

Abstract
Mutations in the presenilin-1 (PS1) gene is a cause of early- onset familial Alzheimer’s disease (AD). Endogenous PS1 is associated with the endoplasmic reticulum in the cell body of undifferentiated SH-SY5Y neuroblastoma cells. At early stages of neuronal differentiation in rat hippocampal culture, PS1 appears in all neuritic processes and in growth cones. In mature differentiated neurons, PS1 is concentrated in the somatodendritic compartment but is also present at lower levels in axons. A similar localization of PS1 is observed in vivoin neurons of the adult human cerebral cortex. In sporadic AD, PS1 appears in the dystrophic neurites of mature amyloid plaques and co-localizes with a subset of intraneuronal neurofibrillary tangles (NFTs). About 30% of hippocampal NFTs are labeled with a highly specific antibody to the PS1 C-terminal loop domain but not with an antibody to the PS1 N terminus. This observation is consistent with a potential association of the PS1 C-terminal fragment with NFTs, because PS1 is constitutively cleaved to N- and C-terminal fragments in neurons. These results suggest that PS1 is highly expressed and broadly distributed during early stages of neuronal differentiation, consistent with a role for PS1 in neuronal differentiation. Furthermore, the co-localization of PS1 with NFTs and plaque dystrophic neurites implicates a role for PS1 in the diverse pathological manifestations of AD.
Keywords: Presenilin, Alzheimer’s disease, differentiation, degeneration, dendritic, cytoskeleton
The identification of specific genes that give rise to Alzheimer’s disease (AD) has provided a powerful tool for the elucidation of the disease mechanism (Yankner, 1996). The majority of familial early-onset AD is caused by mutations in a new gene family, the presenilins (Sherrington et al., 1995). Most familial early-onset AD is caused by mutations in the presenilin-1 (PS1) gene, which is localized to chromosome 14. A much smaller number of cases are caused by mutations in a highly homologous protein, presenilin-2 (PS2), which is localized to chromosome 1 (Levy-Lahad et al., 1995; Li et al., 1995;Rogaev et al., 1995). AD-causing mutations occur in residues that are highly conserved between PS1 and PS2, suggesting that they may alter presenilin function. Presenilins are eight transmembrane domain proteins that are expressed in the brain and other tissues (Doan et al., 1996; Li and Greenwald, 1996). The presenilins are most homologous to the protein sel-12 from Caenorhabditis elegans (Levitan and Greenwald, 1995). Sel-12 may act to facilitate signaling through the Notch/lin-12 pathway that regulates cell fate decisions during early development. The presenilins exhibit a lesser degree of homology to the C. elegans protein spe-4, which may be involved in protein segregation during spermatogenesis (L’Hernault and Arduengo, 1992). However, the biological functions of the presenilins and the mechanism by which they cause AD are unknown.
Analysis by in situ hybridization suggests that in the brain, PS1 and PS2 mRNAs are expressed at the highest levels in neurons (Kovacs et al., 1996). In this report, we show that endogenous PS1 is highly expressed from the earliest stages of neuronal differentiation and is present in all neuritic processes and in growth cones. PS1 exhibits a predominantly somatodendritic localization in more mature neurons in culture and in the adult brain but is still present at lower levels in axons. In the brains of patients with AD, PS1 appears in plaque dystrophic neurites and co-localizes with a subset of intraneuronal neurofibrillary tangles. These findings suggest a role for PS1 in neuronal differentiation and in the diverse pathological manifestations of AD.
MATERIALS AND METHODS
Antibodies. The PS1 antibody 231 (Ab 231) is a rabbit antiserum raised against a synthetic peptide corresponding to residues 2–20 of PS1 (Sherrington et al., 1995) conjugated to keyhole limpet hemocyanin. Antibody R28 is a rabbit antiserum raised against a bacterial glutathione S-transferase GST fusion protein containing residues 263–407 of PS1, corresponding to the loop region between the predicted sixth and seventh transmembrane domains. The GST fusion protein was expressed in Escherichia coli; inclusion bodies were isolated and solubilized in 0.1% SDS and used for immunization of rabbits as described (Harlow and Lane, 1988). Antibody preabsorptions were performed as described (Busciglio et al., 1993) using the PS1 peptide (residues 2–20) for Ab 231 and the PS1 fusion protein (residues 263–407) for Ab R28. The mouse monoclonal antibody to amyloid β protein (β1) has been described (Schrader-Fischer and Paganetti, 1996). Other antibodies used include monoclonal anti-BiP (clone 10C3, 1:50; Stressgen), a polyclonal antibody to the Golgi marker JE4 (Gabuzda et al., 1994), and monoclonal antibodies to phosphorylated tau (PHF-1, 1:500) (Greenberg and Davies, 1990), nonphosphorylated tau (tau-1, 1:200; Boehringer Mannheim), MAP-2 (clone AP20, 1:500; Sigma), and GFAP (clone GA5, 1:1000; Boehringer Mannheim).
Cell culture. Primary cultures of rat hippocampal neurons were established from embryonic day 18 fetuses as described previously (Lorenzo and Yankner, 1994). Primary human cortical cultures were established from 15- to 17-week-gestation fetal abortuses as described previously (Busciglio and Yankner, 1995). Rat and human cortical cultures were plated on poly-l-lysine-coated dishes and maintained in DMEM containing 10% supplemented calf serum (HyClone). Forty-eight hours after plating the medium was changed to DMEM containing N2 supplements (Gibco-BRL). Cells were harvested for Western blotting at day 7 after plating. Primary human skin fibroblasts were obtained from the Coriell Institute for Medical Research and were maintained in DMEM containing 10% fetal bovine serum. Cells were harvested for Western blotting in 4% SDS, 20% glycerol, and 0.1m Tris-HCl, pH 6.8, supplemented with protease inhibitors (Complete tablets, Boehringer Mannheim). COS-1 cells were grown in 35 mm dishes and transfected using LipofectAMINE (Gibco-BRL) by addition of 0.8 μg of PS1 cDNA in the cytomegalovirus promoter-based pRK-7 vector (Gorman et al., 1990). Controls were transfected with pRK-7 without the PS1 insert. Cells were harvested for Western blotting 36 hr after transfection. Detergent-extracted samples were sheared with a 22 gauge needle and centrifuged at 14,000 × g for 20 min, and the protein concentration of the supernatant was determined (Bio-Rad protein assay kit).
Western blotting. Fifteen micrograms of protein per lane were separated by 4–20% PAGE without previous heating and electrotransferred to a polyvinylidene difluoride membrane. Western blotting was performed with antibodies 231 and R28 at dilutions of 1:3000 followed by incubation with peroxidase-conjugated anti-rabbit IgG and detection by enhanced chemiluminescence (Amersham).
Immunocytochemistry. Cell cultures were fixed in 4% paraformaldehyde/120 mm sucrose in PBS for 30 min. For double labeling, coverslips were permeabilized with 0.02% Triton X-100 in PBS and blocked with 5% albumin in PBS for 1 hr. The coverslips were then incubated with both primary antibodies for 12 hr at 4°C, washed three times with PBS, and incubated for 1 hr at room temperature with Texas Red-conjugated anti-rabbit IgG (Jackson Labs) and fluorescein-conjugated anti-mouse IgG (Vector Laboratories). Fluorescence was visualized with a Nikon fluorescence microscope. Formalin-fixed human brain tissue was embedded in paraffin, and 5–8 μm sections were obtained from the frontal and parietal cortices, hippocampus, and parahippocampal gyrus. Sections were pretreated with 2% hydrogen peroxide followed by incubation in Target unmasking fluid (Signet Laboratories) for 10 min at 90°C. After blocking in 5% albumin in PBS for 30 min, sections were incubated in primary antibody overnight at 4°C. The sections were then incubated with biotin-conjugated secondary antibodies and developed with streptavidin–alkaline phosphatase and Fast Red (Signet). For double labeling, sections were incubated with the second primary antibody followed by incubation with a peroxidase-conjugated secondary antibody. The second antibody reaction was developed using a metal-enhanced DAB reaction kit (Pierce). Some sections were lightly counterstained with cresyl violet after immunostaining. For double label immunofluorescence of paraffin sections, the first antibody was visualized using an enzyme-based fluorescence kit (ELF-AP kit, Molecular Probes). Sequential double labeling was performed using a secondary antibody conjugated to Cascade Blue (Molecular Probes). Both secondary antibodies were visualized by UV light with a Hoechst filter. Under these conditions, the green ELF and blue Cascade signals are readily distinguished from the tissue autofluorescence, which is pale white.
RESULTS
Expression of PS1 in transfected cells and primary neurons
The PS1 protein was examined in neuronal and non-neuronal cells using Ab 231 directed against residues 2–20 of the PS1 N terminus and Ab R28 directed against residues 263–407 of the PS1 C-terminal loop region. COS-1 cells were transiently transfected with the PS1 cDNA, and cell lysates were analyzed by Western blotting. Ab 231 detected a 45–50 kDa species corresponding to the predictedMr of full-length PS1. The 45–50 kDa species was markedly increased by transfection and was abolished by preabsorption of the antibody with the antigenic peptide (Fig.1A). In addition, transfected cells showed higher molecular weight PS1 aggregates and lower molecular weight PS1 derivatives. In contrast, untransfected COS cells and primary cultures of human fibroblasts, human cortical neurons, and rat hippocampal neurons showed a major 28–30 kDa N-terminal derivative of PS1 that was abolished by antibody preabsorption (Fig.1A). Western blotting with Ab R28, which is directed against the PS1 C-terminal loop, showed full-length PS1 in transfected COS cells and an additional 20–22 kDa species; both PS1 species were abolished by antibody preabsorption (Fig. 1B). In untransfected COS cells and primary neuronal cultures, the 20–22 kDa band was the predominant species recognized by Ab R28. The full-length PS1 species was detectable at only low levels in primary neuronal and fibroblast cultures using either antibody. These results suggest that PS1 undergoes constitutive proteolytic cleavage to N- and C-terminal fragments in human neurons and in other nontransfected cell types. Antibodies 231 and R28 were not immunoreactive with PS2 in Western blots of COS cells transfected with the PS2 cDNA (data not shown).
Fig. 1.

Expression of PS1 in transfected and primary cell cultures. A, Western blot analysis of cell lysates with Ab 231 directed against the PS1 N terminus. B, Western blot analysis with Ab R28 directed against the PS1 C-terminal loop.Lanes: −, wild-type COS cells; PS1, PS1-transfected COS cells; PS1/Preab, PS1-transfected COS cells blotted with preabsorbed antibody; Fib, primary human fibroblasts; Fib/preab, fibroblasts blotted with preabsorbed antibody; Neurons (Cort), cultured human cortical neurons; Neurons (Cort)/Preab, cultured human cortical neurons blotted with preabsorbed antibody;Neurons (Hipp), cultured rat hippocampal neurons. Note the predominant 28–30 kDa N-terminal and 20–22 kDa C-terminal PS1 fragments in nontransfected cells and primary neurons.
Immunofluorescence microscopy of endogenous PS1 in the human neuroblastoma cell line SH-SY5Y demonstrated a reticular and perinuclear distribution characteristic of the endoplasmic reticulum (ER) (Fig. 2A). The ER localization of endogenous PS1 was confirmed by double labeling for PS1 and BiP, a marker of the rough endoplasmic reticulum. Confocal microscopy demonstrated co-localization of PS1 with the ER marker BiP (Fig.2B,C). Endogenous PS1 did not co-localize with a marker of the Golgi complex (data not shown). These results suggest that endogenous PS1 is localized predominantly to the endoplasmic reticulum in an undifferentiated neuronal cell line.
Fig. 2.

PS1 is a resident protein of the endoplasmic reticulum in SH-SY5Y neuroblastoma cells. A, SH-SY5Y neuroblastoma cells show a reticular pattern of PS1 immunoreactivity (Ab R28) characteristic of the ER. B, C, Localization of PS1 in the endoplasmic reticulum is confirmed by double label immunofluorescence with the PS1 C-terminal loop antibody R28 (B) and anti-BiP (C). Confocal microscopy demonstrates the overlap of both antigens in an image section taken 5 μm above the substrate plane. PS1 does not co-localize with the Golgi marker JE4 in SH-SY5Y cells (data not shown). Scale bars, 10 μm.
Broad distribution of PS1 during neuronal differentiation
To investigate the pattern of appearance of PS1 during neuronal differentiation, the localization of PS1 was determined in primary E18 rat hippocampal cultures at different stages of neuronal differentiation. Immunofluorescence microscopy of rat hippocampal cultures using both the N- and C-terminal loop antibodies showed predominantly neuronal labeling; astrocytes were only very weakly labeled. After 1 d in culture, hippocampal pyramidal neurons exhibited primary neuritic processes that labeled intensely for PS1 (Fig. 3A). PS1 was also detected in growth cones in filopodia-like structures (Fig. 3B). After several days in culture, hippocampal neuritic processes became polarized into axons and dendrites that can be distinguished morphologically and by immunoreactivity for tau and MAP-2, respectively. Double labeling for PS1 and either MAP-2 or tau in 10-d-old cultures demonstrated that PS1 was concentrated in the MAP-2-positive somatodendritic compartment (Fig. 4C,D). However, tau-positive axons were also clearly immunoreactive for PS1, but at lower levels (Fig.4A,B). The specificity of staining was confirmed by preabsorption of the PS1 antibody with the antigenic PS1 peptide or fusion protein, which abolished immunoreactivity (Fig. 3C). A similar pattern of PS1 immunoreactivity was observed in differentiated cultures of primary human cortical neurons (data not shown). These results suggest that PS1 is broadly distributed in neurons during early stages of differentiation and is concentrated in the somatodendritic compartment in mature neurons.
Fig. 3.

Localization of PS1 during the initial stages of neuronal differentiation. A, Rat hippocampal neurons after 1 d in culture show PS1 immunoreactivity homogeneously distributed in the cell body and primary neuritic processes. Immunocytochemistry was performed with Aβ 231 to the PS1 N terminus. Scale bar, 10 μm. B, Examination at a higher magnification shows that PS1 is present in neuritic growth cones. Scale bar, 3 μm. C, Negative immunostaining is obtained after preabsorption of the antibody with the antigenic PS1 peptide.
Fig. 4.

Polarized distribution of PS1 after neuronal differentiation. Rat hippocampal neurons were cultured for 10 d and then double labeled for PS1 using Ab R28 to the PS1 C-terminal loop (A) and tau using Ab tau-1 (B). Arrows show co-localization of PS1 and tau in axons. Double labeling for PS1 (C) and MAP-2 (D) using Ab AP20 shows that PS1 is predominantly localized to the somatodendritic compartment (arrows) in differentiated neurons. Scale bars, 10 μm.
Association of PS1 with neurofibrillary tangles and dystrophic neurites in Alzheimer’s disease
The distribution of PS1 in the adult human brain was examined in paraffin-embedded sections by immunocytochemistry with the antibodies to the N terminus and the C-terminal loop. Using either antibody, PS1 immunoreactivity was detected predominantly in neurons of the cortex and hippocampus (Fig. 5A,B). Astrocytes did not exhibit significant levels of staining (Fig. 5D,E). Immunofluorescence microscopy of hippocampal pyramidal neurons at high magnification demonstrated a somatodendritic localization of PS1, similar to that observed in differentiated neuronal cultures (Fig.5C). However, neuropil staining for PS1 was also observed, consistent with an axonal component at low levels, similar to that detected in differentiated hippocampal cultures (Fig.4A,B). Thus, the localization of PS1 in the adult brain is similar to that observed in differentiated neuronal cultures and suggests that PS1 is broadly distributed in neuritic processes.
Fig. 5.
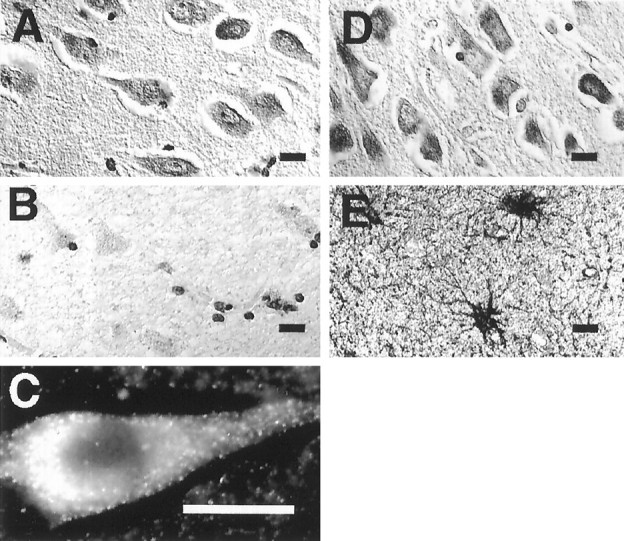
Localization of PS1 in the human brain.A, PS1 immunoreactivity in the perikarya of pyramidal neurons in layer CA1 of the hippocampus. Shown is immunocytochemical staining with the PS1 C-terminal loop antibody R28. B, Preabsorption of Ab R28 with the PS1 fusion protein (PS1 residues 263–407) abolishes neuronal immunoreactivity. Cellular profiles are still detected by the Nissl counterstain. C, Immunofluorescence microscopy with Ab 231 at a higher magnification shows PS1 immunoreactivity in the cytoplasm and primary dendrite of a pyramidal neuron. Adjacent sections of hippocampus labeled with PS1 Ab 231 (D) and anti-GFAP (E) demonstrate the absence of detectable PS1 in astrocytes. Sections were derived from the hippocampus of a normal 60-year-old man. Scale bar, 10 μm.
We then determined whether the localization of PS1 is perturbed in the brain in AD. To determine the spatial relationship of PS1 to neurofibrillary tangles (NFTs), immunocytochemical labeling was performed on adjacent hippocampal sections from cases of sporadic AD with the monoclonal antibody PHF-1 (Greenberg and Davies, 1990), which is specific for tau phosphorylated at Ser-396 and Ser-404, and antibodies to the N terminus or C-terminal loop domain of PS1. The antibody to the PS1 C-terminal loop specifically labeled a subpopulation of intraneuronal NFTs (Fig.6B,C). The specificity of PS1 staining was confirmed by loss of staining after antibody preabsorption with the antigenic fusion protein. Quantitative analysis of adjacent sections stained with PHF-1 or the PS1 antibody R28 showed that 29 ± 2% (mean ± SD) of intraneuronal NFTs were positive for PS1 in three separate cases of sporadic late-onset AD. In contrast, the N-terminal PS1 antibody (Ab 231) showed the normal PS1 somatodendritic staining pattern in AD hippocampal sections but did not clearly label NFTs in sections adjacent to those in which NFTs were labeled by the C-terminal antibody (Fig. 6C,D). To confirm that the structures labeled by the C-terminal PS1 antibody were NFTs, double label immunofluorescence was performed with antibody PHF-1 to phosphorylated tau and either the N- or C-terminal PS1 antibodies. PHF-1-immunoreactive NFTs were not labeled by Ab 231 against the PS1 N terminus, although N-terminal PS1 immunoreactivity was present in perikarya of tangle-bearing neurons (Fig. 7C, arrows). However, when double labeling was performed with PHF-1 and the PS1 C-terminal loop antibody, double-labeled NFTs that stained light blue were clearly apparent (Fig. 7D, arrow). Because PS1 is constitutively processed to 28–30 kDa N-terminal and 20 kDa C-terminal fragments in human neurons (Fig. 1), these results raise the possibility that the PS1 C-terminal fragment may selectively associate with NFTs.
Fig. 6.

Differential labeling of NFTs by antibodies to the N- and C-terminal loop domains of PS1. Paraffin sections of hippocampus from a case of sporadic late-onset AD were immunocytochemically labeled with antibodies to phosphorylated tau and PS1. A, Labeling of an NFT with antibody PHF-1 to phosphorylated tau.B, Same region of an adjacent section immunostained with Ab R28 to the PS1 C-terminal loop labels an NFT with similar morphology. C, Section showing several NFTs (arrows) labeled by PS1 Ab R28. D, Section adjacent to the one shown in C immunostained with Ab 231 to the PS1 N terminus shows somatodendritic staining (arrow). NFTs are not labeled.
Fig. 7.

Association of PS1 with amyloid plaques and NFTs in AD. Shown are sections of hippocampus from an individual with sporadic AD. A, Double labeling with Ab 231 to PS1 (brown) and the monoclonal antibody β1 to Aβ (red) shows that PS1-positive neurons (arrow) surround amyloid plaques (arrowhead). B, Analysis of a single mature plaque by double label immunofluorescence for PS1 (green) and Aβ (dark blue). Note that PS1 appears in dystrophic neurites, which are labeledgreen for PS1 or light blue from the overlap of PS1 and Aβ. A low level of PS1 immunoreactivity appears in the amyloid core (arrowhead), which is staineddark blue for Aβ. C, Double labeling for phosphorylated tau (dark blue, Ab PHF-1) and the N-terminal epitope of PS1 (green, Ab 231). Two neurons with NFTs (arrows) are labeled for both phosphorylated tau and PS1, but the N-terminal PS1 Ab does not double label NFTs, which are labeled with Ab PHF-1. D, Double labeling for phosphorylated tau (dark blue, Ab PHF-1) and the C-terminal loop region of PS1 (green, Ab R28). Shown is an NFT that is double-labeled (arrow, light blue). An example of a neuron showing predominantly PHF-1 labeling (dark blue) is seen in the bottom right corner.
To determine the relationship of PS1 to amyloid deposits in AD, double labeling of cortical sections was performed with antibodies to amyloid β protein (Aβ) and PS1. PS1-positive neurons were detected adjacent to and surrounding amyloid plaques, but significant PS1 immunoreactivity was not detected in most plaque cores (Fig.7A). The spatial relationship of PS1 to plaques was further analyzed by high-resolution double label immunofluorescence for PS1 and Aβ. PS1 was detected in plaque dystrophic neurites and in surrounding cell bodies (Fig. 7B). Very low levels of PS1 immunoreactivity were detected in the region of the amyloid core, which showed strong immunoreactivity for Aβ (Fig. 7B, arrowhead). Thus, PS1 is selectively associated with dystrophic neurites in plaques, where it could potentially play a role in amyloid deposition or in the degenerative process.
DISCUSSION
These experiments suggest that during the early stages of neuronal differentiation PS1 is localized in the cell body and in all neuritic processes. After differentiation, PS1 is concentrated in the somatodendritic region but is still detected at lower levels in axons. These findings provide information on the localization of endogenous PS1 in neurons in culture and in vivo and therefore provide information that is different than that obtained in transfected cells or in transgenic mice, in which PS1 is overexpressed, possibly altering its localization. A previous study suggested that PS1 is restricted to the cell body and dendrites in the NT2N neuronal cell line after infection with a recombinant PS1 Semliki virus (Cook et al., 1996). However, the overexpression of PS1 in this system and its detection by epitope tagging precluded the detection of endogenous PS1. In contrast, our findings suggest that endogenous PS1 appears in both axons and dendrites. Furthermore, the widespread distribution of PS1 in neurites and growth cones during the early stages of neuronal differentiation raises the possibility that PS1 may play a role in neuronal differentiation. This possibility is supported by the observation that proteolytic processing of PS1 is regulated by neuronal differentiation and exhibits a brain-specific pattern (Hartmann et al., 1997).
PS1 is constitutively cleaved to N- and C-terminal fragments in human and rat neurons (Fig. 1). Although full-length PS1 is present at high levels in transfected cells, it is present at low levels in cultured neurons and other nontransfected cell lines. These results are in agreement with recent reports that demonstrated constitutive cleavage of PS1 in cell lines and in the mouse brain (Lee et al., 1996;Thinakaran et al., 1996). Thus, it is likely that PS1 detected in neuritic processes is composed predominantly of the N- and C-terminal proteolytic cleavage products. Because both N- and C-terminal antibodies show identical PS1 recognition patterns in culture and in the normal adult human brain, it is likely that the N- and C-terminal fragments are coordinately transported in neuritic processes. This could occur if the N- and C-terminal fragments form a complex or are complexed with other membrane proteins.
PS1 has previously been localized to the ER and Golgi complex in transfected cells (Kovacs et al., 1996). The results reported here confirm an ER localization for endogenous PS1 in nontransfected SH-SY5Y neuroblastoma cells. However, it remains to be determined whether PS1 in neurons is localized exclusively to the ER. We have observed that the rough ER marker BiP does not co-localize with PS1 in the distal neuritic processes of differentiated neurons (unpublished results). However, PS1 may still be associated with smooth ER in these compartments. Smooth ER has been demonstrated in all neuritic processes in cultured rat hippocampal neurons, as well as in some growth cones (Terasaki et al., 1994; Krijnse-Locker et al., 1995). Additional ultrastructural studies will be required to determine the precise localization of PS1 in neurites. Nevertheless, the presence of PS1 in distal neurites suggests that PS1 may be involved in functions that are unrelated to protein synthesis and processing in the ER.
In the brains of individuals with sporadic AD, PS1 is present in neurons that are adjacent to and surround amyloid plaques. Using a new technique for high-resolution immunofluorescence microscopy on paraffin sections, we have localized PS1 in the dystrophic neurites of mature plaques; much lower levels of PS1 immunoreactivity were detected in plaque cores. It has been reported previously that significant immunoreactivity for PS1 could be detected in amyloid plaque cores (Wisniewski et al., 1995). Although the difference between this result and ours may reflect differences in antibody specificity, we were unable to demonstrate high levels of PS1 immunoreactivity in plaque cores with antibodies to either the PS1 N terminus or C-terminal loop. However, the presence of PS1 in plaque dystrophic neurites raises the possibility that PS1 may be involved in the process of amyloid deposition. This possibility is supported by a recent report, which showed that plasma levels of the highly amyloidogenic 42 amino acid form of Aβ are increased in individuals that harbor PS1 mutations, and that cultured fibroblasts from these individuals secrete higher levels of Aβ42 than controls (Scheuner et al., 1996).
PS1 co-localized with a subset of intraneuronal NFTs in the hippocampi of patients with sporadic late-onset AD. The specificity of this association was suggested by the antibody preabsorption controls. The proportion of labeled NFTs (about 30%) was quite similar in three separate cases of late-onset sporadic AD. It will be of interest to determine whether the proportion of PS1-positive tangles is increased in cases of early-onset familial AD associated with PS1 mutations. Another recent study suggested that NFTs are labeled by an antibody to PS1 (Murphy et al., 1996). These authors did not detect PS1 labeling in any normal brain structures, a result that is difficult to interpret given the widespread distribution of PS1 in the brain, as demonstrated in this report and in other studies (Cribbs et al., 1996; Kovacs et al., 1996; Lee et al., 1996; Giannakopoulos et al., 1997). NFTs were specifically labeled by our antibody to the PS1 C-terminal loop but not by our antibody to the PS1 N terminus. Because PS1 is constitutively cleaved to N- and C-terminal fragments, our results are consistent with the possibility that the C-terminal fragment may selectively associate with cytoskeletal elements. Recent analysis of the membrane topography of PS1 suggests that the large hydrophilic loop is cytoplasmic (Doan et al., 1996; Li and Greenwald, 1996). The hydrophilic loop may therefore be accessible to the cytoskeleton. Further biochemical studies will be required to determine whether PS1 binds directly to NFTs or cytoskeletal elements, and whether PS1 plays a role in cytoskeletal function. The association of PS1 with amyloid plaques and NFTs raises the possibility that PS1 may be involved in the pathogenesis of both of these degenerative lesions.
Footnotes
This work was supported by National Institutes of Health Grants NS33325 and NS30352, Alzheimer’s Association Grant IIRG-95-009, and a grant from Sandoz Pharma Ltd. to B.A.Y. and by National Institutes of Health Mental Retardation Research Center Core Grant P30-HD18655. H.H. is a fellow of the Deutsche Forschungsgemeinschaft. J.B. was supported in part by the University of Connecticut. We thank Pieter Dikkes for assistance and Joseph Rogers, Lucie Sue, and the Sun Health Research Institute for supplying autopsy human brain tissue.
Correspondence should be addressed to Dr. Bruce A. Yankner, The Children’s Hospital, Enders 260, 300 Longwood Avenue, Boston, MA 02115.
Dr. Busciglio’s present address: University of Connecticut Health Center, 263 Farmington Avenue, Farmington CT 06030.
REFERENCES
- 1.Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature. 1995;378:776–779. doi: 10.1038/378776a0. [DOI] [PubMed] [Google Scholar]
- 2.Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of β-amyloid in the secretory pathway in neuronal and non-neuronal cells. Proc Natl Acad Sci USA. 1993;90:2092–2096. doi: 10.1073/pnas.90.5.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook DG, Sung JC, Golde TE, Felsenstein KM, Wolczyk BS, Tanzi RE, Trojanowski JQ, Lee VM-Y, Doms RW. Expression and analysis of presenilin 1 in a human neuronal system: localization in cell bodies and dendrites. Proc Natl Acad Sci USA. 1996;93:9223–9226. doi: 10.1073/pnas.93.17.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cribbs DH, Chen LS, Bende SM, LaFerla FM. Widespread neuronal expression of the presenilin-1 early-onset Alzheimer’s disease gene in murine brain. Am J Pathol. 1996;148:1797–1806. [PMC free article] [PubMed] [Google Scholar]
- 5.Doan A, Thinakaran G, Borchelt DR, Slunt HH, Ratovitsky T, Podlisny M, Selkoe DJ, Seeger M, Gandy SE, Price DL, Sisodia SS. Protein topology of presenilin 1. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 6.Gabuzda D, Busciglio J, Chen LB, Matsudaira P, Yankner BA. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem. 1994;269:13623–13628. [PubMed] [Google Scholar]
- 7.Gorman CM, Gies DR, McCray G. Transient production of proteins using an Adenovirus transformed cell line. DNA Prot Eng Techn. 1990;2:3–10. [Google Scholar]
- 8.Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harlow E, Lane D. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1988. [Google Scholar]
- 10.Hartmann H, Busciglio J, Baumann K-H, Staufenbiel M, Yankner BA (1997) Developmental regulation of presenilin-1 processing in the brain suggests a role in neuronal differentiation. J Biol Chem, in press. [DOI] [PubMed]
- 11.Kovacs DM, Fausett HJ, Page KJ, Kim T-W, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco W. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 12.Krijnse-Locker J, Parton RG, Fuller SD, Griffiths G, Dotti CG. The organization of the endoplasmic reticulum and the intermediate compartment in cultured rat hippocampal neurons. Mol Biol Cell. 1995;6:1315–1332. doi: 10.1091/mbc.6.10.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee MK, Slunt HH, Martin LJ, Thinakaran G, Kim G, Gandy SE, Seeger M, Koo E, Price DL, Sisodia SS. Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J Neurosci. 1996;16:7513–7525. doi: 10.1523/JNEUROSCI.16-23-07513.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levitan D, Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature. 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 15.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu C-E, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu Y-H, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 16.L’Hernault SW, Arduengo PM. Mutation of a putative sperm membrane protein in Caenorhabditis elegans prevents sperm differentiation but not its associated meiotic divisions. J Cell Biol. 1992;119:55–68. doi: 10.1083/jcb.119.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Greenwald I. Membrane topology of the C. elegans SEL-12 presenilin. Neuron. 1996;17:1015–1021. doi: 10.1016/s0896-6273(00)80231-7. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Ma J, Potter H. Identification and expression analysis of a potential familial Alzheimer disease gene on chromosome 1 related to AD3. Proc Natl Acad Sci USA. 1995;92:12180–12184. doi: 10.1073/pnas.92.26.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy GM, Forno LS, Ellis WG, Nochlin D, Levy-Lahad E, Poorkaj P, Bird TD, Jiang Z, Cordell B. Antibodies to presenilin proteins degect neurofibrillary tangles in Alzheimer’s disease. Am J Pathol. 1996;149:1839–1846. [PMC free article] [PubMed] [Google Scholar]
- 21.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, Mar L, Sorbi S, Nacmias B, Placentini S, Amaducci L, Chumakov I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, St. George-Hyslop PH. Familial Alzheimer’s disease in kindred with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 22.Scheuner D, Eckman M, Jensen X, Song M, Citron N, Suzuki TD, Bird J, Hardy M, Hutton W, Kukull E, Larson E, Levy-Lahad M, Viitanen E, Peskind P, Poorkaj G, Schellenberg R, Tanzi W, Wasco L, Lanfelt Selkoe D, Younkin S. Secreted amyloid β-protein similar to that in the senile plagues of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 23.Schrader-Fischer G, Paganetti P. Effect of alkalinizing agents on the processing of the β-amyloid precursor protein. Brain Res. 1996;716:91–100. doi: 10.1016/0006-8993(96)00002-9. [DOI] [PubMed] [Google Scholar]
- 24.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Li G, Holman K, Tsuda T, Mar L, Foncin J-F, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St. George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 25.Terasaki M, Traverse Slater N, Fein A, Schmidek A, Reese TS. Continuous network of endoplasmic reticulum in cerebellar Purkinje neurons. Proc Natl Acad Sci USA. 1994;91:7510–7514. doi: 10.1073/pnas.91.16.7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thinakaran G, Borcheit DR, Lee MK, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey A, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 27.Wisniewski T, Palha JA, Ghiso J, Frangione B. S182 protein in Alzheimer’s disease neuritic plaques. Lancet. 1995;346:1366. doi: 10.1016/s0140-6736(95)92379-9. [DOI] [PubMed] [Google Scholar]
- 28.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]