

Abstract
Studies from several laboratories have generated evidence suggesting that oxidative stress is involved in the pathogenesis of Alzheimer’s disease (AD). The finding that the amyloid β protein (Aβ) has neurotoxic properties and that such effects are, in part, mediated by free radicals has provided insights into mechanisms of cell death in AD and an avenue to explore new therapeutic approaches. In this study we demonstrate that melatonin, a pineal hormone with recently established antioxidant properties, is remarkably effective in preventing death of cultured neuroblastoma cells as well as oxidative damage and intracellular Ca2+ increases induced by a cytotoxic fragment of Aβ. The effects of melatonin were extremely reproducible and corroborated by multiple quantitative methods, including cell viability studies by confocal laser microscopy, electron microscopy, and measurements of intracellular calcium levels. The importance of this finding is that, in contrast to conventional antioxidants, melatonin has a proposed physiological role in the aging process. Secretion levels of this hormone are decreased in aging and more severely reduced in AD. The reported phenomenon may be of therapeutic relevance in AD.
Keywords: Alzheimer’s disease, melatonin, Aβ toxicity, oxidative stress, neuronal cells, antioxidants
Deposition of cerebral amyloid is a primary neuropathological marker of Alzheimer’s disease (AD). The amyloid is composed of a 40–42 amino acid peptide called the amyloid β protein (Aβ) (Glenner and Wong, 1984). Amyloid deposits in AD are found mainly as components of senile plaques and in the walls of cerebral and meningeal blood vessels (Robakis, 1994).
Molecular cloning showed that Aβ comprises a small region of a larger amyloid precursor protein (APP) (Robakis et al., 1987; Weidemann et al., 1989). Briefly, this is a type I integral membrane glycoprotein having a large extracytoplasmic portion, a smaller intracytoplasmic region, and a single transmembranous domain. APP undergoes extensive post-translational modifications (Robakis, 1994; Pappolla and Robakis, 1995) before the secretion of its N-terminal portion (Sambamurti et al., 1992; Robakis, 1994). Physiological processing of APP involves cleavage within the Aβ sequence by an unidentified enzyme, α-secretase (Anderson et al., 1991). Smaller quantities of APP molecules are cleaved at two other sites that potentially could produce amyloidogenic-secreted or membrane-bound APP (Robakis, 1994). Aβ also is produced during normal cellular metabolism (Haass et al., 1992;Shoji et al., 1992).
There is some controversy as to whether amyloid causes AD; however, three main lines of evidence have strengthened the amyloid hypothesis. The first piece of evidence is provided by the identification of several point mutations within the APP gene. These mutations segregate within a subgroup of patients afflicted with a familial form of the disorder and thus suggest a pathogenetic relationship between the APP gene and AD (Chartier-Harlin et al., 1991; Kennedy et al., 1993). Second, amyloid deposition temporally precedes the development of neurofibrillary changes (Pappolla and Robakis, 1995), and this observation is also consistent with a link between amyloid and neuronal degeneration. Finally, it has been shown that Aβ is toxic to neurons (Yankner et al., 1990; Behl et al., 1992, 1994a; Zhang et al., 1994), a finding that also strengthens the notion that the amyloid peptide may contribute to the neuronal pathology in AD.
Several investigators demonstrated that oxygen free radicals (OFRs) are related to the cytotoxic properties of Aβ (Behl, 1992, 1994b;Butterfield et al., 1994; Goodman and Mattson, 1994; Harris et al., 1995). Such findings are important because markers of oxidative injury are associated topographically with the neuropathological lesions of AD (Pappolla et al., 1992, 1996; Smith et al., 1994; Furuta et al., 1995). Because of these observations, antioxidants have been proposed as potential therapeutic agents in AD (Hensley et al., 1994; Mattson, 1994; Pappolla et al., 1996). Interestingly, melatonin exhibits antioxidant properties (Reiter, 1995), but in contrast to conventional antioxidants this hormone has a proposed physiological role in the aging process (Pierpaoli, 1991). In this study we report that melatonin prevented death of cultured neuroblastoma cells exposed to Aβ. Melatonin also averted Aβ-induced increases in intracellular Ca2+ and lipid peroxidation. A striking degree of cytoprotection was observed when melatonin was added simultaneously to the culture medium along with Aβ. The observed phenomenon was confirmed by various independent methodological approaches, including conventional microscopy (trypan blue exclusion method), fluorescent confocal laser microscopy for assessment of cell viability, scanning and transmission electron microscopy, Ca2+ imaging, and measurements of lipid peroxidation.
MATERIALS AND METHODS
Cell viability studies. Most experiments were performed with murine N2a neuroblastoma cells using Aβ(25–35), although a number of confirming experiments were performed with PC12 cells and Aβ(1–40) (see below). We chose N2a cells for most of the experiments because these cells exhibit larger cytoplasmic areas and better attachment to plates than PC12 cells, allowing better morphological analysis of cell damage. N2a cells were exposed to various concentrations of Aβ(25–35), the actively toxic fragment of Aβ (Yankner et al., 1990), or to matching concentrations of a control scrambled sequence KSGNMLGIIAG for various time periods. Aβ(25–35) and the scrambled peptide were obtained from Research Genetics (Huntsville, AL), using identical methods of synthesis for both sequences. Melatonin and Aβ(1–40) were purchased from Sigma (St. Louis, MO). Cells were grown in serum-free DMEM supplemented with 5 μg/ml insulin, 20 μm progesterone, 100 μg/ml transferrin, 40 μm selenium, and 100 μmputrescine. To insure the reliability and reproducibility of our observations, we assessed the cytotoxic effects of Aβ(25–35) on N2a cells and the actions of melatonin by several methodologies. These included fluorescent staining with the probe BODIPY green (Molecular Probes, Eugene, OR), which is a reliable indicator of viability (Poot et al., 1991), dual fluorescent labeling using annexin V-FITC and propidium iodide (R & D Systems, Minneapolis, MN) (Koopman et al., 1994; Vermes et al., 1995), scanning and transmission electron microscopy (Hayat, 1986), and the trypan blue exclusion method (Pike et al., 1993). The rationale to use annexin in our measurements is as follows. During apoptosis cells expose phosphatidylserine of the outer membrane, which dramatically increases binding of annexin V (red fluorescence). Cells undergoing apoptosis characteristically bind annexin V and exclude propidium iodide (Koopman et al., 1994; Vermes et al., 1995). In contrast, staining with both propidium iodide and annexin V has been associated with necrosis (Koopman et al., 1994;Vermes et al., 1995). Although apoptosis is defined by more than one single feature, we used this method as one additional indicator of the phenomenon reported here [whether Aβ causes apoptosis or necrosis is not the subject of this investigation, although it is reported to depend on cell type (Gschwind and Huber, 1995) and/or Aβ concentration (Le et al., 1995)]. Labeling studies with BODIPY green, annexin, and propidium iodide were analyzed by scanning laser confocal microscopy (Koopman et al., 1994; Vermes et al., 1995) with a Molecular Dynamics (Sunnyvale, CA) scanning microscope. Ultrastructural examination was performed because it allowed direct visualization of cell damage, including induction of membrane blebs by Aβ and cell retraction as well as abnormalities in chromatin distribution and karyorrhexis. Cells exhibiting increased membrane blebs and/or shrinking (retraction) were counted at low magnifications and compared with control preparations exposed to the scrambled peptide or melatonin alone. Details on concentrations of Aβ(25–35), melatonin and/or scrambled peptide (control), and incubation times used in the experiments are indicated in the corresponding figures.
At a minimum, all reported experiments, except where indicated, were performed in duplicate and reproduced on different days. However, to ensure reproducibility of the findings further, we used the trypan blue method to measure the viability of PC12 cells exposed to Aβ(25–35) and of N2a and PC12 cells exposed to Aβ(1–40). PC12 cells were handled in a manner identical to that described for N2a cells, except that they were grown on collagen-coated plates. Additional control experiments included treatment with the spin trapN-tert-butyl-α-phenylnitrone (PBN) instead of melatonin as well as adriamycin (see corresponding figures). PBN is an OFR scavenger chemically unrelated to melatonin; because it has been used previously in studies involving Aβ toxicity (Behl, 1994b), it was used here to verify the reliability of the viability measurements. Adriamycin has been used in several studies as a cell-killing agent (Marin et al., 1996), and we decided to include it as an additional control in some experiments.
Intracellular Ca2+ studies. The fluorescent probe fluo-3 was used for measurements of Ca2+ as described (Minta et al., 1985). Control cells (with or without addition of scrambled peptide) and cells exposed to Aβ or Aβ with melatonin were incubated with 2 μm fluo-3 for 15 min (see figures). The cells were scanned for maximum fluorescence by scanning laser confocal microscopy, using a section series. The images with the highest fluorescence were subjected to three-dimensional FishNet modeling to obtain relative fluorescence intensity (RFI) measurements and section line “cutting” for histogram determination of Ca2+ levels with the Silicon Graphics software. Calibration for quantitative measurements of Ca2+ was achieved with a commercially available kit (Molecular Probes).
Lipid peroxidation. To verify that melatonin is a free-radical scavenger in the system under study, we measured the degree of lipid peroxidation in parallel experiments in which N2a cells were either exposed to Aβ(25–35) or to the superoxide dismutase (SOD) inhibitor diethyldithiocarbamic acid (DDTC) (positive control) with or without melatonin. Under these experimental conditions, the degree of lipid peroxidation was estimated by measuring the formation of malondialdehyde acid (MDA) in cell lysates as described (Omar et al., 1987).
RESULTS
Melatonin prevents death of neuroblastoma cells exposed to Aβ
Addition of melatonin to culture plates exposed to Aβ(25–35) showed a striking improvement in cell survival. Figure1 shows cell viability counts as assessed with the fluorescent BODIPY green probe. Figure 2 illustrates representative images obtained with BODIPY green (right panels) and with dual fluorescent labeling with annexin V/propidium iodide. Exposure of cells to Aβ(25–35) induced cell death in over two-thirds of the cells by 24 hr (as assessed by the above-mentioned methods) while simultaneous addition of melatonin to the culture medium prevented cell death. Decreased cell viability with BODIPY green was determined by counting the number of cells exhibiting decreased fluorescent intensity. With the annexin/propidium iodide method, we counted the number of cells showing simultaneous fluorescence staining with annexin V and propidium iodide in the same cells (i.e., cell illustrated in 2A) as well as the number of cells staining with annexin V only (redcells depicted in the left panels of Fig.2B,C). Under the experimental conditions used, we found an almost exclusive increase in red fluorescent cells after exposure to Aβ(25–35). This increase was prevented by the addition of melatonin. The number of cells exhibiting increased fluorescence with propidium iodide after exposure to Aβ alone was comparable to control plates incubated with scrambled peptide, suggesting that apoptosis is the mode of cell death at the indicated concentration of Aβ(25–35) in N2a cells. Cell survival seemed dependent on concentration of Aβ(25–35) and time of exposure (see Figs. 1, 5), as previously noted by several laboratories. By electron microscopy, the effect of Aβ(25–35), as well as the described phenomenon with melatonin, was readily apparent. Exposure of cells to Aβ(25–35) resulted in marked cell damage characterized by diffuse membrane blebbing (Fig. 3B), cell retraction (Fig.3A,B), abnormal distribution of chromatin toward the nuclear membrane (Fig. 3C), and karyorrhexis (defined as fragmentation and condensation of nuclear material into large electron-dense granules) (Fig. 3D). Quantitation of cell retraction and/or increased membrane blebs showed that these toxic effects were prevented by melatonin. Table 1 shows quantitative differences observed between control cells (scrambled peptide) and cells incubated with Aβ(25–35) or Aβ(25–35) plus melatonin.
Fig. 1.

Melatonin prevents death of N2a exposed to Aβ(25–35). N2a cells were plated and after 24 hr, during exponential growth phase, were treated with either scrambled peptide (control), adriamycin (control for apoptotic cell death; Marin et al., 1996), 50 μm Aβ(25–35), or 50 μm Aβ(25–35) with 10 μm melatonin for an additional 24 hr. Live cells were assessed by their fluorescence with BODIPY green (also see Fig. 2,right panels). Results are reported as means ± SD of four experiments (2 duplicate experiments on different days; minimum 100 cells studied per plate). *Measurements significantly different from control (p ≤ 0.02, pairedt test).
Fig. 2.

Representative confocal scanning images of annexin/propidium iodide and BODIPY green studies. The right panels illustrate representative images of one of the experiments plotted in Figure 1. Cultured N2a cells were exposed for 24 hr to either scrambled peptide (1), 50 μmAβ(25–35) (2), or 50 μm Aβ(25–35) plus 10 μm melatonin (3). After exposure to Aβ(25–35) alone (2), many cells showed a marked decrease in fluorescent intensity with BODIPY green, reflecting decreased cell viability (compare with panels 1 and3, which illustrate the fluorescent intensity exhibited by a similar number of cells exposed to either scrambled peptide alone in panel 1 or Aβ plus melatonin in panel 3). The areas photographed are representative fields of typical responses (magnification 1000×). A–C,Left, Representative images obtained from cells exposed to Aβ(25–35) and then stained by a dual fluorescent-tagging method with the probes annexin V-FITC (red) and propidium iodide (green). After examination with the appropriate filters, we counted the number of cells that stained simultaneously with both markers (necrosis) or with annexin V only (B or C, apoptosis). Exposure of cells to 50 μm Aβ(25–35) was followed by an almost exclusive increase in the number cells exhibiting red fluorescence only (annexin V), such as those illustrated in B and C. By 24 hr, 70 ± 25% of the cells exposed to Aβ(25–35) developed strong annexin (red) fluorescence and no increase in propidium iodide (green) fluorescence (means ± SD represent 2 duplicate experiments on different days, 4 experiments total; minimum 300 cells/plate counted). Such effects were prevented by simultaneous addition of melatonin to the culture medium [at 24 hr we counted 20 ± 10% annexin-positive cells in plates containing Aβ(25–35) plus melatonin and 15 ± 10% annexin-positive cells in control plates containing scrambled peptide alone].
Fig. 5.

Cell viability after exposure to Aβ alone or Aβ with various concentrations of melatonin or PBN. N2a cells were plated and after 24 hr, during exponential dividing phase, exposed to the indicated concentrations of Aβ(25–35) for 6 hr and treated with either melatonin or PBN at the indicated concentrations. These experiments were performed at 6 hr because cell death was readily apparent by this time. Viable cells are expressed as a percentage of controls and assessed by their ability to exclude trypan blue. Similar dose– responses were obtained by BODIPY green fluorescence (data not shown). Differences in survival between cells exposed to Aβ alone versus Aβ with melatonin were statistically significant for all concentrations of Aβ and melatonin (i.e., 50 μm Aβ vs 50 μm Aβ + 1.2 μm melatonin, p < 0.002; 50 μm Aβ vs 50 μm Aβ + 10 μmmelatonin, p < 0.001).
Fig. 3.
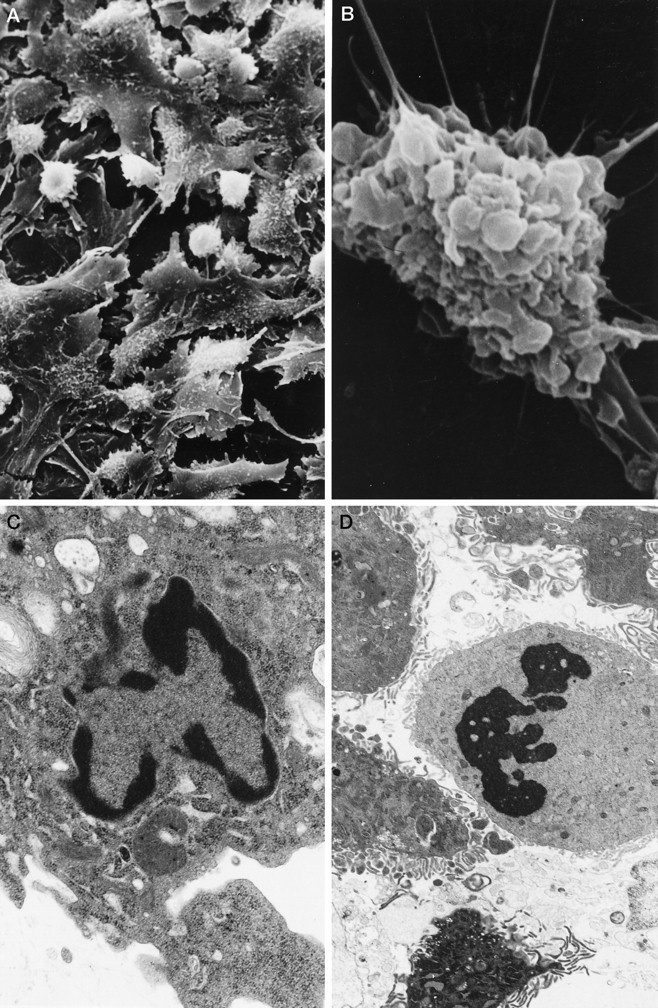
Electron microscopy of N2a cells exposed to Aβ(25–35). Scanning electron microphotographs illustrate conspicuous cell retraction (A, B) induced by the amyloid peptide. Note marked membrane blebbing (B).C, D, Transmission electron microscopy preparations depict chromatin misdistribution and karyorrhexis in cultured N2a induced by Aβ. The prevalence of membrane blebbing (defined as the percentage of cells exhibiting diffuse involvement by large and small blebs on more than one-half of their surface) and cell retraction was quantitated by counting 150 cells per scanning preparation (total of 4 experiments per condition; see Table 1).
Table 1.
Ultrastructural alterations of N2a cells induced by Aβ(25–35)
| Cell blebbing (%) | Cell retraction (%) | |
|---|---|---|
| Scrambled peptide | 11 ± 3 | 16.5 ± 2.5 |
| Aβ(25–35) | 94 ± 3 | 47.5 ± 8.5 |
| Aβ + melatonin | 42 ± 3 | 27.5 ± 2.5 |
Cultured N2a cells were exposed to 50 μmAβ(25–35) or to scrambled peptide (controls) for 12 hr. The number of cells exhibiting conspicuous numbers of large blebs (more than one-half of the cell surface involved) or retraction of their soma was counted (see Fig. 4A,B) and compared with cultures exposed to Aβ plus melatonin. Although both groups treated with Aβ were significantly different (p ≤ 0.005) from the control (scrambled), the group treated with melatonin was also significantly different (p ≤ 0.005) from the group treated with Aβ alone.
Results of the experiments on PC12 cells exposed to Aβ(25–35) (assessed by the trypan blue method) (Fig.4A) and on N2a and PC12 cells exposed to Aβ(1–40) (Fig. 4B,C) corroborated the reproducibility of the findings beyond a particular cell line and with the more physiologically relevant peptide Aβ(1–40). These experiments showed that melatonin prevented cell death after exposure to the above-mentioned peptides in either N2a or PC12 cells. Results were equally striking irrespective of the cell line or peptide used. The viability of cells exposed to Aβ plus melatonin was identical to control cultures.
Fig. 4.

Experiments on PC12 cells exposed to Aβ(25–35) (A) and N2a and PC12 cells exposed to Aβ(1–40) (B and C, respectively). In these experiments cells were plated as described in the previous experiments, except that PC12 cells required 4 d of growth on collagen-coated plates. Cells were exposed to 50 μm Aβ(25–35) (A) or 100 μm Aβ(1–40) (B, C) for 24 hr. Melatonin, where indicated, was at 50 μm. Values represent the means ± SD of four experiments; a minimum of 500 cells was counted per culture plate. Cell viability was assessed by trypan blue exclusion and expressed as a percentage of controls.
We also performed a “checkerboard” dose–response experiment on N2a cells in which the effect of each of two concentrations of Aβ(25–35) was tested in permutation with either two concentrations of melatonin or without the hormone. As a control, we ran another parallel “checkerboard” experiment, but instead of melatonin we used PBN (because PBN previously was reported to enhance the survival of cells exposed to Aβ). The results of these experiments confirmed once again the cytoprotective effects of melatonin and showed a correlation between cell survival and concentrations of Aβ and melatonin (Fig.5), as evaluated by the trypan blue method.
In summary, the reported effects of melatonin on preventing cell death were verified by different experimental approaches and found to be extremely reproducible and statistically significant with all of the methods used.
Melatonin and PBN prevent lipid peroxidation of cultured N2a cells induced by Aβ or inhibition of superoxide dismutase
Exposure of N2a cells to Aβ(25–35) or DDTC resulted in increased lipid peroxidation (Fig.6A,B), and this effect was prevented by melatonin. As noted with Aβ(25–35), control experiments with DDTC also caused cell death in a concentration-dependent manner (Fig.7). These effects were prevented by addition of melatonin to the culture medium (Figs. 6A,B, 7). The experiments with DDTC were designed to provide additional control variables as well as preliminary evidence that melatonin exhibits antioxidant activity in our system. The results from these experiments support the previously reported antioxidant properties of melatonin (Reiter, 1995). PBN, a chemically unrelated free-radical scavenger, also was included as an additional control in the experiments for similar reasons as those discussed on the section on cell survival. This substance was effective in preventing lipid peroxidation induced by Aβ(25–35) (Fig. 6B) and DDTC (data not shown). The cytoprotective effects of melatonin and PBN were concentration-dependent (Figs. 6, 7).
Fig. 6.

Lipid peroxidation induced by Aβ(25–35) is prevented by melatonin or PBN. The byproduct malondialdehyde acid (MDA) was measured in N2a cell lysates as described (Omar et al., 1987) at the indicated concentrations of melatonin (Fig. 5A), PBN (Fig. 5B), and Aβ(25–35). Values are the means of three determinations. SE in all measurements was <20% of the mean. Cells were exposed to Aβ(25–35) for 24 hr with and without melatonin or PBN.
Fig. 7.

Melatonin prevents cell death induced by inhibition of SOD. Cells were plated as previously noted in Figure 1and exposed to DDTC for 24 hr at the indicated concentrations. Melatonin was added at the stated concentrations. Survival was determined by the trypan blue exclusion method and expressed as percentage of controls (no DDTC). Data represent the means ± SD for four experiments (2 duplicated experiments on different days).
Melatonin prevents Aβ-induced intracellular Ca2+ increase
Control cells exhibited an average intracellular Ca2+/fluo-3 fluorescence of 0.3 ± 0.1 RFI units (n = 20 cells), whereas cells exposed to Aβ(25–35) showed a marked increase in intracellular Ca2+ (Fig.8) (at 12 hr, RFI values 0.3 ± 0.09 and 2.2 ± 0.2 control vs Aβ, respectively; n = 12 cells/plate). Inclusion of melatonin in the cultures returned the intracellular Ca2+ levels to near normal (RFI value, 0.55 ± 0.2). Figure 9 shows representative images from each of the experimental groups. Because adriamycin treatment has been used as a model for intracellular Ca2+ increases during apoptosis (Marin et al., 1996), cells treated with 0.03 μg/ml adriamycin were included as a control system for the Ca2+studies.
Fig. 8.

Time course study of fluo-3 fluorescence increase induced by Aβ(25–35) and prevention by melatonin. Cells were exposed to 50 μm scrambled peptide (control), 50 μmAβ(25–35), or 50 μm Aβ(25–35) plus 5 μm melatonin. Aβ alone was significantly different from control and Aβ plus melatonin after 6 hr at all time points (p < 0.002). There were no significant differences between control and Aβ plus melatonin at any time point.
Fig. 9.
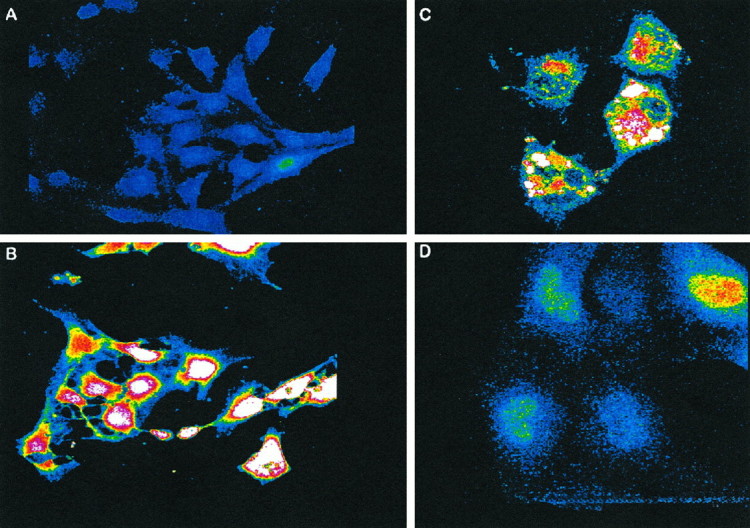
Melatonin prevents intracellular Ca2+increases induced by Aβ. Representative images from various experimental conditions illustrate the characteristic fluorescent patterns exhibited by fluo-3 in cells after 12 hr exposure to 50 μm Aβ(25–35) plus 5 μm melatonin (A), 50 μm Aβ(25–35) (B), 0.03 μg/ml adriamycin (C), and 50 μm scrambled peptide (D). (Final magnifications are 2000× for A and B and 3000× for C and D.)
DISCUSSION
We confirmed by several methods that melatonin prevents death of cultured cells exposed to toxic fragments of Aβ. These methods included conventional light microscopy (trypan blue exclusion method), confocal laser microscopy using various probes for the assessment of cell viability (BODIPY green, annexin V, and propidium iodide), scanning and transmission electron microscopy, fluorescent Ca2+ imaging, and measurements of lipid peroxidation. The importance of the observed phenomenon is twofold. On one hand, melatonin has a proposed physiological role in the aging process (Pierpaoli, 1991; Pierpaoli et al., 1991), and decreased secretion of melatonin with aging is documented (Iguchi et al., 1982; Dori et al., 1994). On the other hand, and perhaps of more relevance to our study, there are reports of more profound reductions of melatonin secretion in populations with dementia than in nondemented controls (Souetre et al., 1989; Mishima et al., 1994). It has been suggested that altered secretion levels of the hormone partially may reflect the loss of daily variation in the concentration of melatonin in the pineals of elderly individuals and AD patients (Skene et al., 1990). These facts regarding melatonin are in sharp contrast with conventional antioxidants, which, despite their reported cytoprotective characteristics, have no comparable correlates with the pathophysiology of human aging.
The effects of melatonin may be complex, and the elucidation of its mechanism(s) of action is outside the scope of this report. In addition to its OFR scavenging properties, melatonin interacts with calmodulin (Benitez-King and Anton-Tay, 1993) microtubular components (Benitez-King and Anton-Tay, 1993) and is reported to increase the activity of the intrinsic cellular antioxidant defenses (Huerto-Delgadillo et al., 1994). Second, the bioavailability of this hormone makes it an ideal candidate for use in therapy. In vivo studies have shown that melatonin rapidly crosses the blood–brain barrier after systemic administration and reaches every neuronal compartment (Menendez-Pelaez et al., 1993). Characterization of the mechanism of action by the use of analogs will be an interesting area to explore in future studies. In this study, the concentrations of melatonin used are supraphysiological, and the potential correlates with human disease as well as the potential therapeutic value are not yet known.
Initial in vitro evidence suggests that the cytoprotective effects of melatonin are related to its antioxidant properties (Reiter, 1995). In line with such an interpretation, melatonin prevented cell death induced by inhibition of superoxide dismutase as well as Aβ-induced lipid peroxidation. Inhibition of SOD by DDTC is a well established model of oxidative injury (Omar and Pappolla, 1993) that has been used previously to induce death of spinal cord neurons via an apoptotic pathway (Rothstein et al., 1994). These observations are also in agreement with the oxidative stress hypothesis of AD.
Melatonin also blocked Aβ-related increases in intracellular Ca2+ levels. Sulfhydryl groups in membrane Ca2+pumps are characteristic targets of oxidative injury (Rohn et al., 1993), and damage to these structures by Aβ has been documented (Mark et al., 1995). It has been proposed that Ca2+ plays an important role in Aβ-mediated cell death (Mattson, 1994). Abnormal efflux of the ion into cellular compartments (Nicotera et al., 1992) causes activation of a number of Ca2+-dependent degradative processes detrimental to the cell. Interestingly, one of the newly discovered “apoptosis-linked genes” encodes a Ca2+ binding protein and shows partial homology to the FAD gene STM2 (Vito et al., 1996).
The finding that Aβ has neurotoxic properties has provided a possible connection between amyloid accumulation and neurodegeneration. After a number of controversial reports, studies from several laboratories now have corroborated this observation and demonstrated that the effects of the peptide are dependent on aggregation (Busciglio et al., 1992; Pike et al., 1993), time of exposure, osmolarity, pH, and concentration (Burdick et al., 1992; Pike et al., 1993). The mechanism of toxicity is not totally understood. In addition to free radicals, increased sensitivity to excitotoxicity (Copani et al., 1995) and/or disruption of Ca2+ homeostasis (Mattson et al., 1992, 1993; Le et al., 1995; Mark et al., 1995) seem to be involved. The magnitude of the damage contributed by each of these factors and the extent of their interaction are unresolved issues (Busciglio et al., 1993; Mattson, 1994; Weiss et al., 1994; Copani et al., 1995). Because of the close association between aging and AD and the similarities in the neuropathology of both conditions, oxidative stress has been proposed to play a role in the pathogenesis of AD lesions. The data reported here are in line with the available evidence suggesting a role for melatonin in oxidative stress and the aging process.
In conclusion, we report that melatonin prevents death of cultured cells induced by Aβ. In AD, the magnitude of the mental impairment correlates better with the severity of neuronal damage rather than with the degree of amyloid accumulation (Hyman et al., 1985). Therefore, improving cell survival has been a primary objective of most therapeutic approaches. The use of melatonin or its derived analogs could be explored as a therapeutic approach in AD.
Note added in proof: The cytoprotective effects of melatonin described in this paper have been, since then, reproduced in rat primary neuronal cultures.
Footnotes
This work was supported by National Institutes of Health Grants AG11130 to M.A.P. and AG08200 to N.K.R.
Correspondence should be addressed to Dr. Miguel A. Pappolla, Department of Pathology and Laboratory Medicine, University of South Alabama Medical Center, 2451 Fillingim Street, Mobile, AL 36617.
REFERENCES
- 1.Anderson JP, Sambamurti K, Esch FS, Keim PS, Lieberburg I, Robakis NK. Exact cleavage site of Alzheimer amyloid precursor in neuronal PC12 cells. Neurosci Lett. 1991;128:126–129. doi: 10.1016/0304-3940(91)90775-o. [DOI] [PubMed] [Google Scholar]
- 2.Behl C, Davis JB, Cole GM, Schubert D. Vitamin E protects nerve cells from amyloid protein toxicity. Biochem Biophys Res Commun. 1992;186:944–950. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- 3.Behl C, Davis JB, Klier FG, Schubert D. Amyloid beta peptide induces necrosis rather than apoptosis. Brain Res. 1994a;645:253–264. doi: 10.1016/0006-8993(94)91659-4. [DOI] [PubMed] [Google Scholar]
- 4.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid protein toxicity. Cell. 1994b;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 5.Benitez-King G, Anton-Tay F. Calmodulin mediates melatonin cytoskeletal effects [Review]. Experientia. 1993;49:635–641. doi: 10.1007/BF01923944. [DOI] [PubMed] [Google Scholar]
- 6.Burdick D, Soregan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman CW, Glabe C. Assembly and aggregation properties of synthetic Alzheimer’s A4/amyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 7.Busciglio J, Lorenzo A, Yankner BA. Methodological variables in the assessment of beta-amyloid neurotoxicity. Neurobiol Aging. 1992;13:609–612. doi: 10.1016/0197-4580(92)90065-6. [DOI] [PubMed] [Google Scholar]
- 8.Busciglio J, Yeh J, Yankner BA. Beta-amyloid neurotoxicity in human cortical culture is not mediated by excitotoxins. J Neurochem. 1993;61:1565–1568. doi: 10.1111/j.1471-4159.1993.tb13658.x. [DOI] [PubMed] [Google Scholar]
- 9.Butterfield DA, Hensley K, Harris M, Mattson M, Carney J. Beta-amyloid peptide free radical fragments initiate synaptosomal lipoperoxidation in a sequence-specific fashion: implications to Alzheimer’s disease. Biochem Biophys Res Commun. 1994;200:710–715. doi: 10.1006/bbrc.1994.1508. [DOI] [PubMed] [Google Scholar]
- 10.Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, Mullan M. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 11.Copani A, Bruno V, Battaglia G, Leanza G, Pellitteri R, Russo A, Stanzani S, Nicoletti F. Activation of metabotropic glutamate receptors protects cultured neurons against apoptosis induced by beta-amyloid peptide. Mol Pharmacol. 1995;47:890–897. [PubMed] [Google Scholar]
- 12.Dori D, Casale G, Solerte SB, Fioravanti M, Migliorati G, Cuzzoni G, Ferrari E. Chrono-neuroendocrinological aspects of physiological aging and senile dementia. Chronobiology. 1994;21:121–126. [PubMed] [Google Scholar]
- 13.Furuta A, Price DL, Pardo C, Troncoso JC, XU Z-S, Taniguchi N, Martin LJ. Localization of superoxide dismutases in Alzheimer’s disease and Down’s syndrome neocortex and hippocampus. Am J Pathol. 1995;146:357–367. [PMC free article] [PubMed] [Google Scholar]
- 14.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 15.Goodman Y, Mattson MP. Secreted forms of beta-amyloid precursor protein protect hippocampal neurons against amyloid beta-peptide-induced oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- 16.Gschwind M, Huber G. Apoptotic cell death induced by amyloid1–42 peptide is cell type-dependent. J Neurochem. 1995;65:292–300. doi: 10.1046/j.1471-4159.1995.65010292.x. [DOI] [PubMed] [Google Scholar]
- 17.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ. Amyloid peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–324. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 18.Harris ME, Hensley K, Butterfield DA, Leedle RA, Carney JM. Direct evidence of oxidative injury produced by the Alzheimer’s beta-amyloid peptide (1–40) in cultured hippocampal neurons. Experimental Neurol. 1995;131:193–202. doi: 10.1016/0014-4886(95)90041-1. [DOI] [PubMed] [Google Scholar]
- 19.Hayat MA. Basic techniques for transmission electron microscopy. Academic; San Diego: 1986. Ion transport ATPases as targets for free radical damage. Protection by an aminosteroid of the Ca2+ pump ATPase of human red blood cell membranes. [Google Scholar]
- 20.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free-radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huerto-Delgadillo L, Anton-Tay F, Benitez-King G. Effects of melatonin on microtubule assembly depend on hormone concentration: role of melatonin as a calmodulin antagonist. J Pineal Res. 1994;17:55–62. doi: 10.1111/j.1600-079x.1994.tb00114.x. [DOI] [PubMed] [Google Scholar]
- 22.Hyman BT, West HL, Gomez-Isla T, Mui S. Quantitative neuropathology in Alzheimer’s disease: neuronal loss in high-order association cortex parallels dementia. In: Iqbal K, Mortimer JA, Windblad B, Wisniewski HM, editors. Research advances in Alzheimer’s disease and related disorders. Wiley; New York: 1985. pp. 453–460. [Google Scholar]
- 23.Iguchi H, Kato K, Ibayashi H. Age-dependent reduction in serum melatonin concentrations in healthy subjects. J Clin Endocrinol Metab. 1982;55:27–29. doi: 10.1210/jcem-55-1-27. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy AM, Newman S, McCaddon A, Ball J, Roques P, Mullan M, Hardy J, Chartier-Harlin MC, Frackowiak RS, Warrington EK. Familial Alzheimer’s disease. A pedigree with a mis-sense mutation in the amyloid precursor protein gene (amyloid precursor protein 717 valine→glycine). Brain. 1993;116:309–324. doi: 10.1093/brain/116.2.309. [DOI] [PubMed] [Google Scholar]
- 25.Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- 26.Le W- D, Colom LV, Xie W-J, Smith G, Alexianu M, Appel SH. Cell death induced by amyloid 1–40 in MES 23.5 hybrid clone: the role of nitric oxide and NMDA-gated channel activation leading to apoptosis. Brain Res. 1995;686:49–60. doi: 10.1016/0006-8993(95)00450-5. [DOI] [PubMed] [Google Scholar]
- 27.Marin MC, Fernandez A, Bick RJ, Brisbay S, Buja LM, Snuggs M, McConkey DJ, von Eschenbach AC, Keating MJ, McDonnell TJ. Apoptosis suppression by bcl-2 is correlated with the regulation of nuclear and cytosolic Ca2+. Oncogene. 1996;12:2259–2266. [PubMed] [Google Scholar]
- 28.Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid-peptide impairs ion-motive ATPase activities: evidence for a role in the loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattson MP. Calcium and neuronal injury in Alzheimer’s disease. Contributions of beta-amyloid precursor protein mis-metabolism, free radicals, and metabolic compromise [Review]. Ann NY Acad Sci. 1994;747:50–76. [PubMed] [Google Scholar]
- 30.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. Amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer’s disease. Trends Neurosci. 1993;16:409–414. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- 32.Menendez-Pelaez A, Poeggeler B, Reiter RJ, Barlow-Walden L, Pablos MI, Tan D. Nuclear localization of melatonin in different mammalian tissues: immunocytochemical and radioimmunoassay evidence. J Cell Biochem. 1993;53:373–382. doi: 10.1002/jcb.240530415. [DOI] [PubMed] [Google Scholar]
- 33.Minta A, Kao JP, Tsien RY. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem. 1985;264:8171–8178. [PubMed] [Google Scholar]
- 34.Mishima K, Okawa M, Hishikawa Y, Hozumi S, Hori H, Takahashi K. Morning bright light therapy for sleep and behavior disorders in elderly patients with dementia. Acta Psychiatr Scand. 1994;89:1–7. doi: 10.1111/j.1600-0447.1994.tb01477.x. [DOI] [PubMed] [Google Scholar]
- 35.Nicotera P, Bellomo G, Orrenius S. Calcium-mediated mechanisms in chemically induced cell death. Annu Rev Pharmacol Toxicol. 1992;32:449–470. doi: 10.1146/annurev.pa.32.040192.002313. [DOI] [PubMed] [Google Scholar]
- 36.Omar RA, Pappolla MA. Oxygen free radicals as inducers of heat shock protein synthesis in cultured human neuroblastoma cells: relevance to neurodegenerative disease. Eur Arch Psychiatry Clin Neurosci. 1993;651:1–6. doi: 10.1007/BF02190384. [DOI] [PubMed] [Google Scholar]
- 37.Omar RA, Yano S, Kikkawa Y. Antioxidant enzymes and survival of normal and SV-10-transformed mouse embryo cells after hyperthermia. Cancer Res. 1987;47:3473–3476. [PubMed] [Google Scholar]
- 38.Pappolla MA, Robakis NK. Neuropathology and molecular biology of Alzheimer’s disease. In: Stein M, Baum M, editors. Perspectives in behavioral medicine, Alzheimer’s disease, and AIDS. Academic; San Diego: 1995. [Google Scholar]
- 39.Pappolla MA, Omar RA, Kim KS, Robakis NK. Immunohistochemical evidence of oxidative stress in Alzheimer’s disease. Am J Pathol. 1992;140:621–628. [PMC free article] [PubMed] [Google Scholar]
- 40.Pappolla MA, Sos M, Omar RA, Sambamurti K. The heat shock/oxidative stress connection. Relevance to Alzheimer’s disease. Mol Chem Neuropathol. 1996;28:21–34. doi: 10.1007/BF02815201. [DOI] [PubMed] [Google Scholar]
- 41.Pierpaoli W. The pineal gland: a circadian or seasonal aging clock? [Editorial]. Aging (Milano) 1991;3:99–101. doi: 10.1007/BF03323985. [DOI] [PubMed] [Google Scholar]
- 42.Pierpaoli W, Dall’Ara A, Pedrinis E, Regelson W. The pineal control of aging. The effects of melatonin and pineal grafting on the survival of older mice. Ann NY Acad Sci. 1991;621:291–313. doi: 10.1111/j.1749-6632.1991.tb16987.x. [DOI] [PubMed] [Google Scholar]
- 43.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptide in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poot M, Kavanagh TJ, Kang HC, Haugland RP, Rabinovitch PS. Flow cytometric analysis of cell cycle-dependent changes in cell thiol level by combining a new laser dye with Hoechst 33342. Cytometry. 1991;12:184–187. doi: 10.1002/cyto.990120214. [DOI] [PubMed] [Google Scholar]
- 45.Reiter RJ. The pineal gland and melatonin in relation to aging: a summary of the theories and of the data [Review]. Exp Gerontol. 1995;30:199–212. doi: 10.1016/0531-5565(94)00045-5. [DOI] [PubMed] [Google Scholar]
- 46.Robakis NK. Amyloid and amyloid precursor protein: chemistry, molecular biology, and neuropathology. In: Terry RD, Katzman R, Bick KL, editors. Alzheimer disease. Raven; New York: 1994. pp. 317–326. [Google Scholar]
- 47.Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci USA. 1987;84:4190–4194. doi: 10.1073/pnas.84.12.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rohn TT, Hinds TR, Vincenzi FF. Ion transport, ATPases a target for free radical damage. Biochem Pharmacol. 1993;46:525–534. doi: 10.1016/0006-2952(93)90530-a. [DOI] [PubMed] [Google Scholar]
- 49.Rothstein JD, Bristol LA, Hosler B, Brown RH, Jr, Kuncl RW. Chronic inhibition of superoxide dismutase produces apoptotic death of spinal neurons. Proc Natl Acad Sci USA. 1994;91:4155–4159. doi: 10.1073/pnas.91.10.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambamurti K, Shioi J, Anderson JP, Pappolla MA, Robakis NK. Evidence for intracellular cleavage of the Alzheimer’s amyloid precursor in PC12 cells. J Neurosci Res. 1992;33:319–329. doi: 10.1002/jnr.490330216. [DOI] [PubMed] [Google Scholar]
- 51.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai X-D, McKay DM, Tinter R, Frangione B, Younkin SG. Production of the Alzheimer amyloid protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 52.Skene DJ, Vivien-Roels B, Sparks DL, Hunsaker JC, Pevet P, Ravid D, Swaab DF. Daily variation in the concentration of melatonin and 5-methoxytryptophol in the human pineal gland: effect of age and Alzheimer’s disease. Brain Res. 1990;528:170–174. doi: 10.1016/0006-8993(90)90214-v. [DOI] [PubMed] [Google Scholar]
- 53.Smith MA, Kutty RK, Richey PL, Yan SD, Stern D, Chader GJ, Wiggert B, Petersen Smith RB, Perry G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am J Pathol. 1994;145:42–47. [PMC free article] [PubMed] [Google Scholar]
- 54.Souetre E, Salvati E, Krebs B, Belugou JL, Darcourt G. Abnormal melatonin response to 5-methoxypsoralen in dementia. Am J Psychiatry. 1989;146:1037–1040. doi: 10.1176/ajp.146.8.1037. [DOI] [PubMed] [Google Scholar]
- 55.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 56.Vito P, Lacana E, D’Adamio L. Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer’s disease gene ALG-3. Science. 1996;721:521–525. doi: 10.1126/science.271.5248.521. [DOI] [PubMed] [Google Scholar]
- 57.Weidemann A, Konig G, Bunke D, Fisher P, Salbaum JM, Masters CL, Beyreuther K. Identification, biogenesis, and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- 58.Weiss JH, Pike CJ, Cotman CW. Ca2+ channel blockers attenuate beta-amyloid peptide toxicity to cortical neurons in culture. J Neurochem. 1994;62:372–375. doi: 10.1046/j.1471-4159.1994.62010372.x. [DOI] [PubMed] [Google Scholar]
- 59.Yankner DA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Z, Drzewiecki GH, Hom JP, May PC, Hyslop PA. Human cortical neuronal (HCN) cell lines: a model for amyloid beta neurotoxicity. Neurosci Lett. 1994;177:162–164. doi: 10.1016/0304-3940(94)90892-3. [DOI] [PubMed] [Google Scholar]