

Abstract
The role of glutamate receptors in regulating programmed neuronal death and deafferentation-induced neuronal death in the brainstem auditory nuclei was studied by in ovo drug administration to chick embryos. The nucleus laminaris (NL) undergoes programmed developmental cell death of 19% between embryonic day 9 (E9) and E17. The AMPA/kainate receptor antagonist CNQX, when administered at doses of 200–300 μg/d from E8 to E15, prevented programmed neuronal death in NL through at least posthatching day 8, without producing anatomical or behavioral abnormalities. 3-((RS)−2-Carboxypiperazin-4-yl)-propyl-1-phos-phonic acid, an antagonist of NMDA receptors, had no effect on normal cell death in the NL. CNQX, given from E8 to E15 or only from E8 to E10, also blocked the 33% neuronal loss in the nucleus magnocellularis (NM) that follows surgical destruction of the otocyst on E3, a procedure that deafferents NM neurons by preventing formation of the cochlear nerve. Treatment either with CNQX or the more highly selective NBQX from E8 to E10, before the onset of synaptic transmission in NM and NL, was also effective in preventing normal neuronal death in NL. Analysis of the effects of CNQX or NBQX on spontaneous embryonic motility at E10 showed that the doses effective in preventing neuronal death suppressed motility for <8 hr. We conclude that periodic blockade of AMPA/kainate receptors can protect CNS neurons against subsequent programmed cell death or deafferentation-induced death.
Keywords: neuroprotection, glutamate, programmed cell death, apoptosis, auditory system, cochlear nuclei, CNQX, NBQX, CPP, nucleus laminaris, nucleus magnocellularis
Modulation of glutamate receptor function can have profound effects on developing neurons. These effects, which are thought to depend on receptor-mediated changes in cytosolic calcium concentration, include changes in neurite outgrowth, synapse formation, expression of neurotrophins and their receptors, expression of the glutamate receptor, and neuronal survival (Mattson, 1996). Because the NMDA subtype of the glutamate receptor is prominent in many CNS regions during key periods in development and is a major source of calcium influx in many neurons (Mori and Mishina, 1995), the role of this receptor subtype in nervous system development has received extensive study (Scheetz and Constantine-Paton, 1994). The non-NMDA receptors, however, are the main mediators of excitatory neurotransmission (Collingridge and Lester, 1989; Bettler and Mulle, 1995) and in a number of CNS regions are also permeable to calcium ions (Jonas and Burnashev, 1995). Calcium-permeable AMPA/kainate receptors have been shown to confer susceptibility to excitotoxic death in cultured spinal cord neurons (Carriedo et al., 1996), and normal neuronal death in the chick spinal cord is increased by treatments purported to raise intracellular calcium levels (Ciutat et al., 1994). It is not known, however, whether non-NMDA receptors are important in controlling cell number during development.
The present study examined the role of AMPA/kainate receptors in regulating neuron number in two developing brainstem auditory nuclei by means of in ovo drug administration to chick embryos. The nucleus laminaris (NL) and nucleus magnocellularis (NM) are well defined, small, and relatively homogeneous nuclei with large neurons that receive excitatory glutamatergic synaptic input from a single major source. For these reasons, NM and NL have been used extensively in studies of the control of neuron survival by afferent synaptic connections and activity (Rubel and Parks, 1988; Rubel et al., 1990). Calcium-permeable AMPA receptors are prominently expressed in NM and NL during development (Zhou et al., 1995) and mediate normal synaptic transmission (Otis et al., 1995). Normal programmed cell death in NL occurs during a period when these neurons receive rhythmic spontaneous excitation via afferent pathways originating in the cochlea and acting via AMPA receptors (Lippe, 1994). In contrast, NM neurons exhibit no normal death but depend on cochlear nerve afferent activity for survival and growth (Parks et al., 1987b; Rubel et al., 1990). We have found that the competitive antagonists CNQX and NBQX can permanently rescue NL neurons from programmed cell death. In addition, we show that CNQX can rescue NM neurons from deafferentation-induced death. Preliminary reports of some of the findings have been made (Zhou et al., 1994; Solum et al., 1996).
MATERIALS AND METHODS
Subjects. Fertilized White Leghorn eggs obtained from a local supplier were maintained in a forced draft incubator at 37°C in the laboratory. At 48–50 hr of incubation, a lateral window was made in the egg with a sanding wheel. In one group of embryos (the “operated” group), the right otocyst was removed with tungsten needles as described previously (Parks, 1981). No surgery was performed on the remaining embryos (the “unoperated” group). For both operated and unoperated embryos, the window was sealed with paraffin and a glass coverslip, and the egg was returned to the incubator. All embryos were then subjected to drug or saline treatments as described below.
In ovo drug injections. Beginning on embryonic day 8 (E8) and continuing daily thereafter until E10, E12, or E15, embryos were given 0.2 ml of either a drug dissolved in vehicle or vehicle alone by lifting the coverslip, directly applying the solution via a 1 ml syringe to the surface of the chorioallantoic membrane, and resealing the coverslip. Drugs used included CNQX, NBQX, and 3-((RS)−2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP), which were obtained from Tocris–Cookson (Essex, UK). CNQX and CPP were dissolved in 0.9% saline, and NBQX was dissolved in 0.9% saline/10% dimethylsulfoxide (DMSO); controls received either saline (n = 7) or saline/DMSO (n = 2). The period during which CNQX was affecting synaptic transmission in the CNS was estimated by measuring embryonic motility as described below. After the series of drug and/or vehicle applications was completed, embryos were allowed to develop until Hamburger–Hamilton stage 43 (E17), at which time the brains of the embryos were prepared for Golgi or Nissl staining as described below.
Embryonic motility. Chick embryos move spontaneously from approximately E6 through hatching (Oppenheim, 1974), and measurements of this endogenous motility can be used to assess drug effects on the developing CNS. Following the method of Oppenheim et al. (1978), the effects on spontaneous motility of a 200 μg dose of CNQX, a 100 μg dose of NBQX, or 200 μl of vehicle given to E10 embryos with windows were examined. At 0.5, 1, 2, 4, 8, and 24 hr after drug administration, the number of gross movements was counted with a dissecting microscope during a 5 min period of observation while the embryo was maintained at normal incubator temperature. The observer was unaware of the treatment received by the embryos. Data were subjected to a repeated measures ANOVA, with post hoc comparisons by Fisher’s protected least significant difference (PLSD) test.
Golgi staining. The effects of in ovo drug treatments on the form of neurons in NL was assessed by preparing E17 embryos for Golgi–RioHortega staining as described previously (Parks et al., 1987a). Photomicrographic negatives of Golgi-stained NL neurons were digitized, and the images were prepared with Photoshop 3.0 software (Adobe) to optimize contrast and brightness, to crop and size the image for the intended figure, and to add scale bars and lettering. Finished figures were printed on a high-resolution digital printer (Pictography 3000; Fuji Corp.).
Nissl staining. Embryos to be Nissl stained were fixed for 1 hr in Carnoy’s fluid, after which the brains were dissected and placed in fresh Carnoy’s fluid for 2 hr. The brains were then dehydrated and embedded in paraffin. Serial coronal sections (10 μm) through the brainstem auditory nuclei were cut on a rotary microtome; alternate sections were mounted on subbed slides and stained with thionin. Photomicrographs were prepared as described above.
Cell counting and statistical analyses. Expert proponents of different techniques for counting neurons in histological sections agree that each method should be calibrated against counts obtained from serial reconstructions. Calibration is necessary to avoid systematic overestimation of cell number because of double counting (Clarke and Oppenheim, 1995; Coggeshall and Lekan, 1996). One method has been shown by such calibration to estimate neuronal number accurately (i.e., with a systematic overestimation of <2%) in all six regions of the developing chick nervous system where calibration has been performed (Clarke and Oppenheim, 1995). Other empirical evidence, as well as theoretical considerations, also support the accuracy of this technique (Clarke and Oppenheim, 1995). We have used this method to estimate neuronal number in NL and NM as follows. In every fourth 10-μm-thick section through NL and NM, only those cellular profiles with all of the following features (when viewed at 400×) were counted: a large cell body, a clear nucleus with an intact nuclear membrane, and a dark round nucleolus. To estimate neuronal number in NL and NM, these counts were multiplied by 4. Numbers determined in this way for the right and left sides of NL were averaged to provide a mean for each animal; these means were used for generating descriptive and inferential statistics. For NM, a percent difference in neuronal number was calculated for each animal as [1 − (O/U)] × 100, where O equals neuronal number in NM on the operated (right) side of the brain andU equals neuronal number on the unoperated (left) side. These individual percent differences were used for generating descriptive and inferential statistics. Within- and between-observer reliability in counting was >95%. For inferential statistical comparisons involving three or more treatment groups, factorial ANOVA (with post hoc comparisons by Fisher’s PLSD test) was performed. Two-group comparisons were made with the Mann–WhitneyU test.
RESULTS
Effects of drug treatments on survival and neuromorphology
Administration of CNQX at 100–300 μg/d or CPP at 200–400 μg/d from E8 to E15 was relatively well tolerated by the embryos; for example, only a 17% increase in mortality occurred in CNQX-treated embryos when compared with saline-treated controls. Embryos treated with CNQX at 200 μg/d often hatched if allowed to do so (see below) and showed no anatomical or behavioral abnormalities. As shown in Figure 1, treatment of embryos from E8 to E15 with 200 μg/d AMPA/kainate receptor antagonist CNQX, a dose that significantly reduces normal neuronal death in NL (see below), produced no detectable change in the gross morphology of the brainstem or in neuronal form in the brainstem auditory nuclei. NL had its characteristic shape, and its neurons showed the anteromedial-to-caudolateral gradient of dendritic form characteristic of this stage of development (Rubel and Parks, 1988). Similarly normal brainstem morphology was observed in embryos treated with the NMDA receptor antagonist CPP and the AMPA/kainate receptor antagonist NBQX (data not shown). In animals with otocyst removal, a portion of the cochlear nucleus angularis (NA) migrates ventromedially to an ectopic position (Parks, 1979); this abnormal migration of NA was not affected by CNQX or CPP treatments (data not shown).
Fig. 1.
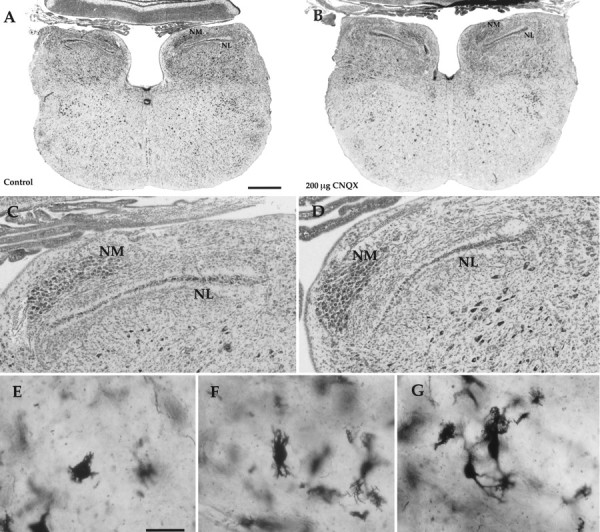
Treatment of chick embryos with 200 μg/d CNQX does not produce gross morphological abnormalities. Photomicrographs show the similar morphology of the brainstem and the auditory nuclei at E17 in control (saline only) and CNQX-treated embryos. A, Coronal section through the brainstem of a control animal. B, Section through the same posterior–anterior level in a CNQX-treated animal. Note the similar sizes and shapes of the brainstem, NL, and NM. C,D, Higher-power views of NL and NM in a control and a CNQX-treated animal, respectively. Medial is to the leftand dorsal is at the top. E–G, Golgi-stained NL neurons from a CNQX-treated animal, revealing the anterior-to-posterior gradient of dendritic form seen in normal animals at E17. E, NL neuron from the anterior part of the nucleus with characteristic multiple short dendrites. F, Neuron more posterior in NL with fewer and longer primary dendrites.G, Two neurons from the posterior NL with characteristic long primary dendrites. Scale bars: A, B, 0.5 mm; E–G, 50 μm.
Normal neuronal death in NL
As shown in Figure 2, neuronal number in NL declines 19% between E9 and E17 in vehicle-treated control animals and remains essentially unchanged thereafter. This developmental decrease in neuronal number is highly reliable [F(3,14)= 9.38; p = 0.0012]. E17 was therefore chosen as the stage at which drug effects on neuronal number in NL would be assessed.
Fig. 2.

Normal developmental neuronal death in NL occurs between E9 and E17. Points and error bars represent the means ± SE of cell counts from three to seven saline-treated control animals at each age.
Effects of drug treatments from E8 to E15 on neuronal number in NL at E17
Figure 3 presents the results of the various treatments from E8 to E15 on neuronal number in NL at E17. A one-factor ANOVA of these data showed a highly significant overall effect of treatment [F(4,26) = 4.58, p = 0.006] and several statistically reliable group differences. Because there were no reliable dose-dependent effects for CPP, data for 100 μg/d (n = 1), 200 μg/d (n = 4), and 400 μg/d (n = 4) treatments were combined for statistical analysis and graphic presentation. As shown in Figure 3, mean NL number in CPP-treated animals did not differ from that in saline-treated controls (n = 9; p = 0.32). Treatment of embryos with CNQX from E8 to E15, however, showed a reliable and dose-dependent rescue of NL neurons otherwise destined to undergo normal neuronal death. At the lowest dose (100 μg/d,n = 3), CNQX had no reliable effect on NL number (p = 0.50), but both 200 μg/d (n = 9) and 300 μg/d (n = 4) CNQX produced statistically reliable increases in NL number over both saline-treated controls and the 100 μg/d CNQX group (p < 0.01). At the higher doses of CNQX, neuronal number in NL was maintained at a level that was not reliably different from that in normal E9 embryos (p = 0.99).
Fig. 3.

Antagonism of AMPA/kainate receptors, but not NMDA receptors, reduces normal neuronal death in NL. Chick embryos were treated with 200 μl/d of saline vehicle or vehicle containing the AMPA/kainate receptor antagonist CNQX or the NMDA receptor antagonist CPP. Bar graphs show means ± SE for NL neuron number at E17. At doses up to 400 μg/d, CPP had no reliable effect. CNQX produced a dose-dependent increase in NL neuron number above control values, with statistically significant effects seen at 200 and 300 μg/d.
Permanence of the rescue of NL neurons by CNQX
We wanted to know whether cessation of drug treatment at E15 would eventually allow NL neurons present at E17 to die. To examine this question, we compared neuronal number in NL at posthatching day 8 (P8) in animals that were treated from E8 to E15 with saline (n = 3) or 200 μg/d CNQX (n = 4) and then allowed to hatch. The number of NL neurons in CNQX-treated animals (2772 ± 43, mean ± SE) was a statistically reliable 28% greater than in controls (2157 ± 133; p = 0.03). Thus, at least through P8, CNQX treatment from E8 to E15 permanently rescues NL neurons from normal death. Prehatching exposure to CNQX did not have any apparent ill effects, because treated chicks seemed anatomically and behaviorally normal throughout posthatching life.
Is suppression of spontaneous activity in the auditory nuclei necessary for the survival-promoting effect of CNQX?
We were also interested in knowing whether the survival-promoting effect of CNQX involved suppression of the spontaneous rhythmic synaptic stimulation that NM and NL receive from the developing cochlea during the period when neuronal death occurs (Lippe, 1994). To assess this, we compared NL number at E17 in embryos treated with 200 μg/d CNQX from E8 to E10 (3 d, n = 3) and from E8 to E12 (5 d, n = 3) with the effects of CNQX treatment from E8 to E15 (8 d, n = 9); synaptic transmission begins in NM at E11 and in NL at E12 (Jackson et al., 1982). Treatment with CNQX for as little as 3 d, from E8 to E10, resulted in a neuronal number in NL at E17 (2829 ± 170, n = 3) that was indistinguishable from the NL number in animals treated with CNQX for 5 (2717 ± 134; n = 3) or 8 (2788 ± 157;n = 7) d (p = 0.80) and also not reliably different from the NL number at E9 (2927 ± 85;n = 3) in normal animals (p = 0.83). To rule out any nonspecific effects of CNQX, we administered NBQX, an AMPA/kainate receptor antagonist that is more potent than CNQX and has no effects at NMDA receptors, at doses of 100–150 μg/d from E8 to E10. As shown in Figure 4, NL number at E17 in NBQX-treated animals (2710 ± 95; n = 7) was also significantly greater than in vehicle-treated controls (2405 ± 48; n = 9). ANOVA showed a highly significant effect of treatment [F(2,16) = 6.48; p = 0.009], with both CNQX (p = 0.008) and NBQX (p = 0.011) showing reliable differences from controls but not from each other (p = 0.43).
Fig. 4.

Treatment from E8 to E10 with CNQX or NBQX results in significant increases in neuronal number in NL at E17. Bar graphs show means ± SE for NL neuron number in animals treated with vehicle only, 200 μg/d CNQX, and 100–150 μg/d NBQX. The drug-treated groups had significantly more NL neurons than the control group but did not differ reliably from each other.
The finding that CNQX or NBQX treatment from E8 to E10, before the onset of afferent-evoked synaptic activity in NM or NL, could prevent normal cell death in NL made it of interest to estimate how long a single dose of these drugs can antagonize non-NMDA receptors. Although quinoxalinedione AMPA/kainate receptor antagonists potently block synaptic transmission in NL in vitro (Zhou and Parks, 1991), a direct in vivo measurement of the effects of CNQX in NL was not feasible. We therefore used the effects of CNQX and NBQX on spontaneous motility, which is mediated by glutamate receptors in the spinal cord (Barry and O’Donovan, 1987), to estimate the length of time that a single dose of the drugs can affect CNS receptors (cf.Vogel and Prittie, 1995). Comparison of motility data from 8 embryos at E10 treated with 200 μg of CNQX, 3 embryos treated with 100 μg of NBQX, and 11 control embryos treated with an equal volume of vehicle showed a highly reliable overall effect of treatment [F(2,13) = 9.97; p = 0.0024], with significant effects for both CNQX (p = 0.005) and NBQX (p = 0.003). Statistically reliable differences for CNQX were seen at 0.5, 1, and 2 hr after treatment, whereas NBQX exerted reliable suppression of motility from 0.5 to 4 hr (Fig. 5). As shown in Figure 5, CNQX and NBQX treatment, at the doses used, produced maximal decreases of ∼60% versus controls, but motility returned to control levels within 4 hr for CNQX (8 hr for NBQX) and remained there until at least 24 hr after treatment. Thus, a dose of quinoxalinedione antagonist that is effective in preventing neuronal death in NL partially suppresses spontaneous motility in chick embryos at E10 for <4 hr after application of the drug.
Fig. 5.

A single dose of CNQX or NBQX suppresses spontaneous motility for <8 hr. The graph shows the mean ± SE numbers of spontaneous movements in E10 embryos observed at intervals after administration of saline (n = 11), 200 μg of CNQX (n = 8), or 100 μg of NBQX (n = 3). CNQX significantly suppressed motility from 0.5 to 2 hr, and NBQX suppressed motility from 0.5 to 4 hr, but motility returned to normal levels thereafter.
Effects of drug treatments on deafferentation-induced neuronal death in the nucleus magnocellularis
By removing the embryonic precursor of the inner ear and acousticovestibular nerve, surgical ablation of the otocyst on E3 prevents innervation of the cochlear nuclei by cochlear nerve axons. Although there is no significant normal neuronal death in NM (Rubel et al., 1976; Parks, 1979), unilateral otocyst removal causes one-third of the neurons in NM to die between E11 and E17 and the survivors to shrink by ∼20% (Parks, 1979). Parks (1979) also showed that neuronal number and size in the normally innervated NM in operated animals did not differ from those in unoperated control animals. In the present study, mean ± SE neuron numbers in NM for the unoperated left sides of control and CNQX-treated animals at E17 were 4124 ± 166 (n = 3) and 4069 ± 207 (n = 8), respectively; these means did not differ reliably. For the operated sides of control and CNQX-treated animals, mean NM numbers were 2724 ± 156 (n = 3) and 3757 ± 157 (n = 8), respectively, a significant difference (p = 0.025).
As illustrated in Figure 6, operated animals that were treated from E8 to E15 with 200 μl of saline vehicle showed a mean ± SE percent difference in NM neuron number of 33.5 ± 5.9% (n = 3), which does not differ significantly from the 30.0 ± 9.2% difference determined previously for E17 animals (Parks, 1979). Treatment of operated animals with 200 μg/d CNQX from E8 to E10 (3 d group) resulted in a mean percent difference of 6.4 ± 3.6% (n = 2), and treatment from E8 to E15 (8 d group) resulted in a mean percent difference in NM neuron number of 6.9 ± 5.5% (n = 6); neither value is reliably >0. A factorial ANOVA comparing percent difference values in the control, 3 d, and 8 d groups showed a significant main effect of CNQX treatment [F(2,8) = 5.41;p = 0.032]. Subsequent comparisons showed that both treatment groups differed significantly from the control group (3 d,p = 0.039; 8 d, p = 0.014) but not from each other. Thus, CNQX treatment for either 3 or 8 d beginning on E10 can rescue essentially all of the NM neurons that would otherwise die after their normal afferent innervation is prevented by removal of the otocyst. Interestingly, although deafferented NM neurons were rescued by CNQX, they remained shrunken (data not shown).
Fig. 6.

CNQX treatment from E8 for 3 or 8 d prevents deafferentation-induced neuronal death in NM. Bar graphs show means ± SE for percent differences between NM neuron number on the operated (O) and unoperated (U) sides of animals that received unilateral removal of the otocyst on E3; percent difference is calculated as [1 − (O/U)] × 100. Control animals (saline only) showed a loss of one-third of the neurons on the operated side of NM, whereas treatment with 200 μg/d CNQX resulted in statistically significant reductions in neuronal loss compared with controls.
DISCUSSION
We have found that CNQX or NBQX given to chick embryos in ovo can prevent essentially all normal neuronal death in NL and that CNQX can prevent the deafferentation-induced death in NM that follows early surgical destruction of the otocyst. CPP, a competitive antagonist of NMDA receptors, had no effect on neuron number in NL. The dose-dependent rescue of neurons by quinoxalinediones is likely to be permanent, because it persists for at least 1 week after hatching. Treatment with CNQX from E8 to E10 is as effective as treatment from E8 to E15 in rescuing NL and NM neurons, and NBQX can also significantly suppress cell death in NL when given from E8 to E10. A single 200 μg dose of CNQX or 100 μg dose of NBQX given at E10 reliably suppresses spontaneous motility for <8 hr.
The minimal effective dose of CNQX (200 μg/d) probably results in drug concentrations <10 times the IC50 for this antagonist in NM and NL. A dose of 200 μg would produce a level of ∼14 μm if distributed uniformly throughout the volume of an average egg. This compares with an IC50 value of 1.5 μm for CNQX in blocking synaptic transmission from the cochlear nerve to NM in vitro at E14 (Zhou and Parks, 1992). The affinity of CNQX for chick brain AMPA/kainate receptors expressed in Xenopus oocytes was studied by Zhou et al. (1993), who found pA2 values of 6.56 for CNQX antagonism of kainate-evoked currents and 6.43 for antagonism of AMPA-evoked currents. Although IC50 values have not been determined for suppression of synaptic transmission in NL by CNQX, the values for the closely related quinoxalinedione AMPA/kainate receptor antagonists DNQX and NBQX in NL from E20 to P3 are 5.4 and 1.4 μm, respectively (Zhou and Parks, 1991). As shown by the motility measurements, 200 μg of CNQX or 100 μg of NBQX has only a transient effect on spinal cord synaptic activity at E10. Because there is no reason to believe that the potency of quinoxalinediones is greatly different at non-NMDA synapses in the spinal cord compared with elsewhere in the nervous system, it is reasonable to assume that the single daily dose of CNQX or NBQX effective in rescuing auditory neurons significantly reduced AMPA/kainate receptor-mediated synaptic transmission throughout the CNS for only a few hours.
The dose-dependent effect of CNQX on neuronal survival seems to depend on its action at AMPA/kainate receptors. CNQX is a potent and relatively specific competitive antagonist of non-NMDA receptors. It has 45- and 80-fold less potent effects, respectively, at the agonist- and glycine-binding sites of NMDA receptors than at the agonist binding site of AMPA receptors (Sheardown et al., 1990; Watkins et al., 1990). The failure of the potent and specific competitive NMDA antagonist CPP to affect neuron number in NL, even at doses well above those affecting neuronal survival in the chick spinal cord (Roberts et al., 1993), suggests that CNQX is not acting via NMDA receptors. This conclusion is strongly supported by the finding that NBQX can reduce programmed neuronal death in NL, because this potent AMPA/kainate receptor antagonist is reported to be inactive at both the agonist- and glycine-binding sites of NMDA receptors and to be completely ineffective in blocking NMDA receptor-mediated synaptic responses (Sheardown et al., 1990; Watkins et al., 1990). NBQX is ∼30-fold selective for AMPA receptors over kainate receptors in binding experiments, compared with a fivefold selectivity for CNQX (Sheardown et al., 1990; Watkins et al., 1990), and approximately twice as potent as CNQX at blocking AMPA receptor-mediated synaptic transmission in NM (Zhou and Parks, 1992). Given the present finding that NBQX is approximately twice as potent as CNQX in preventing normal neuronal death in NL, the survival-promoting effects of these drugs are likely to be exerted primarily or exclusively through AMPA receptors.
Although CNQX and NBQX must act initially on non-NMDA receptors in reducing cell death in NL and NM, the possibility remains that a subsequent signal provides the proximal cause of neuronal survival. That is, the survival-promoting effects of the quinoxalinedione antagonists could occur directly via drug binding to AMPA/kainate receptors on NM and NL neurons or via an intermediate signal, arising elsewhere in the embryo, the expression of which is regulated by the activity of AMPA/kainate receptors. Many potential candidates for such a signal exist. A variety of endogenous ligands (including neurotrophins, growth factors, brain and muscle extracts, and protease inhibitors) can affect both normal and deafferentation-induced neuronal death (Oppenheim, 1991; Qin-Wei et al., 1994; Milligan et al., 1995;Mattson and Furukawa, 1996). There is also increasing evidence that neuronal activity can regulate expression of some of these factors, particularly the neurotrophins (Bonhoeffer, 1996; Ghosh, 1996).
The finding that, when given from E8 to E10, CNQX or NBQX can rescue NL neurons and CNQX can rescue deafferented NM neurons provides a tentative suggestion that the effect of these drugs is mediated indirectly. Neurons in NM and NL depend completely on excitatory auditory afferents to generate spontaneous activity (Rubel et al., 1990; Lippe, 1994). Functional synaptic transmission does not begin in NM until E11 and in NL until E12 (Jackson et al., 1982). The motility experiments reported here support the conclusion that a dose of CNQX or NBQX given on E10 would not be capable of suppressing synaptic transmission on E11 (cf. Vogel and Prittie, 1995). Thus, the E8–E10 drug treatments that prevent death of NM and NL neurons between E9 and E17 probably do not block synaptically mediated depolarizations of these neurons. It is possible that CNQX and NBQX block some interaction of ambient glutamate (Haberecht and Redburn, 1996) with AMPA/kainate receptors on NM and NL neurons before the formation of functioning synapses. At least in NM, such a mechanism could not involve calcium-permeable non-NMDA receptors, however, because they do not appear until after E11 (Zhou et al., 1995). In view of this, we now favor the hypothesis that CNQX and NBQX increase production of one or more diffusible factors that act on auditory neurons to promote their survival. It is of course likely that these factors would also promote survival of other types of neurons.
It is particularly interesting that exposure of the embryo to quinoxalinedione antagonists from E8 to E10 permits NL and NM neurons to resist both normal developmental neuronal death and deafferentation-induced death that would otherwise occur primarily between E11 and E17. It is now appreciated that neuronal death is a process influenced by multiple extracellular signals, including neuronal activity, that are integrated to allow or block expression of a cell death program by neurons (Oppenheim, 1991; Linden, 1994;Bredesen, 1996; Henderson, 1996). There is increasing evidence that neurotrophins mediate important aspects of activity-dependent neuronal survival in some systems (Thoenen, 1995; Henderson, 1996; Mattson, 1996). Although neurotrophins and their receptors are essential to the survival of peripheral auditory neurons (Ernfors et al., 1995), little is known about their role in developing central auditory pathways.Hafidi et al. (1996) have shown recently that TrkB and TrkC receptors are localized at postsynaptic sites on central auditory neurons and that TrkB receptors develop in parallel with auditory function in the brainstem auditory system of the gerbil. These authors suggest that brain-derived neurotrophic factor and neurotrophin-3, the ligands for TrkB and TrkC receptors, respectively, may be regulated by neuronal activity in the auditory pathway and may act as survival signals for auditory neurons. Although it is attractive to hypothesize that such a mechanism might be involved in the survival of NM and NL neurons after CNQX or NBQX treatment, there is significant difficulty with this idea. These drugs reduce neuronal depolarization, and, under current models (Bonhoeffer, 1996; Ghosh, 1996), decreased activity would be expected to cause decreased neuronal responsiveness to neurotrophins. The relationship between neural activity and expression of neurotrophic molecules is likely to be more complex than our current conceptions of it (Mattson, 1996). Therefore, it remains possible that periodic, partial blockade of AMPA/kainate receptors during development, as induced in the present experiments, can result in increased expression of survival-promoting factors.
To our knowledge, this is the first report that an antagonist of non-NMDA receptors can promote neuronal survival in vivoduring normal development or can protect neurons from the effects of embryonic deafferentation. The finding that relatively low doses of a quinoxalinedione antagonist given daily for as little as 3 d can completely and permanently prevent subsequent neuronal death, without producing apparent anatomical or behavioral abnormalities, was surprising. This finding suggests that drugs acting on AMPA/kainate receptors might be used: (1) to prevent programmed cell death in the CNS to understand better what purpose is normally served by neuronal loss (Henderson, 1996); and (2) to prevent transneuronal degeneration after neonatal insults to the CNS or sensory receptors. Several types of AMPA/kainate receptor antagonists, including quinoxalinediones, have been developed as potential clinical neuroprotective agents (Lees, 1996), and at least one such compound is reported to be in clinical trials (Koroshetz and Moskowitz, 1996).
Footnotes
This work was supported by research Grant 5 RO1 DC00144 from the National Institute on Deafness and Other Communicative Disorders to T.N.P. We thank Sheryl A. Scott, Dwan A. Taylor, Ning Zhou, and Jie Zhang for valuable assistance.
Correspondence should be addressed to Dr. Thomas N. Parks, Department of Neurobiology and Anatomy, Room 401, Building 531, University of Utah School of Medicine, Salt Lake City, UT 84132.
REFERENCES
- 1.Barry M, O’Donovan MJ. The effects of excitatory amino acids and their antagonists on the generation of motor activity in the isolated chick spinal cord. Dev Brain Res. 1987;36:271–276. doi: 10.1016/0165-3806(87)90030-7. [DOI] [PubMed] [Google Scholar]
- 2.Bettler B, Mulle C. AMPA and kainate receptors. Neuropharmacology. 1995;34:123–139. doi: 10.1016/0028-3908(94)00141-e. [DOI] [PubMed] [Google Scholar]
- 3.Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- 4. Bredesen DE. Keeping neurons alive: the molecular control of apoptosis. The Neuroscientist 2 1996. 181 190,211–216. [Google Scholar]
- 5.Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1966;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciutat D, Esquerda JE, Caldero J. Evidence for calcium regulation of spinal cord motoneuron death in the chick embryo in vivo. Dev Brain Res. 1994;86:167–179. doi: 10.1016/0165-3806(95)00027-b. [DOI] [PubMed] [Google Scholar]
- 7.Clarke PGH, Oppenheim RW. Neuron death in vertebrate development: in vivo methods. Methods Cell Biol. 1995;46:277–321. [PubMed] [Google Scholar]
- 8.Coggeshall RE, Lekan HE. Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol. 1996;354:6–15. doi: 10.1002/(SICI)1096-9861(19960101)364:1<6::AID-CNE2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 9.Collingridge G, Lester RAJ. Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol Rev. 1989;40:143–210. [PubMed] [Google Scholar]
- 10.Ernfors P, VanDeWater T, Loring J, Jaenisch R. Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron. 1995;14:1153–1164. doi: 10.1016/0896-6273(95)90263-5. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh A. With an eye on neurotrophins. Curr Biol. 1996;6:130–133. doi: 10.1016/s0960-9822(02)00442-6. [DOI] [PubMed] [Google Scholar]
- 12.Haberecht MF, Redburn DA. High levels of extracellular glutamate are present in retina during neonatal development. Neurochem Res. 1996;21:285–291. doi: 10.1007/BF02529146. [DOI] [PubMed] [Google Scholar]
- 13.Hafidi A, Moore T, Sanes DH. Regional distribution of neurotrophin receptors in the developing auditory brainstem. J Comp Neurol. 1996;367:454–464. doi: 10.1002/(SICI)1096-9861(19960408)367:3<454::AID-CNE10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 14.Henderson CE. Programmed cell death. Neuron. 1996;17:579–585. doi: 10.1016/s0896-6273(00)80191-9. [DOI] [PubMed] [Google Scholar]
- 15.Jackson H, Hackett JT, Rubel EW. Organization and development of brain stem auditory nuclei in the chick: ontogeny of postsynaptic responses. J Comp Neurol. 1982;210:80–86. doi: 10.1002/cne.902100109. [DOI] [PubMed] [Google Scholar]
- 16.Jonas P, Burnashev N. Molecular mechanisms controlling calcium entry through AMPA-type glutamate receptor channels. Neuron. 1995;15:987–990. doi: 10.1016/0896-6273(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 17.Koroshetz WJ, Moskowitz MA. Emerging treatments for stroke in humans. Trends Pharmacol Sci. 1996;17:227–233. doi: 10.1016/0165-6147(96)10020-1. [DOI] [PubMed] [Google Scholar]
- 18.Lees GJ. Therapeutic potential of AMPA receptor ligands in neurological disorders. CNS Drugs. 1996;5:51–74. [Google Scholar]
- 19.Linden R. The survival of developing neurons: a review of afferent control. Neuroscience. 1994;58:671–682. doi: 10.1016/0306-4522(94)90447-2. [DOI] [PubMed] [Google Scholar]
- 20.Lippe WR. Rhythmic spontaneous activity in the developing avian auditory system. J Neurosci. 1994;14:1486–1495. doi: 10.1523/JNEUROSCI.14-03-01486.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mattson MP. Calcium and free radicals: mediators of neurotrophic factor and excitatory transmitter-regulated developmental plasticity and cell death. Perspect Dev Neurobiol. 1996;3:79–91. [PubMed] [Google Scholar]
- 22.Mattson MP, Furukawa K. Programmed cell life: anti-apoptotic signaling and therapeutic strategies for neurodegenerative disorders. Restor Neurol Neurosci. 1996;9:191–205. doi: 10.3233/RNN-1996-9401. [DOI] [PubMed] [Google Scholar]
- 23.Milligan CE, Prevette D, Yaginuma H, Homma S, Cardwell C, Fritz LC, Tomaselli KJ, Oppenheim RW. Peptide inhibitors of the ICE protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron. 1995;15:385–393. doi: 10.1016/0896-6273(95)90042-x. [DOI] [PubMed] [Google Scholar]
- 24.Mori H, Mishina M. Structure and function of the NMDA receptor channel. Neuropharmacology. 1995;34:1219–1237. doi: 10.1016/0028-3908(95)00109-j. [DOI] [PubMed] [Google Scholar]
- 25.Oppenheim RW. The ontogeny of behavior in the chick embryo. Adv Stud Behav. 1974;5:133–172. [Google Scholar]
- 26.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 27.Oppenheim RW, Pittman R, Gray M, Maderdrut JL. Embryonic behavior, hatching and neuromuscular development in the chick following a transient reduction of spontaneous motility and sensory input by neuromuscular blocking agents. J Comp Neurol. 1978;179:619–640. doi: 10.1002/cne.901790310. [DOI] [PubMed] [Google Scholar]
- 28.Otis TS, Raman IR, Trussell LO. AMPA receptors with high Ca2+ permeability mediate synaptic transmission in the avian auditory pathway. J Physiol (Lond) 1995;482.2:309–315. doi: 10.1113/jphysiol.1995.sp020519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parks TN. Afferent influences on the development of the brain stem auditory nuclei of the chicken: otocyst ablation. J Comp Neurol. 1979;183:665–678. doi: 10.1002/cne.901830313. [DOI] [PubMed] [Google Scholar]
- 30.Parks TN. Changes in the length and organization of nucleus laminaris dendrites after unilateral otocyst ablation in chick embryos. J Comp Neurol. 1981;202:47–57. doi: 10.1002/cne.902020105. [DOI] [PubMed] [Google Scholar]
- 31.Parks TN, Gill SS, Jackson H. Experience-independent development of dendritic form in the avian nucleus laminaris. J Comp Neurol. 1987a;260:312–319. doi: 10.1002/cne.902600211. [DOI] [PubMed] [Google Scholar]
- 32.Parks TN, Jackson H, Conlee JW. Axon-target cell interactions in the developing auditory system. Curr Top Dev Biol. 1987b;21:309–340. doi: 10.1016/s0070-2153(08)60142-8. [DOI] [PubMed] [Google Scholar]
- 33.Qin-Wei Y, Johnson J, Prevette D, Oppenheim RW. Cell death of spinal motoneurons in the chick embryo following deafferentation: rescue effects of tissue extracts, soluble proteins, and neurotrophic agents. J Neurosci. 1994;14:7629–7640. doi: 10.1523/JNEUROSCI.14-12-07629.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts MA, Brunso-Bechtold J, Oppenheim RW. Both NMDA receptor agonists and antagonists increase cell death in ovo in chick spinal motoneurons. Soc Neurosci Abstr. 1993;19:440. [Google Scholar]
- 35.Rubel EW, Parks TN. Organization and development of the avian brainstem auditory system. In: Edelman GM, et al., editors. Auditory function: neurobiological bases of hearing. Wiley; New York: 1988. pp. 3–92. [Google Scholar]
- 36.Rubel EW, Smith DJ, Miller LC. Organization and development of brain stem auditory nuclei in the chicken: ontogeny of n. magnocellularis and n. laminaris. J Comp Neurol. 1976;166:469–490. doi: 10.1002/cne.901660408. [DOI] [PubMed] [Google Scholar]
- 37.Rubel EW, Hyson RL, Durham D. Afferent regulation of neurons in the brain stem auditory system. J Neurobiol. 1990;21:169–196. doi: 10.1002/neu.480210112. [DOI] [PubMed] [Google Scholar]
- 38.Scheetz AJ, Constantine-Paton M. Modulation of NMDA receptor function: implications for vertebrate neural development. FASEB J. 1994;8:745–752. doi: 10.1096/fasebj.8.10.8050674. [DOI] [PubMed] [Google Scholar]
- 39.Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline: a neuroprotectant for cerebral ischemia. Science. 1990;247:571–574. doi: 10.1126/science.2154034. [DOI] [PubMed] [Google Scholar]
- 40.Solum D, Hughes D, Parks TN. Periodic blockade of AMPA/kainate receptors before the onset of synaptic transmission enhances neuronal survival in the chick brainstem auditory system. Soc Neurosci Abstr. 1996;22:45. [Google Scholar]
- 41.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 42.Vogel MW, Prittie J. Purkinje cell dendritic arbors in chick embryos following chronic treatment with an N-methyl-d-aspartate receptor antagonist. J Neurobiol. 1995;26:537–552. doi: 10.1002/neu.480260407. [DOI] [PubMed] [Google Scholar]
- 43.Watkins J, Krogsgaard-Larsen P, Honore T. Structure–activity relationships in the development of excitatory amino acid receptor agonists and competitive antagonists. Trends Pharmacol Sci. 1990;11:25–33. doi: 10.1016/0165-6147(90)90038-a. [DOI] [PubMed] [Google Scholar]
- 44.Zhou N, Parks TN. Pharmacology of excitatory amino acid neurotransmission in nucleus laminaris of the chick. Hear Res. 1991;52:195–200. doi: 10.1016/0378-5955(91)90199-j. [DOI] [PubMed] [Google Scholar]
- 45.Zhou N, Parks TN. Developmental changes in the effects of drugs acting at NMDA or non-NMDA receptors on synaptic transmission in the chick cochlear nucleus (nuc. magnocellularis). Dev Brain Res. 1992;67:145–152. doi: 10.1016/0165-3806(92)90215-i. [DOI] [PubMed] [Google Scholar]
- 46.Zhou N, Hammerland LG, Parks TN. γ-d-Glutamylaminomethyl sulfonic acid (GAMS) distinguishes kainic acid- from AMPA-induced responses in Xenopus oocytes expressing chick brain glutamate receptors. Neuropharmacology. 1993;32:767–775. doi: 10.1016/0028-3908(93)90185-6. [DOI] [PubMed] [Google Scholar]
- 47.Zhou N, Solum D, Zhang J, Parks TN. Reduction of normal neuronal death in chick nucleus laminaris by in ovo treatment with CNQX, an antagonist of AMPA/kainate receptors. Soc Neurosci Abstr. 1994;20:246. [Google Scholar]
- 48.Zhou N, Taylor DA, Parks TN. Cobalt-permeable non-NMDA receptors in developing chick brainstem auditory nuclei. NeuroReport. 1995;6:2273–2276. doi: 10.1097/00001756-199511270-00002. [DOI] [PubMed] [Google Scholar]