

Abstract
Previous studies have shown that estradiol induces new dendritic spines and synapses on hippocampal CA1 pyramidal cells. We have assessed the consequences of estradiol-induced dendritic spines on CA1 pyramidal cell intrinsic and synaptic electrophysiological properties. Hippocampal slices were prepared from ovariectomized rats treated with either estradiol or oil vehicle. CA1 pyramidal cells were recorded and injected with biocytin to visualize spines. The association of dendritic spine density and electrophysiological parameters for each cell was then tested using linear regression analysis. We found a negative relationship between spine density and input resistance; however, no other intrinsic property measured was significantly associated with dendritic spine density. Glutamate receptor autoradiography demonstrated an estradiol-induced increase in binding to NMDA, but not AMPA, receptors. We then used input/output (I/O) curves (EPSP slope vs stimulus intensity) to determine whether the sensitivity of CA1 pyramidal cells to synaptic input is correlated with dendritic spine density. Consistent with the lack of an estradiol effect on AMPA receptor binding, we observed no relationship between the slope of an I/O curve generated under standard recording conditions, in which the AMPA receptor dominates the EPSP, and spine density. However, recording the pharmacologically isolated NMDA receptor-mediated component of the EPSP revealed a significant correlation between I/O slope and spine density. These results indicate that, in parallel with estradiol-induced increases in spine/synapse density and NMDA receptor binding, estradiol treatment increases sensitivity of CA1 pyramidal cells to NMDA receptor-mediated synaptic input; further, sensitivity to NMDA receptor-mediated synaptic input is well correlated with dendritic spine density.
Keywords: estradiol, dendritic spines, hippocampal slice, CA1 pyramidal cells, biocytin, autoradiography, NMDA receptor
Since the initial anatomical description of dendritic spines by Ramon y Cajal in the late 1800s, many investigators have speculated about dendritic spine function. Spines have been proposed to play primarily connective, electrical, and/or biochemical roles in neuronal physiology (for review, see Koch and Zador, 1992;Horner, 1993; Harris and Kater, 1994). Recent efforts to understand dendritic spine function based on imaging of dye-labeled spiny dendrites have yielded valuable information, particularly with regard to spines’ potential for Ca2+ compartmentalization (Guthrie et al., 1991; Muller et al., 1991). Such findings have been incorporated into proposals for dendritic spine function in synaptic integration (see, for example, Yuste and Denk, 1995) and neuroprotection (Harris and Kater, 1994; Segal, 1995). An additional approach to exploring dendritic spine function, which has not been used to date, is to assess the functional consequences of adding dendritic spines to the dendrites of spiny neurons.
Hippocampal CA1 pyramidal cells are an ideal population of neurons in which to implement this approach. First, these cells have been studied extensively producing a wealth of information regarding their basic properties. Second, in the female rat, the number of spines on the dendrites of CA1 pyramidal cells can be manipulated by altering circulating levels of the ovarian steroid hormone estradiol (Gould et al., 1990; Woolley and McEwen, 1993). The addition of new spines has been quantified as an estradiol-induced increase in the density of spines (number of spines/unit length) on the dendrites of both the apical and the basal trees of CA1 pyramidal cells. This increase in spine density reflects an increase in spine number per se, because no concomitant change in dendritic length or branching pattern is observed (Woolley and McEwen, 1994). Importantly, increased density of dendritic spines is paralleled by increased density of axospinous (but not axodendritic) synapses, indicating that new spines form synaptic contacts (Woolley and McEwen, 1992).
Estradiol-induced changes in dendritic spine and synapse density are paralleled by changes in hippocampal physiology that suggest that increased dendritic spine and synapse density result in increased excitability. With a time course very similar to spine and synapse changes, estradiol treatment facilitates acquisition of kindled seizures in the hippocampus (Buterbaugh and Hudson, 1991), increases severity of kainic acid-induced seizures (Nicoletti et al., 1985), and decreases hippocampal seizure threshold (Terasawa and Timiras, 1968). During the estrous cycle, in which spine and synapse density fluctuate naturally with changing hormone levels (Woolley et al., 1990), the threshold for hippocampal seizure activity decreases (Terasawa and Timiras, 1968) and long-term potentiation (LTP) is enhanced (Warren et al., 1995) as estradiol levels increase.
To address the relationship between estradiol-induced differences in hippocampal physiology and differential dendritic spine density on hippocampal CA1 pyramidal cells, we have recorded CA1 pyramidal cells in in vitro hippocampal slices from ovariectomized control and estradiol-treated animals, labeled the cells with biocytin, and made direct correlations between the electrophysiological (intrinsic and synaptic) properties and dendritic spine density on each cell. These experiments provide information regarding both basic questions of dendritic spine function and the mechanism by which estradiol affects hippocampal physiology.
MATERIALS AND METHODS
Animal surgery and hormone treatments. Adult female Sprague Dawley rats were housed on a 12 hr light/dark cycle with unlimited access to food and water. These animals were left gonadally intact (intact), ovariectomized and treated with estradiol benzoate (OVX+E), or ovariectomized and treated with sesame oil vehicle (OVX+O). The hormone treatment regimen that was used in these experiments has been shown previously to result in differences in CA1 pyramidal cell dendritic spine (Gould et al., 1990) and synapse (Woolley and McEwen, 1992) density. Six days before electrophysiological analysis, animals were ovariectomized under Metofane anesthesia using aseptic surgical procedure. After surgery, animals were housed individually. On days 3 and 4 after surgery, OVX+E animals were injected (s.c.) with 10 μg of 17β-estradiol benzoate in 100 μl of sesame oil vehicle; OVX+O animals received oil vehicle at each injection time. Forty-eight hours after the second estradiol or vehicle injection, animals were killed by decapitation and hippocampal slices were prepared from their brains. Gonadally intact animals were killed at random stages of the estrous cycle.
In a separate experiment, animals were similarly prepared for glutamate receptor autoradiography. This experiment was originally done in conjunction with analysis of dendritic spine density in which we were interested in determining the effects of progesterone in addition to estradiol. As a result, a group of animals that were treated with both estradiol and progesterone was included. Data from the estradiol plus progesterone (OVX+EP) animals are included here because the animals were all part of the same experiment; however, for the purposes of comparison with electrophysiological data, the OVX+O and OVX+E groups are the most relevant. For this experiment, adult female rats were ovariectomized 6 d before killing and were divided into three treatment groups of nine animals each. OVX+E and OVX+O animals were treated as described above; OVX+EP animals were treated with estradiol as above, and then 5 hr before killing they received a single injection (s.c.) of 500 μg of progesterone in 100 μl of sesame oil. All animals were killed by decapitation 48 hr after the second estradiol benzoate or sesame oil injection, and their brains were rapidly removed and frozen on dry ice and stored at −70°C until sectioning.
Preparation of hippocampal slices. After decapitation, brains were quickly removed, cooled briefly with ice-cold, oxygenated (95% O2/5% CO2) sucrose artificial CSF (sACSF; in mm: 220 sucrose, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaCO3, 2 CaCl2, 10 dextrose) and blocked to contain the dorsal hippocampus. Using a Vibroslicer, 400-μm-thick slices transverse to the long axis of the hippocampus were cut into a bath of oxygenated sACSF at 4°C. Sections were then transferred to a holding chamber, where they remained submerged in oxygenated artificial CSF (ACSF; in mm: 124 NaCl, 5 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaCO3, 2 CaCl2, 10 dextrose) at room temperature until used for recording.
Slices were allowed to recover at least 30 min after cutting before they were transferred to a standard interface recording chamber. In the chamber, slices rested on a nylon mesh over a well that was perfused with oxygenated ACSF at ∼1 ml/min; warmed, humidified air was circulated above the slice. Temperature was maintained at 34.5°C. Slices remained undisturbed in the chamber 30 min before recording.
Intracellular recording. Recording electrodes made from borosilicate glass were pulled using a horizontal puller (Sutter Instruments) and filled with 2% biocytin dissolved in 1 mpotassium acetate (100–200 MΩ). Intracellular potentials were recorded using an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA). Bridge balance was continuously monitored on an oscilloscope. Neurons impaled within the CA1 pyramidal cell layer were tentatively identified as CA1 pyramidal cells on the basis of their response to direct current injection (Schwartzkroin, 1975); the identity of these cells was confirmed after visualization of injected biocytin. Neurons were included only if they had a resting potential of at least −55 mV, overshooting action potentials, and input resistance of at least 25 MΩ. Cell activity and response to stimulation were processed on-line and stored on videotape using a PCM device (Neuro-Data Instruments). Data were acquired and analyzed using pClamp software (Axon Instruments). Biocytin was iontophoretically injected with 300 msec, 0.5–1.0 nA hyperpolarizing current pulses delivered every 600 msec for 10–20 min. Slices remained in the recording chamber for 30 min after biocytin injection.
In experiments involving synaptic activation of CA1 pyramidal cells, a bipolar metal stimulating electrode was placed on the surface of the slice in the proximal stratum (st.) radiatum for Schaffer collateral stimulation. Stimuli (100 msec duration) were delivered at 0.1 Hz.
In some experiments, 2 mm kynurenic acid was added to the recording medium to block glutamate receptors nonspecifically. In other experiments, NMDA receptor-mediated EPSPs were pharmacologically isolated from the composite EPSP using the following modifications of recording conditions: (1) Mg2+ in the recording medium was reduced to 0.6 mm; (2) 30 μm6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) to block non-NMDA glutamate receptors and 30 μm bicuculline to block γ-amino-butyric acid A (GABAA) receptors were added to the recording medium; (3) 200 mm QX-314 was included in the recording electrode to block Na+ action potentials; (4) EPSPs were recorded with the cell depolarized to approximately −40 mV.
Extracellular recording. Extracellular recordings were made to verify linearity in the relationship between the intensity of a stimulus delivered to the Schaffer collateral pathway and the amplitude of the presynaptic fiber volley recorded in the CA1 st. radiatum. Recording electrodes made from borosilicate glass pulled using a horizontal puller (Sutter Instruments) were filled with 2m NaCl (5–15 MΩ). Electrodes were placed in the CA1 st. radiatum to record the dendritic EPSP. A bipolar metal stimulating electrode was placed on the surface of the slice in the proximal CA1 st. radiatum for Schaffer collateral stimulation. Extracellular field potentials were recorded using an Axoclamp 2A amplifier (Axon Instruments), processed on-line, and stored on videotape using a PCM device (Neuro-Data Instruments). Data were acquired and analyzed using pClamp software (Axon Instruments).
Tissue processing for visualization of biocytin.Electrophysiological characterization of CA1 pyramidal cells was carried out before biocytin injection. After biocytin injection, slices were removed from the recording chamber and fixed overnight between two pieces of filter paper in 4% paraformaldehyde in 0.1 mphosphate buffer at pH 7.4 (PB) at 4°C. Slices were then rinsed in PB and cryoprotected in 10% sucrose in PB for 30 min followed by 30% sucrose in PB overnight. Each slice was then resectioned (60 μm) on a freezing microtome. Tissue sections were processed to visualize biocytin-filled cells as follows. Sections were first rinsed in 0.1m Tris buffer at pH 7.4 (TB), treated with 0.5–1.0% H2O2 to suppress endogenous peroxidases, incubated in 2% bovine serum albumin in 0.25% DMSO in 0.05m Tris-buffered saline at pH 7.4 (TBS) to block nonspecific staining and permeabilize membranes. Sections were then rinsed in 0.1m TBS and incubated in avidin-biotinylated horseradish peroxidase (Elite ABC kit, Vector Laboratories, Burlingame, CA), diluted 1:500 in the blocking solution for 48 hr at 4°C. After incubation, sections were rinsed in 0.1 m TBS followed by 0.1 m TB, preincubated in 0.025% diaminobenzidine (DAB) and 0.005% NiNH4SO4 for 15 min in the dark. Sections were then reacted with 0.002% H2O2 in DAB/NiNH4SO4 for 60–120 min in the dark. After the reaction, sections were rinsed in 0.1 m TB, mounted onto subbed slides, dehydrated, cleared, coverslipped, and examined for biocytin-filled cells.
Morphological analysis of biocytin-filled cells.Tissue-containing CA1 pyramidal cells that were well filled with biocytin were coded and analyzed (blind to treatment condition) for the density of dendritic spines on lateral branches of the apical dendritic tree; in some cells, the total two-dimensional dendritic length of the apical and basal dendritic trees was measured. Multiple cells from each treatment group were analyzed together, and the code was not broken until the analysis was complete. CA1 pyramidal cells were determined to be well filled if a single dark brown-to-black cell body was present and evenly stained dendrites could be followed into st. lacunosum/moleculare. Cells with patchy or interrupted dendritic staining were excluded from analysis. Dendritic spines were not used as a criterion for the quality of cell filling.
For each cell, dendritic spine density was measured on four to five dendritic segments in the apical dendritic tree within a range of 150–300 μm from the cell body. To be selected for analysis of spine density, dendritic segments had to meet two additional requirements: (1) be relatively thin and of approximately equivalent diameter so as to minimize the effect of spines hidden either above or below the dendrite and to equalize these effects between different cells, and (2) remain in approximately a single plane of focus so that the length of the dendrite projected in two dimensions would approximate the three-dimensional dendritic length. Camera lucida drawings (1000×) were made of each dendritic segment for which spine density was analyzed. The length of each segment was measured from the camera lucida drawing (using NIH Image software), the number of dendritic spines visible along each segment was counted, and spine density was expressed as number of spines/10 μm dendritic length. For cells in which total dendritic length was also measured, reconstructions of the entire dendritic tree were made using a camera lucida (400×), and the length of dendrites was measured from camera lucida drawings using NIH Image software. Dendritic length of the apical tree, basal tree, and combined apical and basal trees was measured for each such cell.
Electrophysiological analysis. Analysis of electrophysiological data using pClamp software was done only for cells that were determined to be well filled with biocytin. The following intrinsic membrane properties were measured: (1) resting membrane potential was determined upon withdrawal from a recorded cell; (2) input resistance was determined by linear regression through the current–voltage relationship. Peak amplitude of the voltage deflection resulting from hyperpolarizing current pulses ranging from 0.2 to 0.6 nA was measured; (3) time constant was taken to be the time required for the membrane to reach 63% of maximum voltage deflection during a 0.3 nA hyperpolarizing current pulse; (4) mV to threshold; (5) action potential amplitude; (6) action potential duration was measured at the base of the spike; (7) spike afterhyperpolarization amplitude; and (8) spike afterhyperpolarization duration.
The relationship between dendritic spine density and sensitivity to synaptic input for each cell was determined on the basis of input/output (I/O) curves in which EPSP initial slope or amplitude was plotted versus stimulus intensity. A bipolar metal stimulating electrode was placed in the st. radiatum ∼1 mm from the recording electrode. In normal recording medium, EPSPs were generated by stimuli delivered embedded within a 100 msec, 0.5 nA hyperpolarizing current pulse. Under recording conditions designed to isolate the NMDA receptor-mediated component of the EPSP (see above), stimuli were delivered from a membrane potential depolarized to approximately −40 mV. In each case, stimulus intensity was increased in 20 μA increments from that which produced no detectable postsynaptic response to a stimulus that produced a maximal postsynaptic response. Ten EPSP measurements were averaged at each stimulus intensity. EPSP slope and amplitude were plotted as percent maximum values versus stimulus intensity. I/O slope was measured by linear regression of the I/O curve through the range from 25 to 75% maximum EPSP slope or amplitude.
Glutamate receptor autoradiography. Sixteen-micrometer-thick coronal sections of OVX+O, OVX+E, and OVX+EP brains were cut on a cryostat and thaw-mounted onto gelatinized slides. These sections were stored at −70°C until used for autoradiography. Some sections were stained for Nissl using cresyl violet; these sections were used for orientation during data analysis. Determination of the NMDA recognition site was performed according to Weiland (1992a). Slide-mounted sections were preincubated in 50 mm Tris acetate buffer, pH 7.4, for 45 min at room temperature to remove endogenous ligand. Sections were then air-dried for 10 min before incubation in 50 mm Tris acetate buffer containing 200 nm[3H]glutamate (17.3 Ci/mmol) in either the presence or the absence of 1 mm NMDA for 30 min at 4°C. After incubation, the sections were washed four times for 3 sec each in buffer at 4°C, fixed in acetone with 2.5% gluteraldehyde, and then dried under a fan.
Determination of the AMPA recognition site was performed according to a modified version of Dewar et al. (1991). Slide-mounted sections were preincubated in 50 mm Tris acetate buffer, pH 7.2, at room temperature for 45 min to remove endogenous ligand. Sections were then air-dried for 10 min before incubation in 50 mm Tris acetate buffer containing 100 mm KCN and 50 nm[3H]AMPA (60 Ci/mmol) in either the presence or the absence of 1 mm glutamate for 45 min at 4°C. After incubation, the sections were washed three times for 3 sec each in buffer at 4°C, fixed in acetone with 2.5% gluteraldehyde, and then dried under a fan.
After binding assays, sections were apposed to either LKB Ultrofilm for 29 d (NMDA-displaceable [3H]glutamate binding) or LKB Hyperfilm for 2 d ([3H]AMPA binding) with3H-labeled brain mash standards. Films were developed using Kodak D-19 developer and fixed with Kodak Rapid-fix.
Quantitative analysis of glutamate receptor binding.Quantitative analysis of films depicting labeled brain sections and standards was performed using the DUMAS image analysis program (Drexel University). This program measures relative optical density and converts the values to concentration of radioligand (fmol/mg protein) using a standard curve generated from 3H-labeled brain mash standards. NMDA receptor binding was taken to be [3H]glutamate binding that was displaced by NMDA; both sides of four sections were measured for each of total [3H]glutamate- and NMDA-displaced binding. AMPA receptor binding was taken to be [3H]AMPA binding that was displaced by glutamate; both sides of four sections were measured for [3H]AMPA binding, and both sides of two sections were used to determine nonspecific binding. For each ligand, separate measurements were made for the CA1 st. radiatum and st. oriens.
Statistical analyses. Dendritic spine density on CA1 pyramidal cells from OVX+O and OVX+E animals was compared using an unpaired, two-tailed Student’s t test. The association of electrophysiological properties and dendritic spine density on CA1 pyramidal cells was determined using simple linear regression analysis of physiological parameters on spine density. The significance of these associations was tested using ANOVA. Mean slope of the NMDA receptor-mediated EPSP I/O curve of cells from OVX+O and OVX+E animals was compared using an unpaired, two-tailed Student’s ttest. For the glutamate receptor binding studies, means were calculated for each animal and the data were subjected to one-way ANOVA with Tukey’s HSD post hoc comparisons.
RESULTS
For this study, 145 CA1 pyramidal cells were recorded and injected with biocytin. Of these 145, 69 cells (48%) were determined to be sufficiently filled with biocytin for analysis of dendritic spine density. In general, biocytin-filled CA1 pyramidal cells (Fig.1) were similar in morphology to Golgi-impregnated CA1 pyramidal cells. Initial experiments indicated no obvious differences in the physiology of CA1 pyramidal cells of different dendritic morphologies (e.g., nonbifurcated vs bifurcated primary dendrite) or between cells located in different portions of the CA1 pyramidal cell layer. Thus, all cells in which a particular electrophysiological parameter was measured were grouped together for analysis.
Fig. 1.
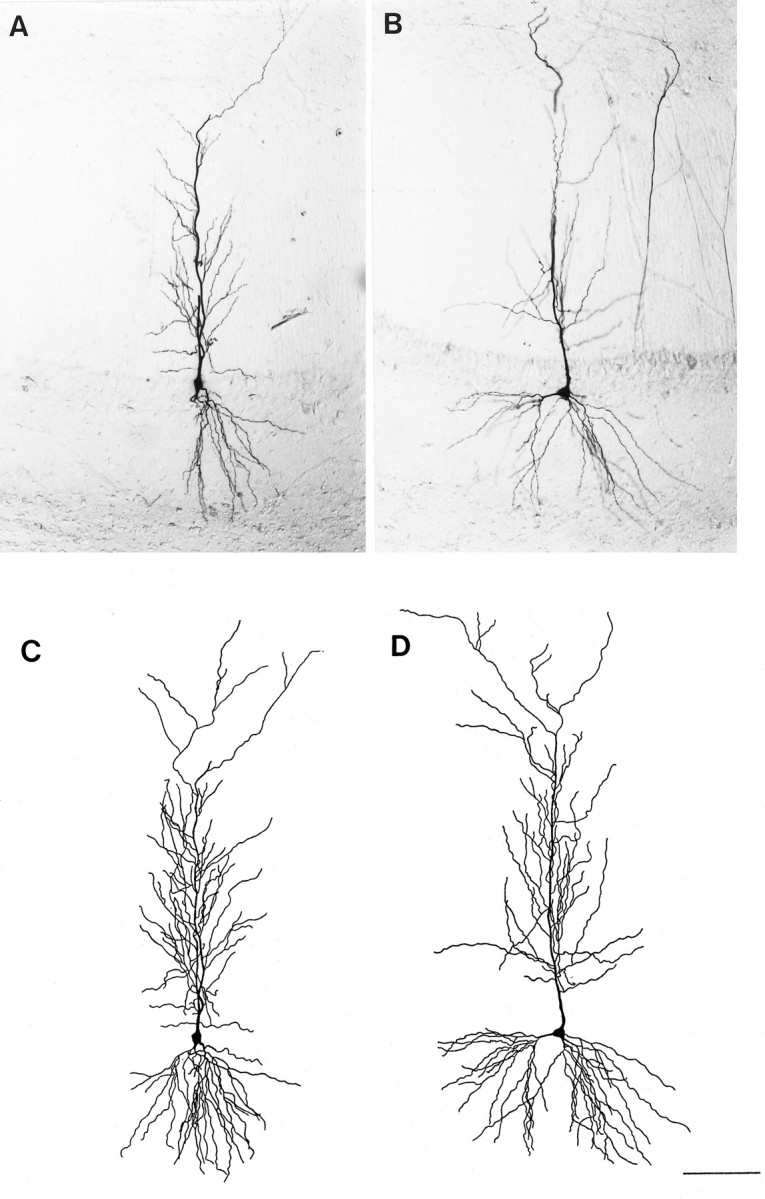
Photomicrographs of single sections containing a representative biocytin-filled CA1 pyramidal cell from an OVX+O (A) and an OVX+E (B) animal. Camera lucida tracings of the cell body and complete dendritic tree reconstructed from all sections containing the same cells from the OVX+O (C) or OVX+E (D) animal are shown in thelower panels. Camera lucida tracings were used to determine total dendritic length of each cell; no differences in total dendritic length were observed between treatment groups. Scale bar, 50 μm (applies to all panels).
Dendritic spine density
The density of dendritic spines on biocytin-filled CA1 pyramidal cells was somewhat higher than previously observed on Golgi-impregnated cells (see, for example, Woolley and McEwen, 1993). The difference in mean spine density between OVX+O and OVX+E cells was statistically significant but of a slightly lower magnitude than previously reported for Golgi-impregnated tissue. On biocytin-filled CA1 pyramidal cells, mean ± SEM spine density was 15.2 ± 1.2 spines/10 μm for OVX+O compared to 19.4 ± 1.1 spines/10 μm for OVX+E (Figs. 2,3; T = −4.655,p < 0.001). Thus, the difference in spine density on biocytin-filled CA1 pyramidal cells from OVX+O versus OVX+E animals was 22% compared to ∼28% previously reported for Golgi-impregnated cells (Woolley and McEwen, 1993). Previous studies based on Golgi-impregnated tissue have shown that estradiol-induced changes in the density of dendritic spines in the lateral branches of the apical dendritic tree are paralleled by similar changes in the dendrites of the basal tree (Gould et al., 1990; Woolley and McEwen, 1994). In general, changes observed in the basal tree are of slightly lower magnitude than in the apical tree.
Fig. 2.
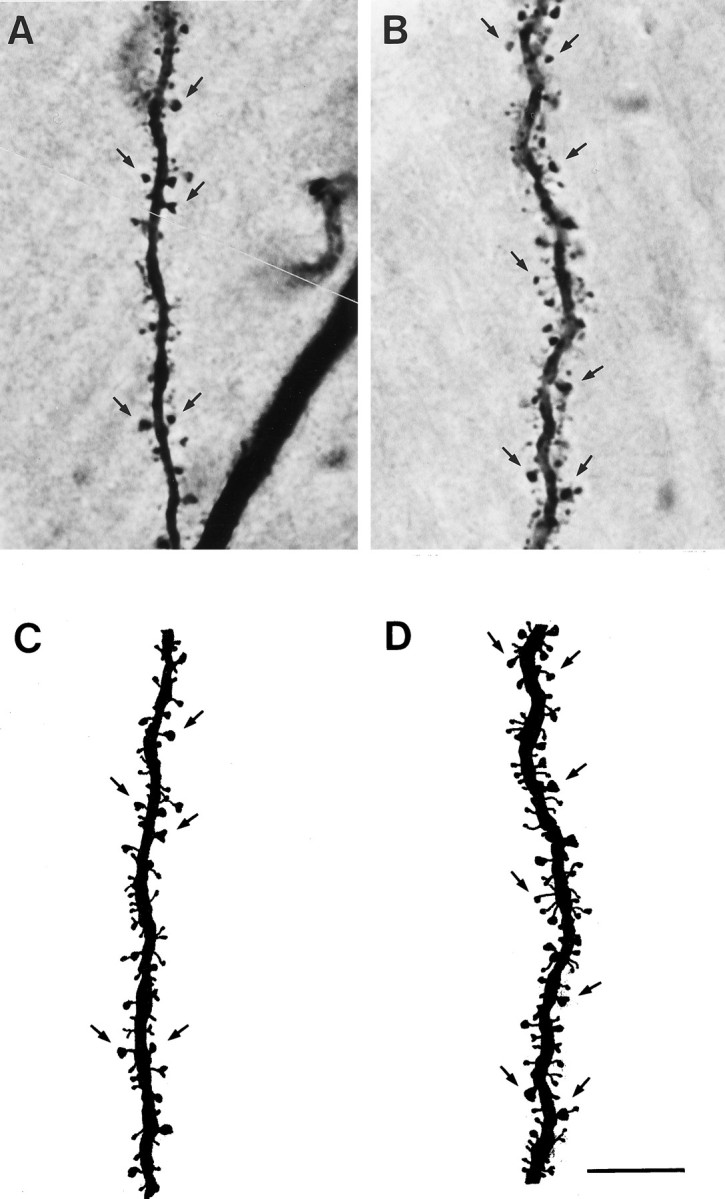
Photomicrographs of representative dendritic segments in the lateral branches of the apical dendritic tree from a CA1 pyramidal cell in an OVX+O (A) and an OVX+E (B) animal. Camera lucida tracings of the segment from the OVX+O (C) and OVX+E (D) cells show all visible dendritic spines. Some dendritic spines are indicated byarrows. Note the increased density of dendritic spines on the dendrites of the cell from an estradiol-treated animal. This estradiol-induced increase in spine density is quantified in Figure 3. Scale bar, 10 μm (applies to all panels).
Fig. 3.
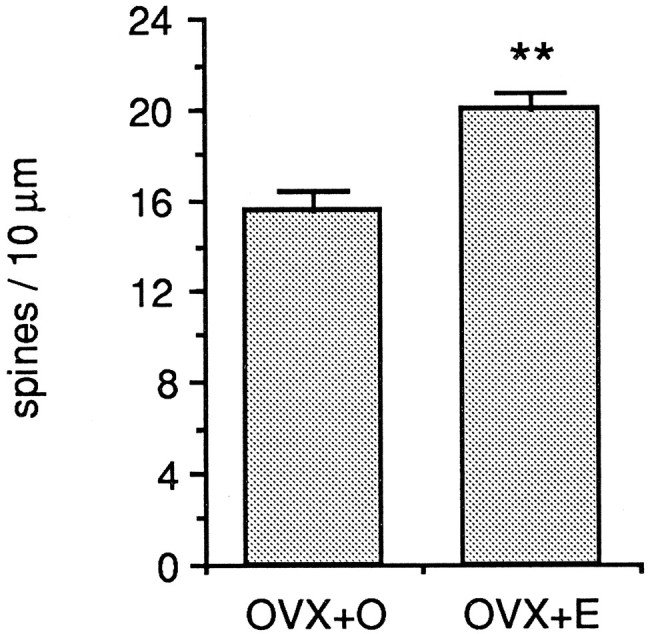
Bar graphs depicting the mean difference in dendritic spine density in the apical dendritic trees of CA1 pyramidal cells from ovariectomized, oil-treated (OVX+O), and ovariectomized, estradiol-treated (OVX+E) animals. All well filled CA1 pyramidal cells in this study were included. The difference in mean dendritic spine density is 22%; **p < 0.001.
Association of dendritic spine density and intrinsic properties
Analysis of the association of CA1 pyramidal cell intrinsic properties with dendritic spine density revealed a correlation only with cellular input resistance. Relating input resistance to dendritic spine density on 31 well filled CA1 pyramidal cells revealed a weak negative association (R = −0.50, p < 0.01; Fig. 4, Table 1). Although spine density was negatively associated with input resistance, and there was a significant difference in mean spine density between cells from OVX+O and OVX+E animals, no significant difference in mean input resistance in cells from OVX+O and OVX+E animals was observed. This finding is consistent with the previous observations of others (Wong and Moss, 1992). In contrast to input resistance, no other intrinsic electrophysiological parameter measured (resting membrane potential, time constant, mV to threshold, action potential amplitude or duration, spike afterhyperpolarization amplitude or duration) was significantly associated with dendritic spine density (Fig. 3, Table 1). However, in cells with increased dendritic spine density, there were statistical trends toward more hyperpolarized resting potential, increased action potential amplitude, and decreased action potential duration (Table 1). Given these trends, we repeated some measurements with cells held at approximately the same membrane potential (−60 mV); in these cells, the significant negative relationship between input resistance and spine density remained (R = −0.46, p< 0.05; data not shown).
Fig. 4.

Representative intracellular recordings from CA1 pyramidal cells in slices from an OVX+O (A) and OVX+E (B) animal. Recordings were made from the resting membrane potential in standard ACSF. In each panel, the top tracesare voltage and the bottom traces are current. Calibration pulse, 10 mV, 10 msec. Note that input resistance is greater in the cell in A (OVX+O), which has a lower density of dendritic spines than the cell in B (OVX+E), which has higher spine density. C shows the negative correlation between input resistance and dendritic spine density in CA1 pyramidal cells. Data from cells in OVX+O (open squares), OVX+E (filled squares), and gonadally intact (open circles) animals are plotted together. Although there is a significant correlation between spine density and input resistance, there is no significant difference in mean input resistance between cells from OVX+O and OVX+E animals (see text).
Table 1.
Association of CA1 pyramidal cell intrinsic properties with dendritic spine density
| Input resistance | R = −0.50 | p < 0.01 |
| Time constant | R = 0.13 | p > 0.10 |
| Resting membrane potential | R = 0.36 | p < 0.10 trend |
| mV to threshold | R = 0.25 | p> 0.10 |
| Action potential amplitude | R = 0.38 | p < 0.10 trend |
| Action potential duration | R = −0.39 | p < 0.10 trend |
| Afterhyperpolarization amplitude | R = 0.07 | p > 0.10 |
| Afterhyperpolarization duration | R = 0.23 | p > 0.10 |
The association of CA1 pyramidal cell intrinsic electrophysiological properties with dendritic spine density was determined by linear regression analysis of each property on spine density for CA1 pyramidal cells. R values and significance levels are shown. A significant negative association between cellular input resistance and dendritic spine density was observed. Additionally, statistical trends toward significant association between spine density and resting membrane potential, action potential amplitude, and action potential duration were detected.
There are at least two possible sources of the negative association between spine density and input resistance: (1) increased dendritic membrane surface area of cells with more spines could contribute to decreased input resistance, and/or (2) increased background synaptic activity onto cells with greater dendritic spine density could result in a lower measured input resistance. The observation that input resistance, but not time constant, was significantly associated with differential spine density suggested that increased membrane surface area was the major contributor to this relationship. To determine the effect of background synaptic activity on input resistance in the slice, 10 additional cells (4 OVX+O, 4 OVX+E, 2 intact) were analyzed in recording medium containing 2 mm kynurenic acid to block glutamate receptors and thereby decrease background synaptic activity. Under these conditions, the negative association between input resistance and dendritic spine density remained (R = −0.55, p < 0.01), indicating that the background synaptic activity was not a major contributor to this negative relationship.
Additionally, no differences in the overall two-dimensional dendritic length of the apical, basal, or combined apical and basal dendritic trees were observed in CA1 pyramidal cells from OVX+O and OVX+E animals, consistent with previous observations from Golgi-impregnated tissue (Woolley and McEwen 1994); nor was any association observed between total dendritic length and dendritic spine density. These two observations indicate that potential differences in dendritic length cannot be responsible for the relationship between input resistance and spine density observed in these experiments.
Glutamate receptor binding
Because dendritic spines are sites of excitatory synaptic input and because a majority of excitatory synapses in CA1 st. radiatum are glutamatergic, we determined whether glutamate receptor binding increases in parallel with dendritic spine and synapse density after ovarian steroid treatment. Quantitative analysis of NMDA-displaced [3H]glutamate binding in the st. radiatum of the CA1 region of the hippocampus revealed a significant overall effect of hormone treatment (F(2,24) = 6.74,p < 0.01). NMDA receptor binding in estradiol-treated rats was 30% greater than that in ovariectomized controls (p < 0.01; Figs. 5,6A). NMDA receptor binding in the st. radiatum of animals treated with estradiol followed by progesterone was intermediate between levels in estradiol-treated and control animals (Fig. 6A). A significant overall effect of hormone treatment was also observed in NMDA receptor binding in the st. oriens (F(2,24) = 15.962,p < 0.01). In this case, estradiol treatment resulted in a 46% increase in NMDA receptor binding ( p < 0.01; Figs. 5, 6A). These results are consistent with previous observations of Weiland (1992a).
Fig. 5.
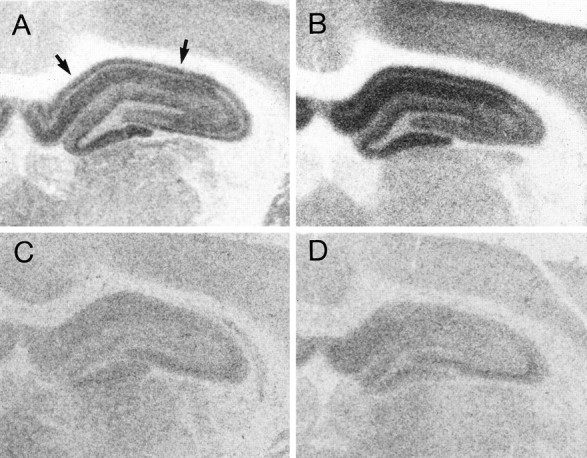
Representative autoradiograms of total [3H]glutamate binding in the hippocampus of ovariectomized rats treated with sesame oil (A) or estradiol (B) and the [3H]glutamate binding that remains after displacement with NMDA in OVX+O (C) or OVX+E (D) animals. Agonist binding to the NMDA receptor was taken to be the amount of total glutamate binding that was displaced by NMDA, i.e., the difference between the top and bottom panels. Note that total glutamate binding is increased by estradiol treatment and that this difference is attributable to enhanced NMDA binding because there is no difference between OVX+O and OVX+E in the binding that remains after displacement with NMDA. NMDA-displaceable [3H]glutamate binding within the CA1 region (indicated by arrows) is quantified in Figure6A.
Fig. 6.

Bar graphs depicting the effect of estradiol and progesterone treatments on NMDA-displaceable [3H]glutamate (A) and [3H]AMPA (B) binding in the regions of the hippocampus containing the apical (str. radiatum) or basal (str. oriens) dendrites of CA1 pyramidal cells. Filled bars represent binding in OVX+O animals; stippled barsrepresent binding in OVX+E animals; open bars represent binding in ovariectomized animals treated with both estradiol and progesterone (OVX+EP). Note that treatment with either estradiol or estradiol plus progesterone increases NMDA receptor binding with no effect on AMPA receptor binding. **, Significant difference from OVX+O (p < 0.01); *, significant difference from OVX+O (p < 0.05).
In alternate sections of the same hippocampi in which these hormone-induced changes in NMDA receptor binding were observed, no changes in [3H]AMPA binding were detected. One-way ANOVA indicated no significant overall effect of hormone treatment in either the st. radiatum or the st. oriens for [3H]AMPA binding (p > 0.1; Fig. 6B).
Association of dendritic spine density with synaptic properties
Anatomical studies suggest that new dendritic spines and synapses induced by estradiol represent functional contacts (Woolley and McEwen, 1992). Furthermore, estradiol treatment increases NMDA receptor binding in parallel with dendritic spine and synapse density. Given these findings, one would expect increased dendritic spine density to be correlated with increased sensitivity to synaptic input. We tested the relationship between dendritic spine density and the efficacy of synaptic input onto CA1 pyramidal cells using full I/O curves. A bipolar stimulating electrode was placed on the surface of the slice to activate the Schaffer collateral fibers. In initial experiments, the relationship between the intensity of a stimulus delivered to the slice and the amplitude of the presynaptic fiber volley evoked in CA1 st. radiatum was tested. This relationship was linear over a wide range of stimulus intensities (data not shown). Linearity of the relationship between stimulus intensity and fiber volley amplitude validated comparison of the slope of the intracellularly recorded EPSP to stimulus intensity in an I/O curve. Intracellular EPSPs were recorded in 11 well filled CA1 pyramidal cells (5 OVX+O, 4 OVX+E, 2 intact) beginning from a stimulus intensity that generated no detectable response to that which generated a maximal EPSP. When dendritic spine density was related to the slope of an I/O curve generated from EPSP initial slope (Fig. 7A,B) or amplitude (data not shown), no consistent relationship was observed (Fig.7C; R = 0.15, p > 0.1 ). Thus, recording in standard medium and from a relatively hyperpolarized membrane potential, we were unable to detect a relationship between dendritic spine density and sensitivity to synaptic input.
Fig. 7.

A shows representative intracellular recordings from a CA1 pyramidal cell recorded in standard ACSF. Calibration pulses in top (voltage) tracesindicate 10 mV, 10 msec, in bottom (current)trace 1.0 nA, 10 msec. Stimuli of increasing intensity (indicated at the left of each trace) were delivered to the CA1 st. radiatum embedded in a 100 msec, 0.5 nA hyperpolarizing current pulse. EPSP (indicated by arrowheads in A) slope was plotted versus stimulus intensity to generate input/output curves (B). The slope of such intracellular input/output curves was taken as a measure of sensitivity to synaptic input. No correlation between input/output curve slope and dendritic spine density was observed under standard recording conditions (C).
The effect of estradiol to increase binding to NMDA receptors, but not to AMPA receptors, suggested that the dendritic spines and synapses induced by estradiol might be specifically enriched in NMDA receptors. If this were the case, then one would not necessarily expect to observe a correlation between spine density and the slope of an I/O curve generated in normal recording medium and from a relatively hyperpolarized membrane potential. Under these standard recording conditions, the CA1 pyramidal cell EPSP is composed of AMPA and NMDA receptor-mediated components and is terminated in part by the GABAA receptor-mediated IPSP. The initial component of the EPSP, as measured above, is dominated by the AMPA component. Because of the voltage-dependent Mg2+ block of the NMDA receptor, this receptor is largely inactivated at hyperpolarized membrane potentials. Because the contribution of NMDA receptors to the CA1 pyramidal cell EPSP is relatively small near the resting membrane potential, any differences in synaptic sensitivity dependent specifically on NMDA receptor-mediated input could easily be missed. To test the possibility that increased dendritic spine density is associated with increased sensitivity specifically to NMDA receptor-mediated synaptic input, we repeated I/O curves under conditions designed to enhance the NMDA receptor-mediated component of the EPSP. For these experiments, Mg2+ in the recording medium was reduced to 0.6 mm, 30 μm CNQX and 30 μmbicuculline were added to the recording medium to block AMPA and GABAA receptors, respectively, and cells were depolarized to approximately −40 mV. To record EPSPs without contamination from action potentials, 200 mm QX-314 was included in the recording electrode to block sodium channels in the recorded cell. Stimulation in the st. radiatum under these recording conditions produced a prolonged EPSP that was blocked by addition of 50 μm 2-amino-5-phosphopentanoic acid (APV; Fig.8A). I/O curves were generated for OVX+O and OVX+E cells from series of NMDA receptor-mediated EPSPs (Fig.8B).
Fig. 8.
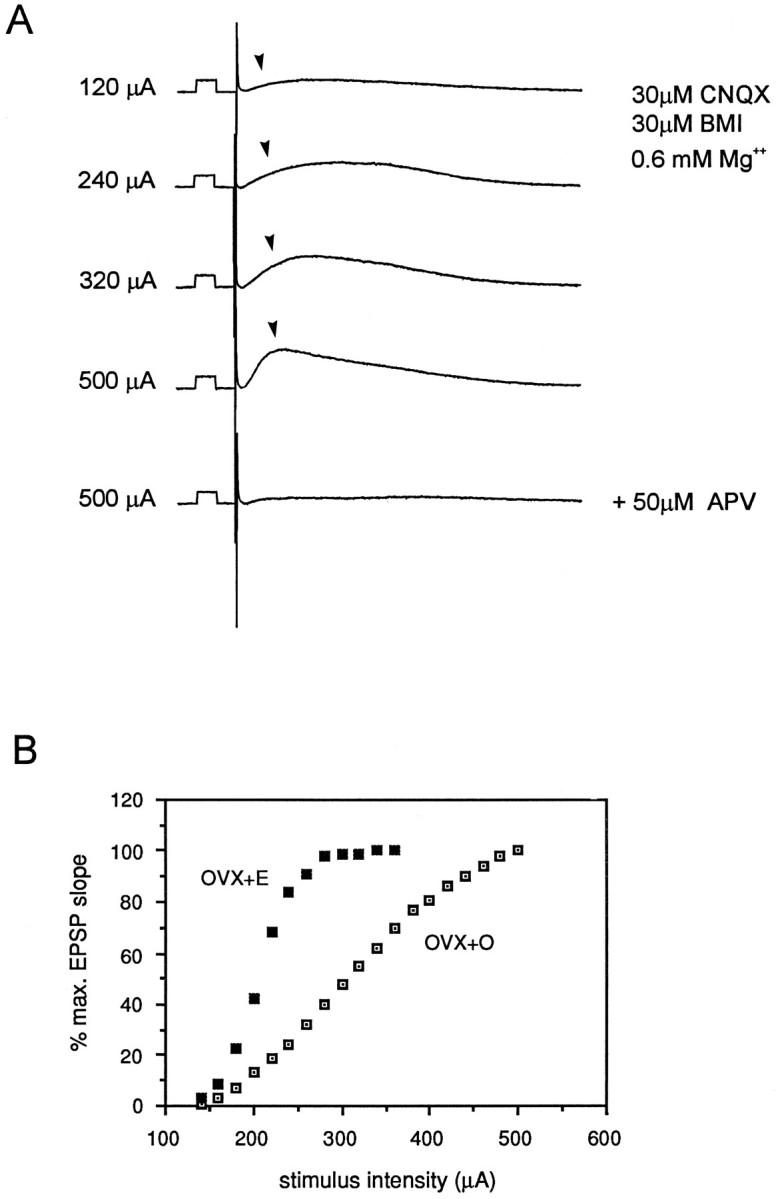
A shows representative intracellular recordings of NMDA receptor-mediated EPSPs (indicated byarrowheads) from a CA1 pyramidal cell. Calibration pulse, 10 mV, 10 msec. Stimuli of increasing intensity were delivered to the st. radiatum to generate input/output curves (B). Stimulus intensities are indicated at the left of the each trace. NMDA receptor-mediated EPSPs were recorded with the following modifications of the recording medium: 30 μm CNQX, 30 μm bicuculline (BMI), Mg2+ reduced to 0.6 mm; in addition, recorded cells were depolarized to approximately −40 mV and 200 mm QX-314 was included in the recording electrode to eliminate Na+ action potentials in the recorded cell. EPSPs generated under these conditions were blocked by addition of 50 μm APV to the recording medium.B shows representative input/output curves from cells in slices from OVX+O and OVX+E animals. Note that the slope of the input/output curve generated from NMDA receptor-mediated EPSPs is greater in the OVX+E cell.
Quantitative analysis of the relationship between dendritic spine density and I/O slope generated from NMDA receptor-mediated EPSPs in 17 cells (7 OVX+O, 7 OVX+E, 3 intact) revealed a significant correlation (R = 0.65, p < 0.01; Fig 9A). Additionally, a significant difference in mean I/O slope between OVX+O and OVX+E cells (T = −3.473, p < 0.01; Fig. 9B) was observed. The mean slope for OVX+O cells was 0.41 compared to 0.92 for OVX+E cells. These results indicate that estradiol treatment results in increased sensitivity of CA1 pyramidal cells to NMDA receptor-mediated synaptic input and that this increase is well correlated with the estradiol-induced increase in dendritic spine density in the apical dendritic tree of these cells.
Fig. 9.

A shows the correlation between the slope of input/output curves generated from NMDA receptor-mediated CA1 pyramidal cell EPSPs and dendritic spine density on each cell. Data from cells in OVX+O (open squares), OVX+E (filled squares), and gonadally intact (open circles) animals are plotted together. B is a bar graph depicting the mean slope of input/output curves generated from NMDA receptor-mediated EPSPs in cells from OVX+O compared to OVX+E animals. Note that the mean slope of the input/output curves generated from NMDA receptor-mediated EPSPs is significantly greater in the cells from OVX+E animals; ** p < 0.01.
DISCUSSION
The results presented in this study demonstrate that differences in numbers of dendritic spines on hippocampal CA1 pyramidal cells are associated with differences in both the intrinsic and the synaptic properties of these cells. These findings represent the first attempt to determine the functional contribution of additional dendritic spines and synapses to the physiology of hippocampal neurons. In this study, we found that cellular input resistance is negatively correlated with dendritic spine density. This negative correlation is consistent with the interpretation that increased numbers of dendritic spines result in increased dendritic surface area and, thus, decreased input resistance; increased background excitatory synaptic activity was ruled out as a possible source of the correlation. Additionally, in agreement with previous binding studies (Weiland, 1992a), our results from glutamate receptor autoradiography indicated that estradiol increases binding to the NMDA but not the AMPA subtype of glutamate receptor. The lack of an estradiol effect on AMPA receptor binding suggested that the sensitivity of CA1 pyramidal cells to synaptic input under normal conditions, in which the AMPA receptor-mediated component of the EPSP is dominant, would not be altered by estradiol treatment. We assessed the sensitivity of CA1 pyramidal cells to synaptic input using full I/O curves. In initial experiments, recording in standard ACSF and at a membrane potential near the resting potential, no correlation was observed between the slope of an I/O curve and dendritic spine density, as predicted from the binding results. However, the estradiol-induced increase in NMDA receptor binding suggested that NMDA receptor-mediated EPSPs might be influenced by estradiol. When recording conditions were altered to enhance the NMDA receptor-mediated component of the EPSP, we observed a significant correlation between I/O slope and dendritic spine density. Furthermore, this relationship translated into a significant increase in mean I/O slope between cells from OVX+O and OVX+E animals. These results confirm that estradiol increases dendritic spine density on CA1 pyramidal cells and further demonstrate that this increase in spine density is well correlated with sensitivity to NMDA receptor-mediated synaptic input.
There are several lines of evidence indicating that estradiol treatment may increase numbers of NMDA receptors in the hippocampal CA1 region. First, it has been shown previously that the estradiol-induced increase in NMDA receptor binding in CA1 reflects an increase inBmax rather than an affinity change in the receptor (Weiland, 1992a). Second, it was reported recently that immunofluorescence for NMDA receptor subunit 1 (NMDAR1), an obligatory subunit of the NMDA receptor complex, is upregulated in the CA1 region by estradiol treatment (Gazzaley et al., 1996). These observations, in combination with our morphological, binding, and electrophysiological results, suggest an increase in NMDA receptor number. However, it should be noted that neither binding nor immunohistochemistry (at the light microscopic level) can distinguish between synaptic and nonsynaptic receptors; thus, it cannot be assumed that all receptors detected histologically contribute to synaptic responses.
There is also evidence to suggest that estradiol may regulate the NMDA receptor and spine/synapse number, at least in part, independently. First, Gazzaley et al. (1996) observed an estradiol-induced increase in NMDAR1 immunofluorescence in the cell body as well as dendritic layers of CA1, and an increase in the dentate gyrus cell body layer (although spine changes are not observed in granule cells). Second, the increase in NMDA receptor binding is of slightly greater magnitude than the morphological change. However, even if the estradiol-induced increases in NMDA receptor binding and NMDAR1 immunofluorescence are relevant to processes other than increased numbers of dendritic spines, the increases in binding and NMDAR1 immunofluorescence in CA1 are likely to be involved in the increased sensitivity of CA1 pyramidal cells to NMDA receptor-mediated synaptic input.
The observations that estradiol specifically increases NMDA receptor binding as well as sensitivity specifically to NMDA receptor-mediated synaptic input suggests the possibility that the new dendritic spines and synapses induced by estradiol treatment could be a specialized subpopulation of contacts in which the NMDA receptor is dominant. This suggestion appears to contradict the generally held notion that NMDA and non-NMDA receptors are colocalized at excitatory synapses in the hippocampus as determined from analysis of miniature EPSCs in cultured hippocampal neurons (Bekkers and Stevens, 1989). However, such colocalization of receptor types may not be the case for all hippocampal spine synapses. Indeed, Bekkers and Stevens (1989) found colocalization of NMDA and non-NMDA receptors at ∼70% of excitatory synapses; 20% of synapses appeared to be non-NMDA and 10% pure NMDA synapses. Second, proportions of synapses in which receptor types are colocalized may differ between cultured cells and cells in the acute hippocampal slice or the hippocampus in vivo. Furthermore, a population of “silent” synapses on CA1 pyramidal cells that, in hippocampal slices, apparently mediate pure NMDA receptor EPSCs has recently been electrophysiologically demonstrated (Isaac et al., 1995;Liao et al., 1995). It seems plausible that the new dendritic spines and synapses induced by estradiol treatment represent such a subpopulation of pure NMDA synapses. However, it should be mentioned that our data cannot distinguish between the possibility that estradiol induces a specialized subpopulation of synapses at which the NMDA receptor dominates the postsynaptic response and, alternatively, uniformly increases the contribution of the NMDA receptor at synapses containing both NMDA and non-NMDA receptor types.
In previous reports, we have hypothesized that estradiol induction of new dendritic spines and synapses could provide a structural mechanism by which estradiol exerts its effects on hippocampal physiology. Estradiol has been shown to increase hippocampal excitability as demonstrated by facilitation of kindled seizure acquisition in the hippocampus (Buterbaugh and Hudson, 1991), increased severity of kainic acid-induced seizures (Nicoletti et al., 1985), and decreased hippocampal seizure threshold (Terasawa and Timiras, 1968). Furthermore, during the estrous cycle, in which spine and synapse density fluctuate naturally with changing hormone levels (Woolley et al., 1990), the threshold for hippocampal seizure activity decreases (Terasawa and Timiras, 1968) and LTP is enhanced (Warren et al., 1995) as spine and synapse density increase. Interestingly, Wong and Moss (1992) previously reported that ∼20% of CA1 pyramidal cells in hippocampal slices from estradiol-treated rats showed prolonged EPSPs in response to Schaffer collateral stimulation. These authors suggested that such prolonged responses could be attributable to enhanced NMDA receptor activation. Because the recordings in that study were made from a membrane potential near the resting potential (i.e., the cells were relatively hyperpolarized), it is possible that some NMDA contribution went undetected because of the voltage dependence of the NMDA receptor.
The possibility that the new spines and synapses induced by estradiol are enriched in NMDA receptors is consistent with the electrophysiological findings mentioned above. The types of stimuli that have been used to detect estradiol’s effects on hippocampal physiology, i.e., seizure- or LTP-inducing stimuli, are those that produce substantial postsynaptic depolarization and thus are likely to be affected by putative pure NMDA receptor synapses. The possibility that ovarian steroids induce synapses at which NMDA receptors dominate has been suggested previously by Warren et al. (1995) to be involved in differences in LTP across the estrous cycle. In this study, animals in different phases of the estrous cycle showed no differences in a pre-LTP I/O curve generated with low-frequency test stimuli. However, animals in the proestrus phase of the cycle, the phase in which spine and synapse density are elevated (Woolley et al., 1990; Woolley and McEwen, 1992), show greater potentiation after a tetanic stimulus than animals in other phases of the cycle (Warren et al., 1995). Warren et al. suggested that, in the proestrus animals, a new population of synapses in which NMDA receptors are the dominant or active receptor type may be available to be potentiated. Our results are consistent with this suggestion.
This effect of estradiol, to enhance sensitivity of CA1 pyramidal cells to NMDA receptor-mediated synaptic input, is not mutually exclusive with other mechanisms by which estradiol treatment may alter excitability of these cells. The most likely additional mechanism by which estradiol may act is through regulation of inhibition of CA1 pyramidal cells. It is the interneurons in the CA1 region that have been shown to concentrate radiolabeled estradiol and, therefore, are most likely to possess classical estradiol receptors (Loy at al., 1988). Furthermore, estradiol has been shown to regulate levels of glutamic acid decarboxylase mRNA in CA1 cells (Weiland, 1992b) as well as GABAA receptor binding in CA1 (Schumacher et al., 1989).
The mechanism by which estradiol induces new dendritic spines and synapses on hippocampal CA1 pyramidal cells is still unclear. Estradiol induction of dendritic spines could be initiated through direct, nongenomic action (see, for example, Wong and Moss, 1992; Gu and Moss, 1996), through a mechanism involving inhibition (or disinhibition), or by some other, as yet unidentified, mechanism. In a recent report,Murphy and Segal (1996) demonstrated estradiol induction of dendritic spines on hippocampal pyramidal cells in vitro. Hippocampal neurons cultured for several days in the presence of estradiol show increased dendritic spine density compared to cells in control cultures. Estradiol induction of dendritic spines in vitromay be similar to estradiol’s effects in vivo because both occur on a similar time scale; further, both the in vitro(Murphy and Segal, 1996) and in vivo (Woolley and McEwen, 1994) effects are blocked by NMDA receptor antagonists. The in vitro induction of dendritic spines by estradiol may provide a useful system in which to address the molecular mechanism(s) by which estradiol induces new dendritic spines.
In summary, the results presented here represent the first attempt to define the functional consequences of estradiol-induced dendritic spines and synapses on hippocampal CA1 pyramidal cells. Our results demonstrate that estradiol treatment increases the sensitivity of CA1 pyramidal cells to NMDA receptor-mediated synaptic input, possibly via induction of a specialized subpopulation of dendritic spines containing synapses at which NMDA rather than non-NMDA glutamate receptors are the dominant receptor type. Although these synapses may not be active under baseline conditions, they could be recruited under conditions of increased postsynaptic depolarization. As such, these synapses may be involved either in learning and memory processes as reflected by LTP or in seizure generation. The significance of such synapses in the normal functioning of the hippocampus remains to be determined.
Footnotes
This research was supported by National Institutes of Health Grants NS 18895 to P.A.S. and NS 09787 to C.S.W.
Correspondence should be addressed to Catherine S. Woolley, Department of Neurological Surgery, Box 356470, University of Washington, Seattle, WA 98195.
REFERENCES
- 1.Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- 2.Buterbaugh GG, Hudson GM. Estradiol replacement to female rats facilitates dorsal hippocampal but not ventral hippocampal kindled seizure acquisition. Exp Neurol. 1991;111:55–64. doi: 10.1016/0014-4886(91)90050-m. [DOI] [PubMed] [Google Scholar]
- 3.Dewar D, Chalmers DT, Graham DI, McCulloch J. Glutamate metabotropic and AMPA binding sites are reduced in Alzheimer’s disease: an autoradiographic study of the hippocampus. Brain Res. 1991;553:58–64. doi: 10.1016/0006-8993(91)90230-s. [DOI] [PubMed] [Google Scholar]
- 4.Gazzaley AH, Weiland NG, McEwen BS, Morrision JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. J Neurosci. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. 1990;10:1286–1291. doi: 10.1523/JNEUROSCI.10-04-01286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu Q, Moss RL. 17β-estradiol potentiates kainate-induced currents via activation of the cAMP cascade. J Neurosci. 1996;16:3620–3629. doi: 10.1523/JNEUROSCI.16-11-03620.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guthrie PB, Segal M, Kater SB. Independent regulation of calcium revealed by imaging dendritic spines. Nature. 1991;354:76–79. doi: 10.1038/354076a0. [DOI] [PubMed] [Google Scholar]
- 8.Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- 9.Horner CH. Plasticity of the dendritic spine. Prog Neurobiology. 1993;41:281–321. doi: 10.1016/0301-0082(93)90002-a. [DOI] [PubMed] [Google Scholar]
- 10.Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 11.Koch C, Zador A. The function of dendritic spines: devices subserving biochemical rather than electrical compartmentalization. J Neurosci. 1992;13:413–422. doi: 10.1523/JNEUROSCI.13-02-00413.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of the hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 13.Loy R, Gerlach JL, McEwen BS. Autoradiographic localization of estradiol-binding neurons in the hippocampal formation and entorhinal cortex in the rat. Dev Brain Res. 1988;39:245–251. doi: 10.1016/0165-3806(88)90028-4. [DOI] [PubMed] [Google Scholar]
- 14.Muller W, Connors JA. Dendritic spines as individual neuronal compartments for synaptic Ca2+ responses. Nature. 1991;354:73–76. doi: 10.1038/354073a0. [DOI] [PubMed] [Google Scholar]
- 15.Murphy DD, Segal M. Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J Neurosci. 1996;16:4059–4068. doi: 10.1523/JNEUROSCI.16-13-04059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicoletti F, Speciale C, Sortino MA, Summa G, Caruso G, Patti F, Canonico PL. Comparative effects of estradiol benzoate, the antiestrogen clomipine citrate and the progestin medroxyprogesterone acetate on kainic acid-induced seizures in male and female rats. Epilepsia. 1985;26:252–257. doi: 10.1111/j.1528-1157.1985.tb05414.x. [DOI] [PubMed] [Google Scholar]
- 17.Schumacher M, Corini H, McEwen BS. Regulation of high affinity GABAA receptors in the dorsal hippocampus by estradiol and progesterone. Brain Res. 1989;487:178–183. doi: 10.1016/0006-8993(89)90955-4. [DOI] [PubMed] [Google Scholar]
- 18.Schwartzkroin PA. Characteristics of CA1 neurons recorded intracellularly in the hippocampal in vitro slice preparation. Brain Res. 1975;85:423–436. doi: 10.1016/0006-8993(75)90817-3. [DOI] [PubMed] [Google Scholar]
- 19.Segal M. Spines for neuroprotection: an hypothesis. Trends Neurosci. 1995;11:468–471. doi: 10.1016/0166-2236(95)92765-i. [DOI] [PubMed] [Google Scholar]
- 20.Terasawa E, Timiras PL. Electrical activity during the estrous cycle of the rat: cyclical changes in limbic structures. Endocrinology. 1968;83:207–216. doi: 10.1210/endo-83-2-207. [DOI] [PubMed] [Google Scholar]
- 21.Warren SG, Humphreys AG, Juraska JM, Greenough WT. LTP varies across the estrous cycle: enhanced synaptic plasticity in proestrus rats. Brain Res. 1995;703:26–40. doi: 10.1016/0006-8993(95)01059-9. [DOI] [PubMed] [Google Scholar]
- 22.Weiland NG. Estradiol selectively regulates agonist binding sites on the N-methyl-D-aspartate receptor complex in the CA1 region of the hippocampus. Endocrinology. 1992a;131:662–668. doi: 10.1210/endo.131.2.1353442. [DOI] [PubMed] [Google Scholar]
- 23.Weiland NG. Glutamic acid decarboxylase messenger ribonucleic acid is regulated by estradiol and progesterone in the hippocampus. Endocrinology. 1992b;131:2697–2702. doi: 10.1210/endo.131.6.1446611. [DOI] [PubMed] [Google Scholar]
- 24.Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woolley CS, McEwen BS. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J Comp Neurol. 1993;336:293–306. doi: 10.1002/cne.903360210. [DOI] [PubMed] [Google Scholar]
- 26.Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N-methyl-D-aspartate receptor-dependent mechanism. J Neurosci. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci. 1990;10:4035–4039. doi: 10.1523/JNEUROSCI.10-12-04035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong M, Moss RL. Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1992;12:3217–3225. doi: 10.1523/JNEUROSCI.12-08-03217.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]