treatment. FeSO4 induced a time-dependent increase in HNE levels in the cells; levels reached ∼1 μm 30 min after exposure, increased to nearly 3 μm 6 hr post-treatment, and then decreased to ∼2 μm by 24 hr post-treatment (Fig. 4B). HNE was not detectable in the medium (<0.1 μm) at any time point after exposure of cultures to FeSO4. When 1 μm HNE was added to the cultures, HNE levels in cells increased to ∼2 μm during the first 2 hr and then decreased somewhat during the next 22 hr; HNE levels in the culture medium remained at ∼1 μm during the first 2 hr and then progressively decreased through 24 hr (Fig. 4B).
Fig. 4.
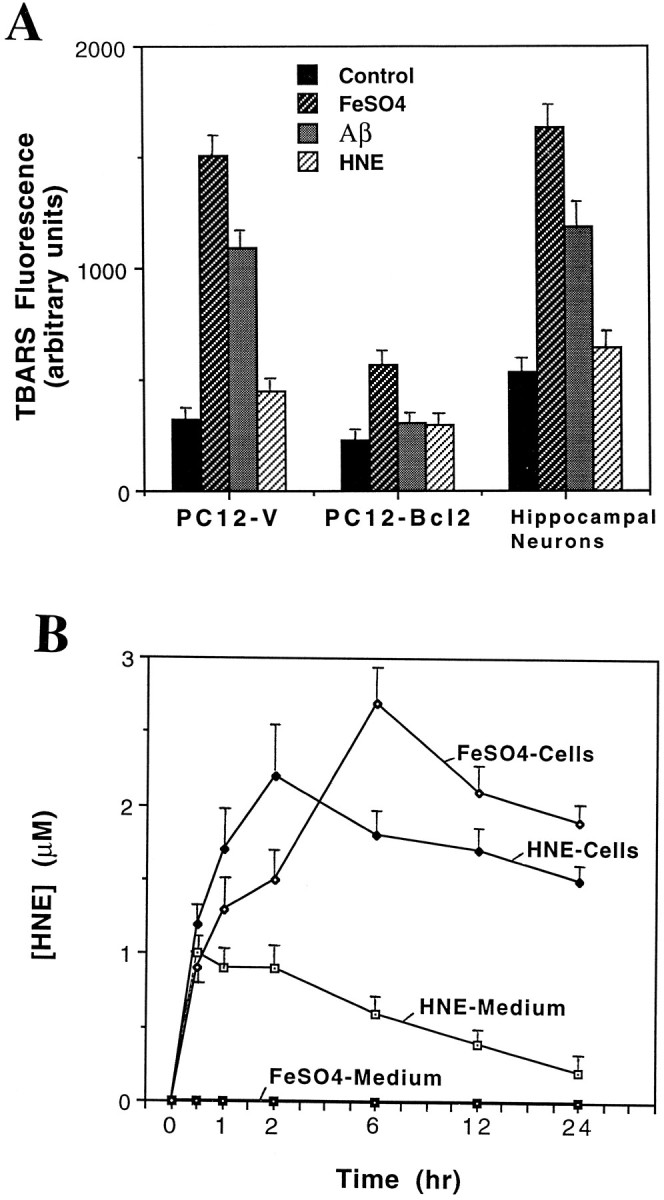
Oxidative insults induce membrane lipid peroxidation in neural cells: suppression by Bcl-2. A, Levels of TBARS fluorescence were quantified in PC12-V cells, PC12-Bcl2 cells, and hippocampal neurons after 4 hr of exposure to vehicle (Control), FeSO4 (1 mm PC12 cells, 2 μm hippocampal neurons),Aβ (50 μm PC12 cells, 10 μm hippocampal neurons), or HNE (10 μm PC12 cells, 2 μm hippocampal neurons). Values represent the mean and SEM of determinations made in four to six separate cultures. B, Hippocampal cell cultures were exposed to 1 μm HNE or 2 μmFeSO4, and HNE levels in the culture medium and cells were quantified at the indicated time points. Values represent the mean and SEM of determinations made in four separate cultures.
Immunocytochemical analyses showed that oxidative insults and HNE induced HNE immunoreactivity in cultured hippocampal neurons and PC12-V cells (Fig. 5). The levels of cellular HNE immunoreactivity after exposure to FeSO4, Aβ, and HNE were reduced greatly in hippocampal cells pretreated with GSH, as compared with control cells (Fig. 5A,D). Exposure of PC12-V cells to FeSO4, Aβ, and HNE for 12 hr resulted in the appearance of HNE immunoreactivity in essentially all cells; in most cells the HNE immunoreactivity appeared to be concentrated in perinuclear regions and at the cell periphery, suggesting plasma membrane and organellar localizations (Fig. 5B,D). In contrast, levels of HNE immunoreactivity in PC12-Bcl2 cells exposed to Aβ, FeSO4, and HNE were reduced markedly, as compared with levels in PC12-V cells exposed to the same insults (Fig.5B,D). To determine whether HNE production occurs in all apoptotic paradigms or is specific for oxidative insults, we exposed PC12-V cells for 2, 6, 12, or 24 hr to an apoptotic concentration of staurosporine (500 nm) and then immunostained the cells with the HNE antibody. No detectable HNE immunoreactivity was observed at any time point after exposure to staurosporine (data not shown).
Fig. 5.
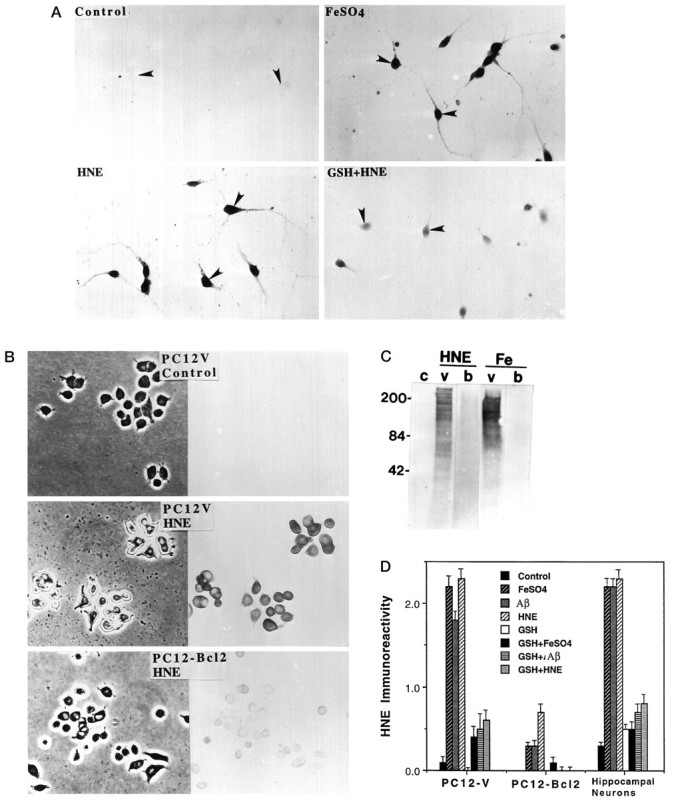
Oxidative insults induce formation of HNE–protein conjugates in neural cells: attenuation by GSH and Bcl-2.A, Hippocampal cultures were exposed for 2 hr to 0.2% ethanol (Control), 2 μmFeSO4, 2 μmHNE, or 1 mm GSH plus 2 μm HNE (GSH+HNE). Then cells were fixed and immunostained with HNE antibody. Arrowheads point to neuronal cell bodies.B, PC12-V cells and PC12-Bcl-2 cells were exposed for 2 hr to vehicle (0.2% ethanol) or 10 μm HNE. Then cells were fixed and immunostained with anti-HNE antibody. Note the much greater level of HNE immunoreactivity in PC12-V cells exposed to HNE, as compared with PC12-Bcl-2 cells exposed to HNE. C, Cell homogenates from untreated control PC12-V cultures (c), PC12-V cultures exposed to 10 μm HNE or 1 mmFeSO4 (v), and PC12-Bcl2 cultures exposed to 10 μm HNE or 1 mm FeSO4(b) were separated by SDS-PAGE (100 μg protein/lane). Then proteins were transferred to a nitrocellulose sheet and immunoreacted with an antibody against HNE–protein conjugates. Note that HNE and FeSO4 induced the appearance of many HNE–protein conjugates in PC12-V cells, but not in the PC12-Bcl2 cells. D, PC12-V cells, PC12-Bcl2 cells, and primary hippocampal neurons were exposed for 2 hr to 0.2% ethanol (Control), FeSO4 (1 mm for PC12 cells and 2 μm for hippocampal neurons), Aβ (50 μm for PC12 cells and 10 μm for hippocampal neurons), or HNE (10 μm for PC12 cells and 2 μm for hippocampal neurons). Then cells were fixed and immunostained with HNE antibody, and relative levels of HNE immunoreactivity were quantified (see Materials and Methods). Values represent the mean and SEM of determinations made in four separate cultures per condition (100 cells scored/culture).
Western blot analysis of PC12-V and PC12-Bcl2 cells exposed for 12 hr to vehicle, FeSO4, or HNE showed that, whereas there were no detectable HNE–protein conjugates in control cultures, there were many proteins immunoreactive with the HNE antibody in PC12-V cells exposed to HNE or FeSO4 (Fig. 5C). The pattern of HNE–protein conjugates in proteins from cells exposed directly to HNE were similar, but not identical, to the pattern seen in cells exposed to FeSO4. Although the basis for the differences in banding was not established, it seems likely that proteins located in the plasma membrane are exposed to locally higher concentrations of HNE in cells exposed to FeSO4 (compared with cells exposed to exogenous HNE) because that is where the endogenous HNE comes from. In contrast to PC12-V cells, only very low levels of HNE–protein conjugates were present in Western blots of proteins from PC12-Bcl2 cells exposed to either FeSO4 or HNE (Fig. 5C). In additional experiments we performed electron paramagnetic spectroscopy analyses with nitroxyl stearate spin labels to quantify lipid peroxidation; the results confirmed that Bcl-2 suppresses lipid peroxidation induced by iron and Aβ in PC12 cells (data not shown; cf. Bruce et al., 1995).
Pretreatment of PC12-V cultures with the antioxidants vitamin E and propyl gallate before exposure to FeSO4 and Aβ resulted in significant protection against apoptosis induced by these oxidative insults (Table 1). In contrast, vitamin E and propyl gallate did not protect PC12-V cells against HNE-induced apoptosis (Table 1), consistent with HNE acting as a downstream effector of lipid peroxidation-induced apoptosis.
Evidence that endogenous GSH serves an antiapoptotic function
The ability of GSH to protect hippocampal neurons against apoptosis induced by oxidative stress and HNE prompted studies of possible antiapoptotic actions of endogenous GSH. When PC12-V cells were pretreated with 1 mm GSH diethyl ester, they were resistant to apoptosis induced by FeSO4, Aβ, and HNE (Fig. 6A). Whereas ∼80% of the cells exhibited apoptotic nuclei after exposure to FeSO4, Aβ, and HNE, only 15–40% of the cells were apoptotic in cultures cotreated with GSH. BSO, an agent that depletes endogenous GSH by inhibiting γ-glutamyl cysteine synthetase (Meister, 1995), induced apoptosis in PC12-V cells, suggesting that depletion of GSH was sufficient to induce apoptosis (Fig.6A). Previous studies have shown that cell expressing Bcl-2 exhibit higher GSH levels than control cells and show less GSH depletion when exposed to oxidative apoptotic insults (Ellerby et al., 1996; Hedley and McCulloch, 1996). GSH levels quantified by the monochlorobimane method were 30–40% greater in PC12-Bcl2 cells, as compared with PC12-V cells (Fig. 6B). After exposure to Aβ, FeSO4, and HNE, levels of GSH were decreased significantly by 40–70% in PC12-V cells (Fig.6B). The levels of GSH in PC12-Bcl2 cells after exposure to Aβ and FeSO4 were significantly greater than in PC12-V cells exposed to the same insults. Similar results were obtained with the enzymatic recycling procedure for quantification of GSH levels (data not shown). As expected, BSO caused a marked depletion of GSH levels in both PC12-V and PC12-Bcl2 cells; GSH levels remained at a higher level in PC12-Bcl2 cells than in PC12-V cells, although the difference was not statistically significant (Fig.6B).
Fig. 6.
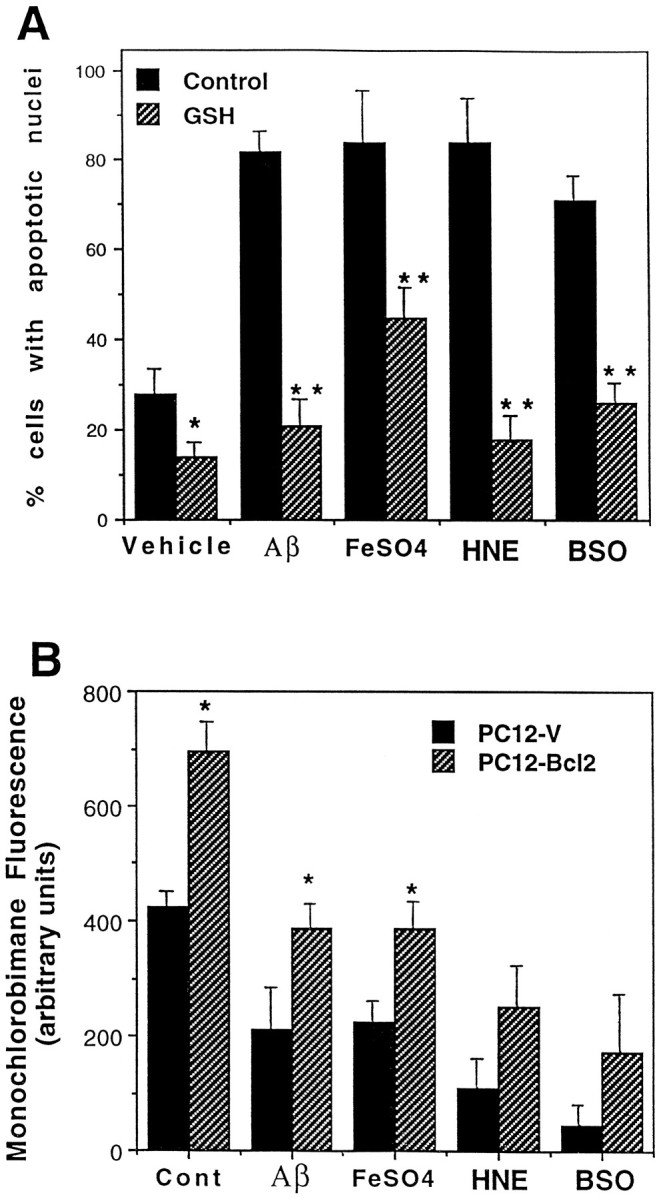
GSH protects PC12 cells against apoptosis induced by oxidative insults and HNE. A, Cultures of PC12-V cells were exposed for 70 hr to Vehicle, 10 μmHNE, 50 μmAβ, or 1 mmFeSO4 in the absence (Control) or presence of 1 mmGSH. Then cultures were fixed and stained with Hoescht dye, and the numbers of cells with condensed and fragmented nuclei were counted. Values are the mean and SEM of determinations made in four separate cultures. *p < 0.01 compared with vehicle control value; **p < 0.01 compared with corresponding control value (ANOVA with Scheffé’s post hoc tests). B, Cultures were exposed for 6 hr to vehicle (Cont), 50 μmAβ, 1 mmFeSO4, 10 μmHNE, or 300 μm buthionine sulfoximine (BSO). Then relative levels of GSH were determined by using the monochlorobimane fluorescence method. Values are the mean and SEM of determinations made in four separate cultures. *p < 0.01 compared with corresponding value for PC12-V cells (ANOVA with Scheffé’s post hoctests).
DISCUSSION
The present findings demonstrate that the lipid peroxidation product HNE can induce apoptosis in PC12 cells and primary rat hippocampal neurons and suggest that HNE is a mediator of oxidative stress-induced apoptosis. HNE induced loss of plasma membrane phosphatidylserine asymmetry (detected by annexin-V binding) and nuclear condensation and DNA fragmentation in PC12 cells and hippocampal neurons. HNE-induced cell death was prevented by cycloheximide, actinomycin-D, and aurintricarboxylic acid, consistent with a role for macromolecular synthesis and endonuclease activity in the cell death process. Although we recently reported that low concentrations of cycloheximide that do not cause sustained blockade of protein synthesis can protect neurons against excitotoxic and oxidative insults by a mechanism that apparently involves stimulation of antioxidant pathways (Furukawa et al., 1997), the concentration of cycloheximide used in the present study (10 μm) causes an essentially complete and sustained blockade of protein synthesis (Furukawa et al., 1997). In addition, PC12 cells expressing the antiapoptotic gene product Bcl-2 were resistant to apoptosis induced by HNE. The concentrations of HNE that induced apoptosis (2–10 μm, estimated from dot-blot analyses) are within the range of concentrations (1–100 μm) known to be generated after exposure of non-neuronal cells (Esterbauer et al., 1991) and neurons (Mark et al., 1997) (present study) to oxidative insults. Among aldehydic products of lipid peroxidation, HNE seems to be a unique inducer of apoptosis, because eight other aldehydes did not induce apoptosis in PC12 cells or hippocampal neurons. These findings are consistent with a recent report showing that HNE, but not other aldehydes, is toxic to cultured human neuroblastoma cells (Mark et al., 1997).
Apoptotic concentrations of FeSO4 and Aβ induced the formation of HNE–protein conjugates, as detected by both Western blot and immunocytochemical analyses. Antioxidants that suppress lipid peroxidation protected PC12 cells against apoptosis induced by FeSO4 and Aβ but did not protect against apoptosis induced by HNE, consistent with HNE acting downstream of lipid peroxidation in the apoptotic pathway induced by oxidative stress. GSH, which was shown previously to detoxify HNE (Spitz et al., 1990;Esterbauer et al., 1991), protected PC12 cells and hippocampal neurons against apoptosis induced by HNE and oxiative insults. Beaver and Waring (1995) reported that decreases in levels of intracellular GSH precede apoptosis in mouse thymocytes exposed to dexamethasone, thapsigargin, and gliotoxin; treatment with 1 mm GSH prevented apoptosis, suggesting a role for GSH depletion in the apoptotic process. Ratan et al. (1994) showed that macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in cultured cortical neurons by increasing GSH levels. We found that GSH levels were higher in PC12 cells expressing Bcl-2 than in PC12-V cells in untreated cultures and after exposure to Aβ, FeSO4, and HNE. Collectively, the data suggest that endogenous GSH may contribute to the resistance of cells expressing Bcl-2 to oxidative insults.
Levels of TBARS fluorescence and HNE–protein conjugates induced by FeSO4 and Aβ were lower in PC12 cells expressing Bcl-2, consistent with data showing that Bcl-2 suppresses lipid peroxidation induced by oxidative insults in a hypothalamic tumor cell line (Kane et al., 1993; Myers et al., 1995) and PC12 cells (present study). On the other hand, levels of HNE–protein conjugates resulting from exposure of cells to HNE also were reduced in PC12 cells expressing Bcl-2, suggesting that Bcl-2 suppresses formation of HNE–protein conjugates. An increased level of GSH in cells expressing Bcl-2 could account for our results because GSH plays a central role in the cellular metabolism of HNE (Ishikawa et al., 1986; Spitz et al., 1990). In studies of spinal cord organotypic cultures, Rothstein et al. (1994) showed that downregulation of Cu/Zn-SOD resulted in neuronal apoptosis, which was inhibited by antioxidants. Troy et al. (1996b) reported that downregulation of Cu/Zn-SOD in PC12 cells induces apoptosis, which can be prevented with inhibitors of nitric oxide synthase, suggesting the involvement of peroxynitrite. When taken together with our data, the latter findings suggest that there may be at least two mechanisms whereby oxidative stress induces apoptosis, one involving nitric oxide production and peroxynitrite formation and the other involving hydroxyl radical formation, lipid peroxidation, and HNE production. However, the two pathways likely converge at some point because Bcl-2 prevents apoptosis in both cases. Because peroxynitrite is known to induce membrane lipid peroxidation, it will be of interest to determine whether HNE plays a role in apoptotic paradigms that involve nitric oxide production and peroxynitrite formation and, conversely, whether there is a role for peroxynitrite in HNE-induced apoptosis.
Loss of cellular ion homeostasis plays a central role in excitotoxic neuronal death and also may contribute to oxidative stress-induced apoptosis. HNE increases neuronal vulnerability to excitotoxicity (Mark et al., 1997), and loss of calcium homeostasis has been implicated in the apoptotic process in a number of paradigms (Nicotera et al., 1992;Takei and Endo, 1994; Le et al., 1995). Recent findings indicate that neuronal death induced by excitotoxins or oxidative insults can manifest as either apoptosis or necrosis, depending on the severity of the insult (Ankarcrona et al., 1995; Bonfoco et al., 1995) (present study). Although not directly tested in the present study, disruption of calcium homeostasis may contribute to oxidative stress-induced apoptosis. FeSO4, Aβ, and HNE each cause impairment of ion-motive ATPase activities in cultured hippocampal neurons, which is linked to subsequent elevation of [Ca2+]i and cell death (Mark et al., 1995, 1997). The ability of ATA to protect PC12 cells against HNE-induced apoptosis is consistent with a role for calcium in that ATA is an inhibitor of calcium-activated endonucleases (Zhivotovsky et al., 1994) and calpains (Posner et al., 1995), although it may have other actions, also (Zeevalk et al., 1995).
Mitochondrial alterations, including decreases in transmembrane potential and decreased levels of MTT reduction, occur at relatively early stages in the apoptotic process (Zamzami et al., 1996). We found that apoptotic concentrations of Aβ, FeSO4, and Aβ each caused a relatively rapid (2–6 hr) small decrease in levels of MTT reduction and mitochondrial transmembrane potential. Shearman et al. (1995) also found that Aβ rapidly decreases levels of MTT reduction in PC12 cells. Interestingly, the initial modest decrease in levels of MTT reduction occurred in both PC12-V and PC12-Bcl2 cells exposed to oxidative insults and HNE, suggesting that this early mitochondrial alteration is not linked causally to subsequent apoptotic death. Indeed, previous data indicate that functioning mitochondria (and resultant ATP) are required for apoptosis (Slater et al., 1995). HNE also impairs mitochondrial function in synaptosomes (Keller et al., 1997), and GSH attenuates oxidative stress-induced disruption of mitochondrial transmembrane potential in lymphocytes (Pieri et al., 1995), suggesting that HNE contributes to oxidative stress-induced mitochondrial dysfunction.
HNE fulfills several criteria for a mediator of oxidative stress-induced apoptosis, including the following: HNE is produced in neurons exposed to apoptotic oxidative insults, HNE itself can induce apoptosis, and an inhibitor of HNE (GSH) protects neurons against oxidative stress- and HNE-induced apoptosis. Because essentially all vertebrate cells are capable of generating HNE in response to oxidative insults, HNE may serve a general role in apoptosis in many different organ systems. Because it can move readily across membranes and through cells, HNE has the ability to induce alterations in subcellular sites affected in cells undergoing apoptosis. Indeed, HNE has been reported to damage both proteins and DNA (Esterbauer et al., 1991; Martelli et al., 1994).
Finally, our data suggest possible roles for HNE in the pathogenesis of neurodegenerative disorders such as AD and Parkinson’s disease, in which apoptosis may occur (Loo et al., 1993; Su et al., 1994; Stern, 1996). Very recent findings indicate that levels of protein-bound HNE are increased selectively in neurons in the substantia nigra of Parkinson’s patients (Yoritaka et al., 1996) and that levels of free HNE are increased more than twofold in CSF of AD patients (M. A. Lovell and W. R. Markesbery, personal communication). Therefore, further studies of roles of HNE in apoptotic neuronal death are warranted to clarify its role in neurodegenerative disorders.
Footnotes
This work was supported by Grants to M.P.M. from the National Institutes of Health (NS30583 and AG10836), the Alzheimer’s Association (Zenith Award), and the Metropolitan Life Foundation; to D.E.B. from National Institutes of Health (AG12282 and NS25554); and to I.K. by a National Institute on Aging training grant fellowship. We thank J. G. Begley, S. Bose, W. Fu, Y. Goodman, and R. Pelphrey for technical assistance.
Correspondence should be addressed to Dr. Mark P. Mattson, 211 Sanders-Brown Building, University of Kentucky, Lexington, KY 40536-0230.
REFERENCES
- 1.Anderson AJ, Pike CJ, Cotman CW. Differential induction of immediate early gene proteins in cultured neurons by β-amyloid (Aβ): association of c-Jun with Aβ-induced apoptosis. J Neurochem. 1995;65:1487–1498. doi: 10.1046/j.1471-4159.1995.65041487.x. [DOI] [PubMed] [Google Scholar]
- 2.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 3.Barhoumi R, Bailey RH, Burghardt RC. Kinetic analysis of glutathione in anchored cells with monochlorobimane. Cytometry. 1995;19:226–234. doi: 10.1002/cyto.990190306. [DOI] [PubMed] [Google Scholar]
- 4.Bastitatou A, Greene LA. Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: correlation with suppression of endonuclease activity. J Cell Biol. 1991;115:461–471. doi: 10.1083/jcb.115.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beal MF, Ferrante RJ, Henshaw R, Matthews RT, Chan PH, Kowall NW, Epstein CJ, Schulz JB. 3-Nitropropionic acid neurotoxicity is attenuated in copper/zinc superoxide dismutase transgenic mice. J Neurochem. 1995;65:919–922. doi: 10.1046/j.1471-4159.1995.65020919.x. [DOI] [PubMed] [Google Scholar]
- 6.Beaver JP, Waring P. A decrease in intracellular glutathione concentration precedes the onset of apoptosis in murine thymocytes. Eur J Cell Biol. 1995;68:47–54. [PubMed] [Google Scholar]
- 7.Behl C, Davis J, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 8.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 10.Bruce AJ, Mark RJ, Bredesen D, Hensley K, Butterfield DA, Mattson MP. Effects of Bcl-2 overexpression on β-amyloid-induced membrane oxidation and ion-motive ATPase activity. Mol Biol Cell. 1995;6:236a. [Google Scholar]
- 11.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 12.Dugan LL, Sensi SL, Canzoniero LMT, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellerby LM, Ellerby HM, Park SM, Holleran AL, Murphy AN, Fiskum G, Kane DJ, Testa MP, Kayalar C, Bredesen DE. Shift of the cellular oxidation-reduction potential in neural cells expressing Bcl-2. J Neurochem. 1996;67:1259–1267. doi: 10.1046/j.1471-4159.1996.67031259.x. [DOI] [PubMed] [Google Scholar]
- 14.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde, and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 15.Evans P. Free radicals in brain metabolism and pathology. Br Med Bull. 1993;49:577–587. doi: 10.1093/oxfordjournals.bmb.a072632. [DOI] [PubMed] [Google Scholar]
- 16.Ferrari G, Yan CY, Greene LA. N-acetylcysteine (d- and l-stereoisomers) prevents apoptotic death of neuronal cells. J Neurosci. 1995;15:2857–2866. doi: 10.1523/JNEUROSCI.15-04-02857.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furukawa K, Estus S, Fu W, Mattson MP. Neuroprotective action of cycloheximide involves induction of Bcl-2 and antioxidant pathways. J Cell Biol. 1997;136:1137–1150. doi: 10.1083/jcb.136.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodman Y, Mattson MP. Secreted forms of β-amyloid precursor protein protect hippocampal neurons against amyloid β-peptide-induced oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- 19.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid β-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 20.Greenlund LJ, Deckwerth TL, Johnson EM., Jr Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 21.Grune T, Siems WG, Zollner H, Esterbauer H. Metabolism of 4-hydroxynonenal, a cytotoxic lipid peroxidation product, in Ehrlich mouse ascites cells at different proliferation stages. Cancer Res. 1994;54:5231–5235. [PubMed] [Google Scholar]
- 22.Hedley DW, McCulloch EA. Generation of reactive oxygen intermediates after treatment of blasts of acute myeloblastic leukemia with cytosine arabinoside: role of bcl-2. Leukemia. 1996;10:1143–1149. [PubMed] [Google Scholar]
- 23.Hockenbery D, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SH. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 24.Ishikawa T, Esterbauer H, Sies H. Role of cardiac glutathione transferase and of the glutathione S-conjugate export system in biotransformation of 4-hydroxynonenal in the heart. J Biol Chem. 1986;261:1576–1581. [PubMed] [Google Scholar]
- 25.Jesberger JA, Richardson JS. Oxygen free radicals and brain dysfunction. Int J Neurosci. 1991;57:1–17. doi: 10.3109/00207459109150342. [DOI] [PubMed] [Google Scholar]
- 26.Johnson EM, Jr, Greenlund LJ, Akins PT, Hsu CY. Neuronal apoptosis: current understanding of molecular mechanisms and potential role in ischemic brain injury. J Neurotrauma. 1995;12:843–852. doi: 10.1089/neu.1995.12.843. [DOI] [PubMed] [Google Scholar]
- 27.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 28.Keller JN, Mark RJ, Bruce AJ, Blanc EM, Rothstein JD, Uchida K, Mattson MP (1997) 4-Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience, in press. [DOI] [PubMed]
- 29.Kovachich GB, Mishra OP. Lipid peroxidation in rat cortical slices as measured by the thiobarbituric acid test. J Neurochem. 1980;35:1449–1452. doi: 10.1111/j.1471-4159.1980.tb09022.x. [DOI] [PubMed] [Google Scholar]
- 30.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 31.Le W-D, Colom LV, Xie W-J, Smith RG, Alexianu M, Appel SH. Cell death induced by β-amyloid 1–40 in MES 23.5 hybrid clone: the role of nitric oxide and NMDA-gated channel activation leading to apoptosis. Brain Res. 1995;686:49–60. doi: 10.1016/0006-8993(95)00450-5. [DOI] [PubMed] [Google Scholar]
- 32.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2008. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 33.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 35.Mah SP, Zhong LT, Liu Y, Roghani A, Edwards RH, Bredesen DE. The proto-oncogene bcl-2 inhibits apoptosis in PC12 cells. J Neurochem. 1993;60:1183–1186. doi: 10.1111/j.1471-4159.1993.tb03275.x. [DOI] [PubMed] [Google Scholar]
- 36.Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid β-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J Neurochem. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 38.Martelli A, Canonero R, Cavanna M, Ceradelli M, Marinari UM. Cytotoxic and genotoxic effects of five N-alkanals in primary cultures of rat and human hepatocytes. Mutat Res. 1994;323:121–126. doi: 10.1016/0165-7992(94)90085-x. [DOI] [PubMed] [Google Scholar]
- 39.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mattson MP. Effects of microtubule stabilization and destabilization on tau immunoreactivity in cultured hippocampal neurons. Brain Res. 1992;582:107–118. doi: 10.1016/0006-8993(92)90323-2. [DOI] [PubMed] [Google Scholar]
- 41.Mattson MP, Zhang Y, Bose S. Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis, and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp Neurol. 1993;121:1–13. doi: 10.1006/exnr.1993.1066. [DOI] [PubMed] [Google Scholar]
- 42.Mattson MP, Lovell MA, Furukawa K, Markesbery WR. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of [Ca2+]i and neurotoxicity, and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem. 1995a;65:1740–1751. doi: 10.1046/j.1471-4159.1995.65041740.x. [DOI] [PubMed] [Google Scholar]
- 43.Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995b;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- 44.Mattson MP, Furukawa K, Bruce AJ, Mark RJ, Blanc EM. Calcium homeostasis and free radical metabolism as convergence points in the pathophysiology of dementia. In: Wasco W, Tanzi RE, editors. Molecular mechanisms of dementia. Humana; Totowa, NJ: 1996. pp. 103–143. [Google Scholar]
- 45.Meister A. Glutathione metabolism. Methods Enzymol. 1995;251:3–7. doi: 10.1016/0076-6879(95)51106-7. [DOI] [PubMed] [Google Scholar]
- 46.Muller U, Krieglstein J. Prolonged pretreatment with alpha-lipoic acid protects cultured neurons against hypoxic, glutamate-, or iron-induced injury. J Cereb Blood Flow Metab. 1995;5:624–630. doi: 10.1038/jcbfm.1995.77. [DOI] [PubMed] [Google Scholar]
- 47.Musser DA, Oseroff AR. The use of tetrazolium salts to determine sites of damage to the mitochondrial electron transport chain in intact cells following in vitro photodynamic therapy with Photofrin II. Photochem Photobiol. 1994;59:621–626. doi: 10.1111/j.1751-1097.1994.tb09666.x. [DOI] [PubMed] [Google Scholar]
- 48.Myers KM, Fiskum G, Liu Y, Simmens SJ, Bredesen DE, Murphy AN. Bcl-2 protects neural cells from cyanide/aglycemia-induced lipid oxidation, mitochondrial injury, and loss of viability. J Neurochem. 1995;65:2432–2440. doi: 10.1046/j.1471-4159.1995.65062432.x. [DOI] [PubMed] [Google Scholar]
- 49.Nicotera P, Bellomo G, Orrenius S. Calcium-mediated mechanisms in chemically induced cell death. Annu Rev Pharmacol Toxicol. 1992;32:449–470. doi: 10.1146/annurev.pa.32.040192.002313. [DOI] [PubMed] [Google Scholar]
- 50.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pieri C, Recchioni R, Marcheselli F, Moroni F, Marra M, Benatti C. The impairment of mitochondrial membrane potential and mass in proliferating lymphocytes from vitamin E-deficient animals is recovered by glutathione. Cell Mol Biol. 1995;41:755–762. [PubMed] [Google Scholar]
- 52.Poli G, Dianzani MU, Chesseman KH, Slater TF, Lang J, Esterbauer H. Separation and characterization of the aldehydic products of lipid peroxidation stimulated by carbon tetrachloride or ADP-iron in isolated rat hepatocytes and rat liver microsomal suspensions. Biochem J. 1985;227:629–638. doi: 10.1042/bj2270629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Posner A, Raser KJ, Hajimohammadreza I, Yuen PW, Wang KK. Aurintricarboxylic acid is an inhibitor of mu- and m-calpain. Biochem Mol Biol Int. 1995;36:291–299. [PubMed] [Google Scholar]
- 55.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rothstein JD, Bristol LA, Hosler B, Brown RH, Jr, Kuncl RW. Chronic inhibition of superoxide dismutase produces apoptotic death of spinal neurons. Proc Natl Acad Sci USA. 1994;91:4155–4159. doi: 10.1073/pnas.91.10.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shearman M, Hawtin S, Taylor V. The intracellular component of cellular 3-(4:5-dimethylthiazol-2-yl)-2:5-diphenyltetrazolium bromide (MTT) reduction is specifically inhibited by β-amyloid peptides. J Neurochem. 1995;65:218–227. doi: 10.1046/j.1471-4159.1995.65010218.x. [DOI] [PubMed] [Google Scholar]
- 60.Simonian NA, Coyle JT. Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 1996;36:83–106. doi: 10.1146/annurev.pa.36.040196.000503. [DOI] [PubMed] [Google Scholar]
- 61.Slater AF, Stefan C, Nobel I, van den Dobbelsteen DJ, Orrenius S. Signaling mechanisms and oxidative stress in apoptosis. Toxicol Lett. 1994;83:149–153. doi: 10.1016/0378-4274(95)03474-9. [DOI] [PubMed] [Google Scholar]
- 62.Spitz DR, Malcolm RR, Roberts RJ. Cytotoxicity and metabolism of 4-hydroxy-2-nonenal and 2-nonenal in H2O2-resistant cells lines. Do aldehyde by-products of lipid peroxidation contribute to oxidative stress? Biochem J. 1990;267:453–459. doi: 10.1042/bj2670453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 64.Stern G. Parkinson’s disease. The apoptosis hypothesis. Adv Neurol. 1996;69:101–107. [PubMed] [Google Scholar]
- 65.Su JH, Anderson AJ, Cummings B, Cotman CW. Immunocytochemical evidence for apoptosis in Alzheimer’s disease. NeuroReport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 66.Takei N, Endo Y. Ca2+ ionophore-induced apoptosis on cultured embryonic rat cortical neurons. Brain Res. 1994;652:65–70. doi: 10.1016/0006-8993(94)90317-4. [DOI] [PubMed] [Google Scholar]
- 67.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 68.Tietze F. Enzymatic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 69.Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML. The contrasting roles of ICE family proteases and interleukin-1β in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc Natl Acad Sci USA. 1996a;93:5635–5640. doi: 10.1073/pnas.93.11.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Troy CM, Derossi D, Prochiantz A, Greene LA, Shelanski ML. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide–peroxynitrite pathway. J Neurosci. 1996b;16:253–261. doi: 10.1523/JNEUROSCI.16-01-00253.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1993;268:6388–6393. [PubMed] [Google Scholar]
- 72.Uchida K, Toyokuni S, Nishikawa K, Kawakishi S, Oda H, Hiai H, Stadtman ER. Michael addition-type 4-hydroxy-2-nonenal adducts in modified low-density lipoproteins: markers for atherosclerosis. Biochemistry. 1994;33:12487–12494. doi: 10.1021/bi00207a016. [DOI] [PubMed] [Google Scholar]
- 73.Ullrich O, Grune T, Henke W, Esterbauer H, Siems WG. Identification of metabolic pathways of the lipid peroxidation product 4-hydroxynonenal by mitochondria isolated from rat kidney cortex. FEBS Lett. 1994;352:84–86. doi: 10.1016/0014-5793(94)00922-8. [DOI] [PubMed] [Google Scholar]
- 74.Van Engeland M, Ramaekers FCS, Schutte B, Reutelingsperger PM. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry. 1996;24:131–139. doi: 10.1002/(SICI)1097-0320(19960601)24:2<131::AID-CYTO5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 75.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 76.Waeg G, Dimsity G, Esterbauer H. Monoclonal antibodies for detection of 4-hydroxynonenal modified proteins. Free Radic Res Commun. 1996;25:149–159. doi: 10.3109/10715769609149920. [DOI] [PubMed] [Google Scholar]
- 77.Whittemore ER, Loo DT, Cotman CW. Exposure to hydrogen peroxide induces cell death via apoptosis in cultured rat cortical neurons. NeuroReport. 1994;5:1485–1488. doi: 10.1097/00001756-199407000-00019. [DOI] [PubMed] [Google Scholar]
- 78.Wiedau-Pazos M, Trudell JR, Altenbach C, Kane DJ, Hubbell WL, Bredesen DE. Expression of bcl-2 inhibits cellular radical generation. Free Radic Res Commun. 1996;24:205–212. doi: 10.3109/10715769609088018. [DOI] [PubMed] [Google Scholar]
- 79.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zamzami N, Susin SA, Marchetti P, Hirsch T, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zeevalk GD, Schoepp D, Nicklas WJ. Excitotoxicity at both NMDA and non-NMDA glutamate receptors is antagonized by aurintricarboxylic acid: evidence for differing mechanisms of action. J Neurochem. 1995;64:1749–1758. doi: 10.1046/j.1471-4159.1995.64041749.x. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Y, Tatsuno T, Carney JM, Mattson MP. Basic FGF, NGF, and IGFs protect hippocampal neurons against iron-induced degeneration. J Cereb Blood Flow Metab. 1993;13:378–388. doi: 10.1038/jcbfm.1993.51. [DOI] [PubMed] [Google Scholar]
- 83.Zhivotovsky B, Cedervall B, Jiang S, Nicotera P, Orrenius S. Involvement of Ca2+ in the formation of high molecular weight DNA fragments in thymocyte apoptosis. Biochem Biophys Res Commun. 1994;202:120–127. doi: 10.1006/bbrc.1994.1901. [DOI] [PubMed] [Google Scholar]
- 84.Zhong LT, Kane DJ, Bredesen DE. Bcl-2 blocks glutamate toxicity in neural cell lines. Mol Brain Res. 1993;19:353–355. doi: 10.1016/0169-328x(93)90139-g. [DOI] [PubMed] [Google Scholar]