

Abstract
Cultured rat hippocampal neurons grown on glass coverslips for 1–3 weeks were loaded with the calcium-sensitive fluorescent dye Fluo-3 and viewed with a confocal laser scanning microscope. Large pyramidal-shaped neurons were found to contain dye-accumulating organelles in their somata, primarily around nuclei and near the base of their primary dendrites. These organelles varied in size and increased in density over weeks in culture, and were not colocalized with the endoplasmic reticulum or with mitochondria. The Fluo-3 fluorescence in these calcium-containing organelles (CCOs) was transiently quenched by exposure to Mn2+, indicating that the dye is a genuine [Ca2+] reporter and is not just a site of accumulating Fluo-3 dye. Recovery of fluorescence in the CCOs after washout of Mn2+ involved activation of a thapsigargin-sensitive process.
CCOs responded to stimuli that evoke a rise of cytosolic [Ca2+] ([Ca]i) in a unique manner; perfusion of caffeine caused a prolonged rise of [Ca] in the CCOs ([Ca]C), whereas it caused only a transient rise of [Ca]i. Pulse application of caffeine also caused a faster effect on [Ca]C than on [Ca]i. Glutamate caused a transient rise of both [Ca]i and [Ca]C, followed by a prolonged fall of only [Ca]C to below rest level. This fall was blocked by preincubation with thapsigargin. Ryanodine blocked the cytosolic effects of caffeine but not its effect on [C]C.
A clear distinction between CCOs and the known calcium stores was seen in digitonin-permeabilized cells; in these, remaining Fluo-3 reported changes in store calcium, i.e., caffeine caused a reduction in Fluo-3 fluorescence in permeabilized cells, whereas it still caused an increase in [Ca]C. A possible role of CCOs in regulation of release of calcium from ryanodine-sensitive stores was indicated by the observation that CCO-containing cells exhibited a larger and faster response to caffeine than cells that did not have them. We propose that CCOs constitute a unique functional compartment involved in release of calcium from calcium-sensitive stores.
Keywords: hippocampus, calcium stores, confocal microscopy, cultured neurons, ryanodine, caffeine
The presence of unique, calcium dye-accumulating organelles in somata of cultured neurons exposed to fluorescent calcium indicators has been noticed ever since the introduction of the membrane-permeable calcium indicators Indo-1-AM, Fura-2 AM, and Fluo-3 AM (Malgaroli et al., 1987; Blatter and Wier, 1990; Gunter et al., 1990; Spurgeon et al., 1990; Wahl et al., 1990, 1992; Kuba et al., 1991). These dyes penetrate the plasma membrane and some membranous structures within the cell and are converted by local esterases to a fluorescent, calcium-sensitive, membrane-impermeant form. This loading procedure results in their accumulation within organelles inside the cell. The functional significance of this apparent accumulation of fluorescence in organelles and the relations of these organelles to the known calciosome (Volpe et al., 1988; Burgoyne et al., 1989; Rossier and Putney, 1991; Spat et al., 1994) are not entirely clear. These organelles are not seen when the cells are loaded with the acid, membrane-impermeant form of the dye (Almers and Nehers, 1985; Connor, 1993; Blatter, 1995), indicating that they constitute closed, membranous structures into which the free dye does not diffuse passively. Do these organelles fluoresce because they have a high concentration of the dye, or do they have a genuine high concentration of calcium, and can be considered as a unique class of “calcium store”? If so, can they be identified with one of the known stores, including mitochondria and endoplasmic reticulum? Whether these organelles accumulate dye or calcium, the presence of dye in them interferes with accurate measurements of real free [Ca]ielsewhere in the cytosol, and so it is important to determine whether there are any systematic changes in fluorescence of these “organelles” that may affect measurements of cytosolic calcium.
Previous studies, done primarily in non-neuronal cells using digital imaging methods, reported that calciosomal calcium rises in the presence of ATP, indicating that the accumulation of calcium in calciosomes is energy-dependent (Connor, 1993). Other stimuli used include drugs that interact with the known calcium stores (Blatter, 1995; Hofer et al., 1995). Differences between cell types can be expected, and an interaction among calciosomes, the novel calcium compartment seen here, and different calcium stores may be unique to the cell types studied. Their roles in neurons remain enigmatic.
Using a confocal laser scanning microscope, which allows a focused, detailed view of living cells, we have studied the behavior of the calcium-containing organelle in conditions expected to produce a change in [Ca]i and report that it does indeed express a unique response pattern to drugs that modify [Ca]i. Although we are unable at the present time to verify that it is enclosed within a specific, known cellular organelle or a restricted compartment of the cytosol, we operationally define it as calcium-containing organelle (CCO) and demonstrate that it is a genuine functional calcium compartment, closely related to, but different from, intracellular calcium stores.
MATERIALS AND METHODS
Cell culture. Primary dissociated cultures of rat hippocampus were prepared from E19 fetuses and grown on 13 mm cover glasses for 1–4 weeks, as described elsewhere (Papa et al., 1995). In brief, cells were plated in DMEM containing 10% horse serum and 10% fetal calf serum, which was replaced, after 1 week, with DMEM containing 10% horse serum. Glia proliferation was blocked by incubation with 5-fluoro-2′-deoxyuridine for 3 d, starting 5 d after plating.
For [Ca]i imaging, cover glasses were washed with recording medium and incubated at room temperature in a shaking bath for 1–1.5 hr with one of the following dyes: Fluo-3 AM (3 μm), Calcium Green-1 AM, Calcium Green-2 AM (both at 2.6 μm; all from Molecular Probes), or FFP-18 AM (7.8 μm; Teflabs), in the presence of pluronic F127 (30 nm). Cultures were then washed for at least 1 hr in the recording medium, and were used during the next 1–4 hr.
Endoplasmic reticulum was visualized with the membrane dye 3,3′-dihexyloxacarbocyanine iodide [DiO6(3), Molecular Probes] (Blatter, 1995). The dye was prepared in a stock solution of 0.5 mg/ml ethanol and was added to the culture dish to a final concentration of 0.25 mg/ml for 15 min. The dye was then washed for 15 additional min, and the culture was mounted on the stage of the confocal laser scanning microscope. Mitochondria were visualized with Rhodamine 6G chloride (Molecular Probes) prepared from stock DMSO solution. The cultures were incubated for 10 min with 1 μm of the dye and washed for 40 min before visualization. The same cells were used for visualization of calcium, using Fluo-3, and for visualization of the organelles, endoplasmic reticulum, or mitochondria. In cases of double labeling, the laser power was adjusted before loading with the second dye, so that Fluo-3 fluorescence was no longer seen.
Solutions and drugs. The recording medium contained (in mm): 129 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 4.2 glucose, and 10 HEPES, along with 0.5 μm tetrodotoxin. pH was adjusted to 7.4 with NaOH, and osmolarity to 320 mOsm by addition of sucrose. In Mn2+-containing solutions, CaCl2 was replaced by MnCl2. In some experiments, EGTA (0.1 mm) was added to the calcium-free medium. Permeabilization medium contained (in mm): 125 KCl, 25 NaCl, 10 HEPES, 3 Na2ATP, 0.1 MgCl2, and 0.1 CaCl2. Osmolarity was adjusted to 320 mOsm with sucrose. [Ca]i was clamped to 150 nm with 0.14 mm EGTA (Tsien and Pozzan, 1989), and pH was adjusted to 7.4 with KOH. Digitonin (10 μm, Merck) was added to the permeabilization medium. Fluo-3 remained trapped in organelles, thereby permitting calcium measurements in these conditions. Drugs were prepared in the recording medium from frozen stocks before use. Glutamate (1 mm), caffeine (5–10 mm), or KCl (200 mm) was loaded in a pressure pipette with a tip diameter of 2 μm placed approximately 50 μm from the cell. The drugs were applied through the pipette with a pressure pulse of 50–500 msec duration. Alternatively, drugs were applied in the perfusion medium [5 mm caffeine (Sigma), 10 μm ryanodine, and 1 μmthapsigargin (Alåmone Lab, Jerusalem, Israel)]. SF-6847 (3,4-ditert, butyl-4-hydroxylbenzylidenemalononitrile), a carbonyl cyanide/p-trifluoro methoyx phenylhydrazone (FCCP)-like mitochondrial uncoupler (White and Reynolds, 1995), was a gift from Dr. Y. Shahak (The Weizmann Institute).
Imaging. After loading of the dye, the glass coverslips were placed in a confocal laser scanning microscope (Leica, Heidelberg, Germany) and superfused with the recording medium at a rate of 3–5 ml/min at room temperature. The confocal laser scanning microscope is equipped with an argon-ion laser for excitation at a wavelength of 488 nm. Laser light was reduced to 1–3% of nominal intensity to avoid photodynamic damage. Images of 256 × 256 or 512 × 512 pixels were taken with a 63× water immersion objective. A complete three-dimensional reconstruction of the cell was made from 15–20 successive 0.5–1.0 μm optical sections taken through the cell. A line was scanned through the center of an image (∼0.8 msec per line) to reveal fast changes in fluorescence during a response to drug application. Fluorescence intensity was quantified using Leica analysis software and Adobe Photoshop (Adobe Systems). Changes in Fluo-3 fluorescence were standardized by dividing the net fluorescence by the pretreatment fluorescence, with background subtracted. Autofluorescence, measured in bis(2-aminophenoxy)ethane-N, N,N′,N′-tetra-acetic acid AM-loaded cells, under identical conditions as used here, was negligible (Korkotian and Segal, 1996) Data are presented as mean ± SEM.
RESULTS
A unique calcium-sequestering compartment
Fluo-3-containing organelles of different sizes and shapes were found primarily in the soma, around the nucleus, and near the base of the main dendrite. Their diameters ranged between 0.3 and 1.5 μm. They tend to cluster, however, single, isolated organelles often could be seen as well. They were clearly absent in nuclei, and few of them were found in secondary dendrites (Fig. 1A). CCOs were stable in shape and fluorescence intensity for at least half an hour of repetitive imaging (e.g., Fig. 5A1,2). In three-dimensional reconstructed neurons stained with both Fluo-3 and DiO, which labels endoplasmic reticulum, the CCOs tended to cluster in the same regions of the cell as the DiO staining [e.g., in perisomatic regions and in the base of the main dendrite (Fig.1A)], but a closer examination of single planes of the three-dimensional reconstructed images (Fig. 1B) using digital subtraction methods revealed that CCOs do not colocalize with endoplasmic reticulum, stained with DiO.
Fig. 1.

A, CCOs are distinct from the endoplasmic reticulum in cultured hippocampal neurons. Cells were initially stained with Fluo-3 and reconstructed in three dimensions using eight optical sections taken at 1 μm intervals (left). The same cells were later stained with DiO, which labels the endoplasmic reticulum (right). Fluo-3 fluorescence was photobleached before the second staining. Note the absence of DiO in the nucleus and the network of labeling in the soma, especially near the base of the primary dendrites. B, An image taken at a single plane through another cell stained with Fluo-3 (left) and DiO (middle).Right, An amplified-difference image between the Fluo-3 and DiO images. Although some organelles are seen also with the DiO staining, they do not overlap with those stained with Fluo-3.
Fig. 5.

CCOs respond to chemical stimulation in cultured hippocampal neurons. A, Reactivity of CCOs to caffeine.Images 1 and 2 were taken 20 min apart to illustrate the stability of the detection of CCOs. Image 3, 10 min after perfusion of the cell with 10 mmcaffeine. Note that at this time, only CCOs respond to caffeine, whereas the rest of the cell is nearly at baseline level. Single-plane optical sections, 16-frame average, 63× water immersion objective. Nucleus is out of the plane of focus. B, Responses to topical application of glutamate. B1, Top, Three-dimensional reconstructed neuron with a line drawn through its soma and a CCO in one of its primary dendrites. This cell was scanned at a rate of 0.8 msec/line. Bottom, Raster display of the line scan, from top to bottom, comprising 512 lines, showing net fluorescence relative to baseline fluorescence (DF/F). Glutamate (1 mm) was applied ∼50 msec from the top and is followed by a rise of fluorescence in the calciosome at about the same time as the rise in the soma of the same cell. B2, Three-dimensional display of the rise of Fluo-3 fluorescence, frombottom to top, illustrating the rise of fluorescence in the CCO and the soma. Intensity of fluorescence is coded in the height/color of the plot with redindicating low intensity and green-white indicating high intensity. C, Responses of a cluster of cells to caffeine and glutamate. Image 1, control; image 2, after 15 min of exposure to caffeine (5 mm), illustrating a rise in [Ca]C; image 3, 15 min wash; image 4, net frame of images 2–1, image amplified to enhance detection of differences in the image; image 5, during response to glutamate, (1 mm, 100 msec pulse). Note that nuclear fluorescence is saturated. Image 6, 1 min later; image 7, 20 min later; image 8, net frame of images 7–6, amplified. Note the decrease in CCOs fluorescence after exposure to glutamate, and the later recovery. Cells are three-dimensionally reconstructed.
CCOs do not colocalize with mitochondria, another small, highly abundant organelle in neurons. Mitochondria were stained with Rhodamine 6G in the same protocol used for staining endoplasmic reticulum (Fig.2B). Analyzing single-plane images, small organelles stained with Rhodamine 6G were seen throughout the cells, but they were smaller in size and were not colocalized with Fluo-3-stained organelles, especially in dendrites rich in mitochondria and poor in CCOs. Another indication that CCOs are not mitochondria comes from experiments in which mitochondrial calcium was released with the proton uncoupler SF-6847, (White and Reynolds, 1995). In these conditions, Fluo-3 fluorescence increased throughout the cell (Fig.2A), as expected, but also in CCOs, which never disappeared in these conditions, indicating that they are, like [Ca]i, affected by release of calcium from the stores, but probably are not mitochondria themselves.
Fig. 2.

CCOs are not colocalized with mitochondria.A, Fluo-3-loaded cell in control condition (left), after onset of exposure to the mitochondria uncoupler SF-6847 (30 μm; middle), and after 10 min exposure and 20 min wash (right). Note the increase in nuclear and [Ca]i but also of [Ca]C. Fluorescence in the right image is saturated. B, Double-labeled cell stained with Fluo-3 AM (left) and Rhodamine 6G (center).Right, Amplified difference between theleft and center images. A single plane taken through the same cell indicates that the two dyes do not stain the same organelles. Rhodamine 6G staining was made in Fluo-3-labeled, and subsequently bleached, cells, as above. Staining was made with 1 μm dye incubated for 10 min, followed by extensive wash for 40 min.
Fluo-3 is not a unique dye in its ability to label CCOs. They are stained also with Calcium Green-1 AM and with the membrane-bound ratio dye FFP-18 AM (data not shown). The more common dye, Fura-2 AM, also labeled CCOs but not as readily as Fluo-3. In most experiments, Fluo-3 was used for its wider dynamic range than that of the other dyes. Membrane-impermeant dyes, Fluo-3 or Fura-2, introduced into cells through a micropipette, did not label CCOs (as in Segal, 1995).
CCOs may constitute a genuine and independent compartment, which is related in some unique way to known free cytosolic or store [Ca]. Alternatively, these may constitute dye-accumulating organelles, rich in esterases that convert Fluo-3 AM to the fluorescent form, but are insensitive to changes in cytosolic [Ca]. In the former case, one would expect a fast and transient change in fluorescence in the presence of a divalent cation that competes with calcium at the binding site of the dye. Such is the case with Mn2+, which competes with calcium and quenches [Ca]i fluorescence in the cytosol (Simpson et al., 1995b), and is used for calibration of [Ca]i (Segal and Manor, 1992). The effects of Mn2+ were examined in 14 cells. First, the cells were exposed momentarily to a high K+-containing medium, which activated voltage-gated calcium channels and raised [Ca]itransiently (Fig. 3A). Next, the cells were perfused for 5 min with medium containing 2 mm Mn2+ replacing extracellular Ca2+. This, by itself, did not cause a change in [Ca]i, because Mn2+ does not permeate the cells freely. After a subsequent pulse application of high K+-containing medium, Mn2+ quenched Fluo-3, and fluorescence level went down momentarily in the cytosol and with a short lag also in the CCOs (Figs. 3B, 4). [Ca]C recovered to about the same level after the potassium/manganese treatment in the two compartments even in the continuous presence of Mn2+ in the extracellular medium. The recovery of [Ca]C is likely to involve an energy-dependent process, as it was prevented by pretreatment of the cells with thapsigargin, which blocks intracellular calcium pumps (Fig.3C, 5 cells). These experiments indicate that CCOs are genuine calcium-sequestering organelles, because such fluorescence transients would not be expected from a dye-accumulating organelle.
Fig. 3.

Transient disappearance of [Ca]Cfluorescence after exposure to Mn2+. A, A Fluo-3-loaded cell shown from left toright, in control condition, immediately after exposure to a pulse application of high K+ (200 mm)-containing medium, 5 sec after K+, and 1 min later. The frame on the right is a net image, subtracting the image taken 5 sec after onset of exposure to K+ from the image taken 1 min later. A andB are the same cell: A, in control medium; B, in a medium containing Mn2+ (2 mm), replacing Ca2+. C, Another cell, preincubated for 15 min in thapsigargin (1 μm), exposed to Mn2+ as in B. The CCOs are clearly visible in B and are nearly absent inC after washout of K+.
Fig. 4.

Time course of changes in Fluo-3 fluorescence after a momentary exposure to high K+ in Mn2+-containing medium, as in Figure 4. Mean of five experiments. Note the log scale of the time base.
CCOs express a distinct response to calcium load
Two chemical stimuli were used routinely to raise [Ca]i: caffeine, which causes release of calcium from intracellular stores (Burgoyne et al., 1989; Sacchetto et al., 1995), and glutamate, which activates ligand-gated calcium channels, causing influx of calcium ions into the cell.
Caffeine was applied by a short pressure pulse on 11 cells (Figs.5C, 6A). It caused a rapid and transient rise in [Ca]i, lasting <10 sec. The latency of the response to caffeine was significantly shorter in the CCOs compared with nuclear and [Ca]i (Fig. 6A,D). On the other hand, the magnitude of change of [Ca]i was largest in the nucleus and smallest in the CCOs. After the initial rise, nuclear calcium ([Ca]N) recovered to below the resting level, whereas [Ca]C returned to baseline level, but slower than [Ca]i. By comparison, a similarly brief application of glutamate (Figs. 5B, 6B) onto 11 cells caused a larger and faster [Ca]i surge, which had the same rise time in the CCOs and the cytosol, indicated in fast line scans through CCOs and cytosolic compartments (Fig.5B). This was followed by a slower recovery in all compartments; [Ca]N did not fully recover, and stabilized at a level slightly above baseline (as in Korkotian and Segal, 1996). By comparison, [Ca]C recovered rapidly to a level below baseline (Figs. 5C5–8, 6C). This recovery happened faster when the initial response to glutamate was higher (compare [Ca]C reduction in Fig. 6B,C). The difference between the effects of caffeine and glutamate on [Ca]C is particularly striking, especially when these effects are related to changes in [Ca]i (Fig.6D).
Fig. 6.

Cellular calcium responses to a brief application of caffeine (A) and glutamate (B andC). A, Exposure to a pulse application of caffeine (5 mm, 300 msec pulse, n = 11 cells) caused a transient rise in [Ca]i, which initially started in CCOs (see the insert amplificationof the first part of the graph) and was larger in the nucleus than in cytoplasm or the CCOs, and a slower recovery to baseline in the latter than in former two compartments. B, Glutamate (1 mm, 100 msec pulse) caused a much larger rise in nuclear [Ca]i than caffeine, but a similar [Ca]Crise and an undershoot in the fluorescence of the latter compartment was clear (glutamate, 1 mm, 50 msec pulse,C). D, Detailed comparison between CCO responses to glutamate and caffeine, relative to the changes in cytoplasmic [Ca]i, at successive time points before and after application of the drug: a, before application;b, the last nonsignificant point on cytoplasmic curve after drug application; c, the first significant point in the cytosolic response; d, at the peak of the response; e, one-half recovery to baseline;f, four-fifths recovery; g, last significant point; h, first point of recovery in cytoplasm. A significant rise of [Ca]C in response to caffeine is seen before [Ca]i changes. [Ca]C remained elevated in response to caffeine compared with the response to glutamate and to caffeine in cytoplasm.Ordinate, Ratio of net/baseline fluorescence.Abscissa (in A, B, and C), time; (in D), points in time relative to a defined [Ca]i state. Abscissa of Cis in a logarithmic scale.
The rise of [Ca]C in response to caffeine and glutamate is not specific to the calcium dye, Fluo-3, used in these experiments. The membrane-bound Fura-2 analog FFP-18 was used under the same conditions as those in the experiments with Fluo-3. Although FFP-18 is used as a ratio dye, like Fura-2, with an optimal calcium-dependent excitation wavelength of 340 nm, it can also be used with the confocal microscope, and fluorescence excited at 488 nm is inversely related to [Ca]i. FFP-18 stains primarily intracellular membranes, including CCOs. A brief application of glutamate caused a transient reduction in FFP-18 fluorescence, indicating an increase in [Ca]C (data not shown, 7 cells tested).
The shorter latency rise of [Ca]C in response to caffeine by comparison to the changes in [Ca]i (Fig.6A, insert) prompted us to try to reach a threshold condition for caffeine effects. Indeed, when a low concentration (1–2 mm), short pulse (30 msec) of caffeine was applied (Fig. 7), there was a transient rise only in [Ca]C, whereas [Ca]i and [Ca]N were slightly reduced. This indicates that the [Ca]C rise is not a consequence of [Ca]irise, and that, in fact, [Ca]i may serve as a source of the calcium rise in the CCOs (i.e., calcium may flow from the cytosol or endoplasmic reticulum to the CCOs).
Fig. 7.

Cell reactivity to short duration (50 msec, 1–2 mm) exposure to caffeine. The response lasted <1 sec. Note the increase in [Ca]C and decrease in nuclear [Ca]i. The same cells responded to a longer exposure to caffeine with an increase in [Ca]i in all compartments, as shown before for other cells.
Caffeine was also applied by continuous perfusion to 15 cells (Figs.5A3, 8). It caused a moderate transient increase in [Ca]N. The maximal response to perfused caffeine was smaller than the response to a fast, pressure application of the drug, probably because caffeine concentration rose slowly during the perfusion, and the response probably desensitized. After the initial rise, and while caffeine was still present, a recovery of [Ca]i was seen in the cytoplasm and the nucleus, but [Ca]C continued to be high, (up to 50 min after onset of perfusion (Fig. 8).
Fig. 8.

Cellular reactivity to prolonged exposure to 5 mm caffeine. Caffeine was perfused in the recording medium (as in Fig. 5C), and changes in Fluo-3 fluorescence were monitored in 15 cells. After an initial rise in [Ca]i, cytosolic and nuclear calcium returned to baseline, whereas [Ca]C continued to rise, and remained high even after removal of caffeine.
CCOs are not calcium stores: pharmacological effects
The effects of glutamate and caffeine on [Ca]i and [Ca]C in the presence of drugs that interact with the calcium pump (thapsigargin) and the calcium-induced calcium release store (ryanodine) were studied to allow a distinction between the CCOs, the calcium stores, and the cytosol.
First, the 10–20 min undershoot of [Ca]C after glutamate (as in Fig. 6B,C) was found to be blocked by a prolonged preincubation of the cell with thapsigargin (Fig.9B), which by itself caused a small and selective rise in [Ca]C (Fig. 9A). Taken together, these observations indicate that the CCOs may be instrumental in pumping calcium into the stores but do not constitute stores (i.e., their fluorescence is not reduced, but increased, by thapsigargin).
Fig. 9.

A, Thapsigargin enhances [Ca]C. Image 1, control; image 2, after 5 min exposure to the drug (0.5 μm);image 3, after 15 min exposure to thapsigargin. Note the increase in [Ca]c in thapsigargin-treated cell.B, Thapsigargin blocks the undershoot of the [Ca]C response to glutamate. CCO responses in control (as in Fig. 6C) and thapsigargin-pretreated cells (15–20 min; n = 13) to same pulse application of glutamate were compared. Unlike the control cases, in which a clear undershoot lasting at least 10 min is seen, the thapsigargin-treated cells do not return to baseline fluorescence after glutamate. Abscissa scale as in Figure 6C.
A striking distinction between cytosolic calcium and [Ca]C was seen after blockade of calcium-induced calcium release stores with ryanodine. In the presence of 10 μmryanodine superfused for 10–15 min (Figs. 10, 11), caffeine was no longer able to cause an increase in [Ca]i, but [Ca]C was still elevated. The response to caffeine could not be recovered after washout of ryanodine (n = 12 cells).
Fig. 10.

Ryanodine blocks cytosolic, but not [Ca]C, reactivity to caffeine. Images were taken before (A), 3 sec after (B), and 6 sec after (C) exposure of the cell to a 300 msec puff of caffeine (5 mm). D–F, Same cell after 15 min of exposure to 10 μm ryanodine (with 2–3 intermediate responses not shown). Single plane/frame images of the cell. Note that the bottom left area, rich in CCOs, increases fluorescence before other cell areas (B), and even after ryanodine, whereas the rest of the cell maintains a low level of fluorescence (E and F).
Fig. 11.
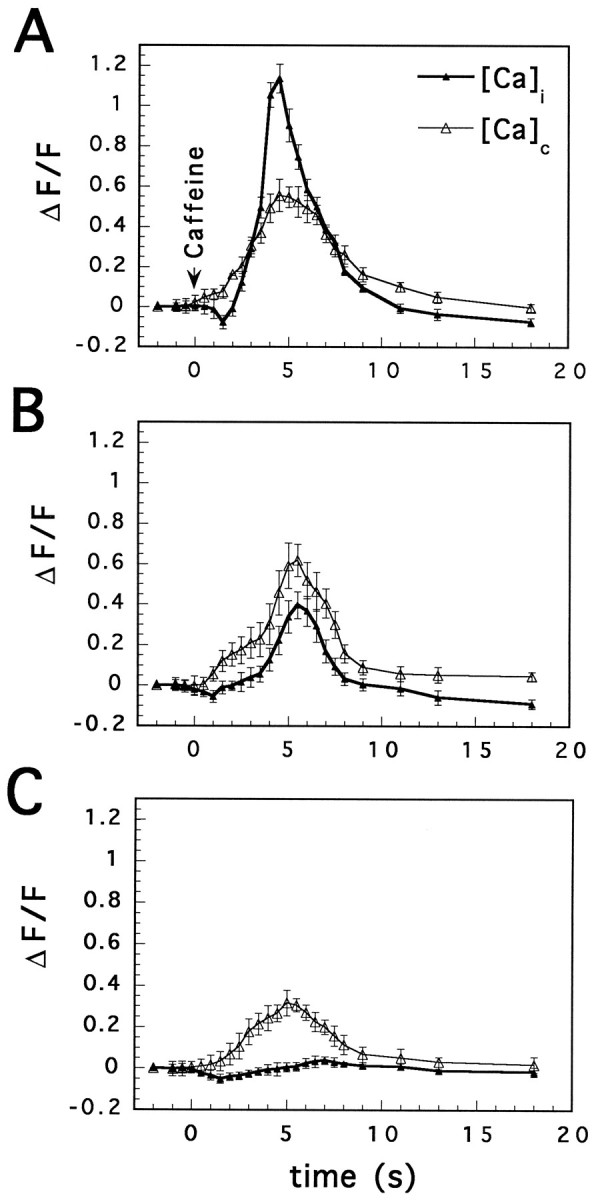
Differential effects of ryanodine on [Ca]i and [Ca]C. Initially, caffeine (5 mm, applied by pressure) caused a larger change in [Ca]i than in [Ca]C (A). After 10–15 min superfusion of 5 μm ryanodine, the baseline fluorescence was not different, the cytosolic response to caffeine was reduced drastically, and [Ca]C was still transiently elevated by caffeine (B). Only 5 min later, and after two to three more responses to caffeine, did [Ca]C response to caffeine attenuate significantly (C).
These experiments indicate that CCOs are genuine calcium-sequestering organelles. They are not likely to constitute “calcium stores,” because their fluorescence increases rather than decreases in the presence of caffeine, which releases calcium from stores. Another distinction between the stores and the CCOs was seen in the following experiment, designed to measure calcium directly in the stores (Hofer and Machen, 1994; Hofer et al., 1995). The membrane was first permeabilized with digitonin (Fiskum, 1985), and [Ca]iwas clamped with EGTA while the cell was perfused with intracellular medium (Fig. 12). Under these conditions, changes in Fluo-3 fluorescence should be attributed to changes in store calcium. Digitonin permeabilizes selectively the cell membrane, leaving intact intracellular membranous structures, including CCOs, during the first few minutes of permeabilization (Malgaroli et al., 1987). At longer exposure times, digitonin can affect stability of intracellular membranes as well (Renard-Kooney et al., 1993). In digitonin-permeabilized cells, caffeine caused a transient reduction of fluorescence (by 21.8 ± 1.3 units) in the clear cytosolic regions of the cell, presumably reflecting a reduction in stored calcium, while it increased [Ca]C (by 14.2 ± 4.3 units, Fig. 12,n = 5 cells).
Fig. 12.

Changes in [Ca]C caffeine are different from those of the calcium stores, measured in permeabilized cells. A–C, Control conditions. A, Baseline; B, response to a pulse of caffeine (10 mm, 300 msec); C, 3 min later. The cell was permeabilized for 2–3 min with intracellular medium containing 10 μm digitonin and ATP. [Ca]i was clamped by EGTA at 150 nm. D–F, Same sequence of exposure to caffeine, 5 min after the cell was permeabilized. Contrast-enhanced images.
Functional implications
A possible function of CCOs in initiating reactivity of neurons to caffeine was studied in cultured neurons of different ages and in cultured neurons of the same age having different concentrations of CCOs. The proportion of cells having distinct Fluo-3-labeled CCOs increased over weeks in culture; few neurons contained them in cultures younger than 2 weeks, whereas most of them were labeled at 3–4 weeks in culture (Fig. 13C).
Fig. 13.
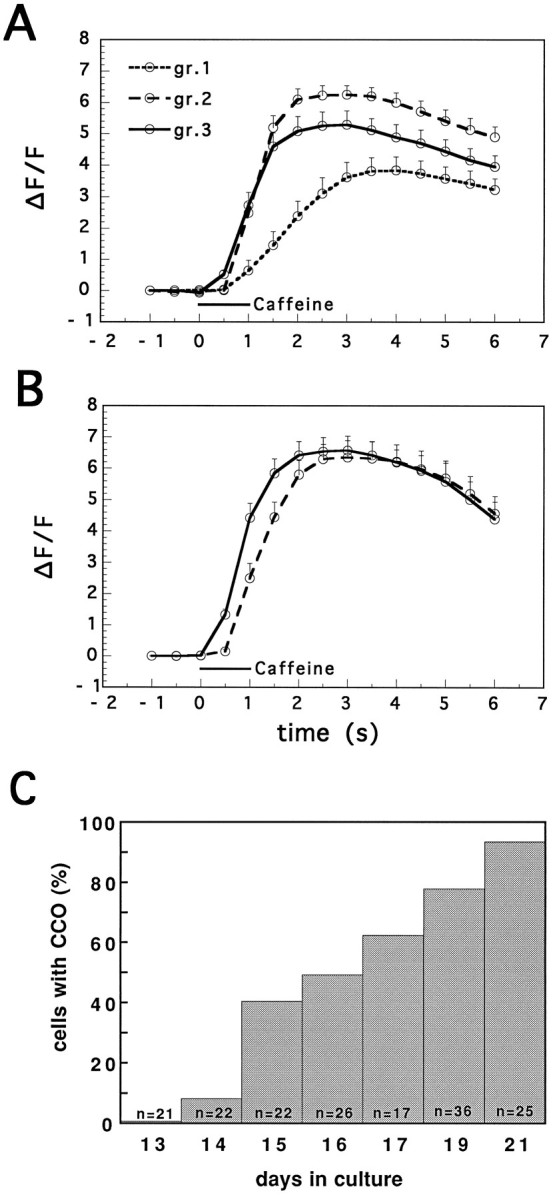
Nuclear reactivity to caffeine is correlated with the presence of calciosomes. The time course of change in nuclear [Ca]i was measured after application of 10 mmcaffeine. Experiments were conducted in young (A) and mature (B) cultures, and the cells were classified into three groups: cells containing no visible CCOs [group (gr.) 1], few CCOs (gr.2), and many CCOs (gr.3). In the older group, no organelle-free cells were found. In both age groups, a shorter latency to caffeine was seen in CCO-containing neurons. A smaller response was seen in CCO-free cells. C, Time course of appearance of CCOs. Cultures were stained with Fluo-3, and the proportion of cells containing these organelles was registered. (The total number of cells counted in each age group is depicted at the bottom of each column.)
Cells in the young cultures were divided into those having low (only few organelles detected in the cell, n = 7 cells), medium (approximately 5–10 organelles, n = 11 cells), and high (many organelles, or regions in the cell loaded with CCOs that cannot be counted, n = 17 cells) density of CCOs. The latency and magnitude of [Ca]N response to caffeine were significantly shorter and larger, respectively, in CCO-containing neurons, compared with organelle-free neurons (Fig.13A). The reason the nucleus was chosen for the measurements was because it is a CCO-free area and so the measurements in the nucleus could not be contaminated by different densities of CCOs. Thus, the nuclear response appears to be clearer than that of the cytosol (Korkotian and Segal, 1996). In the older cultures, only medium (n = 9) and high (n = 8) organelle-containing neurons were found (Fig. 13B). The former group had significantly longer latency than the latter group. It appears that CCOs may be instrumental in reducing the threshold for a rise in [Ca]N during exposure to caffeine. These results are congruent with the effects of low concentration/duration of caffeine effects (Fig. 7), which show a rise of [Ca]Cwith no concomitant changes in [Ca]i.
DISCUSSION
The present results demonstrate that calcium dye-containing organelles, present in cultured rat hippocampal neurons, behave in a characteristic manner in response to chemical stimulation to justify their classification as a unique cellular calcium compartment. These organelles are not colocalized with the endoplasmic reticulum, although the two compartments are found in close proximity. Also, CCOs do not colocalize with mitochondria, indicating that they are not part of any major calcium storage compartment. Their response to drugs that release calcium from stores (caffeine) and that cause influx of calcium through voltage- and ligand-gated channels (glutamate) is unique in that it has a different time course, magnitude, and sensitivity to drugs that interact with stores (e.g., ryanodine) than the response of free cytosolic calcium, or that of presumed calcium stores.
The presence of dye-accumulating organelles has been noted before as a consequence of loading the cell with a membrane-permeable, ester-linked dye (Almers and Nehers, 1985; Malgaroli et al., 1987;Scanlon et al., 1987; Blatter and Wier, 1990). It was argued that the dye traverses membranes and accumulates in acidic organelles (e.g., lysosomes) (Malgaroli et al., 1987) equipped with high esterase activity. In support of this assertion was the observation that cells loaded with the dye in its water-soluble, membrane-impermeable form via a micropipette do not compartmentalize into these organelles (Connor, 1993; Blatter, 1995). Also, it appears that the presence of CCOs is a function of the procedure for dye loading (e.g., temperature, composition of loading medium), as well as the dye and cell type (Malgaroli et al., 1987; Gunter et al., 1990; Wahl et al., 1990;Blatter, 1995).
One of the cellular compartments that may accumulate calcium or calcium dye is the calciosome, first described almost a decade ago (Pozzan et al., 1988; Volpe et al., 1988; Krause et al., 1989) and still not identified at the ultrastructural level. One characterizing property of the calciosome is its colocalization with the calcium-binding protein calsequestrin (Van et al., 1989; Villa et al., 1991) or with calreticulin (Van Delden et al., 1992) In fact, different studies probably call different organelles by the same name, and it is not clear that the calsequestrin-containing calciosome (Pozzan et al., 1988) is the same as our CCOs. At any rate, calreticulin staining of our neurons is not colocalized with CCOs (M. Segal, unpublished observations).
The assertion that calciosomes do exist comes from studies attempting to correlate calsequestrin with markers of endoplasmic reticulum (for review, see Rossier and Putney, 1991). The calciosome is situated close to the endoplasmic reticulum or inositol triphosphate (IP3)-sensitive calcium stores, but is completely distinct from other cell organelles such as mitochondria and lysosomes (Krause et al., 1989; Ross et al., 1989; Van Delden et al., 1992; Spat et al., 1994). However, a distinct difference in distribution of calciosomes and IP3-sensitive stores was found in liver and chromaffin cells (Pozzan et al., 1988; Volpe et al., 1988; Van et al., 1989;Rossier and Putney, 1991; Burgoyne et al., 1992; Spat et al., 1994;Sacchetto et al., 1995).
Later studies on chick Purkinje neurons revealed a molecular heterogeneity of calsequestrin-rich calciosomes with probable sensitivity to both IP3 and ryanodine (Volpe et al., 1991) and a clear difference in ontogenetic development between IP3 and calsequestrin-containing subcompartments. This indicates that calsequestrin is aggregated in calciosomes significantly later than IP3 (Sacchetto et al., 1995). It was proposed that calciosomes representing cisternal stacks of endoplasmic reticulum do not, however, represent a new kind of calcium store, but its functional compartment, probably characterized by different properties (Rossier and Putney, 1991; Villa et al., 1991).
The present CCOs, probably not identical to those described above, constitute a unique compartment, regulating calcium level in a manner different from the surrounding cytosol. However, they cannot be considered as part of a calcium store, hence they are not identical to calciosomes for the following reasons:
(1) CCOs are seen with ester-loaded dyes but not with acid-loaded dyes, indicating that they possess the ability to convert and store the dye in its active form, and that the dye is unable to penetrate these organelles freely. Furthermore, fluorescence of these organelles is quenched rapidly, with a small delay relative to cytosolic Fluo-3, by a brief exposure to Mn2+. This indicates that the CCO is a closed entity that accumulates the dye, but allows fast exchange with cytosolic calcium so that Mn2+ can compete with Ca2+ for the calcium-binding site and quench fluorescence. Furthermore, the CCO is not merely a passive compartment where the dye or dye/calcium enters and accumulates; the blockade of the reappearance of CCOs after washout of Mn2+ by the calcium pump antagonist thapsigargin (Herrington et al., 1996) indicates that the CCO is probably linked to calcium stores, which require energy to be refilled. Our results suggest that calcium moves from the stores to the CCOs because [Ca]C increases rather than decreases during exposure of cells to thapsigargin (Fig. 2E).
(2) CCOs react to glutamate and caffeine in two totally different ways, unlike the response seen in cytosolic [Ca]. In the latter compartment, both glutamate and caffeine evoke a rise of [Ca]i, and the difference between the responses to the two stimuli is in magnitude and time course. Whereas caffeine produces a prolonged rise of [Ca]C, glutamate causes a reduction in [Ca]C to below resting values, and this effect could be blocked by thapsigargin. Glutamate causes an influx of calcium, which induces a large release of calcium from calcium-sensitive stores, and the reduction in [Ca]C may reflect an active refilling of the stores through the CCOs, whereas the action of caffeine is weaker and its prolonged application may not require an active pumping of calcium into the stores. However, repetitive application of high caffeine concentration (10 mm) can bring about a fall in [Ca]C down to their complete disappearance (data not shown). The difference in the response may indicate a role of CCOs in the regulation of calcium release from stores (see below), but it undoubtedly validates the assertion that CCOs are uniquely regulated calcium compartments, and the measured changes in their calcium content do not merely reflect changes in ambient [Ca]i.
Although there is no unequivocal morphological identification of a CCO, it is not part of the endoplasmic reticulum or the mitochondria, judged from DiO and rhodamine staining. Also, it appears to be unique in both its spatial localization and developmental regulation (see alsoMalgaroli et al., 1987; Sacchetto et al., 1995). These organelles are not scattered randomly throughout the cell; they are not found in the nucleus, and they tend to accumulate at the perinuclear area and at the base of the largest dendrite of the cell. Also, they are not found in young cultures, and begin to be prominent in more mature ones. Likewise, the CCO is not unique to a specific dye used; it is best seen with Fluo-3, but can also be seen with Fura-2, although with a lower intensity difference from the cytosole. We have also seen CCOs when using Calcium Green of the high and low affinity varieties and the low affinity dye FFP-18 (data not shown). In the latter case, the dye binds to membranes, thus indicating that CCOs are membranous organelles.
Functions of CCOs
The association of the CCOs with the endoplasmic reticulum, as well as their response to a calcium challenge of the type that evokes release from stores, indicates that these organelles are closely linked to the calcium stores. They are not part of the stores, because they behave differently from what is expected of a store (i.e., their fluorescence increases, rather than decreases, in response to caffeine). Parenthetically, the low level of [Ca]C is not likely to result from the high affinity of Fluo-3 to calcium, because the lower-affinity dye, Calcium Green-2, also reports relatively low [Ca]C and is not saturated by the dye. The CCOs are related to the ability of the cell to respond to caffeine, as indicated in the faster time course of [Ca]N changes in response to caffeine in CCO-containing cells (Fig. 13). Also, in fast imaging series, we found that [Ca]i increases first near a cluster of CCOs, and then spreads to the rest of the cell (e.g., Fig.10), indicating that these organelles are instrumental in initiating the rise of [Ca]i after release from stores, but not after influx from the extracellular space. Furthermore, small amounts of caffeine, or a larger amount applied during exposure to ryanodine, increases only [Ca]C (Fig. 13). This difference may underlie the qualitative difference between reactivity of CCOs to glutamate and caffeine.
If, indeed, the CCOs have the lowest threshold for raising [Ca], what is the source of calcium feeding into these organelles? A partial answer may be seen in Figure 7, in which a fast and small rise in [Ca]C is accompanied by a small fall in [Ca]i and [Ca]N. It is quite likely that both cytoplasmic and store calcium feed into the CCO, and under some conditions (e.g., after glutamate) the opposite direction is taken. Such will be the case when the calcium-induced calcium stores are about to be emptied, and the CCO may provide the initial boost for these calcium stores.
Of the two types of calcium stores reported to exist in cells (for review, see Simpson et al., 1995a), it is likely that the CCOs are associated with the calcium-induced calcium release store, (the ryanodine-sensitive store) and not with the IP3-related store. This is partly because the metabotropic glutamate receptor, known to activate the IP3 store (Sacchetto et al., 1995), does not cause a rise of [Ca]i in our cells (M. Segal and E. Korkotian, unpublished observations). The ryanodine store does exist and contributes to the glutamate-evoked rise in [Ca]i(Segal and Manor, 1992); therefore, it is likely that the CCOs are linked functionally to the ryanodine-sensitive calcium stores. The higher free-calcium concentration in CCOs can be significant for the initiation of calcium-induced calcium release in the resting or weakly stimulated cell, inducing local [Ca]i release from calcium-sensitive stores. Whether the CCO acts as a promoter, helping to raise [Ca]i to a sufficient enough level to activate the calcium release mechanism, remains to be elucidated.
Footnotes
This work was supported by a grant from the Binational U.S.-Israel Science Foundation. We thank Ms. V. Greenberger for the preparation of the cultures and Dr. J. E. Friedman for comments on earlier versions of this manuscript.
Correspondence should be addressed to Menahem Segal at the above address.
REFERENCES
- 1.Almers W, Nehers E. The Ca signal from Fura-2 loaded mast cells depends strongly on the method of dye loading. FEBS Lett. 1985;192:13–18. doi: 10.1016/0014-5793(85)80033-8. [DOI] [PubMed] [Google Scholar]
- 2.Blatter LA. Depletion and filling of intracellular calcium stores in vascular smooth muscle. Am J Physiol. 1995;268:503–512. doi: 10.1152/ajpcell.1995.268.2.C503. [DOI] [PubMed] [Google Scholar]
- 3.Blatter LA, Wier WG. Intracellular diffusion, binding, and compartmentalization of the fluorescent calcium indicators indo-1 and fura-2. Biophys J. 1990;58:1491–1499. doi: 10.1016/S0006-3495(90)82494-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burgoyne RD, Cheek TR, Morgan A, O’Sullivan AJ, Moreton RB, Berridge MJ, Mata AM, Colyer J, Lee AG, East JM. Distribution of two distinct Ca-ATPase-like proteins and their relationships to the agonist-sensitive calcium store in adrenal chromaffin cells. Nature. 1989;342:72–74. doi: 10.1038/342072a0. [DOI] [PubMed] [Google Scholar]
- 5.Burgoyne RD, Cheek TR, Morgan A, O’Sullivan AJ, Moreton RB, Berridge MJ, Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–327. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- 6.Connor JA. Intracellular calcium mobilization by inositol 1,4,5-trisphosphate: intracellular movements and compartmentalization. Cell Calcium. 1993;14:185–200. doi: 10.1016/0143-4160(93)90066-f. [DOI] [PubMed] [Google Scholar]
- 7.Fiskum G. Intracellular levels and distribution of Ca2+ in digitonin-permeabilized cells. Cell Calcium. 1985;6:25–37. doi: 10.1016/0143-4160(85)90032-6. [DOI] [PubMed] [Google Scholar]
- 8.Gunter TE, Restrepo D, Gunter KK. Conversion of esterified Fura-2 and Indo-1 to calcium sensitive forms by mitochondria. Am J Physiol. 1988;255:304–310. doi: 10.1152/ajpcell.1988.255.3.C304. [DOI] [PubMed] [Google Scholar]
- 9.Gunter TE, Zuscik MJ, Puzas JE, Gunter KK, Rosier RN. Cytosolic free calcium concentrations in avian growth plate chondrocytes. Cell Calcium. 1990;11:445–457. doi: 10.1016/0143-4160(90)90077-8. [DOI] [PubMed] [Google Scholar]
- 10.Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- 11.Hofer AM, Machen TE. Direct measurement of free Ca2+ in organelles of gastric epithelial cells. Am J Physiol. 1994;267:442–451. doi: 10.1152/ajpgi.1994.267.3.G442. [DOI] [PubMed] [Google Scholar]
- 12.Hofer AM, Schlue W-R, Curci S, Machen TE. Spatial distribution and quantitation of free luminal [Ca] within the InsP3-sensitive internal store of individual BNK-21 calls: ion dependence of InsP3-induced Ca release and reloading. FASEB J. 1995;9:788–798. doi: 10.1096/fasebj.9.9.7601343. [DOI] [PubMed] [Google Scholar]
- 13.Korkotian E, Segal M. Lasting effects of glutamate on nuclear calcium concentration: regulation by calcium stores. J Physiol (Lond) 1996;496:39–48. doi: 10.1113/jphysiol.1996.sp021663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krause KH, Pittet D, Volpe P, Pozzan T, Meldolesi J, Lew DP. Calciosome, a sarcoplasmic reticulum-like organelle involved in intracellular Ca2+-handling by non-muscle cells: studies in human neutrophils and HL-60 cells. Cell Calcium. 1989;10:351–361. doi: 10.1016/0143-4160(89)90061-4. [DOI] [PubMed] [Google Scholar]
- 15.Kuba K, Mua SY, Nohmi M. Spatial and dynamic changes in intracellular Ca2+ measured by confocal laser scanning microscopy in bullfrog sympathetic ganglion cells. Neurosci Res. 1991;10:245–259. doi: 10.1016/0168-0102(91)90082-a. [DOI] [PubMed] [Google Scholar]
- 16.Malgaroli A, Milani D, Meldolesi J, Pozzan T. Fura-2 measurement of cytosolic Ca2+ in monolayers and suspensions of various types of animal cells. J Cell Biol. 1987;105:2145–2155. doi: 10.1083/jcb.105.5.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papa M, Bundman MC, Greenberger V, Segal M. Morphological analysis of the development of dendritic spines in primary cultures of hippocampal neurons. J Neurosci. 1995;15:1–12. doi: 10.1523/JNEUROSCI.15-01-00001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pozzan T, Volpe P, Zorzato F, Bravin M, Krause KH, Lew DP, Hashimoto S, Bruno B, Meldolesi J. The Ins(1,4,5)P3-sensitive Ca2+ store of non-muscle cells: endoplasmic reticulum or calciosomes? J Exp Biol. 1988;139:181–193. doi: 10.1242/jeb.139.1.181. [DOI] [PubMed] [Google Scholar]
- 19.Renard-Kooney DC, Majnoczky G, Seitz MB, Schnider TG, Thomas AP. Imaging of inositol 1,4,5 trisphosphate-induced Ca2+ fluxes in single permeabilized hepatocytes. J Biol Chem. 1993;268:23601–23610. [PubMed] [Google Scholar]
- 20.Ross CA, Meldolesi J, Milner TA, Satoh T, Supattapone S, Snyder SH. Inositol 1,4,5-trisphosphate receptor localized to endoplasmic reticulum in cerebellar Purkinje neurons. Nature. 1989;339:468–470. doi: 10.1038/339468a0. [DOI] [PubMed] [Google Scholar]
- 21.Rossier MF, Putney JW., Jr The identity of the calcium-storing, inositol 1,4,5-trisphosphate-sensitive organelle in non-muscle cells: calciosome, endoplasmic reticulum or both? Trends Neurosci. 1991;14:310–314. doi: 10.1016/0166-2236(91)90143-i. [DOI] [PubMed] [Google Scholar]
- 22.Sacchetto R, Cliffer KD, Podini P, Villa A, Christensen BN, Volpe P. Intracellular Ca2+ stores in chick cerebellum Purkinje neurones: ontogenetic and functional studies. Am J Physiol. 1995;269:1219–1227. doi: 10.1152/ajpcell.1995.269.5.C1219. [DOI] [PubMed] [Google Scholar]
- 23.Scanlon M, Williams DA, Fay FS. A Ca2+-insensitive form of Fura-2 associated with polymorphonuclear leukocytes. J Biol Chem. 1987;262:6308–6312. [PubMed] [Google Scholar]
- 24.Segal M. Imaging of calcium variations in dendritic spines of cultured hippocampal neurons. J Physiol (Lond) 1995;486:285–296. doi: 10.1113/jphysiol.1995.sp020811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segal M, Manor D. Confocal microscopic imaging of intracellular calcium in cultured hippocampal neurons following exposure to NMDA. J Physiol (Lond) 1992;448:655–676. doi: 10.1113/jphysiol.1992.sp019063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simpson PB, Challiss RAJ, Nahorski SR. Neuronal Ca stores: activation and function. Trends Neurosci. 1995a;18:299–306. doi: 10.1016/0166-2236(95)93919-o. [DOI] [PubMed] [Google Scholar]
- 27.Simpson PB, Challiss RAJ, Nahorski SR. Divalent cation entry in cultured rat cerebellar granule cells measured using Mn2+ quench of fura-2 fluorescence. Eur J Neurosci. 1995b;7:831–840. doi: 10.1111/j.1460-9568.1995.tb01070.x. [DOI] [PubMed] [Google Scholar]
- 28.Spat A, Rohacs T, Hunyady L. Plasmalemmal dihidropiridine receptors modify the function of subplasmalemmal inositol 1,4,5-trisphosphate receptor: a hypothesis. Cell Calcium. 1994;15:431–437. doi: 10.1016/0143-4160(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 29.Spurgeon HA, Stern MD, Baurtz G, Kaffaeli S, Hansford RG, Talo A, Lakatta EG, Capogrossi MC. Simultaneous measurement of Ca2+ contraction and potential in cardiac myocytes. Am J Physiol. 1990;258:574–586. doi: 10.1152/ajpheart.1990.258.2.H574. [DOI] [PubMed] [Google Scholar]
- 30.Tsien R, Pozzan T. Measurement of cytosolic Ca2+ with quin-2. Methods Enzymol. 1989;172:230–262. doi: 10.1016/s0076-6879(89)72017-6. [DOI] [PubMed] [Google Scholar]
- 31.Van PN, Peter F, Soling HD. Four intracellular calcium-binding glycoproteins from rat liver microsomes with high affinity for calcium. No indication for calsequestrin-like proteins in inositol 1,4,5-trisphosphate-sensitive calcium sequestrating rat liver vesicles. J Biol Chem. 1989;264:17494–17501. [PubMed] [Google Scholar]
- 32.Van Delden C, Favre C, Spat A, Cerny E, Krause KH, Lew DP. Purification of an inositol 1,4,5-trisphosphate-binding calreticulin-containing intracellular compartment of HL-60 cells. Biochem J. 1992;281:651–656. doi: 10.1042/bj2810651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villa A, Podini P, Clegg DO, Pozzan T, Meldolesi J. Intracellular Ca2+ stores in chicken Purkinje neurons: differential distribution of the low affinity, high capacity Ca binding protein, calsequestrin, of Ca2+ ATPase and of the ER lumenal protein, Bip. J Cell Biol. 1991;113:779–791. doi: 10.1083/jcb.113.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volpe P, Krause KH, Hashimoto S, Zorzato F, Pozzan T, Meldolesi J, Lew DP. “Calciosome,” a cytoplasmic organelle: the inositol 1,4,5-trisphosphate-sensitive Ca2+ store of nonmuscle cells? Proc Natl Acad Sci USA. 1988;85:1091–1095. doi: 10.1073/pnas.85.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volpe P, Villa A, Damiani E, Sharp AH, Podini P, Snyder SH, Meldolesi J. Heterogeneity of microsomal Ca2+ stores in chicken Purkinje neurons. EMBO J. 1991;10:3183–3189. doi: 10.1002/j.1460-2075.1991.tb04880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wahl M, Lucherini MJ, Gruenstein E. Intracellular Ca2+ measurement with Indo-1 in substrate-attached cells: advantages and special considerations. Cell Calcium. 1990;11:487–500. doi: 10.1016/0143-4160(90)90081-5. [DOI] [PubMed] [Google Scholar]
- 37.Wahl M, Sleight RG, Gruenstein E. Association of cytoplasmic free Ca2+ gradients with subcellular organelles. J Cell Physiol. 1992;150:593–609. doi: 10.1002/jcp.1041500321. [DOI] [PubMed] [Google Scholar]
- 38.White RJ, Reynolds IJ. Mitochondria and Na/Ca exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]