

Abstract
Fragile X syndrome, a leading cause of inherited mental retardation, is attributable to the unstable expansion of a CGG-repeat within the FMR1 gene that results in the absence of the encoded protein. The fragile X mental retardation protein (FMRP) is a ribosome-associated RNA-binding protein of uncertain function that contains nuclear localization and export signals. We show here detailed cellular localization studies using both biochemical and immunocytochemical approaches. FMRP was highly expressed in neurons but not glia throughout the rat brain, as detected by light microscopy. Although certain structures, such as hippocampus, revealed a strong signal, the regional variation in staining intensity appeared to be related to neuron size and density. In human cell lines and mouse brain, FMRP co-fractionated primarily with polysomes and rough endoplasmic reticulum. Ultrastructural studies in rat brain revealed high levels of FMRP immunoreactivity in neuronal perikarya, where it is concentrated in regions rich in ribosomes, particularly near or between rough endoplasmic reticulum cisternae. Immunogold studies also provided evidence of nucleocytoplasmic shuttling of FMRP, which was localized in neuronal nucleoplasm and within nuclear pores. Moreover, labeling was observed in large- and small-caliber dendrites, in dendritic branch points, at the origins of spine necks, and in spine heads, all known locations of neuronal polysomes. Dendritic localization, which was confirmed by co-fractionation of FMRP with synaptosomal ribosomes, suggests a possible role of FMRP in the translation of proteins involved in dendritic structure or function and relevant for the mental retardation occurring in fragile X syndrome.
Keywords: fragile X syndrome, mental retardation, ribosomes, RNA binding proteins, FMRP, fragile X mental retardation protein, trinucleotide repeats, dendritic protein synthesis
Fragile X syndrome is the most frequent form of inherited mental retardation in humans (Warren and Ashley, 1995) and has a phenotype that commonly includes characteristic craniofacial dysmorphisms and macro-orchidism. The mutational mechanism of fragile X syndrome is the unstable expansion of a CGG trinucleotide repeat within the 5′-untranslated region of the FMR1 gene (Fu et al., 1991;Oberle et al., 1991; Verkerk et al., 1991; Ashley et al., 1993). In fragile X syndrome, the FMR1 repeat is massively expanded to an average of 800 triplets, in contrast to the normal mode of 30 triplets (Brown et al., 1993; Kunst and Warren, 1994; Rousseau et al., 1995). When expanded beyond ∼230 repeats, the FMR1 gene becomes aberrantly methylated (Sutcliffe et al., 1992; Hornstra et al., 1993) and transcriptionally silent (Pieretti et al., 1991). Thus, the FMR1 repeat expansion results in the absence of the encoded protein, fragile X mental retardation protein (FMRP), which appears to be responsible for the phenotype (Devys et al., 1993). This has been confirmed by the characterization of rare patients with the clinical phenotype of fragile X syndrome, who harbor intragenic deletions or splice/missense mutations (Meijer et al., 1994; Hirst et al., 1995; Lugenbeel et al., 1995) rather than repeat expansions.
Attention has now focused on the normal function of FMRP. FMR1 mRNA expression has been found to be widespread but not ubiquitous. Within the brain, expression is limited to neurons and has been suggested to be particularly prominent in the hippocampus, nucleus basalis, and granular layer of the cerebellum (Abitbol et al., 1993; Hinds et al., 1993; Hergersberg et al., 1995). FMRP contains amino acid domains that are common among RNA-binding proteins and has been demonstrated to interact with RNA homopolymers as well as with a subset of brain mRNAs, including its own message (Ashley et al., 1993; Siomi et al., 1993). Initial immunocytochemical studies suggested that FMRP localizes predominantly to cytoplasm (Devys et al., 1993; Verheij et al., 1993). Molecular studies, however, have demonstrated the presence of both nuclear localization and nuclear export signals within FMRP, suggesting the potential to shuttle between the nuclear and cytoplasmic compartments (Eberhart et al., 1996). These data, and recent evidence that FMRP co-fractionates with ribosomes (Eberhart et al., 1996;Khandjian et al., 1996; Siomi et al., 1996), suggest that FMRP may be involved in the transport of a subset of nuclear mRNAs, in their subsequent association with ribosomes, and potentially in regulating translation. In the current study, we have taken a combined biochemical and morphological approach to examine whether the subcellular localization of FMRP is consistent with this model and also to examine how FMRP is distributed within neurons. We show that FMRP is cytoplasmic, co-fractionating primarily with free polysomes. Using immunogold electron microscopy (EM), we directly localize FMRP to intraneuronal polysomes and provide the first direct visualization of FMRP in nucleoplasm and nuclear pores. Finally, we find FMRP to be associated with polysomes in dendrites and dendritic spines, suggesting that FMRP may play a role in the translation of proteins related to dendritic function.
MATERIALS AND METHODS
Subcellular fractionation. All fractionation steps in Figure 1 were carried out at 4°C. Cells were disrupted by vacuum cavitation (200 psi for 10 min) in a buffer in 0.25m sucrose, 50 mm Tris, pH 7.5, 25 mm KCl, 5 mm MgCl2, 1 mm PMSF, and 1 μg/ml each aprotinin, pepstatin, and leupeptin (Sigma, St. Louis, MO). The fractionation scheme followed that of Krajewski (1993). The lysate was subjected to 500 ×g centrifugation for 5 min. The pellet was resuspended in 1.6 m sucrose and centrifuged through a 2.1 msucrose pad at 150,000 × g for 1 hr to isolate cytoplasmic-free nuclei. The cytoplasmic supernatant was subjected to 10,000 × g centrifugation for 10 min to yield the heavy membrane pellet and the postnuclear supernatant. The postnuclear supernatant was then centrifuged for 1 hr at 130,000 ×g. The resulting pellet contained light membrane and polysomes, and the supernatant was centrifuged further at 180,000 × g for 3 hr to yield the insoluble and soluble cytoplasmic fractions. Gradient 1 (Frangioni et al., 1992) contained sucrose layers of 0.25 m, 1.35 m, 1.6 m, and 2.0m. After centrifugation at 230,000 × g for 2 hr, the top layer containing cytosol, the low-density membrane at the 0.25/1.35 m interface, the high-density membrane at the 1.6/2.0 m interface, and the polysome pellet at the bottom were collected. Gradient 2 (Krajewski et al., 1993) contained sucrose layers of 0.8 m, 1.23 m, 1.35 m, and 2.1 m. Eighty microliters was taken from each interface, which contained smooth endoplasmic reticulum (ER) (0.8/1.23m interface), light rough ER (RER) (1.23/1.35 minterface), and heavy RER (1.35/2.1 m interface). All the fractions were lysed in 1× Laemmli buffer containing 8 murea (Feng et al., 1995a), and the protein concentration of each fraction was determined by Bradford assay (Bio-Rad, Hercules, CA).
Fig. 1.

FMRP distribution in subcellular fractions of human lymphoblastoid cells. A is a schematic illustration of subcellular fractionation of EBV-transformed human lymphoblastoid cells. The descriptor under key fractions refers to the panels below(B–D), with INT as interface. A detailed description and protocol is provided in Materials and Methods. InB, the left panel shows SDS-PAGE immunoblot analysis of FMRP and P0 in crude subcellular fractions. Total protein (3 μg) from each fraction of B1–B5 was loaded. Densitometric analysis of immunoblot signals was used to calculate the total yield of FMRP in each corresponding fraction, based on the total fraction volume. The relative percentage of FMRP in each fraction was plotted as shown in the right panel. Cshows the SDS-PAGE immunoblot analysis of FMRP and P0 in separated postnuclear supernatant fraction. C1–C4 in sucrose gradient fractionation 1 represent cytosol; low-density membranes (plasma membrane, Golgi, and smooth ER); high-density membranes (RER); and free polysome pellet, respectively. Total protein (1.5 μg) from each fraction was loaded. D shows the SDS-PAGE immunoblot analysis of FMRP and P0 in various ER components.D1–D3 in sucrose gradient fractionation 2 represent smooth ER, light RER, and heavy RER. Total protein (3 μg) from each fraction was loaded.
Fractionation of rat cortex homogenate and synaptosomal lysate.Cerebral cortex from adult male Sprague Dawley rats was rapidly removed after decapitation and placed into 0.32 m sucrose (10% w/v) containing 4 mm HEPES, pH 7.3, 5 mmMgCl2, 200 μg/ml cycloheximide, 1 mm PMSF, and 1 μg/ml each aprotinin, pepstatin, and leupeptin (Sigma). The tissue was incubated for 20 min on ice to arrest polysome migration before gentle homogenization (nine strokes) with a glass homogenizer. The velocity centrifugation procedure was essentially as described by (Huttner et al., 1983) to generate S1, S2, P2, S3, and P3, as illustrated in Figure 2. Each fraction was then subjected to lysis in 1× Laemmli buffer containing 8 murea. Bradford assay was carried out for each fraction to determine the protein concentration, followed by SDS-PAGE analysis. To generate P2 lysate, the pellet was lysed in 600 μl 0.16 m sucrose containing 2.5 mm MgCl2, 10 mmTris-HCl, pH 7.5, 50 mm KCl, and 0.5% NP40. For EDTA lysis, P2 was lysed in the buffer described above, except that MgCl2 was replaced with 30 mm EDTA. The P2 lysate was left on ice for 15 min, followed by microfuge centrifugation to remove insoluble membrane components, and the corresponding supernatant was loaded onto a 20–47% (w/w) sucrose gradient containing 80 mm NaCl, 20 mm Tris, pH 7.5, and 5 mm MgCl2 or 30 mm EDTA, correspondingly. After centrifugation in a Beckman SW41 rotor at 39,000 rpm for 100 min at 4°C, the entire gradient was fractionated by upward displacement into twelve ∼1 ml fractions using a gradient fractionator (Isco, Lincoln, NE). The fractions were subjected to Bradford assay to determine the corresponding protein concentration. Because the top three fractions contained the highest total protein levels, to avoid overloading, an aliquot from fractions 1 and 2 matching the amount of total protein in fraction 3 (70 μg) was subjected to TCA precipitation, whereas for the rest of fractions, the entire 1 ml was subjected to TCA precipitation. The precipitates were resuspended in 10 μl of 0.5 m Tris-HCl, pH 7.5, and 0.5m NaCl, followed by the addition of 30 μl of 1× Laemmli buffer containing 8 m urea before SDS-PAGE analysis.
Fig. 2.
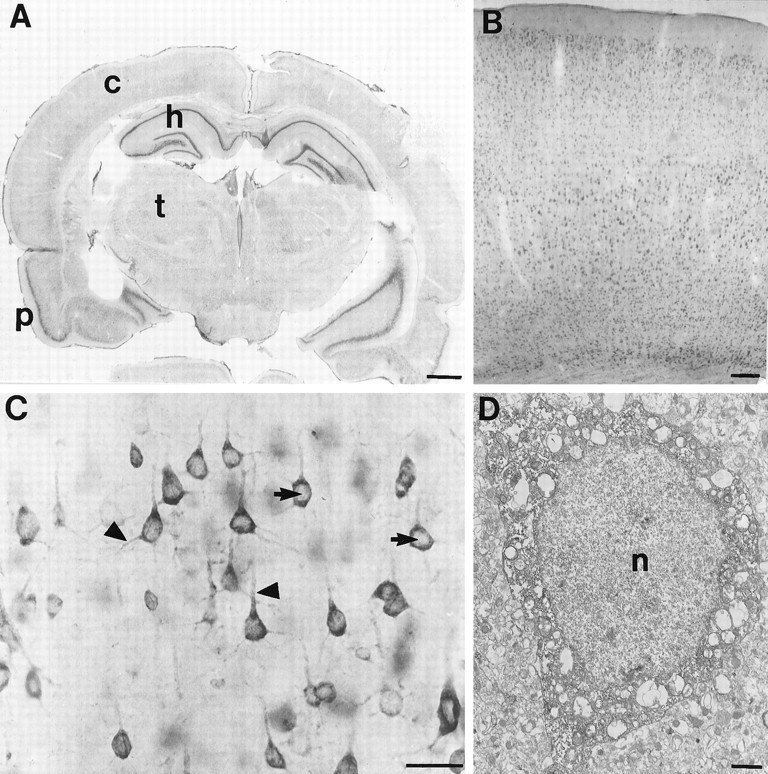
FMRP immunocytochemistry in the rat brain.A is a coronal section demonstrating widespread FMRP labeling. The most intense labeling is in the cellular layers of the hippocampus (h) and pyriform cortex (p), which are regions with extremely high neuronal densities. The deeper layers of the cerebral cortex (c) are also well labeled. B demonstrates FMRP immunoreactivity in the frontal cortex at higher magnification. It appears that most neurons in each cortical layer are FMRP-positive.C illustrates the cellular pattern of FMRP immunoreactivity in layer V pyramidal cells from frontal cortex. Staining is very dense in perikarya and proximal dendrites (triangles). In contrast, nuclear staining (arrows) is uncertain. D is an electron micrograph of the soma of a cerebral cortical pyramidal cell. With immunoperoxidase, dense cytoplasmic staining is evident. Although the nucleus is somewhat dark, FMRP immunoreactivity is not clearly present. Scale bars: A, 50 mm; B, 100 μm;C, 50 μm; D, 1 μm.
Immunoblot analysis and antibodies. Protein samples were resolved on 12% SDS/polyacrylamide gels (Bio-Rad) along with prestained molecular weight markers (Bio-Rad) and were subsequently electroblotted at 30 V overnight onto nitrocellulose membranes (Schleicher and Schuell, Keene, NH). Immunostaining and ECL detection were performed at room temperature according to the manufacturer’s protocol (Amersham, Arlington Heights, IL) with the secondary antibody incubation performed in a buffer containing 0.5× PBS, 0.5% milk, 10 mm Tris, pH 7.5, 75 mm NaCl, 0.5 mmEDTA, 0.5% NP40, 0.25% deoxycholate, 0.05% SDS, and 2.5 mm NaF. The primary antibodies and the concentrations used were as follows: the anti-FMRP monoclonal antibody mAb1a (Devys et al., 1993) at 1:10,000; the anti-P polyclonal human autoantibody (Bonfa et al., 1989) at 1:1000; the monoclonal antibody against the large subunit of wheat germ RNA polymerase II (8WG16, Thompson et al., 1989) at 1:4000; the polyclonal antibody against rabbit mitochondrial branched chain α-ketoacid dehydrogenase E2 (Heffelfinger et al., 1983) at 1:700; the monoclonal antibody against human LDH (Sigma) at 1:4000; and the monoclonal antibody against mouse synaptophysin (Boehringer Mannheim, Indianapolis, IN) at 1:150.
Immunocytochemistry. Four young adult male Sprague Dawley (Harlan, Prattville, AL) rats were deeply anesthetized with chloral hydrate and perfused transcardially with 240 ml of 3% paraformaldehyde and 0.2% glutaraldehyde in 0.1 m phosphate buffer (PB). Brains were removed and sectioned at 40 μm using a vibratome (Technical Products International). Sections were collected in 100 mm PBS and rinsed in 50 mm TBS, pH 7.2. Some sections were processed for immunoperoxidase using DAB as the chromogen. Briefly, sections were incubated for 1 hr at 4°C in TBS containing 4% normal goat serum (NGS) and avidin (Vector Research, Burlingame, CA) at 10 μg/ml, rinsed in TBS, and incubated at 4°C on a shaker for 48 hr in TBS containing 2% NGS, biotin (Vector Research) at 50 μg/ml, and monoclonal mouse anti-FMRP antibodies mAb1a (Devys et al., 1993) at 1/1000. Sections were then rinsed in TBS and incubated overnight in biotinylated goat anti-mouse secondary antibody (Vector Research) in TBS containing 2% NGS. After rinses in TBS, sections were incubated in ABC Elite (Vector) for 4 hr followed by TBS rinses. Final development was done by incubation in 0.05% 3,3′-DAB tetrahydrochloride (Sigma) and 0.01% hydrogen peroxide in 50 mm Tris buffer for 5–15 min. Sections were then rinsed with TBS for another hour.
Immunogold labeling for EM was performed as follows. Rat brain sections were preblocked in TBS containing 4% NGS and 0.05% Triton X-100 and incubated in mouse monoclonal anti-FMRP antibody mAb1a (Devys et al., 1993) at 1/1000 in 2% NGS–TBS for 60 hr at 4°C on a shaker platform. Sections were then rinsed in TBS for a total of 1 hr and incubated overnight in goat anti-mouse secondary antibody (1:50) conjugated to 1.4 nm gold particles (Nanoprobes, Stony Brook, NY) in TBS with 2% NGS. After rinsing in PB, the sections were fixed with 2% glutaraldehyde. After several washes in PB, sections were silver-intensified according to the silver intensification kit from Nanoprobes (Stony Brook, NY). Finally, sections were post-fixed in 1% OsO4 in PB, rinsed, and dehydrated in ascending concentrations of ethanol and propylene oxide (1:1) and embedded in Epon (Ted Pella, Redding, CA). Ultrathin sections (90 μm) were cut using a Leica Ultracut S ultramicrotome. Lead staining was performed on grids by 5 min incubation in 5% aqueous uranyl acetate followed by 10 min incubation in lead citrate. Ultrathin sections were examined using a JEOL 100C electron microscope. Controls for both DAB and immunogold included sections processed in parallel but without exposure to the FMRP antibody and preabsorption controls in which the primary antibody was first incubated with purified FMRP conjugated to CNBr-activated Sepharose. EM was performed on the antibody deletion but not the preabsorption controls, because the latter procedure usually leaves trace residual specific staining at the EM level attributable to absorption not being 100%.
RESULTS
FMRP co-fractionates with nonmembrane polysomes and the RER
To define the distribution of FMRP in various subcellular compartments, normal EBV-transformed human lymphoblastoid cell lysate was fractionated, as illustrated in Figure 1A, followed by immunoblot analysis of the fractions using an anti-FMRP monoclonal antibody (Devys et al., 1993). The cell lysate was initially fractionated by sequential velocity centrifugation, resulting in fractions designated as nuclei, heavy membrane, light membrane/polysome, insoluble cytoplasm, and soluble cytoplasm (Fig.1A, fractions B1–B5). To verify the content of these fractions, immunoblot analysis was performed using antibodies against specific compartmentalized proteins (data not shown). RNA polymerase II was found primarily in the nuclear fraction (B1); mitochondrial branched chain α-ketoacid dehydrogenase E2 subunit was confined to the heavy membrane pellet (B2); ribosomal acidic phosphoprotein P0 (Bonfa et al., 1989) was found primarily in the light membrane/polysome pellet (B3); and lactate dehydrogenase was found in the soluble cytoplasm (B5). These fractions were then examined for the presence of FMRP. Densitometric quantitation of FMRP signal on the immunoblot was used to calculate the percentage of total cellular FMRP in each fraction based on the corresponding fraction volume. Even with a conservative estimate, considering that the most intensive signal in B3 may approach saturation, >80% of FMRP was confined to the light membrane/polysome fraction (Fig. 1,B3). The ∼37 kDa P0 acidic phosphoprotein located on the 60S ribosomal subunit was also confined primarily to this fraction, verifying the expected presence of RER and translating ribosomes. The detection of both FMRP and P0 in the heavy membrane fraction (B2) was most likely attributable to the presence of limited RER in this fraction. Interestingly, a low level of FMRP (∼4%) was detected in the nuclear fraction with no P0 detected even after prolonged exposure. Detection of FMRP in the insoluble cytoplasm required prolonged exposure, and a negligible amount of FMRP was detected in the soluble cytoplasm, indicating that FMRP is rarely present as a free protein. Very similar subcellular distribution of FMRP was observed in mouse brain in a parallel experiment (data not shown).
To determine whether FMRP only co-fractionates with components that carry ribosomes, i.e., RER, the postnuclear supernatant was fractionated through a discontinuous sucrose gradient of 0.25, 1.35, 1.6, and 2.0 m sucrose to separate various membrane components (Frangioni et al., 1992) as well as free polysomes (Fig.1A, sucrose gradient fractionation 1). Four fractions were obtained: free cytosol (C1, 0.25m sucrose layer); low-density membrane containing plasma membrane, Golgi, and smooth ER (C2, 0.25–1.35 msucrose interface); RER (C3, 1.6–2.0 m sucrose interface); and nonmembrane associated polysomes (C4, pellet). Examination of these fractions revealed that all the FMRP was restricted to fractions containing RER and polysomes (Fig.1C, C3, C4). P0 also localized to these fractions, confirming the expected presence of 60S ribosomal subunits. The exclusive fractionation of FMRP and P0 to C3 and C4 was also observed when human fibroblasts, HeLa cells, and mouse brain were examined (data not shown).
The co-fractionation of FMRP with the RER was confirmed by a similar fractionation (Krajewski et al., 1993) of the postnuclear supernatant through a discontinuous sucrose gradient of 0.8, 1.23, 1.35, and 2.1m sucrose (Fig. 1A, sucrose gradient fractionation 2). This fractionation separated smooth ER (D1, 0.8–1.23 m sucrose interface), light RER (D2, 1.23–1.35 m sucrose interface), and heavy RER (D3, 1.35–2.1 m sucrose interface). As shown in Figure 1D, the majority of FMRP localized to the heavy RER (D3), with little FMRP found in the light RER (D2), and none detectable in the smooth ER fraction (D1), even after prolonged film exposure. P0 fractionated to both D2 and D3, as expected. The low level of FMRP in the light RER fraction (D2) as compared with P0 suggests that FMRP is associated with only a subset of ribosomes.
FMRP is highly expressed in neurons
FMRP expression was examined by light microscopy and immunoperoxidase using the FMRP-specific monoclonal antibody, as described above. As described previously in human (Devys et al., 1993), FMRP was highly expressed within neurons throughout the rat brain (Fig.2A), whereas glial staining was minimal. As predicted by in situ hybridization studies (Abitbol et al., 1993;Hinds et al., 1993), hippocampus (Fig. 2A), nucleus basalis of Meynert, and cerebellum showed high levels of FMRP staining at low magnification. When comparing the immunoreactivity of individual neurons, however, many other neuronal types in a variety of forebrain and hindbrain regions were just as intensely stained as the neurons in these regions. Because the EM studies in this paper are in cerebral cortex, its FMRP immunoreactivity will be more fully described. Cerebral cortical pyramidal and nonpyramidal cells were filled with DAB reaction product (Fig. 2B,C). Reaction product densely filled perikarya and proximal dendrites (Fig.2C). Some possible labeling was also apparent in nuclei but always much lighter than in the cytoplasm. Smaller caliber elements were also visible in the neuropil, but their identity could not be resolved at the light microscopic level. All immunoreactivity was abolished by preabsorption of primary antibodies with excess purified FMRP conjugated to CNBr-activated Sepharose. Labeling was also absent when primary antibodies were omitted.
FMRP is present in nuclei and cytoplasm of neurons and co-localizes with RER and somatodendritic polysomes
FMRP localization was examined at the ultrastructural level by immunoperoxidase and immunogold EM. Immunoperoxidase provided greater sensitivity for determining whether neuropil elements were labeled, whereas immunogold provided sufficient spatial resolution to examine the subcellular associations of FMRP. Most neuronal somata encountered appeared to be labeled (Fig. 2D). Because the DAB reaction product was difficult to distinguish from nucleoplasm, however, nuclear localization of FMRP could not be clearly determined in the immunoperoxidase material (Fig. 2D). The most important finding with immunoperoxidase was that many dendrites of all calibers were labeled, indicating that FMRP can be found in relatively distal dendrites.
The immunogold labeling provided much greater spatial resolution. Immunogold particles were found within neuronal nuclei (Fig.3A) but were rare in nuclei of any other cell types including astrocytes (Fig. 3B), oligodendrocytes (Fig.3C), and epithelial cells lining the blood vessels (data not shown). Immunogold particles were visualized in nuclear pores within neurons, most likely labeling FMRP molecules in transit between the cytoplasm and the nucleus (Fig.3D,E). In neuronal perikarya, most immunogold particles were found free in the cytoplasm and appeared to concentrate in regions rich in free ribosomes, particularly near or between cisternae of the RER (Fig. 3F–H). A small proportion of immunogold particles were also found directly in contact with the membranes of RER (Fig.3F,G). Immunogold particles were rarely seen directly associated with the plasma membrane or with other organelles, including mitochondria, transport vesicles, multivesicular bodies, or Golgi apparatus (Fig. 3C). Even though the highest concentrations of immunogold particles were seen in neuronal perikarya, numerous collections of particles were also found in dendrites at sites where ribosomes are customarily found. Specifically, dendritic immunogold particles were frequently visualized near cisternae of smooth ER (Fig.4A,B), at dendritic branch points, and at the origins of dendritic spines (Fig.4C). Label was not confined to proximal dendrites but was also frequently found in relatively small-caliber dendrites. Many dendritic spines also contained immunogold particles that were either free in the cytoplasm (Fig.4E,F) or associated with the spine apparatus (Fig. 4D). In contrast to the intense somatodendritic labeling, most axons and axon terminals were free of immunogold particles. FMRP immunoreactive axon terminals contained only one to three immunogold particles that were cytoplasmic in location (Fig. 4G). When using immunoperoxidase, which is more sensitive than immunogold, more FMRP-immunoreactive axon terminals could be identified (Fig. 4H).
Fig. 3.
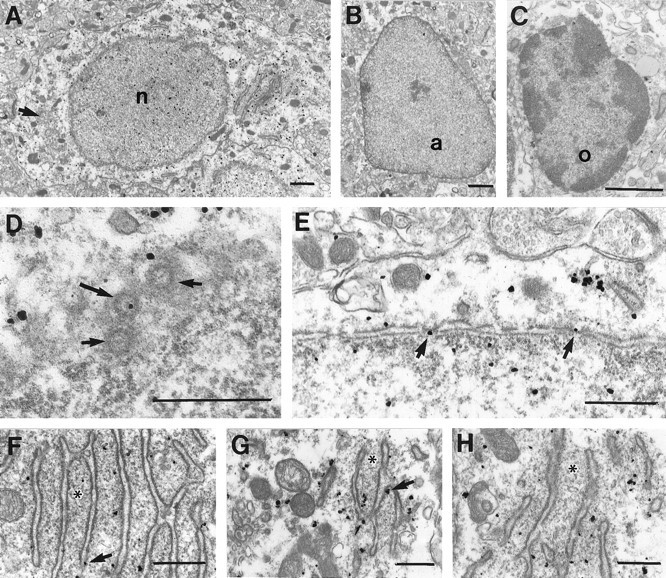
A, Electron micrograph of the soma of a labeled cerebral cortical pyramidal neuron. Immunogold particles are present both in the nucleus (n) and perikaryon. Little of the cytoplasmic label is associated with the plasma membrane, mitochondria, or Golgi apparatus (arrow).B, C, Few particles were found in the nuclei or cytoplasm of astrocytes (a) or oligodendrocytes (o). D, Tangential section through the nuclear envelope of a pyramidal cell showing three nuclear pores (arrows), one of which (longer arrow) contains an immunogold particle. E, Cross-section through the nuclear envelope showing immunogold particles within nuclear pores (arrows).F–H, In perikarya, most immunogold particles are clustered between the cisternae of RER. These regions are especially rich in free ribosomes, visible here as fine electron-dense particles (asterisks). A few immunogold particles are also seen in direct contact with the cisternae (arrows). Scale bars: A–C, 1 μm;D–H, 500 μm.
Fig. 4.

Electron micrographs demonstrating FMRP localization in cellular processes in cerebral cortex.A–C, Dendrites (d) in cross-section and longitudinal section showing that immunogold particles are either free in the cytoplasm or clustered around cisternae of smooth ER (arrows) or at the origins of dendritic spines (triangles).D–F, Dendritic spines (s) containing immunogold particles, which are either free in the cytoplasm or associated with the spine apparatus (arrows).G, Rare axon terminals (a) contain immunogold particles that are cytoplasmic in location.H, FMRP-immunoreactive axon terminals (a) are more easily identified using immunoperoxidase. Scale bars:A–H, 500 nm.
Background immunogold labeling was assessed by examining sections that were processed in parallel but not exposed to FMRP antibody. In these sections, scattered immunogold particles were diffusely present; however, they were at a very low density, did not occur in clusters, and did not show the selectivity described above for neurons, nuclear pores, dendrites, or polysomes.
FMRP co-fractionates with brain polysomes, including those found in synaptosomes
To confirm the microscopic finding that FMRP closely co-localizes with somatodendritic polysomes and RER, rat cortex was gently homogenized and fractionated by velocity centrifugation to generate a low-speed supernatant (Fig. 5, S1), a crude synaptosomal pellet (P2) and corresponding supernatant (S2), and finally a high-speed polysomal pellet (P3) and corresponding supernatant (S3). Various marker antibodies were used in immunoblot analysis to verify contents in the above fractions. As shown in Figure 5A, the majority of synaptophysin, a vesicle membrane protein broadly used as a synaptosomal marker, was fractionated into the synaptosomal pellet. Also as expected, the soluble cytoplasmic protein LDH was confined primarily to the supernatant fractions. FMRP and P0 signals from the cytoplasmic lysate were primarily confined to fraction P3, comparable with what was observed in the light membrane/polysome fraction derived from lymphoblasts. The remaining FMRP and ribosomes fractionated into the crude synaptosomal pellet (P2) and was not detected in the high-speed supernatant S3.
Fig. 5.

Association of FMRP with translating ribosomes in rat cortex. A, Subcellular fractionation of rat cortex by velocity centrifugation. The fractionation procedures are illustrated on the left panel, with Sindicating supernatant and P indicating pellet. A detailed description and protocol are provided in Materials and Methods. The right panel shows SDS-PAGE immunoblot analysis of FMRP and other marker proteins in the corresponding fractions as indicated. Based on Bradford assay, 20 μg of total protein from each fraction was used in this blot. B, Association of FMRP with polysomes in synaptosomal lysate. SDS-PAGE immunoblot analysis was performed using linear sucrose gradient fractions containing synaptosomal lysates with the presence of Mg2+ or EDTA, as described in Materials and Methods. The signals of FMRP and P0 protein are indicated on theright. The sedimentation of ribosomal components in human lymphoblasts monitored at OD254 in a parallel gradient are indicated on top of the corresponding fractions.
To determine whether FMRP associates with polysomes in synaptosomes, the majority of which originate from axospinous synapses, P2 was lysed under mild conditions with a low level of nonionic detergent. After removing the residual membrane component, the synaptosomal lysate was fractionated through a linear sucrose gradient, and each fraction collected was subjected to immunoblot analysis. FMRP co-fractionated with the P0 protein in the fractions containing monosomes and polysomes (Fig. 5B), similar to our previous observation in human lymphoblasts (Eberhart et al., 1996). To provide additional evidence for FMRP–ribosome association, P2 was lysed in the presence of 30 mm EDTA, a condition known to dissociate ribosomes into subunits and release mRNP particles. The disappearance of P0 signals in the polyribosomal fractions, together with the concomitant accumulation of P0 signals in the top fractions (Fig. 5B, fractions 2 and 3), confirmed that EDTA caused ribosome dissociation. FMRP shifted primarily to the top fractions containing ∼60–100S particles (fractions 3 and4). This result was consistent with our previous observation in EDTA-treated lymphoblastoid lysate (Eberhart et al., 1996), suggesting that FMRP associates with polysomes as an mRNP component.
DISCUSSION
We have taken a combined biochemical and morphological approach to study the cellular and subcellular localization and associations of FMRP. Our primary goal was to seek evidence supporting the hypothesis that FMRP shuttles between the nucleus and cytoplasm, carrying mRNAs to ribosomes and perhaps playing a role in the regulation of translation. Furthermore, because we have shown previously that FMRP binds only to a subset of mRNAs (Ashley et al., 1993), our second major goal was to determine whether this selectivity also extends to the ribosomes to which the FMRP–mRNA complexes are presumed to bind. The results of these experiments provide the first visualization of FMRP in neuronal nuclei, in transit within nuclear pores, and in association with polysomes in neuronal perikarya, but also distributed throughout the dendritic tree. We hypothesize that some of the mRNAs carried by FMRP code for proteins important for dendritic functions that are thus compromised in fragile X syndrome, leading to mental retardation.
The cellular localization of FMRP
By immunocytochemistry, we have observed widespread and intense FMRP immunoreactivity in neurons throughout the CNS. Previous studies using in situ hybridization or immunocytochemistry have similarly detected abundant FMRP mRNA and protein and have suggested that it is especially abundant in hippocampal pyramidal cells, giant cholinergic neurons of the nucleus basalis, and the cerebellum (Devys et al., 1993; Hinds et al., 1993). It is our impression, however, that the apparent enrichment of FMRP in these regions is attributable to the high packing density of neurons they contain, because individual neurons do not appear any more immunoreactive than do neurons in many other regions. Immunoperoxidase, however, does not lend itself very well to studying protein abundance of neurons, because the staining process is nonlinear. In addition, neuronal size and packing density are difficult to take into account. Thus, although FMRP expression in the brain is strongly neuronal, it is not yet clear whether there are meaningful differences in expression between neuronal populations.
Nucleocytoplasmic shuttling and ribosome association of FMRP
Several recent studies (Siomi et al., 1993; Burd and Dreyfuss, 1994) have suggested that FMRP may be a member of a new family of RNA binding proteins (RNPs). Expression studies in vitro have shown that FMRP can bind to RNAs (Ashley et al., 1993; Siomi et al., 1993) and that experimental mutations can disrupt RNA binding (Siomi et al., 1994). We recently demonstrated a functional nuclear localization signal and a functional nuclear export signal within FMRP (Eberhart et al., 1996), suggesting that nascent FMRP may be imported into the nucleus for RNP assembly, followed by export to the cytoplasm driven by the FMRP nuclear export signal. Missing, however, has been direct localization of native FMRP within the nucleus, perhaps because of the predominant steady-state localization of FMRP to the cytoplasm. Consistent with this, our cellular fractionation studies showed that only ∼4% of cellular FMRP co-fractionates with the nucleus. FMRP immunocytochemistry using immunoperoxidase suggested nuclear labeling; however, the reaction product was not definitively distinguishable from the normal nuclear contents. Immunogold EM, however, unequivocally localized FMRP within neuronal nucleoplasm. Furthermore, this method was able to label FMRP within nuclear pores, presumably in transit between the nucleus and cytoplasm. These results strongly support the hypothesis that FMRP shuttles from cytoplasm to nucleus, where it associates with RNA and perhaps other proteins before being exported as an mRNP particle (Feng et al., 1995b; Eberhart et al., 1996) to the cytoplasm.
Once in the cytoplasm, >85% of FMRP co-sediments with ribosomes (Khandjian et al., 1996). In the present study, we have defined further the association of FMRP and ribosomes using co-fractionation and immunogold studies. We have determined that FMRP associates most abundantly with a subset of free polysomes and also with some RER-associated ribosomes. These associations were also directly visualized by immunogold EM in rat brain. FMRP was not found to associate with other organelles, including Golgi apparatus, mitochondria, or plasma membrane, by either subcellular fractionation or EM, indicating that FMRP is unlikely to play a role in the corresponding cellular functions. These data directly confirm previous biochemical suggestions that FMRP is chiefly associated with ribosomes in mammalian cells.
FMRP in dendritic and axonal compartments
Neuronal ribosomes are not only present in the perikaryon but are also distributed through their dendrites and dendritic spines (Steward and Levy, 1982; Spacek, 1985; Steward and Reeves, 1988). There is increasing evidence that particular mRNAs are targeted to distinct intraneuronal compartments (Garner et al., 1988; Burgin et al., 1990;Tiedge et al., 1991; Miyashiro et al., 1994; Mohr et al., 1995). This targeting has been proposed as a mechanism to provide and regulate the local synthesis of particular proteins (see Steward, 1994). Our first indication that FMRP could be distributed to pre- or postsynaptic compartments was provided by its co-fractionation with synaptosomal ribosomes from rat cerebral cortex. When synaptosomal ribosomes were dissociated with 30 mm EDTA, FMRP appeared to be present primarily in complexes of 60–100S. This most likely represents FMRP associated with the 60S subunit (Khandjian et al., 1996; Siomi et al., 1996) and large mRNPs (Eberhart et al., 1996). These results also suggest that synaptosomal FMRP is primarily postsynaptic, because ribosomes are not found in axon terminals.
Using immunogold, we localized FMRP not only to neuronal perikarya, but also to large- and small-caliber dendrites and dendritic spines. Because individual immunogold particles are relatively large, direct labeling of individual ribosomes could not be visualized. The immunogold label, however, occurred in clusters at the exact intradendritic sites where ribosomes are usually found, including dendritic branch points, near cisternae of endoplasmic reticulum, at the bases of dendritic spines, and within spine heads. Although FMRP was found everywhere polysomes have been identified, this localization of FMRP in dendrites and spines raises the possibility that FMRP may be involved in regulating synthesis of proteins related to postsynaptic function. Interestingly, synthesis of some postsynaptic proteins can be upregulated by the actions of neurotransmitter receptors or by increased ionic conductances (Weiler and Greenough, 1993). Because FMRP is most abundant in tissues with high levels of protein synthesis, such as CNS neurons, healing tissues, and cells which are induced for proliferation (Devys et al., 1993; Khandjian et al., 1996), it has been suggested that it may play a role in active translation.
Using immunoperoxidase and immunogold, FMRP was also localized in small numbers of axons and axon terminals. Although axons do not contain ribosomes, they do contain many mRNAs (Perrone-Capano et al., 1987). Some mRNA species have been identified that appear to be specifically targeted to axons, including those for β-actin (Olink-Coux and Hollenbeck, 1996), vasopressin (Trembleau et al., 1994; Mohr et al., 1995), oxytocin (Jirikowski et al., 1990), and BC1 (Tiedge et al., 1993). The lack of axonal ribosomes has led to an assumption that translation does not occur in axons, however, some evidence for axonal protein synthesis is beginning to accumulate (reviewed in Van Minnen, 1994). It has also been hypothesized that axonal targeting of mRNA down-regulates protein synthesis by removing these mRNAs from the translating ribosomes (Jirikowski et al., 1990; Mohr et al., 1995). Whether FMRP has any functional significance in the axon still remains to be elucidated; however, it may play a role in the localization or regulation of some axonal mRNAs.
Fragile X syndrome and FMRP
It is now clear that the phenotype associated with fragile X syndrome, chiefly mental retardation, is the consequence of the absence of FMRP. Evidence is now accumulating that the normal function of FMRP is related to some aspect of mRNA transport and/or translation. FMRP clearly does not assume a vital role in translation, given the relatively subtle phenotype occurring in fragile X syndrome. This possibility is also suggested by the general lack of major brain pathology in the fmr1 knockout mouse (Dutch-Belgian Fragile X Consortium, 1994). On the other hand, the general function FMRP serves may be crucial, and its loss in fragile X syndrome may be partially made up for by the presence of other members of its gene family, such as FXR1 (Coy et al., 1995; Siomi et al., 1995) and FXR2 proteins (Zhang et al., 1995).
The most notable neuropathology identified in postmortem brain tissue from patients with fragile X syndrome is that cerebral cortical dendritic spines are lengthened and possess enlarged heads (Rudelli et al., 1985; Hinton et al., 1991). Our localization of FMRP to the spine heads and bases raises the possibility that the spine dysmorphisms occurring in fragile X are related to local alterations in protein translation attributable to the loss of FMRP. It has been proposed that postsynaptic mRNAs and ribosomes may serve to synthesize components required for synapse development and plasticity (Steward and Levy, 1982; Steward, 1983; Steward and Falk, 1986) and for the induction and maintenance of long-term potentiation (Frey et al., 1991; Fazeli et al., 1993). Although direct evidence does not yet exist, we speculate that altered protein synthesis in dendrites and spines occurs in fragile X, leading to synaptic dysfunction and mental retardation.
Footnotes
This work was supported in part by National Institutes of Health Grants HD20521 (S.T.W.) and NS01624 (S.H.). S.T.W. is an investigator of The Howard Hughes Medical Institute. We thank Lisa Lakkis, Janelle Clark, Ryan Berglund, and Priya Naik for assistance in preparation of the manuscript and illustrations and Fuping Zhang for assistance and discussion. We thank Drs. Jean-Louis Mandel, Keith Elkon, Daniel Reines, and Dean Danner for providing antibodies.
Correspondence should be addressed to Dr. Steven M. Hersch, Department of Neurology, Emory University School of Medicine, Woodruff Memorial Building, Suite 6000, Atlanta, GA 30322.
Y.F. and C.-A.G. contributed equally to this work.
REFERENCES
- 1.Abitbol M, Menini C, Delezoide A-L, Rhyner T, Vekemans M, Mallet J. Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat Genet. 1993;4:147–153. doi: 10.1038/ng0693-147. [DOI] [PubMed] [Google Scholar]
- 2.Ashley C, Wilkinson K, Reines D, Warren S. FMR 1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 3.Bonfa E, Parnassa AP, Rhoads DD, Roufa DJ, Wool IG, Elkon KB. Antiribosomal S10 antibodies in humans and MRL/lpr mice with systemic lupus erythematosus. Arthritis Rheum. 1989;32:1252–1261. doi: 10.1002/anr.1780321010. [DOI] [PubMed] [Google Scholar]
- 4.Brown WT, Houck GE, Jeziorowska A, Levinson FN, Ding X, Dobkin C, Zhong N, Henderson J, Sklower Brooks S, Jenkins EC. Rapid fragile X carrier screening and prenatal diagnosis using a nonradioactive PCR test. J Am Med Assoc. 1993;270:1569–1575. [PubMed] [Google Scholar]
- 5.Burd CG, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 6.Burgin K, Waxham M, Rickling S, Westgate S, Mobley W, Kelly P. In situ hybridization histochemistry of Ca++/calmodulin-dependent protein kinase in developing rat brain. J Neurosci. 1990;10:1788–1798. doi: 10.1523/JNEUROSCI.10-06-01788.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coy JF, Sedlacek Z, Bachner D, Hameister H, Joos S, Lichter P, Delius H, Poustka A. Highly conserved 3′ UTR and expression pattern of FXR1 points to a divergent gene regulation of FXR1 and FMR1. Hum Mol Genet. 1995;4:2209–2218. doi: 10.1093/hmg/4.12.2209. [DOI] [PubMed] [Google Scholar]
- 8.Devys D, Lutz Y, Rouyer N, Bellocq J-P, Mandel J-L. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4:335–340. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- 9.Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–35. [PubMed] [Google Scholar]
- 10.Eberhart D, Malter H, Feng Y, Warren S. The fragile X mental retardation protein contains both nuclear localization and nuclear export signals. Hum Mol Genet. 1996;5:1083–1091. doi: 10.1093/hmg/5.8.1083. [DOI] [PubMed] [Google Scholar]
- 11.Fazeli MS, Corbet J, Dunn MJ, Dolphin AC, Bliss TVP. Changes in protein synthesis accompanying long-term potentiation in the dentate gyrus in vivo. J Neurosci. 1993;13:1346–1353. doi: 10.1523/JNEUROSCI.13-04-01346.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Y, Lakkis L, Devys D, Warren S. Quantitative comparison of FMR1 gene expression in normal and premutation alleles. Am J Hum Genet. 1995a;56:106–113. [PMC free article] [PubMed] [Google Scholar]
- 13.Feng Y, Zhang F, Lokey L, Chastain J, Lakkis L, Eberhart D, Warren S. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995b;268:731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- 14.Frey S, Schweigert C, Krug M, Lossner B. Long-term potentiation induced changes in protein synthesis of hippocampal subfields of freely moving rats: time-course. Biomed Biochim Acta. 1991;50:1231–1240. [PubMed] [Google Scholar]
- 15.Frangioni J, Beahm P, Shifrin V, Jost C, Neel B. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid c-terminal sequence. Cell. 1992;68:545–560. doi: 10.1016/0092-8674(92)90190-n. [DOI] [PubMed] [Google Scholar]
- 16.Fu Y, Kuhl D, Pizzuti A, Pieretti M, Sutcliffe J, Richards S, Verkerk A, Holden J, Fenwick R, Warren S, Oostra B, Nelson D, Caskey C. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 17.Garner C, Tucker R, Matus A. Selective localization of messenger RNA for cytoskeletal protein MAP2 in dendrites. Nature. 1988;336:674–677. doi: 10.1038/336674a0. [DOI] [PubMed] [Google Scholar]
- 18.Heffelfinger SC, Sewell ET, Danner DJ. Antibodies to bovine liver branched chain 2-oxoacid dehydrogenase crossreact with this enzyme complex from other tissues and species. Biochem J. 1983;213:339–344. doi: 10.1042/bj2130339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hergersberg M, Matsuo K, Gassman M, Schaffner W, Luscher B, Rulicke T, Aguzzi A. Tissue-specific expression of a FMR1/β-galactosidase fusion gene in transgenic mice. Hum Mol Genet. 1995;4:359–366. doi: 10.1093/hmg/4.3.359. [DOI] [PubMed] [Google Scholar]
- 20.Hinds H, Ashley C, Sutcliffe J, Nelson D, Warren S, Houseman D, Schalling M. Tissue specific expression of FMR-1 provides evidence for a functional role in fragile X syndrome. Nat Genet. 1993;3:36–43. doi: 10.1038/ng0193-36. [DOI] [PubMed] [Google Scholar]
- 21.Hinton V, Brown W, Wisniewski K, Rudelli R. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41:289–294. doi: 10.1002/ajmg.1320410306. [DOI] [PubMed] [Google Scholar]
- 22.Hirst M, Grewal P, Flannery A, Slatter R, Maher E, Barton D, Fryns J-P, Davies K. Two new cases of FMR1 deletions associated with mental impairment. Am J Hum Genet. 1995;56:67–74. [PMC free article] [PubMed] [Google Scholar]
- 23.Hornstra IK, Nelson DL, Warren ST, Yang TP. High resolution methylation analysis of the FMR1 gene trinucleotide repeat region in fragile X syndrome. Hum Mol Genet. 1993;2:1659–1665. doi: 10.1093/hmg/2.10.1659. [DOI] [PubMed] [Google Scholar]
- 24.Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I) a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jirikowski G, Sanna P, Bloom F. mRNA coding for oxytocin is present in axons of the hypothalamo-neurohypophyseal tract. Proc Natl Acad Sci USA. 1990;87:7400–7404. doi: 10.1073/pnas.87.19.7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khandjian E, Corbin F, Woerly S, Rousseau F. The fragile X mental retardation protein is associated with ribosomes. Nat Genet. 1996;12:91–93. doi: 10.1038/ng0196-91. [DOI] [PubMed] [Google Scholar]
- 27.Krajewski S, Tanaka S, Takayama S, Schibler M, Fenton W, Reed J. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- 28.Kunst CB, Warren ST. Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell. 1994;77:853–861. doi: 10.1016/0092-8674(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 29.Lugenbeel KA, Peier AM, Carson NL, Chudley AE, Nelson DL. Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat Genet. 1995;10:483–485. doi: 10.1038/ng0895-483. [DOI] [PubMed] [Google Scholar]
- 30.Meijer H, De Graaf E, Merck D, Jongbloed R, De Die-Smulders C, Engelen J, Fryns J-P, Curfs P, Oostra B. A deletion of 1.6 kb proximal to the CGG repeat of the FMR1 gene causes the clinical phenotype of the fragile X syndrome. Hum Mol Genet. 1994;3:615–620. doi: 10.1093/hmg/3.4.615. [DOI] [PubMed] [Google Scholar]
- 31.Miyashiro K, Dichter M, Eberwine J. On the nature and differential distribution of mRNAs in hippocampal neurites: implications for neuronal functioning. Proc Natl Acad Sci USA. 1994;91:10800–10804. doi: 10.1073/pnas.91.23.10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohr E, Morris J, Richter D. Differential subcellular mRNA targeting: targeting of a single nucleotide prevents the transport to axons but not to dendrites or rat hypothalamic magnocellular neurons. Proc Natl Acad Sci USA. 1995;92:4377–4381. doi: 10.1073/pnas.92.10.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanouer A, Boue J, Bertheas M, Mandel J. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 34.Olink-Coux M, Hollenbeck P. Localization and active transport of mRNA in axons of sympathetic neurons in culture. J Neurosci. 1996;16:1346–1358. doi: 10.1523/JNEUROSCI.16-04-01346.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perrone-Capano C, Giuditta A, Castigli E, Kaplan B. Occurrence and sequence complexity of polyadenylated RNA in squid axoplasm. J Neurochem. 1987;1987:698–704. doi: 10.1111/j.1471-4159.1987.tb00950.x. [DOI] [PubMed] [Google Scholar]
- 36.Pieretti M, Zhang F, Ying-Hui F, Warren ST, Oostra BA, Caskey CT, Nelson DL. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 37.Rousseau F, Rouillard P, Morel M, Khandjian E, Morgan K. Prevalence of carriers of premutation-size alleles of the FMRI gene and implications for the population genetics of the fragile X syndrome. Am J Hum Genet. 1995;57:1006–1018. [PMC free article] [PubMed] [Google Scholar]
- 38.Rudelli R, Brown W, Wisniewski K, Jenkins E, Laure-Kamionowska M, Connell F, Wisniewski H. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol. 1985;67:289–295. doi: 10.1007/BF00687814. [DOI] [PubMed] [Google Scholar]
- 39.Siomi H, Siomi M, Nussbaum R, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74:291–298. doi: 10.1016/0092-8674(93)90420-u. [DOI] [PubMed] [Google Scholar]
- 40.Siomi H, Choi M, Siomi M, Nussbaum R, Dreyfuss G. Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell. 1994;77:33–39. doi: 10.1016/0092-8674(94)90232-1. [DOI] [PubMed] [Google Scholar]
- 41.Siomi M, Siomi H, Sauer WH, Srinivasan S, Nussbaum RL, Dreyfuss G. FXR1, an autosomal homolog of the fragile X mental retardation gene. EMBO J. 1995;14:2401–2408. doi: 10.1002/j.1460-2075.1995.tb07237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siomi M, Zhang Y, Siomi H, Dreyfuss G. Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them. Mol Cell Biol. 1996;16:3825–32. doi: 10.1128/mcb.16.7.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spacek J. Three-dimensional analysis of dendritic spines. II. Sine apparatus and other cytoplasmic components. Anat Embryol. 1985;171:235–243. doi: 10.1007/BF00341418. [DOI] [PubMed] [Google Scholar]
- 44.Steward O. Alterations in polyribosomes associated with dendritic spines during the reinnervation of the dentate gyrus of the adult rat. J Neurosci. 1983;3:177–188. doi: 10.1523/JNEUROSCI.03-01-00177.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steward O. Dendrites as compartments for macromolecular synthesis. Proc Natl Acad Sci USA. 1994;91:10766–10768. doi: 10.1073/pnas.91.23.10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steward O, Falk P. Protein-synthetic machinery at postsynaptic sites during synaptogenesis: a quantitative study of the association between polyribosomes and developing synapses. J Neurosci. 1986;6:412–423. doi: 10.1523/JNEUROSCI.06-02-00412.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steward O, Levy W. Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci. 1982;2:284–291. doi: 10.1523/JNEUROSCI.02-03-00284.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steward O, Reeves T. Protein-synthetic machinery beneath postsynaptic sites on CNS neurons: association between polyribosomes and other organelles at the synaptic site. J Neurosci. 1988;8:176–184. doi: 10.1523/JNEUROSCI.08-01-00176.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sutcliffe J, Nelson D, Zhang F, Pieretti M, Caskey C, Saxe D, Warren S. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- 50.Thompson NE, Steinberg TH, Aronson DB, Burgess RR. Inhibition of in vivo and in vitro transcription by monoclonal antibodies prepared against wheat germ RNA polymerase II that react with the heptapeptide repeat of eukaryotic RNA polymerase II. J Biol Chem. 1989;264:11511–11520. [PubMed] [Google Scholar]
- 51.Tiedge H, Fremeau R, Weinstock P, Arancio O. Dendritic location of neural BC1 RNA. Proc Natl Acad Sci USA. 1991;88:2093–2097. doi: 10.1073/pnas.88.6.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tiedge H, Zhou A, Thorn N, Brosius J. Transport of BC1 RNA in hypothalamo-neurohypophyseal axons. J Neurosci. 1993;13:4214–4219. doi: 10.1523/JNEUROSCI.13-10-04214.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trembleau A, Morales M, Bloom F. Aggregation of vasopressin mRNA in a subset of axonal swellings of the median eminence and posterior pituitary: light and electron microscopic evidence. J Neurosci. 1994;14:39–53. doi: 10.1523/JNEUROSCI.14-01-00039.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Minnen J. RNA in the axonal domain: a new dimension in neuronal functioning? Histochem J. 1994;26:377–391. doi: 10.1007/BF00160050. [DOI] [PubMed] [Google Scholar]
- 55.Verheij C, Bakker C, Graaf ED, Keulemans J, Willemsen R, Verkerk A, Galjaard H, Reuser A, Hoogeveen A, Oostra B. Characterization and localization of the FMR-1 gene product associated with fragile x syndrome. Nature. 1993;363:722–724. doi: 10.1038/363722a0. [DOI] [PubMed] [Google Scholar]
- 56.Verkerk A, Pieretti M, Sutcliffe J, Fu Y-H, Kuhl D, Pizzuti A, Reiner O, Richards S, Victoria M, Zhang F, Eussen B, van Ommen G-JB, Blonden L, Riggins G, Chastain J, Kunst C, Galjaard H, Caskey C, Nelson D, Oostra B, Warren S. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 57.Warren ST, Ashley CT. Triplet repeat expansion mutations: the example of fragile X syndrome. Annu Rev Neurosci. 1995;18:77–99. doi: 10.1146/annurev.ne.18.030195.000453. [DOI] [PubMed] [Google Scholar]
- 58.Weiler I, Greenough W. Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci USA. 1993;90:7168–7171. doi: 10.1073/pnas.90.15.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, O’Connor JP, Siomi MC, Srinivasan S, Dutra A, Nussbaum RL, Dreyfuss G. The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. EMBO J. 1995;14:5358–5366. doi: 10.1002/j.1460-2075.1995.tb00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]