

Abstract
Protein phosphorylation and dephosphorylation are believed to functionally couple neuronal activity and synaptic plasticity. Our previous results indicated that postsynaptic Ca2+/calmodulin (CaM) signaling pathways play an important role in setting synaptic strength, and calcineurin (CaN) activity limits synaptic responses during basal synaptic transmission and long-term potentiation expression. The inhibition of postsynaptic CaN activity by FK-506 or an autoinhibitory peptide induced synaptic potentiation in hippocampal slices, which occludes tetanus-induced LTP. FK-506-induced synaptic potentiation was expressed in adult but not young rats. To elucidate mechanisms underlying CaN-inhibited synaptic potentiation, we co-injected certain agents affecting Ca2+ signaling pathways with CaN inhibitors into CA1 neurons. Synaptic potentiation induced by FK-506 was significantly attenuated by co-injecting BAPTA, heparin/dantrolene (inhibitors of intracellular Ca2+ release), a CaM-binding peptide, or CaM-KII/PKC pseudosubstrate peptides. These results indicate that postsynaptic CaN activity can downregulate evoked synaptic transmission by weakening intracellular Ca2+ signals and downstream protein kinase activities.
Keywords: synaptic potentiation, calcineurin inhibitor, calcium/calmodulin, IP3/ryanodine receptors, positive feedback, postsynaptic manipulation
Synaptic transmission is the basic process of information exchange among neurons in the nervous system, and activity-dependent synaptic plasticity, e.g., long-term potentiation (LTP), is believed to be a cellular model of learning and memory in mammalian brain (Bliss and Lynch, 1988; Gustafsson and Wigstrom, 1988;Bliss and Collingridge, 1993; Lisman, 1994; Eichenbaum, 1995). Much recent experimentation has focused on studying the cellular and molecular mechanisms underlying synaptic transmission and synaptic plasticity. One characteristic of synaptic plasticity is that the strength of synaptic transmission can be enhanced or attenuated by certain electrical or chemical protocols. For example, synaptic strengthening is induced by high-frequency stimulation (Bliss and Gardner-Medwin, 1973; Bliss and Lomo, 1973; Bliss and Collingridge, 1993) or activators of protein kinases (Malenka et al., 1986b; Hu et al., 1987; Wang and Feng, 1992; Wang and Kelly, 1995), and synaptic weakening is induced by low-frequency stimulation (Dunwiddie and Lynch, 1978; Ito, 1982). The molecular mechanisms that reset synaptic strength to new levels after these manipulations may require a shift in balance between protein kinase and phosphatase activities, because many studies have indicated that protein phosphorylation and dephosphorylation regulate the magnitude of many diverse biological activities (Nestler and Greengard, 1984; Krebs, 1994). It is well known that the activities of protein kinases are essential for the induction and maintenance of LTP (Akers et al., 1986; Malinow et al., 1988, 1989;Malenka et al., 1989; O’Dell et al., 1991; Feng and Wang, 1992; Wang and Feng, 1992; Fukunaga et al., 1993; Thomas et al., 1994; Wang and Kelly, 1996a), in which protein phosphorylation is believed to upregulate synaptic strength. The converse of this hypothesis is that protein phosphatases (PrP) may contribute to synaptic weakening, which is supported by results showing that PrP activities are required for long-term depression (LTD) (Mulkey et al., 1993, 1994) and that the inhibition of PrP-1, -2A, or -2B can enhance synaptic transmission (Figurov, 1992; Wyllie and Nicoll, 1994; Wang and Kelly, 1996a).
We have shown that calcium/calmodulin (Ca2+/CaM) signaling pathways are important in regulating the output of synaptic transmission by shifting the balance between postsynaptic Ca2+/CaM-dependent protein kinase II (CaM-KII)/protein kinase C (PKC) and calcineurin (CaN) activities. For example, the injection or perfusion of Ca2+/CaM into postsynaptic neurons induces synaptic potentiation (Wang and Kelly, 1995) (J. Wang and P. Kelly, unpublished observations), the postsynaptic injections of CaN inhibitor (a protocol for inhibiting CaN activity) induces synaptic potentiation (Wang and Kelly, 1996a), which we call CaN-inhibited synaptic potentiation, and the inhibition of postsynaptic CaM-KII and PKC attenuates tetanus-induced LTP (Wang and Kelly, 1996a). Because CaN is preferentially activated by low Ca2+/CaM concentrations (KD = ∼0.1–1 nm) compared with CaM-KII (KD = ∼40–100 nm) (Cohen, 1988;Klee and Cohen, 1988; Schulman and Lou, 1989; Klee, 1991), and oscillations in intracellular Ca2+ range from 10 to 100 nm under resting conditions (Carafoli, 1987; Amundson and Clapham, 1993; Ghosh and Greenberg, 1995), interactions of Ca2+ with CaN β-subunit and CaM result in the possibility that CaN is active and limits synaptic strength at a low and stable level under basal conditions. This can explain why postsynaptic injections of Ca2+/CaM primarily activate protein kinases and induce synaptic potentiation without short-term synaptic depression proceeding potentiation (Wang and Kelly, 1995) as well as why postsynaptic injections of CaN inhibitors induce synaptic potentiation (Wang and Kelly, 1996a).
What is the mechanism(s) underlying synaptic potentiation induced by inhibiting postsynaptic CaN activity? It may be simply understood as shifting the balance between protein kinase and phosphatase activities toward enhancing the phosphorylation of protein substrates responsible for synaptic transmission without increasing the absolute activity of protein kinases. In other words, after CaN is inhibited, the action of basal kinase activities on synaptic substrates becomes dominant, because Ca2+-dependent protein kinases are active under basal conditions in hippocampal slices (Ocorr and Schulman, 1991; Huber, 1995). However, the inhibition of postsynaptic CaM-KII and PKC activities do not significantly alter basal synaptic transmission (Tsien et al., 1990; Feng and Wang, 1992; Wang and Kelly, 1996a), implying that basal CaM-KII and PKC activities are below a threshold for strengthening synaptic transmission. Together with observations that tetanic stimulation produces large increases in protein kinase activities relative to basal conditions (Akers et al., 1986; Fukunaga et al., 1993), it seems a little difficult to understand why CaN-inhibited synaptic potentiation, which presumably requires basal kinase activities, is larger than tetanus LTP (Wang and Kelly, 1996a). We propose that inhibiting postsynaptic CaN enhances positive-feedback cascades that amplify the activities of intracellular signaling pathways and protein kinases more than it shifts the balance between protein kinase and phosphatase activities on synaptic substrates. To better understand these cascades, we examined whether intracellular Ca2+ signaling pathways are involved in CaN-inhibited synaptic potentiation. Experiments were conducted by co-injecting agents that block Ca2+ signaling cascades with CaN inhibitors into hippocampal CA1 neurons.
MATERIALS AND METHODS
Transverse hippocampal slices were prepared from male Harlan Sprague Dawley rats (6–7 or 2–3 weeks old) with a Mcllwain tissue chopper in ice-cold standard medium (gassed with 95% O2/5% CO2), as described previously (Wang and Kelly, 1995, 1996a,b). Slices were incubated at 25°C over 1 hr and transferred to a submersion chamber (31°C; 2 ml/min perfusion rate) for electrophysiological experiments. Standard medium contained (in mm): 124 NaCl, 3 KCl, 1.3 NaH2PO4, 26 NaHCO3, 2.4 MgCl2, 2.4 CaCl2, 10 dextrose, and 10 HEPES, pH 7.25. To reduce the effects of GABAA-mediated inhibitory components on excitable synaptic transmission, experiments were conducted in the presence of bicuculline plus picrotoxin (10 μm each). In addition, orthodromic stimulating electrodes were placed as far away from recording sites as possible to avoid evoking monosynaptic GABAergic synaptic activities caused by directly stimulating interneurons. Certain manipulations were used to prevent neuronal hyperexcitability in the presence of GABAergic antagonists as follows. (1) “Isolation” of area CA1 was achieved by cutting presynaptic axons in stratum radiatum but not in oriens/alveus; this optimizes the integrity of CA1 neurons (Wang and Kelly, 1996a) by preventing the stress of neurons from axon injury during preparation of isolated CA1 slices (Kauer et al., 1988; Malenka et al., 1988); and (2) the concentration of Mg2+ was 2.4 mm to limit hyperactivity of excitatory synapses. Under these conditions, seizure activity was never observed during basal synaptic transmission and postsynaptic injections of agents; complex waveforms only occurred after tetanic stimulation in some experiments.
One bipolar tungsten stimulating electrode (12 MΩ) was positioned in stratum radiatum of CA1 area for orthodromically stimulating Schaffer collateral/commissural axons. The frequency of test stimuli was 0.05 Hz; high-frequency tetanic stimulation was composed of 10 trains with 5 sec intervals in which each train contained 10 pulses with 200 Hz frequency. Glass microelectrodes (60–85 MΩ) filled with 2m potassium acetate (KAc) or 2 m KAc plus various agents [FK-506, rapamycin, CaN autoinhibitory peptide (CaN-AIP), Ca2+/CaM, 1,2-bis(2-aminophenoxy)-ethane-N,N,N′,N′-tetra-acetic acid (BAPTA), a mixture of heparin/dantrolene, a CaM-binding peptide (CBP), or CaM-KII/PKC pseudosubstrate inhibitory peptides] were used for current- or voltage-clamp recordings in CA1 pyramidal neurons; pipettes containing 1 m NaCl were routinely used to record field excitatory postsynaptic potentials (f-EPSPs) in stratum radiatum and were positioned near the intracellular recording site to verify the viability of tissue slices and status of synaptic activity.
Experimental results are based only on neurons in which stable recordings were obtained within the initial 2 min after impalement and that exhibited stable membrane potentials between −70 and −73 mV throughout an experiment. During experiments conducted by voltage-clamp recording with 3 KHz sampling rate, membrane potentials were held at −78 mV (i.e., near the GABAA reversal potential), together with application of GABAergic antagonists, to reduce possible contamination in measuring excitatory postsynaptic currents (EPSCs) from mono- and disynaptic inhibitory activities. Baseline values (the first data point in each figure) of synaptic transmission were averaged from initial slopes of synaptic responses (EPSPs, EPSCs, or f-EPSPs) during the first minute after stable intracellular recordings were established and defined as 100%. Values of CaN-inhibited synaptic potentiation were data points 1–2 min before tetanic stimulation (arrows) compared with baseline. Values of LTP were data points 20 min post-tetanus compared with baseline. Values of synaptic depotentiation were data points 20 min post-tetanus compared with potentiation values. Synaptic responses are represented as mean ± SEM. Series and input resistances were monitored throughout all voltage- and current-clamp experiments by measuring responses to 4 mV and 0.1 nA injections (50 msec). Data were obtained with pClamp 5.5 and analyzed with custom software to compute initial EPSP, EPSC, and f-EPSP slopes.t Tests were used for statistical comparisons. Each waveform was averaged from six consecutive responses, and waveforms in each figure were selected from a representative experiment.
CaN-AIP (3 mm stock in distilled water) was diluted to a final concentration of 300 μm in 2 mKAc. Ca2+/CaM mixtures were prepared from CaCl2 and CaM stock solutions and mixed with CaN-AIP; they were diluted to a final concentrations of 20/80 μm and 300 μm in 2 m KAc, respectively. FK-506 and rapamycin were dissolved in 100% ethanol (50 mm stock solutions) and diluted to a final concentration of 50 μmin 2 m KAc. BAPTA was dissolved in 3 m KOH (400 mm in stock, pH 7.2) and diluted to a final concentration of 20 mm in 2 m KAc plus 50 μmFK-506. Heparin and dantrolene were dissolved in 2 m KAc plus 50 μm FK-506 at final concentrations of 300 and 80 μm, respectively. Stock solutions of CBP, or [Ala286]CaM-KII281–302/PKC19–31, were diluted in KAc to final concentrations of 100 μm or 200 μm/100 μm in 2 m KAc, respectively. These solutions filled the entire microelectrode (i.e., back-filling with 2 m KAc was not performed). Bicuculline and BAPTA were obtained from Sigma (St. Louis, MO), and heparin, dantrolene, D(−)-2-amino-5-phosphonopentanoic acid (D-AP5), and picrotoxin were obtained from RBI (Natick, MA). CaM was a gift from Dr. J. Aronowski; CaN-AIP was a gift from Dr. Randall Kincaid (Veritas, Rockville, MD), and FK-506 and rapamycin were gifts from Dr. Stan Stepkowski (University of Texas Medical School at Houston).
RESULTS
Inhibiting postsynaptic CaN induces greater synaptic potentiation than tetanus LTP
Postsynaptic injections of Ca2+/CaM into CA1 neurons induce synaptic potentiation by activating CaM-KII and PKC (Wang and Kelly, 1995), and postsynaptic injections of FK-506 induce synaptic potentiation by inhibiting basal CaN activity (Wang and Kelly, 1996a). Thus, we predicted that the co-injection of Ca2+/CaM plus CaN inhibitor should induce larger synaptic potentiation than that by Ca2+/CaM alone, especially because partial inhibition of postsynaptic CaN activity facilitates the magnitude of tetanus-induced LTP (Wang and Kelly, 1996a). To test this prediction, microelectrodes were used for both intracellular recordings and postsynaptic co-injections of a CaN-AIP (Hashimoto et al., 1990; Perrino, 1995) plus Ca2+/CaM into CA1 neurons of hippocampal slices from adult rats. One typical experiment is shown in Figure1A,a. Postsynaptic co-injection of Ca2+/CaM and CaN-AIP induced synaptic potentiation (solid circles), whereas synaptic responses in simultaneous field potential recordings were unchanged; tetanic stimulation induced LTP of f-EPSPs (open circles). Figure 1A,b shows the average of group results (n = 7). Synaptic potentiation was induced by co-injecting Ca2+/CaM plus CaN-AIP (184 ± 10% relative to baseline values 100%); tetanic stimulation given at 60 min produced a small decrease in synaptic strength (165 ± 8% relative to baseline values, i.e., 19% depotentiation). Simultaneous f-EPSPs are shown in Figure1A,c; synaptic transmission was stable for 60 min and then tetanic stimulation induced LTP (172 ± 6%, n = 7). Compared with Ca2+/CaM-induced synaptic potentiation (183 ± 9%, n = 8) (Wang and Kelly, 1995) (see bar graphinset in Fig. 1A,b), Ca2+/CaM plus CaN-AIP-induced synaptic potentiation was not significantly different (p = 0.594) 30–35 min after the beginning of injections. This result is different from our prediction. One explanation is that the co-injection of Ca2+/CaM plus CaN-AIP may produce an unknown interaction on CaN activity such that synaptic potentiation is primarily attributable to Ca2+/CaM-stimulated CaM-KII and PKC activities.
Fig. 1.

Postsynaptic injections of Ca2+/CaM and/or CaN-AIP induce synaptic potentiation. A, a, Representative experiment showing potentiated EPSPs induced by postsynaptic injection of Ca2+/CaM plus CaN-AIP (solid symbols) and stable synaptic transmission followed by tetanus-induced LTP during simultaneous f-EPSPs (open symbols). A, b, Average synaptic potentiation induced by injecting Ca2+/CaM plus CaN-AIP (n = 7). Inset waveformsshow potentiated EPSPs and unchanged input resistance. Calibration: 15 mV/30 msec. Inset bar graph compares the magnitude between Ca2+/CaM-induced synaptic potentiation (white bar; value from Wang and Kelly, 1995) and Ca2+/CaM plus CaN-AIP-induced synaptic potentiation (gray bar). A, c, Normalized f-EPSP slopes recorded during injection of Ca2+/CaM plus CaN-AIP (n = 7). Waveforms show tetanus-induced LTP. Calibration: 1 mV/20 msec.B, a, Representative experiment showing synaptic potentiation induced by postsynaptic injection of CaN-AIP (solid symbols) and simultaneous f-EPSPs with stable baseline followed by tetanus-induced LTP (open symbols).B, b, Average synaptic potentiation induced by injection of CaN-AIP (n = 6).Waveforms show potentiated EPSPs and unchanged input resistance. Calibration: 15 mV/30 msec. B,c, Normalized f-EPSP slopes recorded during CaN-AIP injections (n = 6). Waveforms show tetanus-induced LTP. Calibration: 1 mV/20 msec. Vertical arrows at 60 min denote tetanic stimulation.
To better understand the effects of CaN-AIP on synaptic activity, we injected CaN-AIP alone into postsynaptic neurons. One typical experiment is shown in Figure 1B,a. The postsynaptic injection of CaN-AIP induced a gradual increase in EPSP slopes (solid circles); simultaneous f-EPSPs were stable under basal conditions and tetanus LTP (open circles). Average results (n = 6) of intracellular recordings during postsynaptic injections of CaN-AIP and simultaneous f-EPSPs are shown in Figures 1B,band 1B,c, respectively. Synaptic potentiation induced by injecting CaN-AIP was 212 ± 14% relative to baseline. Tetanic stimulation to presynaptic axons making synaptic connections on CaN-AIP-injected neurons after 60 min decreased synaptic potentiation (162 ± 8%, i.e., 50% depotentiation). f-EPSP slopes did not change within the 60 min period of CaN-AIP injections, but increased after tetanic stimulation (177 ± 4%). Interestingly, compared with synaptic potentiation induced by injecting Ca2+/CaM plus CaN-AIP, potentiation induced by injecting CaN-AIP alone was significantly greater (p = 0.00133). These results urged us to study the mechanisms underlying synaptic potentiation induced by inhibiting postsynaptic CaN activity.
CaN-inhibited synaptic potentiation is developmentally regulated
Recent reports have shown that the manipulation of inhibiting postsynaptic CaN activity produces different actions on synaptic plasticity. FK-506 or cypermethrin prevents LTD induction in hippocampal slices from young rats (2–3 weeks old) (Mulkey et al., 1994) or LTP induction in hippocampal slices from young guinea pigs (2 weeks old) (Wang and Stelzer, 1994). In these studies, postsynaptic injections of CaN inhibitors did not affect basal synaptic transmission. These results together with our own findings (Fig. 1) (Wang and Kelly, 1996a) suggest that CaN activity regulates basal synaptic transmission only when hippocampal CaN has reached more mature levels at approximately postnatal day 24 (P24) (Tallant and Cheung, 1983; Polli et al., 1991; Wang and Kelly, 1996a). To examine this hypothesis, we injected FK-506 into CA1 neurons in hippocampal slices from either adult or young rats. FK-506 inhibits CaN activity by binding to FK-506-binding proteins (e.g., FKBP-12) and is a much more potent (IC50 = ∼30 nm) (Wiederrecht et al., 1989; Schreiber and Crabtree, 1992; Steiner et al., 1992; Dawson et al., 1994; MacKintosh and MacKintosh, 1994) than CaN-AIP (IC50 = ∼10 μm) (Hashimoto et al., 1990). Under voltage-clamp recordings with pipettes containing 50 μm FK-506, synaptic responses (EPSC slope) gradually increased over 32 min (220 ± 16%; n = 6) (Fig.2A, solid circles) in adult CA1 neurons; subsequent tetanic stimulation produced an attenuation of synaptic potentiation (189 ± 17%, i.e., 31% depotentiation;p = 0.021). Simultaneous extracellular f-EPSPs were stable during the initial 32 min and expressed LTP after tetanic stimulation (155 ± 5%; n = 6) (Fig.2B). In control experiments using 50 μmrapamycin (in 0.1% ethanol/2 m KAc), which combines with the CaN anchoring protein FKBP-12 but does not inhibit CaN activity (Wiederrecht et al., 1989; Schreiber and Crabtree, 1992; MacKintosh and MacKintosh, 1994), synaptic responses were unchanged for 60 min (Fig.2A, gray circles) and were potentiated by tetanic stimulation (184 ± 10%; n = 5, 3 from 22- to 28-d-old rats and 2 from adult rats). These controls indicate that postsynaptic injections of 50 μm rapamycin in 0.1% ethanol into CA1 neurons from young and adult slices did not affect basal synaptic transmission or tetanus LTP.
Fig. 2.

Synaptic potentiation induced by postsynaptic injections of FK-506 is age-dependent. A, Increases in EPSC slopes induced by postsynaptic injections of FK-506 and partial depotentiation after tetanic stimulation (solid symbols;n = 6); gray symbols show stable synaptic transmission and subsequent tetanus-induced LTP during postsynaptic injections of rapamycin (n = 5).Waveforms show FK-506-induced synaptic potentiation (left) and tetanus LTP in rapamycin controls (right). Calibration: 250 pA/30 msec. B, Stable f-EPSP slopes recorded during FK-506 injections and subsequent tetanus-induced LTP. Waveforms show tetanus LTP. Calibration: 1 mV/20 msec. Results in A andB were obtained using slices from adult rats.C, Postsynaptic injections of FK-506 into CA1 neurons in young slices (14–21 d) did not significantly change synaptic transmission and prevented tetanus LTP (n = 6).Inset shows representative waveforms. Calibration: 200 pA/30 msec. D, f-EPSPs during FK-506 injections in young slices show stable synaptic transmission and tetanus LTP.Waveforms show tetanus LTP. Calibration: 1 mV/20 msec.Vertical arrows denote tetanic stimulation.
Next we examined the effect of postsynaptic injections of FK-506 into CA1 neurons on synaptic transmission in hippocampal slices from young rats (2–3 weeks old). As shown in Figure 2C, EPSC slopes did not significantly change during voltage-clamp recordings with pipettes containing 50 μm FK-506, and tetanic stimulation at 32 min induced no synaptic potentiation (n = 6). Simultaneous f-EPSPs showed tetanus-induced LTP (138 ± 7% relative to baseline, n = 6) (Fig.2D). The block of tetanus LTP by FK-506 in young slices was not attributable to the ethanol solvent (see end of preceding paragraph). These results support the notion that the inhibition of postsynaptic CaN activity enhances basal synaptic transmission only in adult animals; i.e., synaptic potentiation induced by inhibiting postsynaptic CaN activity is age-dependent. Our results also support a previous study in which postsynaptic injections of cypermethrin (a CaN inhibitor with IC50 = ∼40 pm) prevented LTP induction in hippocampal slices from young guinea pigs (Wang and Stelzer, 1994), although another study showed that bath application of FK-506 did not affect LTP induction in young rats (Mulkey et al., 1994). It is noteworthy that the magnitude of tetanus LTP (f-EPSP slopes) in slices from adult (155–184%) was significantly greater than that from young rats (138%;p < 0.01). This indicates that mechanisms underlying LTP magnitude are still developing during this early stage.
Synaptic potentiation induced by inhibiting postsynaptic CaN requires Ca2+
One explanation for CaN-inhibited synaptic potentiation is that the basal activity of protein kinases that strengthen synaptic transmission becomes dominant during the inhibition of CaN. We have shown that inhibiting postsynaptic CaM-KII and PKC activities under basal conditions does not alter synaptic strength (Wang and Kelly, 1996a), which suggests that basal CaM-KII and PKC activities are not high enough to modulate basal synaptic transmission, or their function is masked by high CaN activity. Because CaM-KII and PKC activities increase after LTP induction (Akers et al., 1986; Fukunaga et al., 1993) and are required for tetanus-induced LTP (Malinow et al., 1988,1989; Malenka et al., 1989; Feng and Wang, 1992; Wang and Feng, 1992;Wang and Kelly, 1996a), it would seem logical that tetanus-induced LTP should be greater than synaptic potentiation induced by inhibiting postsynaptic CaN. This prediction was not supported by our results, which showed that CaN-inhibited synaptic potentiation is greater in magnitude than tetanus LTP (Wang and Kelly, 1996a) (see also Figs. 1,2A).
Alternatively, inhibiting postsynaptic CaN activity may trigger an amplification cascade (positive feedback) that produces greater CaN-inhibited synaptic potentiation compared with tetanus LTP. Because synaptic potentiation induced by inhibiting postsynaptic CaN activity appears in a few minutes (Figs. 1, 2), it seems likely that the primary mechanisms underlying this potentiation rely on intracellular second messenger pathways rather than on the activation of gene expression. To examine the possibility that increases in intracellular Ca2+ serve as this amplification cascade, we co-injected BAPTA with FK-506 into postsynaptic CA1 neurons. Figure3A shows the time course of synaptic responses (EPSC slopes) observed while injecting 50 μmFK-506 plus 20 mm BAPTA, or 50 μm FK-506 alone. Compared with FK-506-induced synaptic potentiation at 60 min (207 ± 11%; n = 6), the co-injection of BAPTA significantly attenuated FK-506-induced potentiation (117 ± 5%;n = 6, p = 0.00079) and prevented subsequent tetanus-induced LTP (119 ± 8% relative to baseline at 20 min post-tetanus). Simultaneous f-EPSPs showed stable synaptic transmission and tetanus-induced LTP (167 ± 6%). This result indicates that postsynaptic Ca2+ is involved in CaN-inhibited synaptic potentiation. Because the dissociation constant of BAPTA (Kd) for Ca2+is 107 nm (Tsien, 1980), and postsynaptic injection of BAPTA does not affect basal synaptic transmission (Wang and Kelly, 1996b), BAPTA may not efficiently chelate intracellular Ca2+ under resting conditions (10–100 nm). Thus, the block of CaN-inhibited synaptic potentiation by BAPTA indicates that an increase in intracellular Ca2+ may be associated.
Fig. 3.

Postsynaptic co-injection of BAPTA attenuates FK-506-induced synaptic potentiation. A, Gray symbols show FK-506-induced synaptic potentiation without tetanic stimulation (this data also present in Figs. 4C, 5A, 5C) (n = 6); solid symbols show results from co-injections of BAPTA with FK-506, which attenuates FK-506-induced potentiation and blocks tetanus LTP. Waveforms show FK-506-induced synaptic potentiation (left) and its attenuation by BAPTA (right). Calibration: 250 pA/30 msec.B, f-EPSP slopes recorded during co-injection of BAPTA with FK-506 are stable and exhibit tetanus LTP.Waveforms show LTP. Calibration: 1 mV/20 msec.Vertical arrows denote tetanic stimulation.
Ca2+ release by IP3R and RyR is required for FK-506-induced synaptic potentiation
What are potential sources of intracellular Ca2+ required for FK-506-induced synaptic potentiation? The increase of cytoplasmic Ca2+ may come from influx via NMDA receptor channels or voltage-gated calcium channels and/or release from intracellular stores via inositol-1,4,5-triphosphate (IP3) and ryanodine receptor channels (Ghosh and Greenberg, 1995). Our results suggest that voltage-gated calcium channels may not play an important role in FK-506-induced potentiation, because they should not be significantly activated during our experiments under the conditions of voltage-clamp recordings and low frequency (0.05 Hz) of test stimuli without depolarizing pulses (Figs. 2, 3, 4). Reports have shown that phosphatase inhibitors enhanced currents through NMDA receptor channels (Lieberman and Mody, 1994; Wang and Salter, 1994; Wang et al., 1994), and CaN activated by Ca2+ influx shortens opening time (Lieberman and Mody, 1994). It seems reasonable that the inhibition of postsynaptic CaN activity could enhance NMDA receptor currents and produce increases in intracellular Ca2+. To test this possibility, we conducted experiments injecting FK-506 into postsynaptic neurons in the presence of 40 μm D-AP5 (an NMDA receptor antagonist). Hippocampal slices were perfused with D-AP5-containing standard solution starting 30 min before recordings. Figure 4A shows the effect of D-AP5 on postsynaptic injections of FK-506 (50 μm in pipettes). Robust synaptic potentiation was routinely induced by FK-506 (211 ± 12% at 40 min; n = 5) (Fig. 4A). It is interesting that FK-506-induced synaptic potentiation in the presence of D-AP5 appeared not to be attenuated by tetanic stimulation (237 ± 20% at 72 min; p = 0.0383). This is different from our results in the absence of D-AP5 in that tetanic stimulation produced synaptic depotentiation (19–50%, Figs. 1, 2). Simultaneous f-EPSPs showed stable synaptic transmission, and tetanic stimulation failed to induce LTP (127 ± 7% relative to 111 ± 9% before tetanus; p = 0.11) (Fig.4B) in the presence of D-AP5. These results indicate that the postsynaptic Ca2+ requirement for FK-506-induced synaptic potentiation does not depend on NMDA receptor activity; however, the depotentiation of FK-506-induced synaptic potentiation after tetanic stimulation seems to require NMDA receptor activity.
Fig. 4.

FK-506-induced synaptic potentiation does not require NMDA receptor activation and is attenuated by co-injection of heparin/dantrolene. A, Postsynaptic injections of FK-506 in the presence of 40 μm D-AP5; D-AP5 and tetanic stimulation at 40 min did not affect FK-506-induced synaptic potentiation (n = 5). Waveforms show potentiated EPSCs. Calibration: 250 pA/30 msec. B, Stable f-EPSP slopes during FK-506 injections, and tetanic stimulation at 40 min produced no LTP in the presence of D-AP5.Inset shows representative waveforms. Calibration: 1 mV/20 msec. C, Co-injections of heparin/dantrolene with FK-506 attenuate FK-506-induced potentiation and block tetanus-induced LTP (solid symbols, n = 8);gray symbols show FK-506-induced synaptic potentiation (n = 6). Waveforms show FK-506 potentiation (left) and its attenuation by heparin/dantrolene (right). Calibration: 250 pA/30 msec.D, f-EPSP slopes recorded during co-injections of heparin/dantrolene with FK-506 are stable and exhibit tetanus-induced LTP. Inset shows representative waveforms. Calibration: 1 mV/20 msec. Vertical arrows denote tetanic stimulation.
Next we examined the possibility that Ca2+ release from intracellular stores via IP3 and ryanodine receptor channels contributes to FK-506-induced synaptic potentiation. Heparin and dantrolene, which block activities of IP3 and ryanodine receptors, respectively (Ohta, 1990; Smith and Gallacher, 1994), were co-injected with FK-506 into CA1 neurons. Figure 4C shows the time course of synaptic responses (EPSC slopes) while injecting 50 μm FK-506 plus 300 μm heparin/80 μm dantrolene, or FK-506 alone. Compared with FK-506-induced synaptic potentiation at 60 min (207 ± 11%;n = 6), postsynaptic co-injections of heparin/dantrolene significantly attenuated FK-506-induced synaptic potentiation (124 ± 6%; n = 8, p= 0.0009) and prevented additional potentiation by subsequent tetanic stimulation (119 ± 8%). Simultaneous f-EPSPs showed stable synaptic transmission and tetanus-induced LTP (164 ± 9%) (Fig.4D). Postsynaptic injections of heparin plus dantrolene did not affect basal synaptic transmission but did prevent the induction of tetanus LTP (n = 5; data not shown). These results indicate that postsynaptic Ca2+required for FK-506-induced synaptic potentiation is primarily released from intracellular stores via IP3 and ryanodine receptor channels.
Ca2+/CaM signaling pathways and CaM-KII/PKC activities are required for FK-506-induced synaptic potentiation
Because CaM is an important and ubiquitous target of free calcium, i.e., Ca2+/CaM formation during Ca2+ increases, the role of Ca2+/CaM signal pathways in FK-506-induced synaptic potentiation was examined. We first used a high-affinity CBP (Hanley et al., 1988; Kelly et al., 1989) to block intracellular cascades initially triggered by Ca2+/CaM. Microelectrodes containing 100 μm CBP plus 50 μm FK-506 in 2 m KAc were used for intracellular recordings and postsynaptic co-injections into CA1 neurons. Figure5A shows the time course of synaptic responses (EPSC slopes) while injecting FK-506 plus CBP, or FK-506 alone. Compared with FK-506-induced synaptic potentiation after 60 min of injection (207 ± 11%; n = 6), postsynaptic co-injections with CBP significantly eliminated FK-506-induced synaptic potentiation (105 ± 14%; n = 7,p = 0.00004) and prevented subsequent tetanus-induced LTP (97 ± 6%). Simultaneous f-EPSPs showed stable synaptic transmission and tetanus-induced LTP (165 ± 5%) (Fig.5B). These results indicate that biologically active Ca2+/CaM complexes serve as functional elements in FK-506-induced synaptic potentiation.
Fig. 5.

CaM antagonist (CBP) or autoinhibitory peptides of CaM-KII/PKC attenuate FK-506-induced synaptic potentiation. A, Co-injections of CBP with FK-506 block FK-506-induced potentiation and tetanus LTP (solid symbols, n = 7); gray symbols show FK-506-induced synaptic potentiation (n = 6). Waveforms show FK-506 potentiation (left) and its inhibition by CBP (right). Calibration: 250 pA/30 msec. B, f-EPSP slopes recorded during co-injections of CBP with FK-506 are stable and exhibit tetanus-induced LTP. Inset shows representative waveforms. Calibration: 1 mV/20 msec. C, Co-injections of [Ala286]CaM-KII281–302 and PKC19–31 with FK-506 attenuate FK-506-induced potentiation and block tetanus LTP (solid symbols,n = 7); gray symbols show FK-506-induced synaptic potentiation (n = 6).Waveforms show FK-506 potentiation (left) and its attenuation by [Ala286]CaM-KII281–302 and PKC19–31 (right). Calibration: 250 pA/30 msec. D, f-EPSP slopes recorded during co-injections of [Ala286]CaM-KII281–302 and PKC19–31 with FK-506 are stable and exhibit tetanus-induced LTP. Inset shows representative waveforms. Calibration: 1 mV/20 msec. Vertical arrowsdenote tetanic stimulation.
We have shown that Ca2+/CaM-induced synaptic potentiation requires CaM-KII and PKC activities (Wang and Kelly, 1995). In addition, neuronal IP3 and ryanodine receptors, which mediate the mobilization of intracellular Ca2+stores, are phosphorylated by PKC and CaM-KII, and the distinct PKC phosphorylation site on IP3 receptors is dephosphorylated by CaN (Cameron et al., 1995; Furuichi and Mikoshiba, 1995; Hain et al., 1995; Snyder and Sabatini, 1995). The phosphorylation of IP3 receptors by either of these kinases appears to increase their sensitivity to IP3 (Cameron et al., 1995). We speculated that inhibiting postsynaptic CaN activity could result in increased release of Ca2+ from intracellular stores, and subsequent increases in Ca2+/CaM levels and CaM-KII and PKC activities, which then enhance protein phosphorylation and induce synaptic potentiation. To test this possibility, we co-injected autoinhibitory peptides of CaM-KII and PKC, [Ala286]CaM-KII281–302 (100 μm) and PKC19–31 (100 μm), together with 50 μm FK-506 into postsynaptic neurons. Figure 5C shows the time course of synaptic responses (EPSC slopes) while injecting FK-506 plus [Ala286]CaM-KII281–302 and PKC19–31, or FK-506 alone. Compared with FK-506-induced synaptic potentiation at 60 min (207 ± 11%; n = 6), CaM-KII and PKC inhibitors significantly attenuated FK-506-induced synaptic potentiation (123 ± 11%; n = 7,p = 0.0004) and prevented subsequent tetanus-induced LTP (109 ± 11%). Simultaneous f-EPSPs showed stable synaptic transmission and tetanus-induced LTP (173 ± 13%) (Fig.5D). These results indicate that postsynaptic CaM-II and PKC activities are required for synaptic potentiation induced by inhibiting postsynaptic CaN activity.
DISCUSSION
Our results clearly show that the inhibition of postsynaptic CaN activity by injecting CaN-AIP or FK-506 induces synaptic potentiation at CA1 synapses. This potentiation appears to require Ca2+ release from intracellular stores, the participation of Ca2+/CaM, and the activities of CaM-KII and PKC in postsynaptic neurons. Together with biochemical studies showing that IP3 and ryanodine receptors are phosphorylated by PKC and CaM-KII and dephosphorylated by CaN (Cameron et al., 1995; Furuichi and Mikoshiba, 1995; Hain et al., 1995; Snyder and Sabatini, 1995) and the phosphorylation of IP3receptors appears to increase their sensitivity to IP3(Cameron et al., 1995), we outline a scheme responsible for CaN-inhibited synaptic potentiation (Fig. 6). The inhibition of postsynaptic CaN increases the ratio of active CaM-KII and PKC to CaN and facilitates the action of CaM-KII and PKC in phosphorylating protein substrates that contribute to synaptic potentiation (e.g., glutamate receptors) and Ca2+increases from intracellular stores by IP3 and ryanodine receptor channels. Increased intracellular Ca2+promotes the formation of Ca2+/CaM complexes and further activation of CaM-KII and PKC. This network constitutes an amplification cascade (positive feedback) that is triggered by inhibiting CaN activity and may lead to unlimited potentiation of synaptic transmission. However, IP3 and ryanodine receptor-mediated mobilization of intracellular Ca2+is stimulated in a Ca2+-dependent manner with a bell-shaped range of Ca2+ concentrations; i.e., high Ca2+ inhibits Ca2+ release (Bootman and Berridge, 1995; Ehrlich, 1995; Furuichi and Mikoshiba, 1995; Simpson et al., 1995). This may explain why synaptic potentiation appeared to be saturated in our experiments (Figs. 1, 2, 3, 4, 5).
Fig. 6.
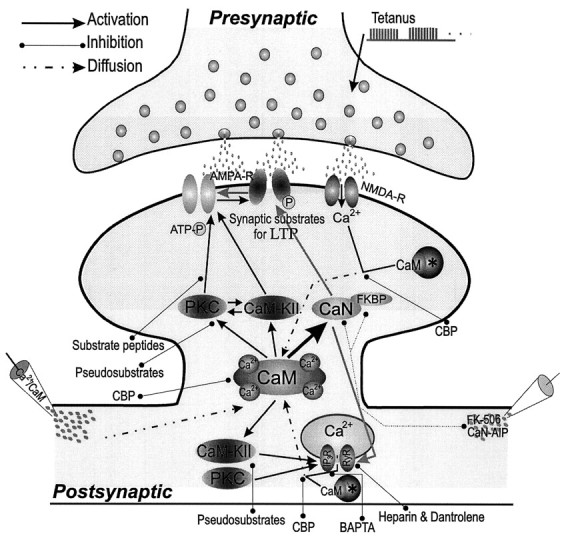
Postsynaptic Ca2+/CaM pathways play key roles in regulating synaptic strength. Ca2+/CaM is involved in three forms of synaptic potentiation, including tetanus LTP, Ca2+/CaM-induced potentiation, and CaN-inhibited potentiation. (1) NMDA receptor (NMDA-R) activation by tetanus permits Ca2+ influx into postsynaptic neuron and increases Ca2+/CaM complexes. (2) Postsynaptic injection of Ca2+/CaM (bottom left) elevates Ca2+/CaM concentrations. (3) Postsynaptic injection of CaN inhibitors (CaN-AIP orFK-506; bottom right) reduce basal CaN activity, facilitates the action of CaM-KII and PKC activities on IP3 and ryanodine receptors, produces more Ca2+ release, and elevates Ca2+/CaM concentrations. The Ca2+and Ca2+/CaM concentrations under basal conditions make CaN highly active (thick arrow), whereas Ca2+/CaM increased by LTP-inducing protocols primarily stimulates CaM-KII and/or PKC. Synaptic substrates phosphorylated by CaM-KII and PKC contribute to synaptic potentiation; the phosphorylation of IP3 and ryanodine receptors by CaM-KII and PKC further facilitate Ca2+ release from intracellular stores and activation of Ca2+/CaM pathways. The decreased dephosphorylation of synaptic substrates during CaN inhibition shifts the equilibrium to phosphorylation for synaptic potentiation. Asterisks, Endogenous CBP;AMPA-R, AMPA receptors; light gray arrows, dephosphorylation. This scheme is supported by our results using postsynaptic co-injections of inhibitors that block these cascades (see text) (Wang and Kelly, 1995, 1996a).
One could argue that CaN-inhibited synaptic potentiation only results from basal CaM-KII/PKC activities (Ocorr and Schulman, 1991; Huber, 1995) instead of from an absolute increase in their activities by positive feedback. Because inhibiting postsynaptic CaM-KII/PKC activities does not significantly alter basal synaptic transmission (Tsien et al., 1990; Feng and Wang, 1992; Wang and Kelly, 1996), basal CaM-KII/PKC activities are not high enough to strengthen synaptic transmission. In addition, tetanus LTP is associated with increases of protein kinase activities (Akers et al., 1986; Fukunaga et al., 1993), and CaN-inhibited synaptic potentiation, which requires CaM-KII/PKC activities, is larger than tetanus LTP (Wang and Kelly, 1996). We suggest that basal CaM-KII and PKC activities may not be the major contributor to the large synaptic potentiation induced by inhibiting CaN activity. Instead, our results suggest that inhibiting CaN not only shifts the equilibrium between protein kinase and phosphatase activities to favor the phosphorylation of substrates responsible for synaptic transmission but, more importantly, triggers positive-feedback cascades that produce long-term increases in synaptic strength.
Postsynaptic application of CaN inhibitors prevented the induction of LTD (Mulkey et al., 1993, 1994), LTP, and synaptic disinhibition (Wang and Stelzer, 1994, 1996) in young animals without changing basal synaptic transmission. Why do postsynaptic injections of CaN inhibitors not induce synaptic potentiation in young animals? Examining the effect(s) of inhibiting CaN on excitatory synaptic transmission in CA1 neurons of hippocampal slices from young rats (14–21 d old), we observed that postsynaptic injections of FK-506 did not induce synaptic potentiation and prevented tetanus LTP (Fig. 2B). The inability of FK-506 to induce potentiation at this age may be attributable to CaN and/or FKBPs being functionally immature, because CaN expression (activity and protein mass) dramatically increases during P10–21 and reaches plateau levels by P24 Polli et al., 1991;Tallant and Cheung, 1983), and the developmental expression of FKBP-12 parallels CaN (P. Kelly and J.-H. Wang, unpublished observations). Therefore, we postulate that CaN activity limits synaptic transmission at basal levels only after CaN and FKBP expression is mature and they are functionally co-localized. Why does inhibiting CaN activity in young animals produce multiple results, i.e., prevent induction of LTD, LTP, and synaptic disinhibition (LTD of inhibitory synapses by tetanus)? We propose that although the developing pattern of Ca2+-dependent protein kinases in brain is similar to that for CaN, their expression is not synchronous (Turner et al., 1984; Kelly and Vernon, 1985; Kelly et al., 1987; Hashimoto, 1988;Yoshida et al., 1988; Hirata et al., 1991) such that shifting the equilibrium between kinases and CaN in postnatal versus adult brain produces different effects on synaptic plasticity. CaN activity may play a dual role in synaptic plasticity during postnatal development and is required for LTD and LTP. In terms of physiological significance, the role of CaN activity in excitatory synaptic potentiation and inhibitory synaptic depression may facilitate the excitatory output of neurons.
Our results indicate that CaN-inhibited synaptic potentiation requires Ca2+ release from intracellular stores by IP3 and ryanodine receptor channels, but not influx via NMDA receptor channels. Because tetanus LTP at CA1 synapses requires NMDA receptor activation (Collingridge et al., 1983a,b), one could argue that CaN-inhibited synaptic potentiation is mechanistically different from tetanus LTP. Because CaN-inhibited synaptic potentiation occludes LTP (Figs. 1, 2A), we suggest that they share common mechanisms. This suggestion is supported in that postsynaptic injections of heparin and dantrolene attenuate CaN-inhibited synaptic potentiation (Fig. 4C) and prevent LTP induction (data not shown), and bath application of dantrolene blocked tetanus LTP (Obenaus et al., 1989). At mechanistic levels, how do we explain the similarities and differences between CaN-inhibited synaptic potentiation and tetanus LTP? Although positive results from occlusion experiments suggest that two forms of synaptic potentiation share common mechanisms (Malenka et al., 1986; Gustafsson et al., 1988;Muller et al., 1988; Wang and Feng, 1992; Wang and Kelly, 1995), this does not mean that they are identical at every step in sequential and/or parallel pathways. The best understanding of occlusion results is that the mechanisms underlying two forms of synaptic potentiation are “common” only in the final key expression element(s). CaN-inhibited synaptic potentiation starts at Ca2+and Ca2+/CaM and employs downstream targets (e.g., CaM-KII, PKC and IP3 receptors), which are essential for tetanus LTP. Thus, CaN-inhibited synaptic potentiation and tetanus LTP share common signaling pathways beginning with increased intracellular Ca2+ and Ca2+/CaM to their final expression element(s) through multiple postsynaptic protein kinase cascades.
Tetanic stimulation given 32–60 min after CaN-inhibited synaptic potentiation does not induce additional synaptic potentiation (Figs. 1,2A). However, tetanus after 4 min of injecting CaN inhibitor increases the magnitude of tetanus LTP (Wang and Kelly, 1996a). We postulate that the postsynaptic injection of FK-506 for 4 min results in partial inhibition of CaN activity and small synaptic potentiation, which is not sufficient to initiate secondary mechanisms such as Ca2+ release from intracellular stores. Under this condition, tetanic stimulation activates protein kinases and induces larger synaptic potentiation than tetanus LTP. The injection of FK-506 over 30 min produces substantial inhibition of CaN and secondary activation of protein kinases to induce large synaptic potentiation. Under this situation, additional kinase activation by tetanus does not enhance synaptic potentiation but causes depotentiation. Thus, the different ratios of postsynaptic protein kinase and phosphatase activities may determine the efficiency of an individual synapse as well as the percentage of sampled synapses that actually express plasticity (potentiation or depotentiation).
Our model may also explain why CaN-inhibited synaptic potentiation is greater than tetanus LTP (Wang and Kelly, 1996a), although one could argue that tetanus LTP induced by our protocols does not reach saturation (i.e., the percentage of sampled synapses that are potentiated by tetanus is lower than that by inhibiting CaN activity). Based on the understandings that protein kinase activities play a critical role in synaptic potentiation and that the percentage of potentiated synapses is determined by the phosphorylation of synaptic substrates, the higher ratio of CaM-KII and PKC to CaN activities produced by inhibiting CaN relative to tetanus increases the proportion of potentiated synapses for a larger synaptic potentiation. However, if the percentage of potentiated synapses in tetanus LTP or CaN-inhibited synaptic potentiation is constant, then higher kinase activities during CaN inhibition enhance synaptic strength to a greater degree at each synapse.
Tetanic stimulation produced partial depotentiation of CaN-inhibited synaptic potentiation (Figs. 1, 2A); this depotentiation did not occur in D-AP5 (Fig. 4A). This indicates that NMDA receptor activation by tetanus during CaN-inhibited synaptic potentiation is responsible for the synaptic depotentiation. Because NMDA receptor activation is essential for induction of tetanus LTP under basal conditions (Collingridge et al., 1983), why does tetanus induce synaptic depotentiation during CaN-inhibited synaptic potentiation? Studies showed that phosphatase inhibitors enhance current through a single NMDA receptor channel (Lieberman and Mody, 1994; Wang and Salter, 1994; Wang et al., 1994) and CaN activated by Ca2+ influx shortens opening time of NMDA channels (Lieberman and Mody, 1994). Although our results show that CaN-inhibited synaptic potentiation does not depend on NMDA receptor channels for Ca2+, inhibiting postsynaptic CaN activity may facilitate NMDA receptor activity. Thus, tetanic stimulation during CaN-inhibited synaptic potentiation may evoke excessive NMDA receptor activity and massive Ca2+influx. Such a great increase in intracellular Ca2+may produce Ca2+-dependent inhibition of Ca2+ release by IP3 and ryanodine receptor channels (Bootman and Berridge, 1995; Ehrlich, 1995; Furuichi and Mikoshiba, 1995; Simpson et al., 1995) and/or neurotoxicity attributable to the superactivities of protein kinases (Meldrum and Garthwaite, 1990), which contribute to the depotentiation of CaN-inhibited synaptic potentiation.
CaN also dephosphorylates type II regulatory subunits of cAMP-dependent protein kinase, inhibitor-1 (I-1) of PrP-1, microtubule-associated proteins, and neuromodulin (Klee, 1991), which may indirectly affect synaptic plasticity. Thus, the equilibrium between CaN and CaM-KII/PKC activities may constitute a pivotal balance in regulating synaptic transmission. Phosphorylated I-1 actively inhibits PrP-1, whereas the dephosphorylation of I-1 by CaN increases PrP-1 activity, which dephosphorylates substrate proteins at synapses (Shields et al., 1985). Thus, inhibiting CaN will also decrease PrP-1 activity, and the attenuation of two phosphatase pathways during CaN-inhibited synaptic potentiation may synergistically facilitate the phosphorylation of substrate proteins that contribute to very large synaptic potentiation.
Footnotes
This study was supported by Nation Institute of Neurological Disorders and Stroke Grant NS-32470. We thank Dr. J. Aronowski for CaM, Dr. Randall Kincaid for CaN-AIP, and Dr. Stan Stepkowski for FK-506 and rapamycin. We also thank Drs. Neal Waxham and Robert Malenka for critical reading and helpful comments on this manuscript before submission.
Correspondence should be addressed to Dr. Paul T. Kelly, Department of Neurobiology and Anatomy, University of Texas Medical School at Houston, P.O. Box 20708, Houston, TX 77225.
REFERENCES
- 1.Akers R, Lovinger D, Colley P, Linden D, Routtenberg A. Translocation of protein kinase C activity may mediate hippocampal long-term potentiation. Science. 1986;231:587–589. doi: 10.1126/science.3003904. [DOI] [PubMed] [Google Scholar]
- 2.Amundson J, Clapham D. Calcium waves. Curr Opin Neurobiol. 1993;3:375–382. doi: 10.1016/0959-4388(93)90131-h. [DOI] [PubMed] [Google Scholar]
- 3.Bliss TVP, Collingridge GL. A synaptic model of memory: LTP in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 4.Bliss TVP, Gardner-Medwin AR. Long-lasting potentiation of synaptic transmission in the dentate area of the unanaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:357–374. doi: 10.1113/jphysiol.1973.sp010274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bliss TVP, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bliss TVP, Lynch MA. Long-term potentiation of synaptic transmission in the hippocampus: properties and mechanisms. In: Landfield PW, Deadwyler SA, editors. Long-term potentiation: from biophysics to behavior. Liss; New York: 1988. pp. 3–72. [Google Scholar]
- 7.Bootman MD, Berridge MJ. The elemental principles of calcium signaling. Cell. 1995;83:675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 8.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. Calcineurin associated with the inositol 1,4,5-triphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell. 1995;83:463–472. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 9.Carafoli E. Intracellular calcium homeostasis. Annu Rev Biochem. 1987;56:395–433. doi: 10.1146/annurev.bi.56.070187.002143. [DOI] [PubMed] [Google Scholar]
- 10.Cohen P. The calmodulin-dependent multiprotein kinase. In: Cohen P, Klee CB, editors. Calmodulin. Elsevier; Amsterdam: 1988. pp. 145–193. [Google Scholar]
- 11.Collingridge GL, Kehl SJ, McLennan H. The antagonism of amino acid-induced excitations of rat hippocampal CA1 neurons in vitro. J Physiol (Lond) 1983a;334:19–31. doi: 10.1113/jphysiol.1983.sp014477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol (Lond) 1983b;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dawson TM, Steiner JP, Lyons WE, Fotuhi M, Blue M, Snyder SH. The immunophilin, FK506 binding protein and cyclophilin, are discretely localized in the brain: relationship to calcineurin. Neuroscience. 1994;62:569–580. doi: 10.1016/0306-4522(94)90389-1. [DOI] [PubMed] [Google Scholar]
- 14.Dunwiddie T, Lynch G. Long-term potentiation and depression of synaptic responses in the hippocampus: localization and frequency dependency. J Physiol (Lond) 1978;276:353–361. doi: 10.1113/jphysiol.1978.sp012239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehrlich BE. Functional properties of intracellular calcium-release channels. Curr Opin Neurobiol. 1995;5:304–309. doi: 10.1016/0959-4388(95)80042-5. [DOI] [PubMed] [Google Scholar]
- 16.Eichenbaum H. The LTP-memory connection. Nature. 1995;378:131. doi: 10.1038/378131a0. [DOI] [PubMed] [Google Scholar]
- 17.Feng TP, Wang J-H. PKC(19–31) and CaMKII(273–302) given together intracellularly to the postsynaptic neuron synergistically block LTP in hippocampal CA1 region. Soc Neurosci Abstr. 1992;18:760. [Google Scholar]
- 18.Figurov A, Boddeke H, Muller D. Enhancement of AMPA-mediated synaptic transmission by protein phosphatase inhibitor Calyculin A in rat hippocampal slices. Eur J Neurosci. 1992;5:1035–1041. doi: 10.1111/j.1460-9568.1993.tb00956.x. [DOI] [PubMed] [Google Scholar]
- 19.Fukunaga K, Stoppini L, Miyamoto E, Muller D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1993;268:7863. [PubMed] [Google Scholar]
- 20.Furuichi T, Mikoshiba K (1995) Inositol 1,4,5 triphosphate receptor-mediated Ca2+ signaling in the brain. J Neurochem 953–960. [DOI] [PubMed]
- 21.Ghosh A, Greenberg ME. calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 22.Gustafsson B, Wigstrom H. Physiological mechanisms underlying long-term potentiation. Trends Neurosci. 1988;11:156–162. doi: 10.1016/0166-2236(88)90142-7. [DOI] [PubMed] [Google Scholar]
- 23.Gustafsson B, Huang YY, Wigstrom H. Phorbol ester-induced synaptic potentiation differs from long-term potentiation in the guinea pig hippocampus in vitro. Neurosci Lett. 1988;85:77–81. doi: 10.1016/0304-3940(88)90432-6. [DOI] [PubMed] [Google Scholar]
- 24.Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem. 1995;270:2074–2081. doi: 10.1074/jbc.270.5.2074. [DOI] [PubMed] [Google Scholar]
- 25.Hanley RM, Means AR, Kemp BE, Shenolikar S. Mapping of calmodulin-binding domain of Ca2+/calmodulin-dependent protein kinase II from rat brain. Biochem Biophys Res Commun. 1988;152:122–128. doi: 10.1016/s0006-291x(88)80688-0. [DOI] [PubMed] [Google Scholar]
- 26.Hashimoto T, Ase K, Sawamura S, Kikkawa U, Saito N, Tanaka C, Nishizuka Y. Postnatal development of a brain-specific subspecies of protein kinase C in rat. J Neurosci. 1988;8:1678–1683. doi: 10.1523/JNEUROSCI.08-05-01678.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimoto Y, Perrino BA, Soderling TR. Identification of an autoinhibitory domain in calcineurin. J Biol Chem. 1990;265:1924–1927. [PubMed] [Google Scholar]
- 28.Hirata M, Saito N, Kono M, Tanaka C. Differential expression of the BI- and BII-PKC subspecies in the postnatal developing rat brain: an immunocytochemical study. Dev Brain Res. 1991;62:229–238. doi: 10.1016/0165-3806(91)90170-n. [DOI] [PubMed] [Google Scholar]
- 29.Hu GY, Hvalby O, Walaas SI, Albert KA, Skjeflo P, Andersen P, Greengard P. PKC injected into hippocampal pyramidal cells elicits features of long term potentiation. Nature. 1987;328:426–429. doi: 10.1038/328426a0. [DOI] [PubMed] [Google Scholar]
- 30.Huber KM, Mauk MD, Thompson C, Kelly PT. A critical period of protein kinase activity after tetanic stimulation is required for the induction of long-term potentiation. Learn Memory. 1995;2:81–100. doi: 10.1101/lm.2.2.81. [DOI] [PubMed] [Google Scholar]
- 31.Ito M, Sakurai M, Tongroach P. Climbing fiber induced depression of both mossy fiber responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J Physiol (Lond) 1982;324:113–134. doi: 10.1113/jphysiol.1982.sp014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kauer JA, Malenka RC, Nicoll RA. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- 33.Kelly P, Vernon P. Changes in the subcellular distribution of calmodulin-kinase II during brain development. Dev Brain Res. 1985;18:211–224. doi: 10.1016/0165-3806(85)90265-2. [DOI] [PubMed] [Google Scholar]
- 34.Kelly PT, Shields S, Conway K, Yip R, Burgin K. Developmental changes in calmodulin-kinase II activity at brain synaptic junctions: alterations in holoenzyme composition. J Neurochem. 1987;49:1927–1940. doi: 10.1111/j.1471-4159.1987.tb02456.x. [DOI] [PubMed] [Google Scholar]
- 35.Kelly PT, Honeycutt T, Weinberger R, Blumenthal D, Yip R, Waxham N. Functional analysis of the calmodulin (CaM)-binding domain of CaM-Kinase II (CK-II) using synthetic peptides and site-directed mutagenesis. Soc Neurosci Abstr. 1989;15:381.7. [Google Scholar]
- 36.Klee CB. Concerted regulation of protein phosphorylation and dephosphorylation by calmodulin. Neurochem Res. 1991;16:1059–1065. doi: 10.1007/BF00965851. [DOI] [PubMed] [Google Scholar]
- 37.Klee CB, Cohen P. The calmodulin-regulated protein phosphatase. In: Cohen P, Klee CB, editors. Calmodulin. Elsevier; Amsterdam: 1988. pp. 225–248. [Google Scholar]
- 38.Krebs EG. The growth of research on protein phosphorylation. Trends Biochem Sci. 1994;19:439. doi: 10.1016/0968-0004(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 39.Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca2+-dependent phosphatase. Nature. 1994;369:235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- 40.Lisman J. The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci. 1994;17:406–412. doi: 10.1016/0166-2236(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 41.MacKintosh C, MacKintosh RW. Inhibitors of protein kinases and phosphatases. Trends Biochem Sci. 1994;12:444–448. doi: 10.1016/0968-0004(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 42.Malenka RC, Madison DV, Andrade R, Nicoll RA. Phorbol esters mimic some cholinergic actions in hippocampal pyramidal neurons. J Neurosci. 1986a;6:475–480. doi: 10.1523/JNEUROSCI.06-02-00475.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986b;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- 44.Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- 45.Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- 46.Malinow R, Madison DV, Tsien RW. Persistent protein kinase activity underlying long-term potentiation. Nature. 1988;335:820–824. doi: 10.1038/335820a0. [DOI] [PubMed] [Google Scholar]
- 47.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 48.Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- 49.Mulkey RM, Herron C E, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- 50.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calmodulin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 51.Muller D, Turnbull J, Baudry M, Lynch G. Phorbol ester-induced synaptic facilitation is different than long-term potentiation. Proc Natl Acad Sci USA. 1988;85:6997–7000. doi: 10.1073/pnas.85.18.6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nestler EJ, Greengard P. Introduction. In: Nestler E, Greengard P, editors. Protein phosphorylation in the nervous system. Wiley; New York: 1984. [Google Scholar]
- 53.Obenaus A, Mody I, Baimbridge KG. Dantrolene-Na (Dantrium) blocks induction of long-term potentiation in hippocampal slices. Neurosci Lett. 1989;98:172–178. doi: 10.1016/0304-3940(89)90505-3. [DOI] [PubMed] [Google Scholar]
- 54.Ocorr KA, Schulman H. Activation of multifunctional Ca2+/calmodulin-dependent kinase in intact hippocampal slices. Neuron. 1991;6:907–914. doi: 10.1016/0896-6273(91)90231-n. [DOI] [PubMed] [Google Scholar]
- 55.O’Dell TJ, Kandel ER, Grant SG. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature. 1991;353:558–560. doi: 10.1038/353558a0. [DOI] [PubMed] [Google Scholar]
- 56.Ohta EA. Inhibitory action of dantrolene on Ca2+-induced Ca2+ release from sarcoplasmic reticulum in guinea pig skeletal muscle. Eur J Pharmacol. 1990;178:11–17. doi: 10.1016/0014-2999(90)94788-y. [DOI] [PubMed] [Google Scholar]
- 57.Perrino BA, Ng LY, Soderling TR. Calcium regulation of calcineurin phosphatase activity by its B subunit and calmodulin. J Biol Chem. 1995;270:340–346. doi: 10.1074/jbc.270.1.340. [DOI] [PubMed] [Google Scholar]
- 58.Polli JW, Billingsley ML, Kincaid RL. Expression of the calmodulin-dependent protein phosphatase, calcineurin, in rat brain: developmental patterns and the role of nigrostriatal innervation. Dev Brain Res. 1991;63:105–119. doi: 10.1016/0165-3806(91)90071-p. [DOI] [PubMed] [Google Scholar]
- 59.Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- 60.Schulman H, Lou LL. Multifunctional Ca2+/calmodulin-dependent protein kinase: domain structure and regulation. Trends Biol Sci. 1989;14:62–66. doi: 10.1016/0968-0004(89)90045-5. [DOI] [PubMed] [Google Scholar]
- 61.Shields SM, Ingebritsen TS, Kelly PT. Identification of protein phosphatase 1 in synaptic junctions: dephosphorylation of endogenous calmodulin-dependent kinase II and synapse-enriched phosphoproteins. J Neurosci. 1985;5:3414–3422. doi: 10.1523/JNEUROSCI.05-12-03414.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Simpson PB, Challiss RAJ, Nahorski SR. Neuronal Ca2+ store: activation and function. Trends Neurosci. 1995;18:299–306. doi: 10.1016/0166-2236(95)93919-o. [DOI] [PubMed] [Google Scholar]
- 63.Smith P, Gallacher DV. Thapsigargin-induced Ca2+ mobilization in acutely isolated mouse lacrimal acinar cells is dependent on a basal level of Ins(1,4,5) P3 and is inhibited by heparin. Biochem J. 1994;299:37–40. doi: 10.1042/bj2990037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Snyder SH, Sabatini DM. Immunophilins and the nervous system. Nature Med. 1995;1:32–37. doi: 10.1038/nm0195-32. [DOI] [PubMed] [Google Scholar]
- 65.Steiner JP, Dawson TM, Fotuhi M, Glatt CE, Snowman AM, Cohen N, Snyder SH. High brain densities of the immunophilin FKBP colocalized with calcineurin. Nature. 1992;358:584–587. doi: 10.1038/358584a0. [DOI] [PubMed] [Google Scholar]
- 66.Tallant EA, Cheung WY. Calmodulin-dependent protein phosphatase: a developmental study. Biochemistry. 1983;22:3630–3635. doi: 10.1021/bi00284a014. [DOI] [PubMed] [Google Scholar]
- 67.Thomas KL, Laroche S, Errington ML, Bliss TVP, Hunt SP. Spatial and temporal changes in signal transduction pathways during LTP. Neuron. 1994;13:737–745. doi: 10.1016/0896-6273(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 68.Tsien RW, Schulman H, Malinow R. Peptide inhibitors of PKC and CaMKII block induction but not expression of long-term potentiation. Adv Second Messenger Phosphoprotein Res. 1990;24:101–107. [PubMed] [Google Scholar]
- 69.Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry. 1980;19:2396–2404. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- 70.Turner RS, Raynor RL, Mazzei GJ, Girard PR, Kuo JF. Developmental studies of phospholipid-sensitive Ca2+-dependent protein kinase and its substrates and of phosphoprotein phosphatases in rat brain. Proc Natl Acad Sci USA. 1984;81:3143–3147. doi: 10.1073/pnas.81.10.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J-H, Feng DP. Postsynaptic protein kinase C essential to induction and maintenance of long-term potentiation in the hippocampal CA1 region. Proc Natl Acad Sci USA. 1992;89:2576–2580. doi: 10.1073/pnas.89.7.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J-H, Kelly PT. Postsynaptic injection of Ca2+/CaM induces synaptic potentiation requiring CaM-KII and PKC activity. Neuron. 1995;15:443–452. doi: 10.1016/0896-6273(95)90048-9. [DOI] [PubMed] [Google Scholar]
- 73.Wang J-H, Kelly PT. Balance between postsynaptic Ca2+-dependent protein kinase and phosphatase activities controlling synaptic strength. Learn Memory. 1996a;3:170–181. doi: 10.1101/lm.3.2-3.170. [DOI] [PubMed] [Google Scholar]
- 74.Wang J-H, Kelly PT. Regulation of synaptic facilitation by postsynaptic Ca2+/CaM pathways in hippocampal CA1 neurons. J Neurophysiol. 1996b;76:276–286. doi: 10.1152/jn.1996.76.1.276. [DOI] [PubMed] [Google Scholar]
- 75.Wang J-H, Stelzer A. Inhibition of phosphatase 2B prevents expression of hippocampal long-term potentiation. NeuroReport. 1994;5:2377–2380. doi: 10.1097/00001756-199411000-00041. [DOI] [PubMed] [Google Scholar]
- 76.Wang J-H, Stelzer A. Shared calcium signaling pathways in the induction of long-term potentiation and synaptic disinhibition in CA1 pyramidal cell dendrites. J Neurophysiol. 1996;75:1687–1701. doi: 10.1152/jn.1996.75.4.1687. [DOI] [PubMed] [Google Scholar]
- 77.Wang L-Y, Orser BA, Brautlgan DL, MacDonald JF. Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature. 1994;369:230–232. doi: 10.1038/369230a0. [DOI] [PubMed] [Google Scholar]
- 78.Wang Y-T, Salter MW. Regulation of NMDA receptors by tyrosine kinase and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 79.Wiederrecht G, Lam E, Shirley H, Martin M, Sigal N. The mechanism of action of FK-506 and cyclosporin A. Ann NY Acad Sci. 1989;104:9–19. doi: 10.1111/j.1749-6632.1993.tb17137.x. [DOI] [PubMed] [Google Scholar]
- 80.Wyllie DJA, Nicoll RA. A role for protein kinases and phosphatases in the Ca2+-induced enhancement of hippocampal AMPA receptor-mediated synaptic responses. Neuron. 1994;13:635–643. doi: 10.1016/0896-6273(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 81.Yoshida Y, Huang FL, Nakabayashi H, Huang K-P. Tissue distribution and developmental expression of protein kinase C isozymes. J Biol Chem. 1988;263:9868–9873. [PubMed] [Google Scholar]