

Abstract
The prominent symptoms associated with central demyelinating diseases such as multiple sclerosis (MS) are primarily caused by conduction deficits in affected axons. The symptoms may go into remission, but the mechanisms underlying remissions are uncertain. One factor that could be important is the restoration of conduction to affected axons, but it is not known whether demyelinated central axons resemble their peripheral counterparts in being able to conduct in the absence of repair by remyelination. In the present study we have made intra-axonal recordings from central axons affected by a demyelinating lesion, and then the axons have been labeled ionophoretically to permit their subsequent identification. Ultrastructural examination of 23 labeled preparations has established that some segmentally demyelinated central axons can conduct, and that they can do so over continuous lengths of demyelination exceeding several internodes (2500 μm). Such segmentally demyelinated central axons were found to conduct with the anticipated reduction in velocity and a refractory period of transmission (RPT) as much as 34 times the value obtained from the nondemyelinated portion of the same axon; the RPT was typically prolonged to 2–5 times the normal value. We conclude that some segmentally demyelinated central axons can conduct, and we propose that the restoration of conduction to such axons is likely to contribute to the remissions commonly observed in diseases such as MS.
Keywords: demyelination, multiple sclerosis, axon, conduction properties, glia, ionophoresis
Multiple sclerosis (MS) is a disease of the CNS in which affected central axons often lose one or more internodes of their myelin sheath; the continuity of the axon through the lesion is frequently maintained, although degeneration becomes more prominent as the disease progresses (McDonald, 1994). The disease is associated with symptoms such as paralysis, blindness, and numbness, which can be explained by conduction block in the relevant pathways. Such symptoms may spontaneously go into remission; however, the mechanisms underlying such remissions are not well understood. It is now clear that some demyelinated lesions are partially repaired by remyelination and that this phenomenon can be quite common and extensive in early lesions (Prineas et al., 1993a). Remyelination is known to restore secure conduction to central (Smith et al., 1979,1981; Blight and Young, 1989; Felts and Smith, 1992; Honmou et al., 1996) and peripheral (Saida et al., 1980; Smith and Hall, 1980; Sedal et al., 1983; Shrager, 1988) demyelinated axons, and it is reasonable to believe that conduction in remyelinated central axons will contribute to remissions; however, even where remyelination occurs in MS, it may be temporary (Prineas et al., 1993b), and persistently demyelinated lesions are a common feature of the disease. It is known that some of these persistently demyelinated lesions are clinically silent, i.e., they do not produce symptoms (Ghatak et al., 1974; Phadke and Best, 1983; for review, see Matthews, 1991). Such silent lesions may sometimes be explained by the utilization or unmasking of alternative pathways that avoid the demyelinated site, but such a phenomenon cannot explain recovery when lesions occur in structures where no alternative pathway exists, such as within the optic nerves (Wisniewski et al., 1976). Such observations could be explained if the demyelinated axons were able to acquire the ability to conduct even if not repaired by remyelination, but experimental studies have so far failed to provide any conclusive direct evidence that conduction can occur in central demyelinated axons, although there is good evidence that conduction can occur in their peripheral counterparts (McDonald, 1963; Mayer and Denny-Brown, 1964; Rasminsky and Sears, 1972; Bostock and Sears, 1976, 1978; Smith and Hall, 1980; Smith et al., 1982;Shrager et al., 1987; Shrager and Rubinstein, 1990). In the CNS, the classic studies of McDonald and Sears (1969a,b, 1970a,b) established that some axons that passed through an experimental demyelinating lesion could conduct and did so with abnormal conduction properties [e.g., decreased conduction velocity and increased refractory period of transmission (RPT)], but it was not clear whether these axons were segmentally demyelinated. As the authors noted, axons affected by the lesion displayed an array of myelin abnormalities (such as paranodal demyelination, myelin thinning, and remyelination) in addition to segmental demyelination, and so the state of myelination of the conducting axons was uncertain. It was also possible that the observed conduction deficits were the direct effect of the diphtheria toxin (used to induce the demyelination) on otherwise normal axons. Several subsequent studies have similarly demonstrated that some central axons affected by demyelinating lesions were able to conduct (albeit insecurely) (Carroll et al., 1985; Kaji et al., 1988; Heininger et al., 1989; Black et al., 1991); however, as before, it was not possible to conclude with certainty that the observed conduction occurred in segmentally demyelinated axons. We have sought therefore to determine the conduction properties of single, central axons affected by an experimental demyelinating lesion and then to label these particular axons so that subsequent histological examination could reveal the extent of their myelin abnormality.
MATERIALS AND METHODS
Lesion induction. Experimental demyelinating lesions were induced in the dorsal columns of male Sprague Dawley rats (285–610 gm, mean 467 gm) as described previously (Felts and Smith, 1992). Briefly, under general anesthesia (2% halothane/30% N2O, balance O2) the spinal cord was exposed via a partial laminectomy at the T-11 vertebral level using sterile techniques. Two injections of ethidium bromide (EBr) (each of 0.5 μl of 0.5 mg/ml in saline) were made along a single track of a drawn-glass micropipette (tip lumen <10 μm) inserted into the dorsal column through a small hole in the dura. EBr is a nucleic acid chelating agent that kills those cells which have their nuclei in the vicinity of the injection (Yajima and Suzuki, 1979; Blakemore, 1982). Because the cell bodies of the dorsal column axons are remote from the site of injection, the axons are spared and undergo segmental demyelination attributable to the death of their surrounding glial cells.
Electrophysiological examination. After 17–37 d the rats were reanesthetized, a laminectomy was performed from vertebral levels T-7 to L-5, and the dura was opened longitudinally. The renal arteries, renal veins, internal carotid arteries, and jugular veins were occluded by electrocautery, and the animal was decapitated while under anesthesia (this precaution was to ensure that consciousness could not be regained during the following procedure). The vasculature was rinsed by perfusion via the left ventricle with warmed, oxygenated artificial cerebrospinal fluid (csf) that contained (in mm): NaCl 123, KCl 3, CaCl2 1.5, NaH2PO41.2, MgCl2 1, glucose 10, HEPES 10, pH 7.4. Such perfusion was continued during the rapid dissection of a 3-cm-length of dorsal column, centered around the lesion, which was then placed in a modified “interface” brain slice recording chamber (Digitimer Ltd., Wellwyn Garden City, UK). Great care was taken to minimize tension or torsion of the tissue during dissection and subsequent handling. The dorsal column was orientated across three lanes of warmed, oxygenated artificial csf, separated by thin wax walls; the walls included slots to enable the dorsal column to cross without distortion. The dorsal column was loosely sealed into the slots with petroleum jelly, such that the lesion was located entirely within the central lane. The temperature of the artificial csf flowing in each lane could be varied individually but was maintained at 35°C ± 0.3C using an electrical heater unless noted otherwise. The portion of the dorsal column in each outside lane rested across a pair of platinum–iridium wire electrodes (Fig. 1), which could be used either to apply electrical stimuli (DS2 isolated stimulator, Digitimer Ltd.) or to make recordings of the evoked compound action potential (CAP) via a high impedance AC amplifier (Digitimer Ltd.). During the first hour in the chamber the CAP evoked by supramaximal stimulation gradually increased in magnitude (5–100%) and then remained stable for the duration of the experiment (>6 hr). After the first hour, in some preparations the gross electrophysiological properties of the lesion were examined by determining the RPT spectrum (Smith, 1980) and the response of the CAP to cooling. In this latter test only the center lane of the chamber (containing the lesion) was cooled; the outside lanes (containing the stimulating and recording sites) were maintained at 35°C. Intra-axonal recordings were then made using micropipettes (average resistance 140 MΩ) filled with a filtered (0.45 μm) solution containing 10% (w/v) horseradish peroxidase (HRP) (Grade 1; Boehringer, Lewes, Sussex, UK), 0.25m KCl, and 0.05 m HEPES, pH 7.4, positioned ∼2 mm rostral to the visible rostral margin of the lesion (delineated by a more translucent appearance of the tissue). The micropipette was advanced into the dorsal column in 8 μm steps using a stepping microdrive with high acceleration (Nanostepper, Scientific Precision Instruments, Oppenheim, Germany). The micropipette was advanced until a stable intra-axonal record was obtained in which action potentials could be recorded in response to stimulation at either end of the dorsal column. Stimulation at the caudal stimulating site included the lesion in the conduction pathway, whereas stimulation at the rostral site excluded the lesion and provided a measure of the properties of the normal part of the same axon.
Fig. 1.

Diagram illustrating the intra-axonal electrophysiological and labeling techniques used. Starting at thebottom of the figure, the diagrams show the excision from the spinal cord of the dorsal columns containing the lesion, the arrangement of the two pairs of stimulating electrodes and the recording micropipette (which is shown inserted into an axon that is demyelinated in the lesion), and (top) the filled axon after ionophoresis of HRP. Note that the caudal pair of electrodes was also used for recording CAPs.
The RPT (McDonald and Sears, 1970a) was determined in both the normal and lesioned portions of the axon by stimulation at the rostral and caudal stimulating sites, respectively. The axon was considered to be potentially demyelinated if the RPT through the lesion was more than twice the RPT with rostral stimulation, and axons fulfilling this criterion were labeled by the ionophoresis of HRP using positive current pulses (1 Hz, 500 msec duration, 1–5 nA, 3.5–26 nA · min total current) applied to the recording pipette. It is known that not all dorsal column axons will ascend the length of the dorsal column, and so not all axons present at the recording site would be expected to ascend to the level of the rostral stimulating electrodes. Correspondingly, in a few axons no action potential could be evoked from the rostral site (i.e., the conduction pathway omitting the lesion), but these axons were sometimes accepted as probably demyelinated if the RPT through the lesion was >2.2 msec. In some preparations only a single axon was labeled to ensure the lack of any ambiguity over the correlation of the electrophysiological properties, with the particular axon found to be labeled on subsequent microscopic examination; however, because not every ionophoresis resulted in the recovery of a labeled axon at microscopy (recovery rate was ∼60%), in some preparations as many as five axons were ionophoresed,providing that each axon exhibited similar conduction properties. Typically, two to three axons were labeled in any one preparation.
Morphological examination. After the last ionophoresis the tissue remained in the chamber for 2–4 hr to allow the HRP to pass along the axon. The dorsal column was then fixed by immersion in 3% glutaraldehyde (in 0.1 m cacodylate buffer with 0.6 mm CaCl2, pH 7.4) for 30 min and stored overnight in buffer at 4°C. The dorsal column was then embedded in agar and cut longitudinally into sections 200 μm thick using a vibratome. The HRP was visualized by exposure of the sections to a preincubation solution containing Hanker–Yates reagent (Hanker et al., 1977) (Sigma, Poole, Dorset, UK; 0.5 mg/ml in 0.1 mcacodylate buffer) for 15 min, followed by a 15 min exposure to the same solution with the addition of 0.015% H2O2. If a stained axon was observed within the lesion on light microscopic examination, the lesion was excised, osmicated, dehydrated through ascending ethanols, passed through propylene oxide, and embedded in Epon 812 (Polysciences, Warrington, PA) using standard techniques. Serial transverse sections were taken through the lesion and mounted on glass slides, using a protocol of 10 sections 2 μm thick, followed by a single section 1 μm thick. The 1 μm sections were stained (0.5% azure II, 0.5% methylene blue in 0.5% borax) and coverslipped for light microscopic examination. If more than one labeled axon was present, the path of each axon within the tissue, across different tissue sections, was drawn using a camera lucida, so that each axon could be followed without ambiguity. Distances in the rostrocaudal plane were calculated from the numbers and thicknesses of intervening sections, as measured by the microtome (Ultracut S, Leica, Milton Keynes, UK). Regions where the axon appeared to be demyelinated were identified along the length of the axon using light microscopic examination of the 1 μm sections, and then the neighboring length of axon was examined by skip-serial investigation of the adjacent 2 μm sections. These sections were trimmed, reembedded in Epon, and cut for examination beneath the electron microscope (Hitachi H7000), without additional staining. The type of cellular contacts made with the demyelinated axon, and the surface area of the axon contacted, were then determined from morphometric examination of the electron micrographs.
RESULTS
Description of the lesion
The lesion was lentiform, with its long axis arranged rostrocaudally along the dorsal column for ∼3–5 mm. Although the sectioning technique prevented the determination of the transverse extent of the lesion in these animals, other animals receiving identical injections in our laboratory exhibited lesions that occupied an average of 45% of the transverse area of the whole dorsal columns. The lesions consisted mainly of demyelinated axons, but some axons were undergoing remyelination, which in this lesion is accomplished primarily by Schwann cells; the extent of remyelination increased with lesion age. The progression of the lesion is described in greater detail elsewhere (Blakemore, 1982; Felts and Smith, 1992).
Electrophysiological findings
CAP recordings
The CAPs recorded from dorsal columns containing demyelinated lesions were substantially smaller than those recorded either from dorsal columns with similar lesions that had been allowed to repair by remyelination (i.e., examined 10 months after lesion induction) (Fig.2) or from control (data not shown) dorsal columns. This decrease indicates a substantial degree of conduction block during the period of extensive demyelination. Cooling of the demyelinated region produced a substantial increase in the area of the CAP (Fig. 2A), in contrast to the only slight increase observed with cooling of a lesion repaired by remyelination (Fig. 2A). In addition to the presence of substantial numbers of fibers with blocking temperatures below 37°C, examination of the spectrum of RPTs of the constituent fibers revealed the presence of many fibers with long RPTs, as has been described previously as characteristic of conduction in axons passing through a demyelinated lesion (Fig. 2B). In a previous study, it was demonstrated that the range of RPTs of the remyelinated fibers (Fig.2B) is practically indistinguishable from that of nonlesioned control animals (Felts and Smith, 1992).
Fig. 2.

CAPs recorded from dorsal columns with either a predominantly demyelinated lesion [left(A) 21 d and (B) 28 d after EBr injection] or a remyelinated lesion (right, 300 d after EBr injection, bothtop and bottom illustrations are from the same preparation). Stimulus artifacts are indicated byarrows. In A, the segment of the dorsal columns containing the lesion was held at the temperatures indicated, whereas the stimulating and recording sites remained at 35°C. Note the large relative increase in the CAP after cooling of the demyelinated lesion. The range of RPTs of the fibers can be deduced from the records in B, which show the CAPs resulting from the second of two impulses separated by the interstimulus intervals indicated (the interval scale is not linear) (for further explanation, see Smith, 1980). The CAP resulting from the first stimulus has been subtracted from each trace, and the artifacts from the second stimulus have been aligned. Note the following: (1) many fibers in the demyelinated lesion exhibit a prolonged RPT, as illustrated by the rapid growth of the CAP further back in the array; (2) the CAP amplitude in the demyelinated lesion is smaller, indicating a degree of conduction block; and (3) there is an increased latency of most fibers in the demyelinated lesion.
Intra-axonal recordings: refractory period of transmission and conduction velocity
Recordings were made from many axons, but only a limited number fulfilled the criteria described in Materials and Methods (stable resting potential, ability to conduct through the lesion, prolonged RPT). The RPTs of the ionophoresed axons are presented graphically in Figure 3. This figure includes the RPTs only from the axons that were selected for ionophoresis, and only from animals in which subsequent morphological analysis revealed the presence of labeled, demyelinated axons. The RPT of the lesioned portion of the conduction pathway was on average 516 ± 707% (mean ± SD) of the RPT of the nonlesioned portion of the same axon. It should be noted, however, that much of the apparently large variation in RPT values derives from just one axon that had the unusually long RPT through the lesion of 27 msec (RPT in the unlesioned segment was 0.8 msec) (Fig. 3A). If this axon is excluded from the analysis, then the prolongation of the RPT was 357 ± 150%. In all cases, the RPT through the lesion was, of course, at least double the RPT excluding the lesion, because this was one of the criteria by which axons were selected for ionophoresis. There was no obvious relationship between the time after EBr injection and the relative increase in the RPT (Fig. 3B), although relatively young lesions (<20 d after EBr injection) had axons that all exhibited RPT differentials (RPT including the lesion/RPT excluding the lesion) well above the twofold criterion. Such elevated RPT differentials were still present in some axons at 32 d after injection (Fig.3B). RPTs recorded from axons contralateral to the lesion were within the normal range with both rostral and caudal stimulation (data not shown).
Fig. 3.

RPT values recorded intra-axonally from preparations with demyelinating lesions. Only recordings from dorsal columns in which at least one labeled axon segment was recovered are included. A scatter plot showing the RPTs of the conduction pathway through the lesion, plotted as a function of the RPT in the normal nonlesioned portion of the axon, is shown in A. Note the large difference in axis scales; the dotted linerepresents equal RPTs in both pathways. In B, the proportional increase in RPT through the lesion is plotted versus the time after EBr injection. Note the break in the y-axis in each graph.
It was apparent that the axons with prolonged RPTs through the lesion also had longer latencies of conduction through the lesion, when compared with their supposedly normal neighboring axons. The latency through the lesion was also often noticeably longer than the latency when conducting from the rostral stimulating site, excluding the lesion; however, the use of only a single stimulating site for each conduction pathway and the short conduction distances precluded a precise determination of the degree of reduction in conduction velocity.
Twenty-two percent (6 of 27) of the axons that were labeled by ionophoresis did not conduct action potentials from the rostral stimulating site (i.e., in the unlesioned segment), although they conducted through the lesion with abnormally long RPTs (>2.2 msec). The absence of conduction with rostral stimulation probably occurred in axons that left the dorsal column before reaching the rostral stimulating electrodes.
Morphological findings
On light microscopic examination, labeled axons were distinguished in the wet, vibratome sections by their appearance within the lesion as continuous thin lines of dark HRP reaction product (Fig.4A,B). Not surprisingly, individual labeled axons sometimes appeared to pass from one vibratome section to an adjacent section; however, even when only one axon was ionophoresed in a particular experiment, we did not assume that lengths of labeled axon in adjacent sections were contiguous. Rather, to ensure that our measurements of the length of continuous demyelination along the axon were not overestimates, we have treated the two lengths as separate, shorter specimens. Furthermore, to ensure confidence in the measurements, in a few axons in which labeling within a vibratome section was not both dense and continuous, we have considered the clearly labeled segments as being separate lengths of axons. With these restrictions, we recovered 23 labeled lengths of axon from 11 different experiments. Of these, 16 axon segments from seven animals were found to include lengths of continuous demyelination of >200 μm. Three axon segments were demyelinated for short lengths of only 60, 90 and 130 μm, and the remaining four axon segments contained only remyelinated internodes along the portion of their length that was labeled. These remyelinated axons will not be considered further. When labeled axons observed within the lesions in vibratome sections were followed rostrally to the unlesioned tissue, the continuous labeling was usually replaced by short (5–25 μm) labeled segments that were separated from each other by lengths similar to the presumed internodal spacing.
Fig. 4.

Light micrographs of labeled axons within the lesion. A, Low magnification view of a 200-μm-thick longitudinal section through the dorsal columns after exposure to Hanker–Yates reagent, viewed with transmitted light. The lesion, which is 23 d after EBr injection, is the translucent, lenticular region. The area indicated by the box contains a darkly labeled axon, which can be seen coursing through the section at higher magnification in B. In C, a 200-μm-thick vibratome section from a lesion 28 d after EBr injection has been embedded in plastic, sectioned in the transverse plane at 1 μm thickness, and stained (see Materials and Methods). This section contains a region of Schwann cell remyelination just beneath the pial surface (arrowheads); ventral to this region there is a large area of demyelinated axons that includes the labeled axon (arrow). The region including the demyelinated axon (arrow) is enlarged inD. This axon had an RPT of 1.4 msec in the portion of the axon excluding the lesion and 3.8 msec in the portion of the axon traversing the lesion. A portion of the labeled length of this axon is illustrated in Figure 7C. Scale bar (shown inC): A, 2000 μm; B, 160 μm; C, 70 μm; D, 20 μm.
When the labeled axons were embedded in plastic and viewed at higher resolution in 1 μm transverse sections, they were again clearly identified by their dark, intra-axonal reaction product (Fig.4C,D). Labeled axons that appeared on light microscopic examination under oil immersion to exhibit significant lengths of continuous demyelination in the 1 μm sections were also examined in the electron microscope in skip-serial ultrathin sections cut from the adjacent 2 μm sections. Initially, sections with long intervening distances were examined to allow confirmation of demyelination without the need for cutting excessive numbers of blocks. If these sections confirmed that the axon was demyelinated, then progressively shorter intervals were examined. Labeled axons were again distinguished by their dark reaction product, and it was clear that most of the labeled axons were cleanly demyelinated (Fig.5).
Fig. 5.
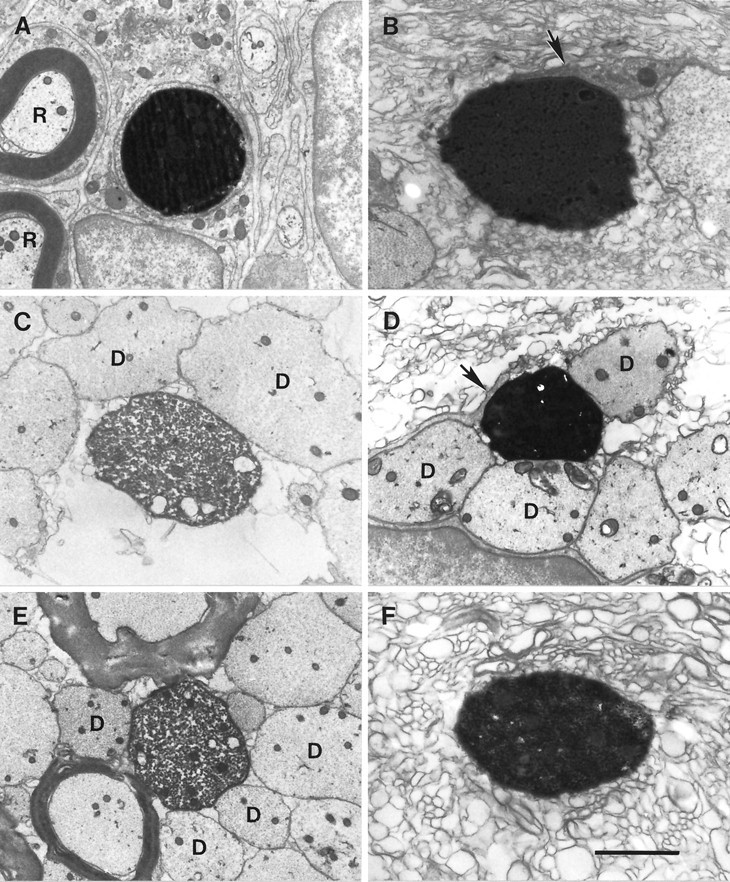
Electron micrographs of transverse sections through several labeled, demyelinated axons illustrating some of the various axonal environments observed. In each micrograph, the electron-dense reaction product within the axoplasm distinguishes the labeled axon. Labeled axons included examples in which (A) the axon was completely ensheathed by nonaxonal cellular processes [nearby are two central axons (R) remyelinated by Schwann cells]; (B) part of the circumference was adjacent to a nonaxonal cellular process (arrow); (C) part of the circumference was adjacent only to other demyelinated (D) axons; (D) portions of the circumference were adjacent to both a nonaxonal cellular process (arrow) and other demyelinated (D) axons; (E) portions of the circumference were adjacent to both demyelinated axons (D) and the myelin sheaths of nearby fibers; and (F) the axonal circumference was free of any cellular processes, but the axon was surrounded by vesicular myelin debris. The lesions are of the following ages: A, 27 d; B, D, andF, 21 d; C and E, 19 d. Scale bar (shown in F): A, C, D, E, 1.7 μm; B, F, 1 μm.
Of the 16 labeled axon segments that were determined at the light microscope level to have continuous lengths of demyelination >200 μm (average, 775 μm; range, 310–2450 μm), 12 were examined in ultrathin sections under the electron microscope at intervals of 200 μm or less. The longest continuous region of demyelination, the maximum and average intervals between ultrathin sections examined along the demyelinated axon, and the average axonal diameters for each of these axons are shown in Table 1. The axonal diameters (range, 1.6–3.8 μm) are substantially larger than those reported previously for unmyelinated axons in the dorsal columns (McNeill et al., 1988), and this establishes beyond a doubt that the filled axons were indeed demyelinated rather than unmyelinated.
Table 1.
Characteristics of filled axon segments
| Axon segment | Longest length of continuous demyelination (μm) | Maximum interval between EM sections (μm) | Average interval between EM sections (μm) | Average axonal diameter1-a (μm) |
|---|---|---|---|---|
| 1 | 2452 | 80 | 30 | 2.2 |
| 2 | 1325 | 200 | 102 | 2.0 |
| 3 | 890 | 190 | 110 | 2.3 |
| 4 | 750 | 140 | 108 | 2.1 |
| 5 | 740 | 100 | 62 | 3.8 |
| 6 | 670 | 130 | 83 | 3.3 |
| 7 | 480 | 100 | 69 | 2.2 |
| 8 | 380 | 130 | 127 | 2.7 |
| 9 | 350 | 90 | 69 | 2.3 |
| 10 | 340 | 60 | 38 | 2.5 |
| 11 | 315 | 130 | 105 | 1.6 |
| 12 | 310 | 50 | 34 | 2.8 |
The longest lengths of continuous demyelination observed in the filled, conducting axons are shown, along with the average axonal diameter. Only segments with >200 μm of continuous demyelination are included. Axon segments were observed in skip-serial sections under the electron microscope. The maximum and average distances between EM sections are also shown. EM, Electron microscopic.
Average of diameters measured near each end, and near the middle, of the demyelinated segment. Each measurement was the average of the maximum diameter and the diameter at right angles to it.
The perimeter of the demyelinated axons was either completely naked or in contact with different cellular elements for various extents of its circumference (Figs. 5A–E). Along much of their length, demyelinated axons exhibited either partial (Fig. 5B) or complete (Fig. 5A) ensheathment by cellular processes other than axons. The labeled axons were also frequently in contact with other demyelinated axons, either solely (Fig. 5C) or in combination with other cellular processes (Fig. 5D), or with the myelin sheaths of nearby axons (Fig. 5E). The nonaxonal cellular contact could usually be identified as emanating from astrocytes, oligodendrocytes, Schwann cells, or macrophages, but identification was not possible in all cases, because sometimes only a thin tongue of cytoplasm was observed (e.g., Fig. 5B). The profile of cellular contact for the segment with the longest continuous length of demyelination, including its contacts with other demyelinated axons, is presented in Figure 6. This axon also exhibited the longest continuous length lacking contact with any cellular processes other than demyelinated axons: 126 μm (from locations 1743 to 1869 μm in Fig. 6; observed ultrastructurally at intervals of 29 μm or less). Four other axonal segments exhibiting only contact with demyelinated axons for lengths ranging from 45 to 90 μm were also observed. Some labeled axonal segments were totally devoid of any cellular contacts, including contacts with other demyelinated axons, but they were only short in length (<60 μm; e.g., in Fig. 6, from locations 1278–1328 μm, with five sections observed over this length). These axons were surrounded by vesicular myelin debris (e.g., Fig. 5F).
Fig. 6.

Graph illustrating the axolemmal contacts made by a single labeled axon segment, chosen because it had the longest continuous length of demyelination (2452 μm). Thex-axis shows the distance along the axon; the first section in which HRP reaction product could be identified is defined as 0 μm. The y-axis shows the percentage of the circumference of the labeled axon that abuts either cellular processes or other demyelinated axons, or which lacks contact with cellular elements (although often contacted by vesicular myelin debris).
Seven of the 16 axon segments with demyelinated regions longer than 200 μm also exhibited regions of remyelination. Unlike the four axon segments mentioned above as being remyelinated over their entire labeled extent, these seven axons exhibited patchy remyelination, with intervening regions of demyelination. In several axons, these regions of remyelination were isolated short patches. For example, in one segment only a single region of remyelination was noted within the 1670 μm length of axonal labeling, and sections 150 and 120 μm on either side of the section containing remyelination exhibited no nonaxonal cellular contacts; thus the region of remyelination was limited to a length of <270 μm. In our conducting axons, no remyelinated segment with fewer than five lamellae of remyelination was observed. Typically the remyelinated segments exhibited ∼5–16 turns of compact myelin.
Morphological and electrophysiological correlates
All of the axons described above were capable of conducting single impulses through the lesion; this was one of the selection criteria for ionophoresis. Similarly, a prolonged RPT was used as an additional qualifying characteristic. For several axons, the RPT is either known, because only a single axon was ionophoresed, or is known to be in a very narrow range of values. The ensheathment characteristics and RPT values of three such axons are illustrated in Figure7.
Fig. 7.

Diagram illustrating the type of ensheathment surrounding the labeled portion of three axons with known RPTs; the RPTs through the lesion are shown below each axon. The type of ensheathment was measured from electron micrographs; theNaked category includes axons surrounded by vesicular myelin debris (e.g., Fig. 5F). Lesion ages wereA, 21 d; B, 19 d; andC, 28 d.
DISCUSSION
For the first time it has been possible to correlate the conduction properties of individual axons passing through a central demyelinated lesion with their precise morphological characteristics. Our results demonstrate conclusively that some central axons can conduct even if they are segmentally demyelinated for lengths of several internodes, and even if they are entirely devoid of glial contacts for lengths up to 126 μm. Conduction is with a reduced velocity and a prolonged RPT, as predicted from previous studies of axons passing through regions of central demyelination (McDonald and Sears, 1969a,b, 1970a,b; Smith et al., 1979, 1981; Kaji et al., 1988; Black et al., 1991; Felts and Smith, 1992), and in common with the conduction properties of peripheral demyelinated axons (McDonald, 1963; Mayer and Denny-Brown, 1964;Rasminsky and Sears, 1972; Bostock and Sears, 1976, 1978; Smith and Hall, 1980; Smith et al., 1982; Pender and Sears, 1984; Shrager et al., 1987; Pender, 1988; Shrager and Rubinstein, 1990).
It should be noted that although we have observed conduction in segmentally demyelinated central axons, it has not been possible from this study to conclude whether such conduction is a rare event or whether it is typical. We recorded from many axons that were unresponsive to stimulation caudal to the lesion, but it is possible that the normal course of these axons meant that they were not present at the stimulating site, and so it was not possible to conclude that conduction was blocked because of demyelination. Given the fact that such impalements were very common, however, it is our belief that conduction in segmentally demyelinated axons is likely to be the exception rather than the rule, at least in the EBr lesion at 17–37 d after injection.
Studies of conduction in demyelinated peripheral axons have demonstrated that in addition to continuous conduction (Bostock and Sears, 1976, 1978), conduction can also proceed by a microsaltatory (Bostock et al., 1980; Smith et al., 1982) method. This microsaltation occurs before remyelination via new regions of high inward current conductance that have been termed “phi-nodes” (Bostock et al., 1980; Smith et al., 1982). A determination of the precise mode of conduction in the demyelinated axons observed in the current experiments will require further study.
It is possible that the likelihood of conduction in any particular demyelinated central axon may correlate with the presence around it of ensheathing glial processes. This possibility is supported by the finding by Shrager and Rubinstein (1990) that demyelinated frog sciatic axons are only rarely capable of conduction before ensheathment by Schwann cells. In view of the known interactions between axons and glial cells, with respect to both the reestablishment of high densities of putative sodium channels (Blakemore and Smith, 1983; Rosenbluth et al., 1985) and improvements in axonal cable properties (Shrager and Rubinstein, 1990), it is probable that conduction may be more likely in demyelinated axons invested with glial processes. It is interesting in this respect that the axons in this study, with the exception of relatively short segments, have been at least partially ensheathed by cellular processes (Figs. 6, 7). It is notable, however, that conduction was also observed in fibers which for short lengths (<60 μm) were entirely without cellular contacts or were in contact only with other demyelinated axons (up to 126 μm in length). Thus continuous glial contact is not a prerequisite for successful conduction in demyelinated central axons. Whether conduction is favored in lesions in which axoglial contact is common warrants further investigation.
Although conduction was observed in axons that were thinly remyelinated, it was never observed in axons with internodes of fewer than five lamellae of compact myelin. This observation adds weight to previous theoretical studies (Koles and Rasminsky, 1972; Halter and Blight, 1996), which have suggested that very thin sheaths may preclude successful conduction. A preliminary examination of axons undergoing remyelination has revealed very few sheaths of only a few lamellae (our unpublished observations), and it may be that during myelinogenesis myelin compaction is delayed until a relatively thick sheath can be made quite rapidly. In this way the duration of any potential period of conduction block would be minimized.
The RPTs of the ionophoresed axons, as illustrated in Figure 3, were much longer in the lesioned than in the unlesioned portion of the axon, in agreement with previous studies (McDonald and Sears, 1970a,b;Rasminsky and Sears, 1972; Smith et al., 1979, 1981; Smith and Hall, 1980; Felts and Smith, 1992). It should be noted, however, that axons were selected for study on the basis of a prolonged RPT, and so any demyelinated axon with a short RPT (theoretically unlikely) will have been excluded. Four axon segments were recovered that exhibited remyelination along their entire labeled length, and yet they still had prolonged RPTs. Because axons were never labeled across the entire length of the lesion, however, and because complete remyelination of the central EBr lesion has been shown to restore the RPT to within normal limits (Felts and Smith, 1992), it seems likely that these axons may have been demyelinated along an unlabeled portion of their length.
The discontinuous labeling at regular intervals observed in the portion of the axons within the normally myelinated dorsal column adjacent to the lesions suggests that labeling occurred only at the nodes of Ranvier, and indeed in some cases nodes could be visualized at the site of labeling. This restricted labeling could have resulted from the HRP being present only at the node of Ranvier, or from restricted access of the Hanker–Yates reagent and/or H2O2 to the internode, presumably attributable to the myelin sheath. In an unrelated series of experiments we have found that myelinated axons labeled with HRP generally appear continuously labeled when the tissue is exposed to a solubilizing agent such as Triton X-100 before incubation in the chromogen solution, suggesting that in the present experiments the nodal labeling can be attributed to restricted access of the reagents.
Clinical implications
MS is characterized by the presence within the CNS of lesions containing many demyelinated axons (Charcot, 1877). The lesions are often several millimeters long, and so the axons must lose one or more internodes of myelin, i.e., they are often segmentally demyelinated. In experimental lesions, the onset of segmental demyelination is seemingly routinely accompanied by conduction block (attributable to an initial paucity of sodium channels along the newly exposed axolemma) (for review, see Waxman and Ritchie (1993), but the lesion in MS can sometimes become, or remain, asymptomatic (Ghatak et al., 1974; Phadke and Best, 1983). In some cases the asymptomatic nature of the lesion can be explained by its location in a clinically silent area of the brain, or in a location where function can be subserved by an alternative pathway, but these possibilities cannot always explain the presence of continued function. For example, alternative pathways cannot explain a patient who has vision despite demyelination affecting a substantial portion of the optic nerve (Wisniewski et al., 1976). In this case vision was abnormal; however, a number of studies have found lesions that produce conduction deficits such as reduced conduction velocity, without causing symptoms (Halliday et al., 1972; Hume and Waxman, 1988). Our observation that segmentally demyelinated central axons can conduct for distances equivalent to several internodal lengths, with variable degrees of conduction deficit, provides a sufficient explanation for both residual, abnormal, function, and clinically silent lesions that nevertheless exhibit conduction deficits.
Ultrastructural study of MS lesions has established that although the axons of some lesions in MS are embedded in a dense astroglial matrix (“closed lesions”), in other lesions the axons are largely free of glial contacts (“open lesions”) (Barnes et al., 1991). The open and closed lesions can be distinguished using magnetic resonance imaging (MRI) criteria, and it appears that the open type of lesion is quite common in MS (perhaps even more common than the closed type). Because there are good reasons to suppose that the axons in the open type of lesion may not be able to conduct (Blakemore and Smith, 1983;Rosenbluth et al., 1985), it follows that it may be possible using MRI criteria to distinguish lesions likely to be symptomatic from those likely to be asymptomatic. The present findings show, however, thatcontinuous glial ensheathment is not necessary for conduction and indeed that conduction is possible when at least 88% of the axonal surface area is free of glial contacts for at least 1608 μm (axon representing the region from 844 to 2452 μm on thex-axis in Fig. 4). It is therefore unlikely that MRI can be reliably used to distinguish symptomatic from asymptomatic lesions, even when the lesions are located in pathways where conduction block would be anticipated to cause symptoms.
Because we have shown that conduction can occur in central demyelinated axons with different types of ensheathment, it follows that conduction might be possible in most demyelinated axons in MS. On theoretical grounds a small axonal diameter should favor successful conduction in demyelinated axons (Bostock, 1994), but the conducting axons in this study were mostly of 2–3 μm diameter. Because axons are considered to shrink by 30–40% on demyelination (Prineas and McLeod, 1976), the normal axonal diameter of these axons would be ∼4–5.5 μm. It would be incorrect to interpret this finding as indicating that smaller diameter axons are unlikely to conduct because the intra-axonal recording techniques used in this study will have selected for larger axons, since these are more easily penetrated. The observation that axons of at least 5.5 μm in diameter can conduct when demyelinated, however, suggests that conduction should be possible in most axons affected by MS, because most myelinated human central axons are smaller than 5.5 μm in diameter. It does not necessarily follow that conduction would typically occur in most axons, because other factors may well impair conduction in MS lesions. Factors such as inflammation (McDonald, 1994; Redford et al., 1997) or an extracellular fluid that either contains putative conduction blocking factors (Brinkmeier et al., 1996) or is unregulated in composition because of defects in the blood–brain barrier may act to hold conduction in abeyance. Because conduction is possible in principle, attempts are warranted to promote conduction, perhaps by reducing inflammation (McDonald, 1994), removing deleterious factors (Brinkmeier et al., 1996) from the csf (perhaps by removing them from the serum), blocking potassium channels (Sherratt et al., 1980; Bostock et al., 1981), or promoting repair of the blood–brain barrier (Felts and Smith, 1996).
Footnotes
This work was supported by grants from the Multiple Sclerosis Society of Great Britain and Northern Ireland and Guy’s Special Trustees. P.A.F. was supported by a Hitchings–Elion Fellowship from the Wellcome Trust. We thank Ms. Sarah Fielding for her excellent technical assistance.
Correspondence should be addressed to Dr. Paul A. Felts, Department of Neurology, United Medical and Dental Schools, Guy’s Campus, London SE1 9RT UK.
REFERENCES
- 1.Barnes D, Munro PM, Youl BD, Prineas JW, McDonald WI. The longstanding MS lesion. A quantitative MRI and electron microscopic study. Brain. 1991;114:1271–1280. doi: 10.1093/brain/114.3.1271. [DOI] [PubMed] [Google Scholar]
- 2.Black JA, Felts P, Smith KJ, Kocsis JD, Waxman SG. Distribution of sodium channels in chronically demyelinated spinal cord axons: immuno-ultrastructural localization and electrophysiological observations. Brain Res. 1991;544:59–70. doi: 10.1016/0006-8993(91)90885-y. [DOI] [PubMed] [Google Scholar]
- 3.Blakemore WF. Ethidium bromide induced demyelination in the spinal cord of the cat. Neuropathol Appl Neurobiol. 1982;8:365–375. doi: 10.1111/j.1365-2990.1982.tb00305.x. [DOI] [PubMed] [Google Scholar]
- 4.Blakemore WF, Smith KJ. Node-like axonal specializations along demyelinated central nerve fibres: ultrastructural observations. Acta Neuropathologica. 1983;60:291–296. doi: 10.1007/BF00691879. [DOI] [PubMed] [Google Scholar]
- 5.Blight AR, Young W. Central axons in injured cat spinal cord recover electrophysiological function following remyelination by Schwann cells. J Neurol Sci. 1989;91:15–34. doi: 10.1016/0022-510x(89)90073-7. [DOI] [PubMed] [Google Scholar]
- 6.Bostock H. The pathophysiology of demyelination. In: Herndon RM, Seil FJ, editors. Multiple sclerosis: current status of research and treatment. Demos Publications; New York: 1994. pp. 89–112. [Google Scholar]
- 7.Bostock H, Sears TA. Continuous conduction in demyelinated mammalian nerve fibers. Nature. 1976;263:786–787. doi: 10.1038/263786a0. [DOI] [PubMed] [Google Scholar]
- 8.Bostock H, Sears TA. The internodal axon membrane: electrical excitability and continuous conduction in segmental demyelination. J Physiol (Lond) 1978;280:273–301. doi: 10.1113/jphysiol.1978.sp012384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bostock H, Hall SM, Smith KJ. Demyelinated axons can form “nodes” prior to remyelination. J Physiol (Lond) 1980;308:21–23. [Google Scholar]
- 10.Bostock H, Sears TA, Sherratt RM. The effects of 4-aminopyridine and tetraethylammonium ions on normal and demyelinated mammalian nerve fibres. J Physiol (Lond) 1981;313:301–315. doi: 10.1113/jphysiol.1981.sp013666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brinkmeier H, Seewald MJ, Wollinsky KH, Rudel R. On the nature of endogenous antiexcitatory factors in the cerebrospinal fluid of patients with demyelinating neurological disease. Muscle Nerve. 1996;19:54–62. doi: 10.1002/(SICI)1097-4598(199601)19:1<54::AID-MUS7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Carroll WM, Jennings AR, Mastaglia FL, Levick WR. Conduction in single nerve fibres in experimental demyelinative optic neuropathy. Electroencephalogr Clin Neurophysiol. 1985;61:S178. [Google Scholar]
- 13.Charcot JM. Lectures on the diseases of the nervous system. (English translation by G. Sigerson). New Sydenham Society; London: 1877. [Google Scholar]
- 14.Felts PA, Smith KJ. Conduction properties of central nerve fibers remyelinated by Schwann cells. Brain Res. 1992;574:178–192. doi: 10.1016/0006-8993(92)90815-q. [DOI] [PubMed] [Google Scholar]
- 15.Felts PA, Smith KJ. Blood-brain barrier permeability in astrocyte-free regions of the central nervous system remyelinated by Schwann cells. Neuroscience. 1996;75:643–655. doi: 10.1016/0306-4522(96)00282-5. [DOI] [PubMed] [Google Scholar]
- 16.Ghatak NR, Hirano A, Lijtmaer H, Zimmerman HM. Asymptomatic demyelinated plaque in the spinal cord. Arch Neurol. 1974;30:484–486. doi: 10.1001/archneur.1974.00490360060012. [DOI] [PubMed] [Google Scholar]
- 17.Halliday AM, McDonald WI, Mushin J. Delayed visual evoked response in optic neuritis. Lancet. 1972;1:982–985. doi: 10.1016/s0140-6736(72)91155-5. [DOI] [PubMed] [Google Scholar]
- 18.Halter JA, Blight AR. Pathophysiology of incomplete remyelination in the axon: a study using an electrodiffusion model. Soc Neurosci Abstr. 1996;22:776.9. [Google Scholar]
- 19.Hanker JS, Yates PE, Metz CB, Rustioni A. A new specific, sensitive and non-carcinogenic reagent for the demonstration of horseradish peroxidase. Histochem J. 1977;9:789–792. doi: 10.1007/BF01003075. [DOI] [PubMed] [Google Scholar]
- 20.Heininger K, Fierz W, Schafer B, Hartung HP, Wehling P, Toyka KV. Electrophysiological investigations in adoptively transferred experimental autoimmune encephalomyelitis in the Lewis rat. Brain. 1989;112:537–552. doi: 10.1093/brain/112.2.537. [DOI] [PubMed] [Google Scholar]
- 21.Honmou O, Felts PA, Waxman SG, Kocsis JD. Restoration of normal conduction properties in demyelinated spinal cord axons in the adult rat by transplantation of exogenous Schwann cells. J Neurosci. 1996;16:3199–3208. doi: 10.1523/JNEUROSCI.16-10-03199.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hume AL, Waxman SG. Evoked potentials in suspected multiple sclerosis: diagnostic value and prediction of clinical course. J Neurol Sci. 1988;83:191–210. doi: 10.1016/0022-510x(88)90068-8. [DOI] [PubMed] [Google Scholar]
- 23.Kaji R, Suzumura A, Sumner AJ. Physiological consequences of antiserum-mediated experimental demyelination in CNS. Brain. 1988;111:675–694. doi: 10.1093/brain/111.3.675. [DOI] [PubMed] [Google Scholar]
- 24.Koles AJ, Rasminsky M. A computer simulation of conduction in demyelinated nerve fibres. J Physiol (Lond) 1972;227:351–364. doi: 10.1113/jphysiol.1972.sp010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthews WB. Pathophysiology. In: Matthews WB, editor. McAlpine’s multiple sclerosis. Churchill-Livingstone; Edinburgh: 1991. pp. 231–250. [Google Scholar]
- 26.Mayer RF, Denny-Brown D. Conduction velocity in peripheral nerve during experimental demyelination in the cat. Neurol. 1964;14:714–726. doi: 10.1212/wnl.14.8_part_1.714. [DOI] [PubMed] [Google Scholar]
- 27.McDonald WI. The effects of experimental demyelination on conduction in peripheral nerve: a histological and electrophysiological study. II. Electrophysiological observations. Brain. 1963;86:501–524. doi: 10.1093/brain/86.3.501. [DOI] [PubMed] [Google Scholar]
- 28.McDonald WI. Rachelle Fishman-Matthew Moore Lecture. The pathological and clinical dynamics of multiple sclerosis. J Neuropathol Exp Neurol. 1994;53:338–343. doi: 10.1097/00005072-199407000-00003. [DOI] [PubMed] [Google Scholar]
- 29.McDonald WI, Sears TA. Effect of demyelination on conduction in the central nervous system. Nature. 1969a;221:182–183. doi: 10.1038/221182a0. [DOI] [PubMed] [Google Scholar]
- 30.McDonald WI, Sears TA. The effects of demyelination on conduction in the central nervous system. Trans Am Neurol Assoc. 1969b;94:168–173. [PubMed] [Google Scholar]
- 31.McDonald WI, Sears TA. The effects of experimental demyelination on conduction in the central nervous system. Brain. 1970a;93:583–598. doi: 10.1093/brain/93.3.583. [DOI] [PubMed] [Google Scholar]
- 32.McDonald WI, Sears TA. Effect of a demyelinating lesion on conduction in the central nervous system studied in single nerve fibres. J Physiol (Lond) 1970b;207:53P–54P. [PubMed] [Google Scholar]
- 33.McNeill DL, Chung K, Carlton SM, Coggeshall RE. Calcitonin gene-related peptide immunostained axons provide evidence for fine primary afferent fibers in the dorsal and dorsolateral funiculi of the rat spinal cord. J Comp Neurol. 1988;272:303–308. doi: 10.1002/cne.902720212. [DOI] [PubMed] [Google Scholar]
- 34.Pender MP. The pathophysiology of myelin basic protein-induced acute experimental allergic encephalomyelitis in the Lewis rat. J Neurol Sci. 1988;86:277–289. doi: 10.1016/0022-510x(88)90105-0. [DOI] [PubMed] [Google Scholar]
- 35.Pender MP, Sears TA. The pathophysiology of acute experimental allergic encephalomyelitis in the rabbit. Brain. 1984;107:699–726. doi: 10.1093/brain/107.3.699. [DOI] [PubMed] [Google Scholar]
- 36.Phadke JG, Best PV. Atypical and clinically silent multiple sclerosis: a report of 12 cases discovered unexpectedly at necropsy. J Neurol Neurosurg Psychiatry. 1983;46:414–420. doi: 10.1136/jnnp.46.5.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prineas JW, McLeod JG. Chronic relapsing polyneuritis. J Neurol Sci. 1976;27:427–458. doi: 10.1016/0022-510x(76)90213-6. [DOI] [PubMed] [Google Scholar]
- 38.Prineas JW, Barnard RO, Kwon EE, Sharer LR, Cho ES. Multiple sclerosis: remyelination of nascent lesions. Ann Neurol. 1993a;33:137–151. doi: 10.1002/ana.410330203. [DOI] [PubMed] [Google Scholar]
- 39.Prineas JW, Barnard RO, Revesz T, Kwon EE, Sharer L, Cho ES. Multiple sclerosis. Pathology of recurrent lesions. Brain. 1993b;116:681–693. doi: 10.1093/brain/116.3.681. [DOI] [PubMed] [Google Scholar]
- 40.Rasminsky M, Sears TA. Internodal conduction in undissected demyelinated nerve fibres. J Physiol (Lond) 1972;227:323–350. doi: 10.1113/jphysiol.1972.sp010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Redford EJ, Kapoor R, Smith KJ. Nitric oxide donors cause a reversible block of conduction in rat central and peripheral demyelinated axons. J Physiol (Lond) 1997;499P:13P. doi: 10.1093/brain/120.12.2149. [DOI] [PubMed] [Google Scholar]
- 42.Rosenbluth J, Tao-Cheng J-H, Blakemore WF. Dependence of axolemmal differentiation on contact with glial cells in chronically demyelinated lesions of cat spinal cord. Brain Res. 1985;358:287–302. doi: 10.1016/0006-8993(85)90973-4. [DOI] [PubMed] [Google Scholar]
- 43.Saida K, Sumner AJ, Saida T, Brown MJ, Silberberg DH. Antiserum-mediated demyelination: relationship between remyelination and functional recovery. Ann Neurol. 1980;8:12–24. doi: 10.1002/ana.410080103. [DOI] [PubMed] [Google Scholar]
- 44.Sedal L, Ghabriel MN, He F, Allt G, Le Quesne PM, Harrison MJG. A combined morphological and electrophysiological study of conduction block in peripheral nerve. J Neurol Sci. 1983;60:293–306. doi: 10.1016/0022-510x(83)90070-9. [DOI] [PubMed] [Google Scholar]
- 45.Sherratt RM, Bostock H, Sears TA. Effects of 4-aminopyridine on normal and demyelinated mammalian nerve fibres. Nature. 1980;283:570–572. doi: 10.1038/283570a0. [DOI] [PubMed] [Google Scholar]
- 46.Shrager P. Ionic channels and signal conduction in single remyelinating frog nerve fibres. J Physiol (Lond) 1988;404:695–712. doi: 10.1113/jphysiol.1988.sp017314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shrager P, Rubinstein CT. Optical measurement of conduction in single demyelinated axons. J Gen Physiol. 1990;95:867–890. doi: 10.1085/jgp.95.5.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shrager P, Chiu SY, Ritchie JM, Zecevic D, Cohen LB. Optical recording of action potential propagation in demyelinated frog nerve. Biophys J. 1987;51:351–355. doi: 10.1016/S0006-3495(87)83342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith KJ. A sensitive method for the detection and quantification of conduction deficits in nerve. J Neurol Sci. 1980;48:191–199. doi: 10.1016/0022-510x(80)90200-2. [DOI] [PubMed] [Google Scholar]
- 50.Smith KJ, Hall SM. Nerve conduction during peripheral demyelination and remyelination. J Neurol Sci. 1980;48:201–219. doi: 10.1016/0022-510x(80)90201-4. [DOI] [PubMed] [Google Scholar]
- 51.Smith KJ, Blakemore WF, McDonald WI. Central remyelination restores secure conduction. Nature. 1979;280:395–396. doi: 10.1038/280395a0. [DOI] [PubMed] [Google Scholar]
- 52.Smith KJ, Blakemore WF, McDonald WI. The restoration of conduction by central remyelination. Brain. 1981;104:383–404. doi: 10.1093/brain/104.2.383. [DOI] [PubMed] [Google Scholar]
- 53.Smith KJ, Bostock H, Hall SM. Saltatory conduction precedes remyelination in axons demyelinated with lysophosphatidyl choline. J Neurol Sci. 1982;54:13–31. doi: 10.1016/0022-510x(82)90215-5. [DOI] [PubMed] [Google Scholar]
- 54.Waxman SG, Ritchie JM. Molecular dissection of the myelinated axon. Ann Neurol. 1993;33:121–136. doi: 10.1002/ana.410330202. [DOI] [PubMed] [Google Scholar]
- 55.Wisniewski HM, Oppenheimer D, McDonald WI. Relation between myelination and function in MS and EAE. J Neuropathol Exp Neurol. 1976;35:327. [Google Scholar]
- 56.Yajima K, Suzuki K. Demyelination and remyelination in the rat central nervous system following ethidium bromide injection. Lab Invest. 1979;41:385–392. [PubMed] [Google Scholar]