

Abstract
Mice are the preferred mammalian species for genetic investigations of the role of proteins. The normal function of the prion protein (PrP) is unknown, although it plays a major role in the prion diseases, including fatal familial insomnia. We investigated its role in sleep and sleep regulation by comparing baseline recordings and the effects of sleep deprivation in PrP knockout mice (129/SV) and wild-type controls (129/SV × C57BL/6), which are the mice used for most gene targeting experiments and whose behavior is not well characterized. Although no difference was evident in the amount of vigilance states, the null mice exhibited a larger degree of sleep fragmentation than the wild-type with almost double the amount of short waking episodes. As in other rodents, cortical temperature closely reflected the time course of waking. The increase of slow-wave activity (SWA; mean EEG power density in the 0.25–4.0 Hz range) at waking to nonrapid eye movement (NREM) sleep transitions was faster and reached a lower level in the null mice than in the wild-type. The contribution of the lower frequencies (0.25–5.0 Hz) to the spectrum was smaller than in other rodents in all three vigilance states, and the distinction between NREM sleep and REM sleep was most marked in the theta band. After the sleep deprivation, SWA was increased, but the changes in EEG power density and SWA were more prominent and lasted longer in the PrP-null mice. Our results suggest that PrP plays a role in promoting sleep continuity.
Keywords: prion protein, mice, sleep, sleep regulation, EEG spectral analysis, sleep deprivation, brain temperature, knockout mice, transgenic mice
The genetic factors determining sleep are still unresolved, although in mice several sleep variables appear to be inherited (Valatx et al., 1972, 1980; Friedman, 1974; Valatx and Bugat, 1974; Cespuglio et al., 1975). Gene targeting allows a selective approach to investigate inheritance (Capecci, 1994) and is usually performed in the inbred mouse strain “129” backcrossed with C57BL (Gerlai, 1996). The behavior of the mouse strains and their hybrids, including sleep, needs to be characterized (Crawley, 1996) to provide a basis for the investigation of sleep in mice subjected to gene targeting.
Prion protein (PrP) gene mutations lead to prion diseases including fatal familial insomnia (FFI) (Lugaresi et al., 1986; Goldfarb et al., 1991; Galassi et al., 1992; Medori et al., 1992; Monari et al., 1994;Aguzzi and Weissmann, 1996). In FFI, there is a severe reduction of sleep, a gradual disappearance of spindle activity in the sleep EEG (Gambetti et al., 1995; Reder et al., 1995; Sforza et al., 1995), and a disruption of hormone rhythms (Portaluppi et al., 1994, 1995; Montagna et al., 1995). The main neurological features include hypometabolism, loss of neurons, and astrogliosis, with the first and most severe neuropathology appearing in the thalamus (Gambetti et al., 1995). Thus, PrP may be important for sleep regulation, because the thalamus is implicated in the regulation of sleep spindles and EEG slow waves (Steriade et al., 1993). PrP is a glycoprotein localized on neuronal membranes and in astrocytes (Collinge et al., 1994; Moser et al., 1995) that may promote neuronal cell differentiation and maintain neuronal function (Clinton et al., 1993), but its function is not yet understood (Estibeiro, 1996). There are indications that the loss of the natural PrP function may be responsible for the pathology in prion diseases (Collinge et al., 1995; Gambetti et al., 1995).
The investigation of the prion diseases and the normal function of PrP involved the generation of mice devoid of PrP (for review, seePrusiner, 1991, 1996; Weissmann, 1996). They exhibited no abnormal behavior or learning ability (Büeler et al., 1992; Manson et al., 1994), although ataxia and loss of Purkinje cells have been observed in older mice with a larger deletion (Sakaguchi et al., 1996). Moreover, PrP may be necessary for synaptic function and sleep and rhythm regulation, because long-term potentiation was impaired (Collinge et al., 1994; Manson et al., 1995; Whittington et al., 1995), the period of circadian motor activity was more stabilized, and sleep fragmentation and the response to sleep deprivation (SDEP) were more prominent in PrP-null mice (Tobler et al., 1996).
Our purpose was to compare in detail sleep in the hybrid mouse strain used for the deletion, with the PrP-deficient mice. Because SDEP has served to uncover differences in sleep regulation between species (Tobler, 1995), the effects of SDEP on the two genotypes were compared.
MATERIALS AND METHODS
Animals. Adult mice devoid of prion protein (Prn-p0/0) (Büeler et al., 1992) and wild-type controls (Prn-p+/+; inbred strains C57BL-6J/129SV), mean age 80.6 ± 4.3 d (SEM) and weighing 26.69 ± 0.93 gm at the onset of recordings, served as experimental animals. Both genotypes had a genetic background derived from two inbred mouse strains, 129/SV (ev) backcrossed with C57BL/6J (Bühler et al., 1992). In addition, three mice transgenic for the PrP protein (TG) were recorded. For their generation, a PrP minigene comprising 5.5 kb of the PrP promotor, exon 1, intron 1, exon 2, exon 3, and 2.3 kb of a 3′-nontranscribed sequence was constructed and introduced into the germline of a Prn-p+/0 mouse. Breeding with Prn-p0/0 mice gave rise to transgenic mice that were TG+/0;Prn-p0/0 (line20) (Fischer et al., 1996). The mice were individually kept in Macrolon cages (36 × 20 × 35 cm) and placed in sound-attenuated chambers. Food and water were available ad libitum. The animals were maintained under a 12 hr light/dark cycle (light from 10:00 A.M. to 10:00 P.M.; daylight-type fluorescent tubes, 18 W, ∼150 lux 5 cm above the floor level). The animals were adapted to these conditions for a minimum of 3 weeks before the experiment. Mean values of ambient temperature continuously recorded for 4 sec epochs in two of the four boxes was in the range of 21.8–22.4°C during the three recording days.
Surgery. EEG and EMG electrodes and an epidural thermistor were implanted under deep anesthesia (50 mg/kg pentobarbital sodium, i.p.). Two gold-plated, round-tipped miniature screws (φ = 1.19 mm) served as EEG electrodes and were screwed through the skull onto the dura over the right cortex (2.0 mm lateral to the midline, 3.5 mm posterior to bregma) and the cerebellum (at the midline, 1.5 mm posterior to lambda). Two entwined wires (φ = 0.2 mm) were inserted into the neck muscle tissue to record the EMG. A thermistor (Thermometrics, P20, R(25°C) = 1 kΩ,ICONST. = 1 mA, maximum φ = 0.5 mm) inserted between the skull and dura through a hole in the skull over the left frontal cortex (the tip lying over the occipital cortex) was used to measure cortical temperature (TCRT). The electrodes and the thermistor were anchored to the skull with dental cement.
Experimental protocol and data acquisition. The mice were connected to counterbalanced recording leads for habituation at least 8 d before the experiment. Three consecutive 24 hr recordings of the EEG, EMG, and TCRT were obtained. The first 2 d served as baseline (1 Prn-p+/+ mouse was recorded for 1 baseline only), and on day 3 the mice were subjected to 6 hr SDEP and recorded for the remaining 18 hr. Sleep deprivation began at light onset and was attained by disturbing the animals acoustically and by introducing objects into the cage whenever the animals looked drowsy, attempted to engage in a sleeping posture, or the EEG showed signs of low-frequency activity. The EEG and EMG signals were amplified (amplification factor ∼ 2000), conditioned by analog filters (high-pass filter: −3 dB at 0.016 Hz; low-pass filter: −3 dB at 40 Hz; less than −35 dB at 128 Hz), sampled with 256 Hz, digitally filtered (EEG: low-pass filter FIR filter 25 Hz; EMG: band-pass FIR filter 20–50 Hz), stored with a resolution of 128 Hz, and displayed on a PC monitor. Subsequently, EEG power spectra were computed for consecutive 4 sec epochs by an FFT routine within the frequency range of 0.25–25.0 Hz (between 0.25 and 5.0 Hz, the values were collapsed into 0.5 Hz bins, between 5.25 and 25.0 Hz into 1 Hz bins). The amplified EMG signal was AD-converted, full-wave-rectified, and integrated over 4 sec epochs. TCRT and the ambient temperature inside the chambers were recorded at 4 sec intervals. All data were recorded simultaneously and stored on optical disks. Before the beginning of the recordings, a calibration signal (10 Hz sine wave, 300 μVPP) was recorded on the EEG channels.
Vigilance states and analysis. The vigilance states were determined as follows. For each epoch, the EEG power density in the delta (0.75–4.0 Hz) and theta band (6.25–9.0 Hz), the integrated EMG value, and TCRT were displayed on a PC monitor. Three vigilance states—(1) waking (high EMG and low EEG amplitude and high theta activity concomitant with highest EMG values), (2) NREM sleep (low EMG and high EEG amplitude, high delta activity), and (3) REM sleep (low EMG and low EEG amplitude, high theta activity)—were determined for 4 sec and the scores were entered into the PC via the keyboard. Epochs the vigilance state of which could not be identified, and epochs containing two different vigilance states within a 4 sec epoch, were given the score of the predominant state but not used for spectral analysis (0.18 ± 0.05%). Epochs containing EEG artifacts were marked and omitted from further analysis of the power spectra (6.55 ± 0.91% of total recording time). The vigilance states were expressed as a percentage of artifact-free recording time.
To allow the comparison of the duration and frequency of episodes with other rodents, the same minimal interruption criteria were applied as used previously for the rat and Djungarian hamster (Franken et al., 1991; Deboer et al., 1994). The algorithm is based on the frequency and the duration of episode interruptions.
Differences between the baselines and between baseline 2 and recovery from SDEP were compared by t tests. Overall effects on the 2 hr mean values within a genotype were analyzed by two-way ANOVA with the factors Time (2 hr intervals) and Condition (baseline 2 vs recovery). Whenever significant effects were present, contrasts were assessed by t tests. The 2 hr spectral values of the recovery day were expressed for each individual relative to the corresponding 2 hr reference value of baseline 2, and statistics were performed after log transformation. Comparisons between Prn-p+/+ and Prn-p0/0 were performed bypost hoc t tests and Bonferroni corrections for multiple tests if significance was reached in an ANOVA for the factor Genotype or interaction Genotype × Interval. To avoid multiple tests, the 2 hr intervals and the 30 frequency bins were comprised to 6 or 12 hr values and three frequency bands, respectively.
RESULTS
Baseline vigilance states, TCRT, and slow-wave activity
All mice exhibited a marked diurnal preference for sleep (Figs.1, 2; Table 1), and no significant difference was found between baseline 1 and 2 within either genotype. The mice spent ∼70% of the light period in sleep, and a considerable amount occurred also in the dark period (30%). The Prn-p0/0 mice did not differ from the wild-type in the 24 hr values of the baseline vigilance states (Table 1, Fig. 2), with the exception of a higher value for REM sleep per total sleep time (TST) in the Prn-p0/0 mice (Table 1). A difference in the distribution of waking and NREM sleep was apparent only in the second half of the dark period, in which waking was more prominent and NREM sleep lower in the Prn-p0/0 mice in both baselines (Fig. 2, Table 2). REM sleep was more abundant in the first half of the dark period in the null mice. In all mice, REMS/TST exhibited a minor, significant increase in the course of the light period (data not shown).
Fig. 1.
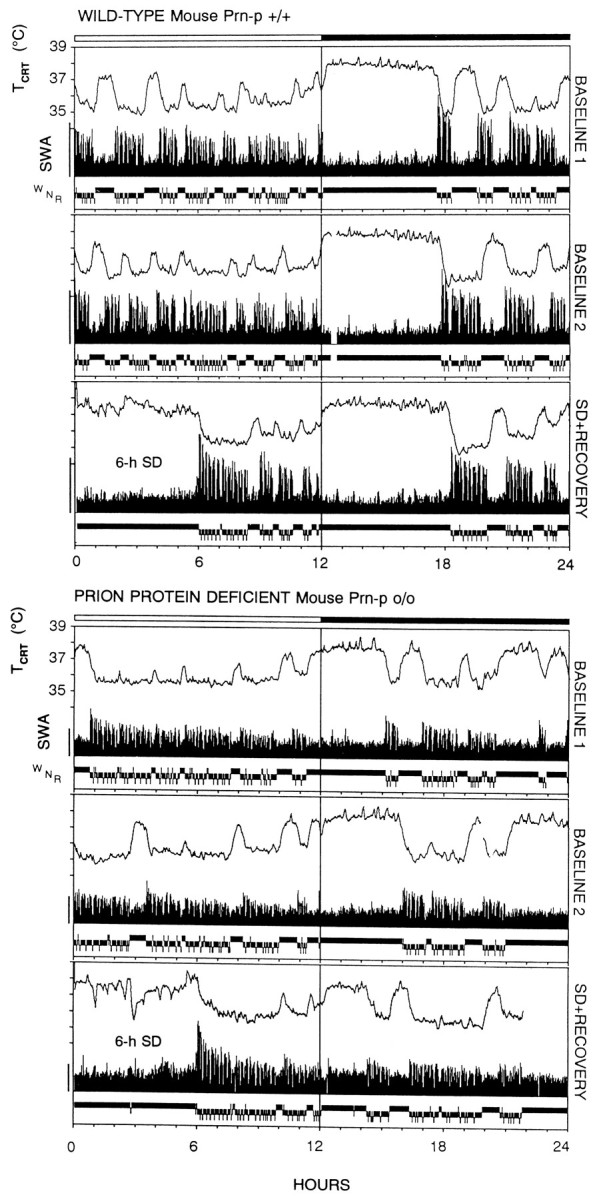
Individual 24 hr sleep and brain temperature records of the two genotypes. Individual 3 d records (baseline 1, baseline 2, and day 3 consisting of sleep deprivation and recovery) of a wild-type (Prn-p+/+) and a prion protein-deficient mouse (Prn-p0/0). Cortical temperature (TCRT), slow-wave activity (SWA; EEG power density 0.75–4.0 Hz) in NREM sleep (N), and the vigilance states waking (W), N, and REM sleep (R). The bars at the top mark the 12 hr light/dark cycle. The calibration mark on theleft corresponds to 50 μV2.
Fig. 2.

Distribution of the vigilance states and brain temperature across the two baselines. Vigilance states (W, waking; N, NREM sleep; R, REM sleep), slow-wave activity (SWA; power density 0.75–4.0 Hz inN), and cortical temperature (TCRT) for the 48 hr baseline recording in Prn-p+/+ and Prn-p0/0 mice. Mean 2 hr values ± 2 SEM, n = 8 for each strain (1 Prn-p+/+ mouse contributed with 1 baseline). ANOVA factor “genotype” (Prn-p+/+ vs Prn-p0/0) was not significant for all 12 and 24 hr variables.
Table 1.
Vigilance states in the two baselines
| WAKING | NREMS | REMS | TST | REMS/TST | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Light | Dark | Light | Dark | Light | Dark | Light | Dark | Light | Dark | |
| Prn-p+/+ | ||||||||||
| BL1 | 33.6 | 64.3 | 54.5 | 30.8 | 11.8 | 4.9 | 66.4 | 35.7 | 17.8 | 13.8 |
| (2.1) | (2.7) | (1.9) | (2.5) | (0.9) | (0.5) | (2.1) | (2.8) | (1.2) | (1.2) | |
| BL2 | 31.3 | 64.4 | 57.3 | 30.8 | 11.4 | 4.8 | 68.7 | 35.6 | 16.5 | 13.9 |
| (1.4) | (3.8) | (1.6) | (3.5) | (0.9) | (0.5) | (1.4) | (3.8) | (1.3) | (1.3) | |
| Prn-p0/0 | ||||||||||
| BL1 | 32.3 | 66.6 | 54.4 | 27.5 | 13.3 | 5.9 | 67.7 | 33.4 | 19.6 | 17.8* |
| (2.2) | (1.7) | (1.8) | (1.5) | (0.7) | (0.4) | (2.2) | (1.7) | (0.8) | (1.3) | |
| BL2 | 32.1 | 65.0 | 55.0 | 29.3 | 13.0 | 5.7 | 67.9 | 35.0 | 19.1 | 16.6 |
| (2.6) | (2.4) | (2.1) | (2.3) | (0.7) | (0.4) | (2.6) | (2.4) | (0.7) | (1.5) | |
Values are means ± SEM for the 12 hr intervals of the light and dark period; n = 8 for each strain (except for BL1 wild-type, where n = 7). BL1, BL2, Baselines 1 and 2; REMS, rapid eye movement sleep; NREMS, non-rapid eye movement sleep. REMS is also expressed as a percentage of total sleep time (REMS/TST). Comparisons BL1 vs BL2 within each genotype, and Prn-p+/+vs Prn-p0/0, were not significant for all variables, except for REMS/TST in BL1, dark (
p < 0.04 Prn-p+/+vs Prn-p0/0; two-tailed t test). ANOVA factor “LD” within each genotype, p < 0.02 for each vigilance state.
Table 2.
Vigilance states and cortical temperature for 6 h intervals
| WAKING | NREMS | REMS | REMS/TST | TCRT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Light | Dark | Light | Dark | Light | Dark | Light | Dark | Light | Dark | ||
| Prn-p+/+ | |||||||||||
| Hours | |||||||||||
| BL2 | 0 –6 | 28.7 | 77.2 | 60.3 | 20.9 | 11.0 | 2.5 | 15.4 | 11.8 | 35.3 | 36.9 |
| (2.1) | (7.0) | (2.0) | (6.3) | (1.9) | (0.0) | (1.2) | (1.3) | (0.2) | (0.3) | ||
| 7 –12 | 33.9 | 52.0 | 54.4 | 41.0 | 11.7 | 7.0 | 17.7 | 14.3 | 35.5 | 35.9 | |
| (1.0) | (2.9) | (1.5) | (2.3) | (1.0) | (0.9) | (1.5) | (1.4) | (0.2) | (0.3) | ||
| SD+REC | 0 –6 | 97.0 | 64.0 | 3.0 | 30.6 | 0.0 | 5.5 | 0.0 | 13.1 | 37.22-a | 36.6 |
| (0.9) | (8.4) | (0.9) | (7.2) | (0.0) | (1.4) | (0.0) | (2.4) | (0.2) | (0.3) | ||
| 7 –12 | 25.0 | 45.0 | 62.2 | 46.6 | 12.7 | 8.4 | 17.0 | 15.2 | 35.5 | 35.8 | |
| (1.7) | (1.9) | (1.6) | (1.3) | (1.1) | (0.7) | (1.4) | (0.9) | (0.2) | (0.2) | ||
| Prn-p0/0 | |||||||||||
| Hours | |||||||||||
| BL2 | 0 –6 | 25.9 | 66.1 | 60.6 | 28.7 | 13.5 | 5.3b | 18.2 | 15.6 | 35.4 | 36.8 |
| (3.2) | (4.1) | (2.6) | (3.6) | (0.8) | (1.0) | (0.6) | (2.3) | (0.1) | (0.1) | ||
| 7 –12 | 38.4 | 63.9b | 49.3 | 30.0b | 12.4 | 6.1 | 19.9 | 16.7 | 35.8 | 36.7b | |
| (4.1) | (4.3) | (3.2) | (3.5) | (1.1) | (1.0) | (1.2) | (1.5) | (0.1) | (0.1) | ||
| SD+REC | 0 –6 | 97.6 | 58.1 | 2.4 | 34.2 | 0.0 | 7.7 | 0.0 | 18.8 | 37.42-a | 36.6 |
| (0.5) | (4.1) | (0.5) | (3.4) | (0.0) | (0.9) | (0.0) | (1.3) | (0.1) | (0.1) | ||
| 7 –12 | 23.1 | 54.7 | 61.4 | 37.6 | 15.52-a | 7.7 | 20.32-a | 17.0 | 35.8 | 36.5 | |
| (1.9) | (3.9) | (1.8) | (3.1) | (0.4) | (0.9) | (0.6) | (0.8) | (0.1) | (0.1) | ||
Values are means ± SEM expressed as percentage of recording time; n = 8. Baseline 2 (BL2), sleep deprivation (SDEP), recovery (REC), REMS, rapid eye movement sleep; NREMS, non-rapid eye movement sleep. REMS, REM sleep is also expressed as a percentage of total sleep time (REMS/TST).
p < 0.04 BL2 vs recovery within a genotype. bp < 0.04, Prn-p+/+ vs Prn-p0/0 (two-tailed ttest).
Slow-wave activity (SWA) encompasses the frequency band between 0.75 and 4.0 Hz and is computed within NREM sleep, because in this vigilance state slow waves are most prominent in mammals (Tobler, 1995). SWA in NREM sleep decreased progressively in the course of the 12 hr light period and increased in the dark, where most values were higher than in the light (Fig. 2). In the wild-type mice, a decrease of SWA began already in the second half of the dark period, reflecting the differences in waking and NREM sleep between the genotypes in this interval. The larger amount of NREM sleep in the wild-type mice allowed an earlier dissipation of SWA, compared to the null mice, which were awake more.
TCRT largely reflected the time course of waking, reaching its peak value at dark onset (Figs. 1, 2; Table 2). The only difference in TCRT between the genotypes was a significantly higher TCRT in the second half of the dark period in the null mice, which corresponds to their larger amount of waking in that interval (Table 2). The difference in TCRT between the genotypes remained when TCRT was determined for waking alone (p < 0.05, two-tailed t test), indicating that the null mice may also have been more active during this interval.
Because we had found previously a marked difference between the genotypes in the amount of brief awakenings (Tobler et al., 1996), we analyzed this aspect of sleep in more detail (Fig. 3, Table 3). The 12 hr values revealed that within both the light and the dark periods of the baselines waking episodes shorter than 4, 8, and 16 sec invariably were significantly more prominent in the null mice. The episode histogram of the frequency of the three vigilance states illustrates that the Prn-p+/+ mice exhibited less brief waking episodes lasting 4 sec and less brief NREM sleep episodes of 32–124 sec, concomitant with a higher amount of longer NREM sleep episodes. The analysis of the 24 hr time course of brief waking episodes < 16 sec showed that the brief awakenings were consistently more abundant in the Prn-p0/0 than in the Prn-p+/+ mice throughout the entire recording (data not shown). Although there were no LD differences in episode duration, the frequency of NREM sleep and REM sleep episodes was significantly lower in the dark period compared with the light period in both genotypes. However, there were differences between the genotypes. Thus, REM sleep episodes were more abundant (episodes per hr: Prn-p0/0, 4.3 ± 0.6; Prn-p+/+, 2.6 ± 0.5;p < 0.05, two-tailed t test) and REM sleep and NREM sleep episode duration was shorter in the dark period in the Prn-p0/0 mice (REM sleep: Prn-p0/0, 0.9 ± 0.1 min; Prn-p+/+, 1.2 ± 0.1 min; NREM sleep: 5.8 ± 0.5 min, 7.6 ± 0.6 min, respectively;p < 0.05, two-tailed t test).
Fig. 3.
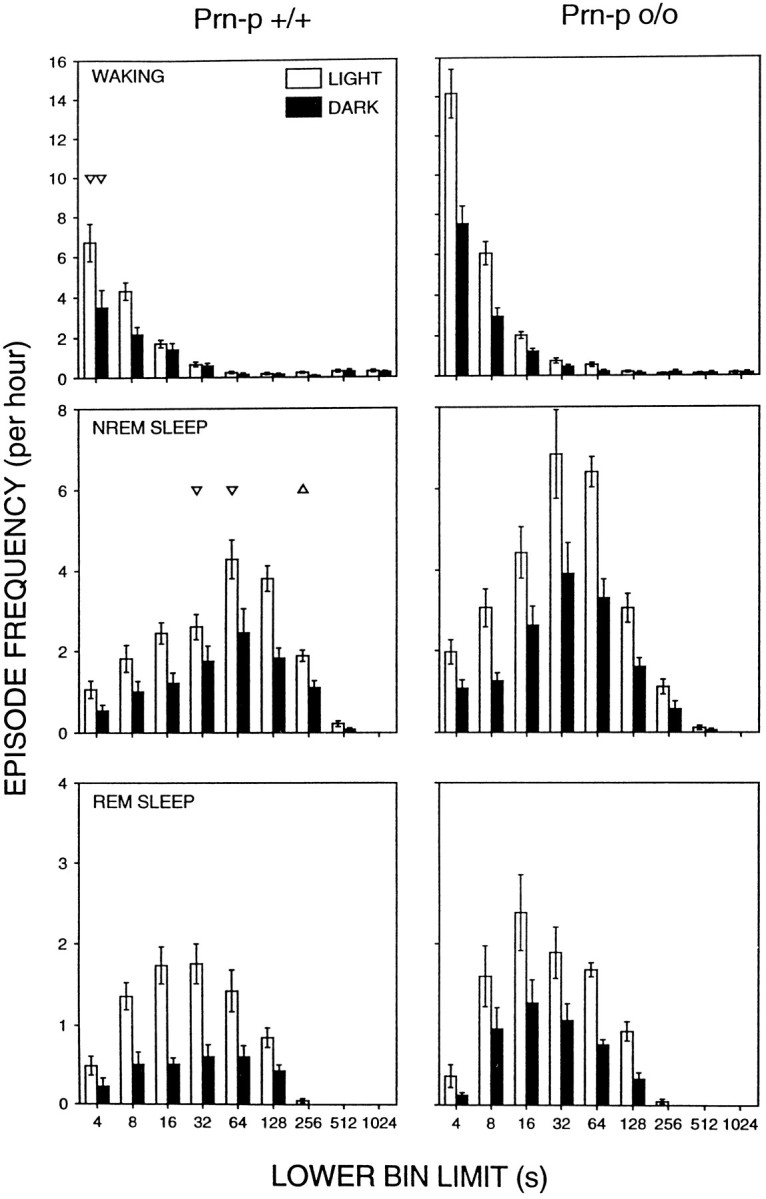
Episode frequency histogram of the three vigilance states. Waking, NREM sleep, and REM sleep episodes during the light (open bars) and dark period (black bars) of baseline 2 for the Prn-p+/+ and Prn-p0/0 mice. Time bins are shown with logarithmically increasing size. The inclusive range of eight consecutive bins was: 4, 8–12, 16–28, 32–60, 64–124, 128–252, 512–1020, and 1024–2048 sec. Bars represent means ± SEM of 8 animals. The abscissae denote lower bin limits.Triangles indicate significant differences between the two genotypes (p < 0.005, two-tailed ttest after Bonferroni correction). Orientation of triangles indicates the direction of deviation.
Table 3.
Brief awakenings as a measure of sleep fragmentation
| BASELINE 1 | BASELINE 2 | RECOVERY | ||||
|---|---|---|---|---|---|---|
| Light | Dark | Light | Dark | Light | Dark | |
| Shorter than 16 sec | ||||||
| Prn-p+/+ | 21.0 | 19.0 | 17.5 | 18.1 | 16.7 | 17.4 |
| (0.8) | (1.9) | (1.5) | (2.6) | (2.7) | (1.6) | |
| Prn-p0/0 | 35.23-150 | 35.13-150 | 33.13-150 | 33.83-150 | 30.4 | 32.5 |
| (4.9) | (4.9) | (4.0) | (4.2) | (3.1) | (3.5) | |
| Shorter than 8 sec | ||||||
| Prn-p+/+ | 17.0 | 15.0 | 14.4 | 14.5 | 13.9 | 13.8 |
| (1.1) | (1.7) | (1.4) | (2.4) | (2.4) | (1.6) | |
| Prn-p0/0 | 31.53-150 | 30.83-150 | 29.03-150 | 29.63-150 | 26.3 | 28.7 |
| (4.7) | (4.5) | (3.8) | (4.0) | (2.9) | (3.3) | |
| Shorter than 4 sec | ||||||
| Prn-p+/+ | 11.9 | 10.5 | 10.0 | 10.0 | 9.7 | 9.3 |
| (1.1) | (1.5) | (1.3) | (2.0) | (1.9) | (1.5) | |
| Prn-p0/0 | 24.73-150 | 24.13-150 | 22.23-150 | 23.13-150 | 20.1 | 22.7 |
| (3.8) | (3.5) | (2.6) | (2.9) | (2.2) | (2.6) | |
Brief awakenings are expressed per hour of total sleep time. Values are 12 hr means ± SEM (n = 8, except baseline, 1 Prn-p+/+, n = 7). No significant differences between the light and the dark period (baseline 1 and 2) and between baseline 2 and recovery after 6 hr sleep deprivation were present. Comparison of baselines between Prn-p+/+ and Prn-p0/0,
F3-150: p < 0.02, two-tailed ttest.
Time course of SWA within NREM sleep episodes
The time course of SWA within NREM sleep episodes may reflect the dynamics underlying the generation of SWA. If the normal prion protein were involved in the thalamocortical generation of SWA, the null mice would be expected to attain less SWA than the wild-type mice or to reach the maximum more slowly. For the analysis of SWA within NREM sleep, all NREM sleep episodes > 4 min within baseline 2 were pooled for each mouse. Statistical comparisons were performed on the mean values of the last minute before the waking-to-NREM sleep transition and for 4 min after the transition. The null mice reached the highest SWA value significantly faster than the wild-type mice (Fig. 4). Thus, the time course of the SWA increase differed significantly between the genotypes for the first minute after the transition, whereas they did not differ in the minute before the transition. Moreover, when SWA was expressed as percentage of the first 4 sec NREM sleep epoch after the transition within each genotype, the SWA values attained after 4 min were as follows: 151.0 ± 5.5% for Prn-p+/+ and 101.3 ± 3.7% for Prn-p0/0 (p < 0.05, two-tailedt test).
Fig. 4.

Time course of slow-wave activity within NREM sleep episodes. SWA, Mean EEG power density in the 0.75–4.0 Hz band. All NREM sleep episodes of the light period of baseline 2 lasting at least 4 min were pooled for each mouse. The curves connect mean 4 sec bins for 1 min before and 4 min after the transition from waking to NREM sleep separately for each genotype (n = 8 mice per genotype). The curves are expressed as a percentage of the 24 hr baseline value for each genotype. The genotypes differed significantly in the first 1 min interval after the transition (p = 0.004, two-tailed t test), whereas the 1 min interval before the transition was not significant (p = 0.083).
Baseline EEG power spectra
The absolute EEG power density values (0.25–25.0 Hz) computed for each of the three vigilance states (pooled values of the two baselines) did not differ significantly between the two genotypes (data not shown). Clear differences were seen in the EEG power spectra between the three vigilance states within each genotype (Fig.5). Thus, NREM sleep exhibited higher values than both REM sleep and waking in the 0.5–4.0 Hz band, whereas REM sleep values in the theta band (6.25–9.0 Hz) were above those in NREM sleep and waking in both phenotypes. Moreover, NREM sleep power density was above the values of both waking and REM sleep in a broad frequency band (11.25–25 Hz) in the null mice, whereas this difference was not significant in the wild-type.
Fig. 5.

Spectral distribution of EEG power density in the three vigilance states. Waking (W), NREM sleep (N), and REM sleep (R) for Prn-p+/+ and Prn-p0/0 mice computed for pooled 24 hr values of Baseline 1 and 2. The curves represent logarithmic mean values of absolute power densities (log μV2/0.25 Hz,n = 8 for each genotype, except n = 7 for Baseline 1, Prn-p+/+). Lines below the abscissa indicate frequency bands that differ significantly between two vigilance states (p < 0.05, two-tailedt test).
The dynamics of the EEG power spectrum in NREM sleep in the course of the light period is illustrated in a broad frequency range (0.5–25 Hz) for consecutive 2 hr intervals. EEG power density showed a decreasing trend in the low frequencies (0.25–4.5 Hz) in the null mice, whereas only one bin in this range reached significance in the wild-type (Fig.7, top; ANOVA factor “2 hr interval”). In contrast, most frequencies above ∼7 Hz increased progressively in the course of the light period in both genotypes, reaching a maximum in the last 2 hr interval of the light period.
Fig. 7.

Time course of EEG power density in NREM sleep.Top, Light period of baseline 2 (six 2 hr intervals;1–6). Bottom, Light period after sleep deprivation (three 2 hr intervals; 1–3) for the Prn-p+/+ and Prn-p0/0 mice. The curves connect geometric means of relative EEG power density for consecutive 2 hr intervals (n = 8 for each genotype). Values are plotted at the upper limit of each bin. The baseline data are expressed relative to the first 2 hr interval of the light period (=100%). Consecutive 2 hr intervals of recovery are expressed relative to the first three consecutive 2 hr intervals of the light period of Baseline 2 (=100%). Lines below the abscissa indicate frequency bands that differed significantly from 100% (p< 0.05, top, one-way ANOVA “2 hr intervals”;bottom, frequency bands: 0.5–4.0, 6.25–9.0, 11.25–25 Hz; two-tailed t test).
Effects of sleep deprivation
No difference was found in the effect of SDEP on the vigilance states between the wild-type and null mice. After SDEP, NREM sleep showed a minor, significant increase in the first 2 hr in the wild-type, but the 6 hr interval did not differ between the genotypes (Fig. 6, Table 2). Also, SWA was significantly enhanced above baseline and decreased progressively, but the magnitude of the increase differed significantly between Prn-p+/+ and Prn-p0/0 (ANOVA “genotype” for the three 6 hr recovery intervals vs baseline 2; p < 0.02). The increase of SWA in all three 6 hr recovery intervals was significantly larger in the null mice (p < 0.04, two-tailedt test). These differences were not attributable to differences between the genotypes during the deprivation because the amount of NREM sleep the mice obtained during the deprivation was similar (Prn-p+/+: 3.0 ± 0.9%; Prn-p0/0: 2.4 ± 0.5%). Furthermore, the minimum criterion of 6 min NREM sleep per 2 hr interval of SDEP to compute SWA reliably was not attained by either genotype.
Fig. 6.

Effects of sleep deprivation on sleep and brain temperature. Vigilance states, slow-wave activity (SWA; mean EEG power density in the 0.75–4.0 Hz band), and cortical temperature (TCRT) for the Prn-p+/+ and Prn-p0/0 mice. The curves represent 2 hr mean values ± 2 SEM (n = 8 for each genotype) for Baseline 2 (dashed lines), 6 hr sleep deprivation, and recovery (solid lines). SWA is expressed as 95% of mean 24 hr value (=100%). Triangles indicate significant differences within a genotype between recovery and baseline (p < 0.05; two-tailed t test, after Bonferroni correction). For the vigilance states and SWA, the 2 hr recovery intervals of the light period were compared with consecutive intervals of baseline beginning with lights on. For TCRT, all intervals were compared with baseline intervals corresponding to the time of day.
TCRT was above baseline during the SDEP in both genotypes, and in the Prn-p0/0 it remained above baseline in the first 2 hr recovery interval (Table 2, Fig. 6).
In the first 2 hr after SDEP, a significant increase of EEG power density was observed in the three frequency bands—delta, beta, and sigma—in both genotypes (Fig. 7). In the wild-type, a gradual decrease and a narrowing of the frequencies in the low-frequency range were observed in the course of the recovery, whereas in the null mice the highest values in the low frequencies were present in interval 1, followed by a decrease from interval 1 to 2. In interval 3, the values were still above baseline in the low- and high-frequency band in both genotypes.
Transgenic mice
The differences between the PrP-null mice and wild-type mice could indicate a specific role for the prion protein in sleep, or the differences may stem from the different genetic background of the mice (Gerlai, 1996). The recording of transgenic mice, in which the prion gene has been reintroduced into the genome of null mice and thereby expression of prion protein is reestablished, allowed us to investigate whether the reintroduction of PrP would also restore the sleep characteristics of the wild-type mice. The results show that the transgenic mice were more similar to the wild-type than to the null mice both in sleep fragmentation and in the response to SDEP. Thus, the number of brief awakenings (<16 sec) per hr total sleep time in the transgenic mice was as follows: 12 hr baseline, 17.6 ± 6.1; 6 hr recovery, 19.5 ± 8.3 (PrP+/+: 17.5 ± 1.5 and 16.7 ± 2.7; PrP0/0: 33.1 ± 4.0 and 30.4 ± 3.1). SWA was increased by 9.9 ± 10.2% in the first 2 hr interval after SDEP compared to the first 2 hr interval of baseline. The corresponding values were as follows: Prn-p+/+, 23.9 ± 4.3%; Prn-p0/0, 41.1 ± 5.7%. The mean values ± SEM of NREM sleep and REM sleep for the 12 hr light period in the transgenic mice were 42.0 ± 2.2% and 12.6 ± 1.0% of recording time, respectively (n = 3). The amount of NREM sleep during the 6 hr recovery interval was increased (50.9 ± 1.5%) compared to the corresponding 6 hr baseline value (37.0 ± 2.3%).
DISCUSSION
The comparison of knockout mice lacking the prion protein with the corresponding wild-type controls showed a remarkably similar daily amount of NREM sleep and REM sleep that was comparable to previous studies (Mitler et al., 1973, 1977; Oliverio and Malorni, 1979; Faradji et al., 1980; Ibuka et al., 1980; Dazuta et al., 1983; Richardson et al., 1985; Edgar et al., 1991; Fang et al., 1995, 1996). The genotypes did differ in the distribution of NREM sleep in the dark period, which was reflected also in the time course of waking and inTCRT. The use of a running wheel increased total sleep time and enhanced the amplitude of the circadian sleep–wake rhythm (Welsh et al., 1988), but our mice were not provided with running wheels; thus, the nocturnal sleep patterns were not a consequence of different running wheel activity.
Mice are similar to other rodents in that they exhibit little quiet wakefulness, and the activity during waking results in highTCRT. Thus, the curves for waking andTCRT were similar, as has been observed in the rat and the Djungarian hamster (Franken et al., 1992; Deboer et al., 1994).
A remarkable difference between the two genotypes was the larger sleep fragmentation encountered in the PrP0/0 mice. The 4 sec episodes included events that were equal to or shorter than 4 sec. Such events were not necessarily accompanied by a behavioral change, and their significance is unknown. Their occurrence was almost double in the null mice irrespective of their duration (4–16 sec), reflecting a diminished capacity to sustain NREM sleep but not REM sleep. The build-up of SWA within a NREM sleep episode was not affected by an interruption of NREM sleep by a waking episode < 8 sec in the rat (Trachsel et al., 1988). Moreover, the number of brief awakenings correlated negatively with the amount of SWA, reflecting a behavioral measure of sleep intensity (rat: Franken et al., 1991; Tobler et al., 1994; guinea pig: Tobler and Franken, 1993). The build-up of SWA within NREM sleep was unhindered by the larger amount of brief awakenings in the null mice, because it was faster than in the wild-type.
The analysis of the EEG power spectrum revealed a remarkable difference between the mice and other mammals in the lower frequencies. The relative contribution of slow waves to the spectrum, and especially to NREM sleep, is much smaller compared to other species (rat:Borbély et al., 1984; Djungarian hamster: Deboer et al., 1994; Syrian hamster: Tobler and Jaggi, 1987; rabbit: Tobler et al., 1990), which makes it more difficult to distinguish the vigilance states. Slow waves and sleep spindles are closely related to cellular changes in the thalamic and cortical neurons (Steriade et al., 1993, 1994), but it is not known which cellular characteristics are responsible for the changes in the amount or amplitude of slow waves. It is possible that the cortical layer of the mice lacks the capacity to produce a substantial amount of slow waves. This notion is confirmed by the relatively similar, low values of absolute EEG power in mice and the Djungarian hamster, another small rodent (Deboer and Tobler, 1996), compared to the rat. However, despite this similarity, SWA in the Djungarian hamster (Deboer et al., 1994) and the Syrian hamster (Tobler and Jaggi, 1987) is more prominent than in the mice in all vigilance states and is in this respect more similar to the rat (Borbély et al., 1984). The most distinctive feature discriminating NREM sleep from REM sleep was the prominent theta band peak in REM sleep, which was absent in NREM sleep.
Overall, the differences in the EEG power spectra between the vigilance states were similar in the null mice and the wild-type. Because mice strains differ in the occurrence of spindles in NREM sleep (Valatx et al., 1972; Valatx and Bugat, 1974), and spindles are more prominent in a frontal or sensorimotor derivation than in the parietal or occipital region (rat: Terrier and Gottesmann, 1978; cat: Ursin and Sterman, 1981), comparisons of the EEG spectra derived from different electrode localizations are necessary to resolve the problem whether spindle activity is affected by the absence of PrP.
There is a large body of literature demonstrating that sleep is regulated as a function of prior wakefulness (human: Borbély et al., 1981; Achermann et al., 1993; Borbély 1994; rat: Tobler and Borbély, 1986, 1990). In particular, SWA is used as an index for sleep intensity (Borbély et al., 1981; Franken et al., 1991). This is supported by the higher arousal threshold when SWA is high in the rat (Nekelmann and Ursin, 1993). SWA closely reflected the previous sleep–waking history in both mouse genotypes. Because the mice differed in the distribution of sleep and waking in the dark period, it was not surprising that SWA also differed in its time course: the earlier SWA decrease in the dark period in the wild-type mice was attributable to the earlier occurrence of sleep in the later part of the dark period, which allowed the dissipation of SWA. This difference was seen also in the more prominent decrease of EEG power density in the lower frequencies in the null mice. Furthermore, the effects of 6 hr SDEP were evident in the increase of SWA at the beginning of recovery sleep in both genotypes. Thus, also in mice sleep appears to be regulated as a function of prior wakefulness.
A remarkable difference was the larger increase of SWA after SDEP in the Prn-p0/0 mice than in the wild-type mice. It is consistent with the more prominent sleep fragmentation encountered in the Prn-p0/0 mice. We suggest the following explanation: the null mice have a low sleep pressure, which leads to (1) more frequent interruptions of sleep, and (2) low amounts of SWA. This interpretation is supported by the lower amount of SWA reached within NREM sleep episodes in the null mice. As a consequence, the propensity for SWA can increase more during SDEP in these mice. This notion is consistent with the findings in humans, in which habitual long sleepers displayed a larger increase in SWA after SDEP than habitual short sleepers (Aeschbach et al., 1996). It was concluded that the short sleepers live at a higher SWA pressure and, therefore, that SDEP cannot enhance SWA to the same extent as in the long sleepers. Because TST in the mice was not different, it is possible that cellular mechanisms responsible for the increase of SWA are affected by the lack of PrP. The absence of PrP, therefore, seems to affect sleep via two mechanisms that may be interrelated: (1) a decrease in sleep pressure, and (2) a diminished capacity to sustain NREM sleep. In addition, the faster increase of SWA at the transitions from waking to NREM sleep in the null mice may reflect a difference in the underlying cellular mechanisms leading to this transition because of the lack of PrP.
The SWA increase after SDEP was relatively small in both genotypes (20 and 40%). To investigate whether the larger response to SDEP was merely a consequence of the larger amount of waking preceding the SDEP in the Prn-p0/0 mice, correlations of the initial SWA increase after SDEP with the amount of wakefulness in the second half of the dark period preceding SDEP were computed. No difference in the correlation coefficients was found between the genotypes (data not shown). Furthermore, the initial SWA values before the SDEP did not differ. This raises the question why 6 hr of wakefulness did not elicit a similar degree of compensation as in other rodents. The NREM sleep and REM sleep increase does indicate that sleep pressure was enhanced by the deprivation. Moreover, the progressive decrease and the narrowing of the lower-frequency band are indications for a recovery process. It should be noted that the mice, in contrast to all other rodents previously recorded, can exhibit spontaneous bouts of waking (even in the absence of running wheels) that may last up to 6 hr. It is intriguing that such a small species has the capacity to remain spontaneously awake for so long. Thus, it appears that the relatively small increase of SWA after SDEP is a reflection of limitations at the cellular level. Studies prolonging SDEP are needed to clarify further the SWA dynamics in mice.
Thus, several differences were found between the genotypes that may be related to the absence of PrP. It is important to note that only a fine analysis of the vigilance states and the imposition of an SDEP revealed differences between the genotypes that otherwise may have gone unnoticed. The transgenic mice were more similar to the wild-type than to the null mice in sleep fragmentation and in the response to SDEP, indicating that the phenotype was rescued by the insertion of PrP in null mice. More extensive studies in transgenic mice and in other genotypes are needed to investigate whether genes adjacent to the PrP gene exerted a modulating effect.
The prion protein is a well conserved protein present in many mammalian species (Westaway and Prusiner, 1986), with a large degree of homology between species (Krakauer et al., 1996); therefore, it may have a very basic biological function. Sleep and circadian rhythms are extremely basic functions of all mammals. The detailed analysis of sleep in the two mice genotypes supports our previous notion that the prion protein may be involved not only in the regulation of circadian motor activity but also in the maintenance of sleep continuity and its regulation after sleep deprivation (Tobler et al., 1996).
Footnotes
This study was supported by Swiss National Science Foundation Grants 31.32574.91 and 3100-042500.94. The mice were obtained from Prof. Ch. Weissmann. We thank S. Gaus, F. Morgenthaler, and B. Schwierin for technical assistance and scoring, and Dr. P. Achermann and Prof. A. A. Borbély for critical comments on this manuscript.
Correspondence should be addressed to Prof. Irene Tobler, Institute of Pharmacology, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland.
REFERENCES
- 1.Achermann P, Dijk DJ, Brunner DP, Borbély AA. A model of human sleep homeostasis based on EEG slow-wave activity: quantitative comparison of data and simulations. Brain Res Bull. 1993;31:97–113. doi: 10.1016/0361-9230(93)90016-5. [DOI] [PubMed] [Google Scholar]
- 2.Aguzzi A, Weissmann C. Sleepless in Bologna: transmission of fatal familial insomnia. Trends Microbiol. 1996;4:129–131. doi: 10.1016/0966-842x(96)30012-7. [DOI] [PubMed] [Google Scholar]
- 3.Aeschbach D, Cajochen C, Landolt HP, Borbély AA. Homeostatic sleep regulation in habitual short sleepers and long sleepers. Am J Physiol. 1996;270:41–53. doi: 10.1152/ajpregu.1996.270.1.R41. [DOI] [PubMed] [Google Scholar]
- 4.Borbély AA. Sleep homeostasis and models of sleep regulation. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine, 2nd Ed. Saunders; Philadelphia: 1994. pp. 309–320. [Google Scholar]
- 5.Borbély AA, Baumann F, Brandeis D, Strauch I, Lehmann D. Sleep deprivation: effect on sleep stages and EEG power density in man. Electroencephalogr Clin Neurophysiol. 1981;51:483–493. doi: 10.1016/0013-4694(81)90225-x. [DOI] [PubMed] [Google Scholar]
- 6.Borbély AA, Tobler I, Hanagasioglu M. Effect of sleep deprivation on sleep and EEG power spectra in the rat. Behav Brain Res. 1984;14:171–182. doi: 10.1016/0166-4328(84)90186-4. [DOI] [PubMed] [Google Scholar]
- 7.Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 8.Capecchi MR. Targeted gene replacement. Sci Am. 1994;100:34–41. doi: 10.1038/scientificamerican0394-52. [DOI] [PubMed] [Google Scholar]
- 9.Cespuglio R, Musolino R, Debilly G, Jouvet M, Valatx JL. Organisation des mouvements oculaires rapides du sommeil paradoxal chez deux souches consanguines de Souris. CR Acad Sci (Paris) 1975;280:2681–2684. [PubMed] [Google Scholar]
- 10.Clinton J, Forsyth C, Royston MC, Roberts GW. Synaptic degeneration is the primary neuropathological feature in prion disease: a preliminary study. NeuroReport. 1993;4:65–68. doi: 10.1097/00001756-199301000-00017. [DOI] [PubMed] [Google Scholar]
- 11.Collinge J, Whittington MA, Sidle KCL, Smith CJ, Palmer MS, Clarke AR, Jefferys JGR. Prion protein is necessary for normal synaptic function. Nature. 1994;370:295–297. doi: 10.1038/370295a0. [DOI] [PubMed] [Google Scholar]
- 12.Collinge J, Palmer MS, Sidle KC, Gowland I, Medori R, Ironside J, Lantos P. Transmission of fatal familial insomnia to laboratory animals. Lancet. 1995;346:569–570. doi: 10.1016/s0140-6736(95)91405-6. [DOI] [PubMed] [Google Scholar]
- 13.Crawley JN. Unusual behavioral phenotypes of inbred mouse strains. Trends Neurosci. 1996;19:181–182. doi: 10.1016/s0166-2236(96)20021-9. [DOI] [PubMed] [Google Scholar]
- 14.Dazuta A, Gambarelli F, Ternaux JP. Sleep variations in C57BL and BALBc mice from 3 weeks to 14 weeks of age. Dev Brain Res. 1983;7:87–96. doi: 10.1016/0165-3806(83)90084-6. [DOI] [PubMed] [Google Scholar]
- 15.Deboer T, Tobler I. Shortening of the photoperiod affects sleep distribution, EEG and cortical temperature in the Djungarian hamster. J Comp Physiol [A] 1996;179:483–492. doi: 10.1007/BF00192315. [DOI] [PubMed] [Google Scholar]
- 16.Deboer T, Franken P, Tobler I. Sleep and cortical temperature in the Djungarian hamster under baseline conditions and after sleep deprivation. J Comp Physiol [A] 1994;174:145–155. doi: 10.1007/BF00193782. [DOI] [PubMed] [Google Scholar]
- 17.Edgar DM, Kilduff TS, Martin CE, Dement WC. Influence of running wheel activity on free-running sleep/wake and drinking circadian rhythms in mice. Physiol Behav. 1991;50:373–378. doi: 10.1016/0031-9384(91)90080-8. [DOI] [PubMed] [Google Scholar]
- 18.Estibeiro JP. Multiple roles for PrP in the prion diseases. Trends Neurosci. 1996;19:257–258. doi: 10.1016/S0166-2236(96)30010-6. [DOI] [PubMed] [Google Scholar]
- 19.Fang J, Sanborn CK, Renegar KR, Majde J, Krueger J. Influenza viral infections enhance sleep in mice (43945). PSEMB. 1995;210:242–252. doi: 10.3181/00379727-210-43945. [DOI] [PubMed] [Google Scholar]
- 20.Fang J, Tooley D, Gatewood C, Renegar KB, Majde JA, Krueger JM. Differential effects of total and upper airway influenza viral infection on sleep in mice. Sleep. 1996;19:337–342. [PubMed] [Google Scholar]
- 21.Faradji H, Cespuglio R, Rondont G, Paut L, Jouvet M. Absence of light-dark entrainment on the sleep-waking cycle in mice with intact visual perception. Brain Res. 1980;202:41–49. [PubMed] [Google Scholar]
- 22.Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino terminal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 23.Franken P, Dijk DJ, Tobler I, Borbély AA. Sleep deprivation in the rat: effects of electroencephalogram power spectra, vigilance states, and cortical temperature. Am J Physiol. 1991;261:R198–R208. doi: 10.1152/ajpregu.1991.261.1.R198. [DOI] [PubMed] [Google Scholar]
- 24.Franken P, Tobler I, Borbély AA. Cortical temperature and EEG slow-wave activity in the rat: analysis of vigilance state related changes. Eur J Physiol. 1992;420:500–507. doi: 10.1007/BF00374625. [DOI] [PubMed] [Google Scholar]
- 25.Friedmann J. A diallel analysis of genetic underpinnings of mouse sleep. Physiol Behav. 1974;12:169–175. doi: 10.1016/0031-9384(74)90169-3. [DOI] [PubMed] [Google Scholar]
- 26.Gallassi R, Morreale A, Montagna P, Gambetti P, Lugaresi E. Fatal familial insomnia: neuropsychological study of a disease with thalamic degeneration. Cortex. 1992;28:175–187. doi: 10.1016/s0010-9452(13)80046-9. [DOI] [PubMed] [Google Scholar]
- 27.Gambetti P, Parchi P, Petersen RB, Chen SG, Lugaresi E. Fatal familial insomnia and familial Creuzfeldt-Jakob disease: clinical, pathological and molecular features. Brain Pathol. 1995;5:43–51. doi: 10.1111/j.1750-3639.1995.tb00576.x. [DOI] [PubMed] [Google Scholar]
- 28.Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 29.Goldfarb LG, Haltia M, Brown P, Nieto A, Kovanen J, McCombie WR, Trapp S, Gadjusek DC. New mutation in scrapie amyloid precursor gene (at codon 178) in Finnish Creuzfeldt-Jakob kindred. Lancet. 1991;337:425. doi: 10.1016/0140-6736(91)91198-4. [DOI] [PubMed] [Google Scholar]
- 30.Ibuka N, Nihonmatsu I, Sekiguchi S. Sleep-wakefulness rhythms in mice after suprachiasmatic nucleus lesions. Waking and Sleeping. 1980;4:167–173. [PubMed] [Google Scholar]
- 31.Krakauer DC. Phylogenesis of prion protein. Nature. 1996;25:675. doi: 10.1038/380675a0. [DOI] [PubMed] [Google Scholar]
- 32.Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A, Tinuper P, Zucconi M, Gambetti P. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med. 1986;315:997–1003. doi: 10.1056/NEJM198610163151605. [DOI] [PubMed] [Google Scholar]
- 33.Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- 34.Manson JC, Hope J, Clarke AR, Johnston A, Black C, MacLeod N. PrP Gene dosage and long term potentiation. Neurodegeneration. 1995;4:113–115. doi: 10.1006/neur.1995.0014. [DOI] [PubMed] [Google Scholar]
- 35.Medori R, Tritschler H-J, LeBlanc A, Villare F, Manetto V, Chen HY, Xue R, Leal S, Montagna P, Cortelli P, Tinuper P, Avoni P, Mochi M, Baruzzi A, Hauw JJ, Ott J, Lugaresi E, Autilio-Gambetti L, Gambetti P. Fatal familial insomnia, a prion disease with a mutation at Codon 178 of the prion protein gene. N Engl J Med. 1992;326:444–449. doi: 10.1056/NEJM199202133260704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitler MM, Cohen HB, Grattan J, Dominic J, Degushi T, Barchas JD, Dement WC, Kessler S. Sleep and serotonin in two strains of Mus musculus. Pharmacol Biochem Behav. 1973;1:501–507. doi: 10.1016/0091-3057(73)90073-7. [DOI] [PubMed] [Google Scholar]
- 37.Mitler MM, Lund R, Sokolove PG, Pittendrigh CS, Dement WC. Sleep and activity rhythms in mice: a description of circadian patterns and unexpected disruptions in sleep. Brain Res. 1977;131:129–145. doi: 10.1016/0006-8993(77)90033-6. [DOI] [PubMed] [Google Scholar]
- 38.Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J, Gray F, Cortelli P, Montagna P, Ghetti B, Goldfarb LG, Gadjusek DC, Lugaresi E, Gambetti P, Autilio-Gambetti L. Fatal Familial Insomnia and familial Creuzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci USA. 1994;91:2839–2842. doi: 10.1073/pnas.91.7.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montagna P, Cortelli P, Gambetti P, Lugaresi E. Fatal familial insomnia: sleep, neuroendocrine and vegetative alterations. Adv Neuroimmunol. 1995;5:13–21. doi: 10.1016/0960-5428(94)00042-m. [DOI] [PubMed] [Google Scholar]
- 40.Moser M, Colello RJ, Pott U, Oesch B. Developmental expression of the prion protein gene in glial cells. Neuron. 1995;14:509–517. doi: 10.1016/0896-6273(95)90307-0. [DOI] [PubMed] [Google Scholar]
- 41.Neckelmann D, Ursin R. Sleep stages and EEG power spectrum in relation to acoustical stimulus arousal threshold in the rat. Sleep. 1993;16:467–477. [PubMed] [Google Scholar]
- 42.Oliverio A, Malorni W. Wheel running and sleep in two strains of mice: plasticity and rigidity in the expression of circadian rhythmicity. Brain Res. 1979;163:121–133. doi: 10.1016/0006-8993(79)90156-2. [DOI] [PubMed] [Google Scholar]
- 43.Portaluppi F, Cortelli P, Avoni P, Vergnani L, Maltoni P, Pavani A, Sforza E, Degli Uberti EC, Gambetti P, Lugaresi E. Progressive disruption of the circadian rhythm of melatonin in fatal familial insomnia. J Clin Endocrinol Metab. 1994;78:1075–1078. doi: 10.1210/jcem.78.5.8175963. [DOI] [PubMed] [Google Scholar]
- 44.Portaluppi F, Cortelli P, Avoni P, Vergnani L, Maltoni P, Pavani A, Sforza E, Manfredini R, Montagna P, Roiter I, Gambetti P, Fersini C, Lugaresi E. Dissociated 24 hr patterns of somatotropin and prolactin in Fatal Familial Insomnia. Neuroendocrinol. 1995;61:731–737. doi: 10.1159/000126901. [DOI] [PubMed] [Google Scholar]
- 45.Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 46.Prusiner SB. Prion biology and diseases: laughing cannibals, mad cows, and scientific heresy. Med Res Rev. 1996;16:487–505. doi: 10.1002/(SICI)1098-1128(199609)16:5<487::AID-MED4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 47.Reder AT, Mednick AS, Brown P, Spire JP, Van Cauter E, Wollmann RL, Cervenàkova L, Goldfarb LG, Garay A, Ovsiew F, Gadjusek DC, Roos RP. Clinical and genetic studies of fatal familial insomnia. Neurology. 1995;45:1068–1075. doi: 10.1212/wnl.45.6.1068. [DOI] [PubMed] [Google Scholar]
- 48.Richardson GS, Moore-Ede M, Czeisler CA, Dement WC. Circadian rhythms of sleep and wakefulness in mice: analysis using long-term automated recording of sleep. Am J Physiol. 1985;248:R320–R330. doi: 10.1152/ajpregu.1985.248.3.R320. [DOI] [PubMed] [Google Scholar]
- 49.Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Kataoka Y, Houtani T, Shirabe S, Okada H, Hasegawa S, Miyamoto T, Noda T. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature. 1996;380:528–531. doi: 10.1038/380528a0. [DOI] [PubMed] [Google Scholar]
- 50.Sforza E, Montagna P, Tinuper P, Cortelli P, Avoni P, Ferrillo F, Petersen R, Gambetti P, Lugaresi E. Sleep-wake cycle abnormalities in fatal familial insomnia: evidence for the role of the thalamus in sleep regulation. EEG Clin Neurophysiol. 1995;94:398–405. doi: 10.1016/0013-4694(94)00318-f. [DOI] [PubMed] [Google Scholar]
- 51.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 52.Steriade M, Contreras D, Amzica F. Synchronized sleep oscillations and their paroxismal developments. Trends Neurosci. 1994;17:199–208. doi: 10.1016/0166-2236(94)90105-8. [DOI] [PubMed] [Google Scholar]
- 53.Terrier G, Gottesmann CL. Study of cortical spindles during sleep in the rat. Brain Res Bull. 1978;3:701–706. doi: 10.1016/0361-9230(78)90021-7. [DOI] [PubMed] [Google Scholar]
- 54.Tobler I. Is sleep fundamentally different between mammalian species? Behav Brain Res. 1995;69:35–41. doi: 10.1016/0166-4328(95)00025-o. [DOI] [PubMed] [Google Scholar]
- 55.Tobler I, Borbély AA. Sleep EEG in the rat as a function of prior waking. Electroencephalogr Clin Neurophysiol. 1986;64:74–76. doi: 10.1016/0013-4694(86)90044-1. [DOI] [PubMed] [Google Scholar]
- 56.Tobler I, Borbély AA. The effect of 3 hr and 6 hr sleep deprivation on sleep and EEG spectra of the rat. Behav Brain Res. 1990;36:73–78. doi: 10.1016/0166-4328(90)90161-7. [DOI] [PubMed] [Google Scholar]
- 57.Tobler I, Franken P. Sleep homeostasis in the guinea pig: similar response to sleep deprivation in the light and dark period. Neurosci Lett. 1993;164:105–108. doi: 10.1016/0304-3940(93)90868-l. [DOI] [PubMed] [Google Scholar]
- 58.Tobler I, Jaggi K. Sleep and EEG spectra in the Syrian hamster (Mesocricetus auratus) under baseline conditions and following sleep deprivation. J Comp Physiol [A] 1987;161:449–459. doi: 10.1007/BF00603970. [DOI] [PubMed] [Google Scholar]
- 59.Tobler I, Franken P, Scherschlicht R. Sleep and EEG spectra in the rabbit under baseline conditions and following sleep deprivation. Physiol Behav. 1990;48:121–129. doi: 10.1016/0031-9384(90)90272-6. [DOI] [PubMed] [Google Scholar]
- 60.Tobler I, Franken P, Alföldi P, Borbély AA. Room light impairs sleep in the albino rat. Behav Brain Res. 1994;63:205–211. doi: 10.1016/0166-4328(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 61.Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rülicke T, Moser M, Oesch B, McBride PA, Manson JC. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996;380:639–642. doi: 10.1038/380639a0. [DOI] [PubMed] [Google Scholar]
- 62.Trachsel L, Tobler I, Borbély AA. Electroencephalogram analysis of non-rapid eye movement sleep in rats. Am J Physiol. 1988;255:27–37. doi: 10.1152/ajpregu.1988.255.1.R27. [DOI] [PubMed] [Google Scholar]
- 63.Ursin R, Sterman MB. A manual for standardized scoring of sleep and waking states in the adult cat. Brain Information Service/Brain Research Institute; Los Angeles: 1981. [Google Scholar]
- 64.Valatx JL, Bugat R. Facteurs génétiques dans le déterminisme du cycle veille-sommeil chez la souris. Brain Res. 1974;69:315–330. doi: 10.1016/0006-8993(74)90009-2. [DOI] [PubMed] [Google Scholar]
- 65.Valatx JL, Bugat R, Jouvet M. Genetic studies of sleep in mice. Nature. 1972;238:226–227. doi: 10.1038/238226a0. [DOI] [PubMed] [Google Scholar]
- 66.Valatx JL, Cespuglio R, Paut L, Bailey DW. Etude génétique du sommeil paradoxal chez la souris. Waking and Sleeping. 1980;4:175–183. [PubMed] [Google Scholar]
- 67.Welsh DK, Richardson GS, Dement WC. Effect of running wheel availability on circadian patterns of sleep and wakefulness in mice. Physiol Behav. 1988;43:771–777. doi: 10.1016/0031-9384(88)90375-7. [DOI] [PubMed] [Google Scholar]
- 68.Westaway D, Prusiner SB. Conservation of the cellular gene encoding the scrapie prion protein. Nucleic Acids Res. 1986;14:2035–2044. doi: 10.1093/nar/14.5.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Whittington MA, Sidle KCL, Gowland L, Meads J, Hill AF, Palmer MS, Jefferys JGR, Collinge J. Rescue of neurophysiological phenotype seen in PrP-null mice by transgene encoding human prion protein. Nature Genet. 1995;9:197–201. doi: 10.1038/ng0295-197. [DOI] [PubMed] [Google Scholar]
- 70.Weissmann C. Molecular biology of transmissible spongiform encephalopathies. FEBS Lett. 1996;389:3–11. doi: 10.1016/0014-5793(96)00610-2. [DOI] [PubMed] [Google Scholar]