

Abstract
To assess the contribution of PKA to injury-induced inflammation and pain, we evaluated nociceptive responses in mice that carry a null mutation in the gene that encodes the neuronal-specific isoform of the type I regulatory subunit (RIβ) of PKA. Acute pain indices did not differ in the RIβ PKA mutant mice compared with wild-type controls. However, tissue injury-evoked persistent pain behavior, inflammation of the hindpaw, and ipsilateral dorsal horn Fos immunoreactivity was significantly reduced in the mutant mice, as was plasma extravasation induced by intradermal injection of capsaicin into the paw. The enhanced thermal sensitivity observed in wild-type mice after intraplantar or intrathecal (spinal) administration of prostaglandin E2 was also reduced in mutant mice. In contrast, indices of pain behavior produced by nerve injury were not altered in the mutant mice. Thus, RIβ PKA is necessary for the full expression of tissue injury-evoked (nociceptive) pain but is not required for nerve injury-evoked (neuropathic) pain. Because the RIβ subunit is only present in the nervous system, including small diameter trkA receptor-positive dorsal root ganglion cells, we suggest that in inflammatory conditions, RIβ PKA is specifically required for nociceptive processing in the terminals of small-diameter primary afferent fibers.
Keywords: peripheral and central sensitization, inflammatory pain, plasma extravasation, prostaglandins, spinal cord plasticity, PKA RIβ expression
Tissue or nerve injury not only activates unmyelinated primary afferent nociceptors to produce acute pain but also initiates changes in the properties of the nociceptors and of spinal cord nociresponsive neurons (see Dubner and Basbaum, 1994). These changes can establish a persistent pain state in which there are enhanced responses to noxious stimuli (hyperalgesia) and non-noxious stimuli can produce pain (allodynia). Although many studies have focused on NMDA-mediated long-term changes in the firing of dorsal horn neurons (Woolf and Thompson, 1991), other studies provided evidence for a contribution of specific second messenger pathways to the establishment of these prolonged changes in the excitability of nociceptive afferents and of dorsal horn neurons (Taiwo et al., 1989;Taiwo and Levine, 1991; Cerne et al., 1992, 1993; Mao et al., 1993;Palecek et al., 1994; Lin et al., 1996).
cAMP-dependent protein kinase (PKA) has been implicated in prolonged changes in synaptic efficacy, notably long-term potentiation in the hippocampus (Frey at al., 1993; Huang et al., 1995). Because brief, high-frequency electrical stimulation of primary afferent fibers evokes comparable changes in dorsal horn neurons of the spinal cord (Randic et al., 1993; Lozier and Kendig, 1995), it is conceivable that PKA also contributes to long-term changes in nociceptive processing at the spinal cord level. In fact, injection of cAMP or the catalytic subunit of PKA enhances the response of dorsal horn neurons to glutamate-gated ion channel activation (Cerne et al., 1992, 1993). Other studies have provided evidence that cAMP- and PKA-dependent mechanisms underlie the sensitization of primary afferent neurons in the setting of inflammation (England et al., 1996; Kress et al., 1996).
Although PKA has been implicated in both central and peripheral nociceptive processing, it is not known whether the different isoforms of PKA, which have distinct patterns of expression in the brain (Cadd and McKnight, 1989), are involved in specific signaling events. The PKA holoenzyme is a tetramer composed of a regulatory subunit dimer, which contains the cAMP binding sites, and a single catalytic subunit bound to each regulatory subunit. At least four regulatory (RIα, RIβ, RIIα, and RIIβ) and two catalytic (Cα and Cβ) subunits have been characterized. The α isoforms of the PKA subunits are ubiquitously expressed in neural and non-neural tissues, but the β isoforms are more restricted and are highly expressed in the nervous system (Cadd and McKnight, 1989). Importantly, because selective inhibitors of the PKA isoforms are not available, previous pharmacological studies could not implicate specific isoforms in nociceptive processing. An alternative to using selective PKA inhibitors is to study animals in which the function of one of the isoforms is absent. In the present study, we analyzed mice that carry a null mutation in the gene that encodes the neuronal-specific isoform of the type I regulatory subunit (RIβ) of PKA. This mutation produces viable mice with general behavior that cannot be distinguished from that of their wild-type controls (Brandon et al., 1995).
We report that the processing of acute nociceptive information is normal in the mice with a targeted deletion of RIβ, but that there is a profound decrease of nociceptive processing in the setting of tissue injury. By contrast, neither the behavioral nor the anatomical consequences of peripheral nerve injury are altered in these mice. We suggest that the changes in nociceptive processing that arise with inflammation depend on a PKA-mediated phosphorylation of substrate proteins in the central and peripheral terminals of small-diameter primary afferent fibers.
MATERIALS AND METHODS
Generation of mutant mice. PKA RIβ mutant mice were produced as described previously (Brandon et al., 1995). Wild-type and homozygous mutant littermates were used for breeding; for experiments we used age-matched (10–14 weeks) and sex-matched mutant and wild-type mice. In all studies the experimenter was blind to the genotype of the mice. A behavioral analysis of these mice was reported in an earlier study (Huang et al., 1996).
Thermal and mechanical stimulation. We assessed thermal sensitivity by measuring paw withdrawal latencies to a radiant heat stimulus (Hargreaves et al., 1988). Mechanical sensitivity was determined with calibrated von Frey hairs using the up–down paradigm of Chaplan et al. (1994). Both the thermal and mechanical stimuli were applied to the plantar surface of the paw; the mice were not restrained during these tests.
The formalin test. To study nociceptive pain behavior, we used the paw formalin test. The mice received a 10 μl intraplantar injection of 2% formalin solution, and then we monitored the amount of time that the mice licked the injected paw. The incidence of licking was measured in 2 min periods at 5 min intervals for 60 min. To quantify the magnitude of the inflammatory response, we measured the paw diameter with a spring-loaded caliper (Mitutoyo) 90 min after the formalin injection.
Plasma extravasation. To test the integrity of the peripheral terminals of the primary afferent nociceptors directly, we assessed the magnitude of capsaicin-evoked neurogenic inflammation, an inflammatory response that results from release of neuropeptides from the small, unmyelinated (C-fiber) primary afferent terminals (Lembeck and Holzer, 1979). To study capsaicin-induced plasma extravasation, we anesthetized the mice with 50 mg/kg pentobarbital and made an intravenous injection of 10 mg/kg Evans blue into a tail vein. Five minutes later, capsaicin (8-methyl-N-vanillyl-6-noneaneamide; Sigma, St. Louis, MO) or vehicle (10% ethanol, 10% Tween 80, and 80% saline) was injected intradermally into the dorsal part of the hindpaw, and punches of skin were sampled 30 min later. The Evans blue was extracted from the tissue samples by incubation in formamide; extravasated protein was measured spectrophotometrically as described previously (Coderre et al., 1989).
Prostaglandin E2-evoked thermal allodynia: peripheral and central mechanisms. Because there is evidence that prostaglandin E2 (PGE2) sensitizes primary afferent nociceptors and produces allodynia via a cAMP-dependent pathway (Ferreira and Nakamura, 1979; Taiwo et al., 1989; Taiwo and Levine, 1991), we studied the effect of intraplantar injection of PGE2 on thermal and mechanical nociceptive thresholds. We also examined the effect of spinal administration of PGE2 in mutant and wild-type mice, because it has been shown that spinal injections of PGE2 evokes a profound hyperalgesia and allodynia (Taiwo and Levine, 1986, 1988; Uda et al., 1990; Minami et al., 1994a,b; Malmberg et al., 1995). A total dose of 0.1 μg of PGE2 delivered in a volume of 5.0 μl was administered either into the plantar surface of the paw or intrathecally, by lumbar puncture, according to the method of Hylden and Wilcox (1980). Thermal sensitivity was assessed by measuring paw withdrawal latency to a radiant heat stimulus as described above. In the intrathecal injection study, the paw withdrawal latency was measured on both the right and the left paw, and the mean was calculated. PGE2 (Sigma) was dissolved in 100% ethanol, stored at −20°C, and diluted in saline just before the experiment. The final concentration of ethanol was 1.0%. Neither intraplantar nor intrathecal injection of 1.0% ethanol changed thermal withdrawal latencies. The PGE2 dose (0.1 μg) that we used for intraplantar and intrathecal injections was based on dose–response studies and represents a just maximally effective dose (data not shown).
Nerve injury model. Nerve injury was produced by tying a tight ligature around approximately one-third to one-half of the diameter of the sciatic nerve, similar to the approach described in rats by Seltzer et al. (1990). The surgery was performed under halothane (2.0–3.0%) anesthesia. Through a small skin incision, the biceps femoralis was bluntly dissected to expose the sciatic nerve. Next, we inserted a 9–0 silk suture into the sciatic nerve, just distal to the sciatic notch, and then tightly ligated the nerve. The responses of the mice to thermal and mechanical stimulation were tested before and up to 14 d after the nerve injury.
Immunocytochemistry. For immunocytochemistry, the mice were deeply anesthetized with pentobarbital (100 mg/kg) and perfused with a 10% phosphate-buffered formalin fixative. The spinal cords were removed and post-fixed for 4 hr in the same fixative and then cryoprotected in 30% sucrose overnight. The spinal cord was sectioned transversely at 30 μm on a freezing microtome. Immunostaining was performed according to the avidin–biotin–peroxidase method (Hsu et al., 1981). We used a nickel-intensified diaminobenzidine protocol with glucose oxidase to localize the horseradish peroxidase immunoreaction product. Fos protein immunoreactivity was examined on spinal cord sections from mice that were studied in the formalin test. Substance P (SP) and substance P receptor (SPR) immunostaining were performed on spinal cord sections from nerve-injured mice. We used the following dilutions of antisera: 1:30,000 for Fos (kindly provided by Dr. Dennis Slamon, University of California at Los Angeles, Los Angeles, CA), 1:30,000 for SP (Peninsula Laboratories, Belmont, CA), and 1:20,000 for SPR (kindly provided by Dr. Steve Vigna, University of North Carolina, Chapel Hill, NC). Reacted sections were mounted on gelatin-coated slides, dried, and then coverslipped with Eukitt (Calibrated Instruments Inc.).
PKA RIβlac expression. To examine PKA RIβ expression in areas involved in nociceptive processing and peripheral inflammation, we used transgenic mice with the RIβ promoter fused to the lacZ gene and assayed for β-galactosidase (β-gal) expression in the spinal cord, dorsal root ganglion (DRG), and superior cervical ganglion. The transgenic mice were generated as described previously (Rogers et al., 1992). Two mice were deeply anesthetized and perfused with a 4% paraformaldehyde fixative. The spinal cord, DRG, and superior cervical ganglion were removed, post-fixed in the same fixative, and then cryoprotected in 30% sucrose overnight. The spinal cord was sectioned transversely at 30 μm on a freezing microtome; the DRG and superior cervical ganglia were cut at 12 μm on a cryostat. Staining for β-gal activity was performed with X-Gal as the substrate. The sections were counterstained with neutral red. To determine whether PKA RIβ is expressed in small-diameter nociceptive neurons, we performed double-label studies for the high-affinity NGF receptor trkA (Averill et al., 1995) and for β-gal. TrkA expression was examined by immunocytochemistry (1:10,000 dilution; kindly provided by Dr. L. F. Reichardt, University of California San Francisco, San Francisco, CA). To estimate the percentage of β-gal- and trkA-positive cells in the DRG, counts were made on four to six sections of L5 ganglia from the two transgenic mice.
Electron microscopy of dorsal roots. Two wild-type and two mutant mice were deeply anesthetized with pentobarbital and perfused with a mixture of 3% glutaraldehyde, 3% formaldehyde, and 0.1% picric acid (Langford and Coggeshall, 1979). After the perfusion, the spinal cord with attached dorsal roots was removed and post-fixed in the same fixative overnight. The following day, L5 dorsal roots were dissected, rinsed in 0.1 m phosphate buffer followed by post-fixation for 1 hr in 1% OsO4, and en bloc-stained in 2% aqueous uranyl acetate for 30 min. The dorsal roots were then dehydrated in ascending concentrations of ethanol, passed into propylene oxide, and embedded in Durcupan resin (Fluka, Buchs, Switzerland). Finally, the ultrathin sections were placed on single-hole grids coated with butvar and stained with uranyl acetate and lead citrate. The thin sections of the dorsal roots were examined in the electron microscope, and photographs were taken at an original magnification of 1600×. Myelinated and unmyelinated axons from the dorsal roots were counted from montages of electron micrographs.
Data and statistical analyses. To quantitate the number of Fos-like immunoreactive neurons, we took photographs of 5–10 sections from L4–L5 at low (4×) power on a Nikon Microphot-FXA microscope. The photographs were divided into four segments: laminae I–II, II–VI, and V–VI, and the ventral horn. A person blinded to the groups counted the number of Fos-immunoreactive neurons. Five to 10 spinal cord sections were counted per mouse and averaged so that each mouse had a mean value for the regional Fos immunoreactivity.
We quantified changes in SP or SPR immunoreactivity in the superficial dorsal horn of the spinal cord with a computer-assisted image analysis system (NIH Image software). Images of the spinal cord were captured with a 4× objective and a CDD camera and converted to a digital image with a gray value ranging from 0 to 255. To quantify staining density, we established a threshold above which the number of pixels was counted; the threshold was the same for each spinal cord section. Nerve injury-evoked changes in staining were based on a ratio of the density of the injured to the noninjured side, which was calculated for each spinal cord section. In each mouse, we measured three to eight sections from the L1–S1 segments of the spinal cord.
For statistical analyses of parametric data we used Student’st test or ANOVA followed by the Fisher’s PLSD test for multiple comparisons, when appropriate. Nonparametric data (mechanical withdrawal thresholds; see Chaplan et al., 1994) were analyzed using the Friedman test for within-group comparisons and the Mann–Whitney test for comparisons between groups. Critical values that reached thep < 0.05 level were considered statistically significant.
RESULTS
General behavior and nociceptive pain responses
There was no difference in open field behavior or general motor function between the wild-type and mutant mice. When we tested acute thermal and mechanical nociceptive thresholds, we also found no difference between wild-type and mutant, uninjured mice. The two groups of mice, however, differed considerably in a test of persistent pain, namely the formalin test. The injection of 2% formalin solution into the paw reliably evoked two phases of paw licking in the wild-type mice (Fig. 1A). The first phase, which is presumed to result from direct activation of primary afferent nociceptors, was similar in wild-type and mutant mice. In contrast, the second, prolonged phase of paw licking, which is partly driven by a delayed inflammation of the paw, was significantly reduced in mutant mice compared with wild-type mice (Fig.1A,B). Mutant mice also showed significantly reduced swelling of the formalin-injected paw (Fig. 1C). Consistent with the reduced inflammation resulting in decreased nociceptive inputs to the spinal cord, ipsilateral to the formalin-injected paw, we recorded significantly fewer Fos-immunoreactive neurons, a marker of neuronal activity in the L4–L5 dorsal horn of mutant mice compared with wild-type mice (p < 0.05, ANOVA; Figs.2, 3). The decreased Fos immunoreactivity was observed in laminae I–II and V–VI of the dorsal horn (Figs. 2, 3), which are areas that contain the majority of nociresponsive neurons in the dorsal horn (Menétrey et al., 1977).
Fig. 1.
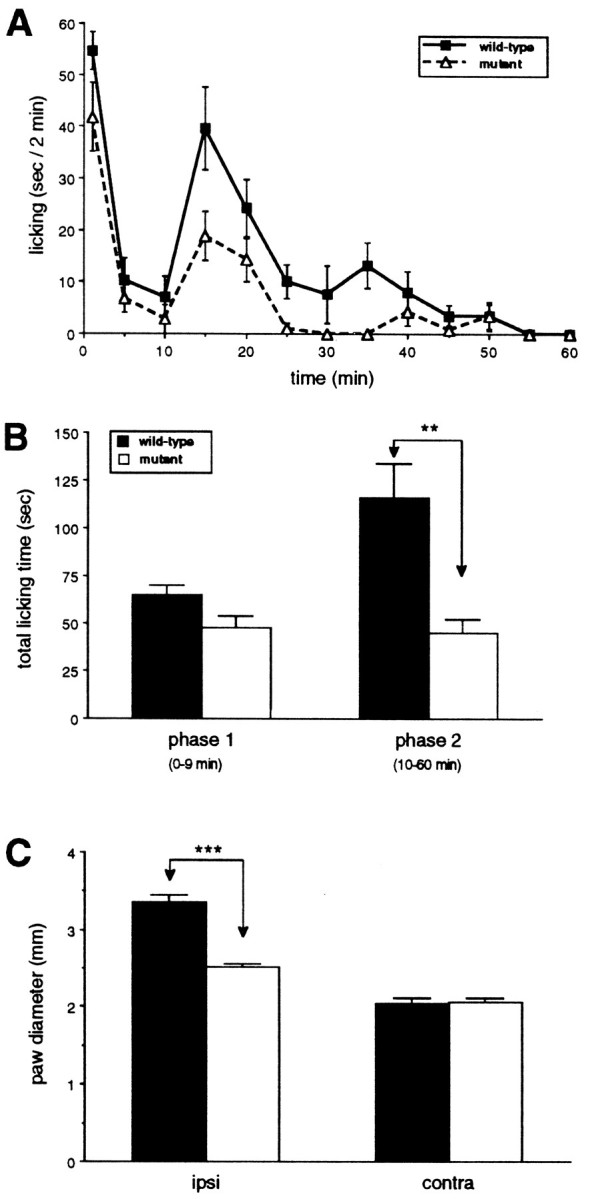
Formalin-evoked paw-licking behavior and paw swelling in wild-type and mutant mice. A, Time course of licking behavior of the formalin-injected paw presented as the mean licking time ± SEM per 2 min; n = 5 per group. B, The formalin-evoked paw licking behavior was divided into two phases: phase 1, 0–9 min; andphase 2, 10–60 min. Data are presented as the mean total licking time ± SEM of the first and second phases after formalin injection into the paw. **p < 0.01,t test, comparing wild-type and mutant mice.C, Data are presented as the mean paw diameter ± SEM in millimeters of the formalin-injected and noninjected paws. **p < 0.01, t test, comparing wild-type and mutant mice. ipsi, Ipsilateral;contra, contralateral.
Fig. 2.

Photomicrographs illustrating formalin-evoked Fos immunoreactivity at the L4 spinal segment of wild-type (A, B) and mutant (C, D) mice contralateral (A, C) and ipsilateral (B, D) to the paw that received formalin. Scale bar, 150 μm.
Fig. 3.

Formalin-evoked Fos immunoreactivity. Data are presented as mean number of Fos-immunoreactive neurons ± SEM on the formalin-injected side at the L4–L5 level; n = 5 mice per group. *p < 0.05; **p < 0.01, Fisher’s PLSD test, comparing the two groups.
Capsaicin-evoked plasma extravasation
We found that an intradermal injection of capsaicin on the dorsal surface of the hindpaw produced a dose-dependent increase in neurogenic inflammation, as indicated by the magnitude of extravasated Evans blue in the skin of wild-type mice. Plasma extravasation in response to the capsaicin injection was not eliminated in the mutant mice, but it was significantly reduced compared with wild-type mice (Fig. 4). Vehicle (10% ethanol/Tween 80) injections in both wild-type and mutant mice produced minimal plasma extravasation. These data suggest that RIβ PKA is important for full expression of capsaicin-evoked neurogenic inflammation.
Fig. 4.

Capsaicin-evoked plasma extravasation. Mutant mice displayed significantly reduced capsaicin-evoked plasma extravasation. Data are presented as mean micrograms per sample extravasated Evans blue ± SEM; n = 5 per group.Asterisks indicate significant differences between wild-type and mutant mice (**p < 0.01,t test).
PGE2-evoked changes in thermal nociceptive thresholds
Baseline paw withdrawal latencies were similar in mutant and wild-type mice. Although PGE2 injection resulted in a significant lowering of the thermal nociceptive thresholds in both wild-type and mutant mice, the magnitude of the thermal allodynia was significantly smaller (p < 0.01, repeated measures ANOVA) in the mutant mice compared with wild-type mice (Fig.5A). Similar to what we recorded after intraplantar injection, we found that intrathecal administration of PGE2 produced thermal allodynia in both wild-type and mutant mice, compared with baseline values (Fig.5B), but the mutant mice displayed significantly less thermal allodynia (p < 0.05, repeated measures ANOVA) compared with wild-type mice (Fig. 5B). In neither wild-type nor mutant mice did the intrathecal dose of PGE2evoke behaviors indicative of pain, such as vocalization and/or biting and scratching of the lower back.
Fig. 5.

Paw withdrawal latencies before and after intraplantar (A) or intrathecal (B) injection of 0.1 μg of PGE2 in wild-type and mutant mice. Mutant mice showed significantly less thermal allodynia compared with wild-type mice after both intraplantar (p < 0.01, repeated measures ANOVA) or intrathecal (p < 0.05) administration of PGE2. Data are presented as the mean latency in seconds ± SEM; n = 5 per group.Asterisks indicate significant differences between the groups (*p < 0.05; **p < 0.01, Fisher’s PLSD test).
Nerve injury-evoked changes of mechanical and thermal nociceptive threshold
As noted above, before the injury, there were no differences in paw withdrawal latencies to heat and no difference in the threshold for withdrawal to noxious mechanical stimulation between the two groups of mice (Fig. 6). Furthermore, in both wild-type and mutant mice, partial injury to the sciatic nerve produced a significant increase in thermal and mechanical sensitivity of the ipsilateral paw 1–14 d after the injury (Fig. 6). This was manifest as a significantly decreased latency to respond to the heat stimulus and a decreased intensity of mechanical stimulation required to elicit a paw withdrawal. Neither thermal nor mechanical stimulation were altered in the noninjured hindpaw. Importantly, the magnitude of the threshold changes did not differ in the two groups of mice.
Fig. 6.

Effect of nerve injury on withdrawal responses to thermal and mechanical stimulation in wild-type and mutant mice.A, Paw withdrawal latencies to thermal stimulation. Data are presented as the mean time in seconds ± SEM to paw withdrawal of the injured (ipsilateral) and the noninjured (contralateral) sides;n = 3 per group. Both groups developed thermal allodynia of comparable magnitude on the injured side.B, Paw withdrawal thresholds to mechanical stimulation. Data are presented as the mean von Frey hair threshold in grams ± SEM of the injured (ipsilateral) and the noninjured (contralateral) sides. There was no difference between the groups in the response to acute thermal or mechanical stimulation (day 0), and the threshold did not change in the nerve-injured animals.
Anatomical correlates of the nerve injury were also comparable in the two different groups of mice and were consistent with previous studies in the rat (Hökfelt et al., 1994; Abbadie et al., 1996). Thus, in both groups nerve injury produced a significant decrease in SP immunoreactivity (L4–L6, 20–30%) and a significant increase in SPR immunoreactivity (L4–L6, 50–70%) in the dorsal horn of the spinal cord on the injured side (Figs. 7,8). The magnitude of the anatomical changes did not differ in wild-type and mutant mice. Although it is not certain that the anatomical changes are pathophysiological in the development of nerve injury-induced pain, these results indicate that RIβ PKA is not required for the normal development and persistence of neuropathic pain.
Fig. 7.

Photomicrographs illustrating the density of SP (A, B) and SPR (C, D) immunoreactivity at the L4 spinal segment of wild-type mice contralateral (A, C) and ipsilateral (B, D) to the partial injury of the sciatic nerve. Scale bar, 150 μm.
Fig. 8.

Nerve injury-induced decreases of SP immunoreactivity (A) and increases of SPR immunoreactivity (B) in the L4–L6 spinal segments in wild-type and mutant mice. The magnitude of nerve injury-induced changes in SP or SPR immunoreactivity was similar for the two groups (ANOVA, p > 0.05). The density of labeling was quantified after digitizing images of the immunostaining. Data are presented as mean percent of the density ± SEM on the injured versus the control side; n = 3 per group.
PKA R1βlac expression
Previous studies demonstrated that the pattern of expression of the RIβlac transgene is comparable to the endogenous RIβ expression in neural tissue (Rogers et al., 1992). Although the RIβ subunit is primarily expressed in the nervous system, it is not expressed in all neurons. In the spinal cord, we found RIβlac expression in all laminae; however, neither the gray nor white matter contained labeled glial cells (Fig. 9A). We found that ∼35% of all DRG cells showed β-gal expression. The staining was observed in both small and large DRG neurons (Fig.9B). Although the nonuniform staining may have resulted from a mosaic expression of the transgene, it raises the possibility that RIβ is expressed by a distinct subpopulation of DRG neurons. Because small-diameter, nociceptive neurons express the high-affinity NGF and trkA receptors (Averill et al., 1995), we performed double-label studies for trkA and β-gal. Double-label studies revealed β-gal activity in both trkA-positive and trkA-negative DRG neurons (Fig.9C). In the two transgenic mice studied, we found that ∼57% of β-gal-positive cells showed trkA immunoreactivity. There were also both small and large DRG cells that showed neither trkA immunoreactivity nor β-gal activity (Fig. 9C). Finally, because there is evidence of a sympathetic contribution to plasma extravasation (Coderre et al., 1989), we also examined postganglionic sympathetic neurons for β-gal staining. In contrast to the DRG, we found no staining of neurons in the superior cervical ganglion (data not shown).
Fig. 9.
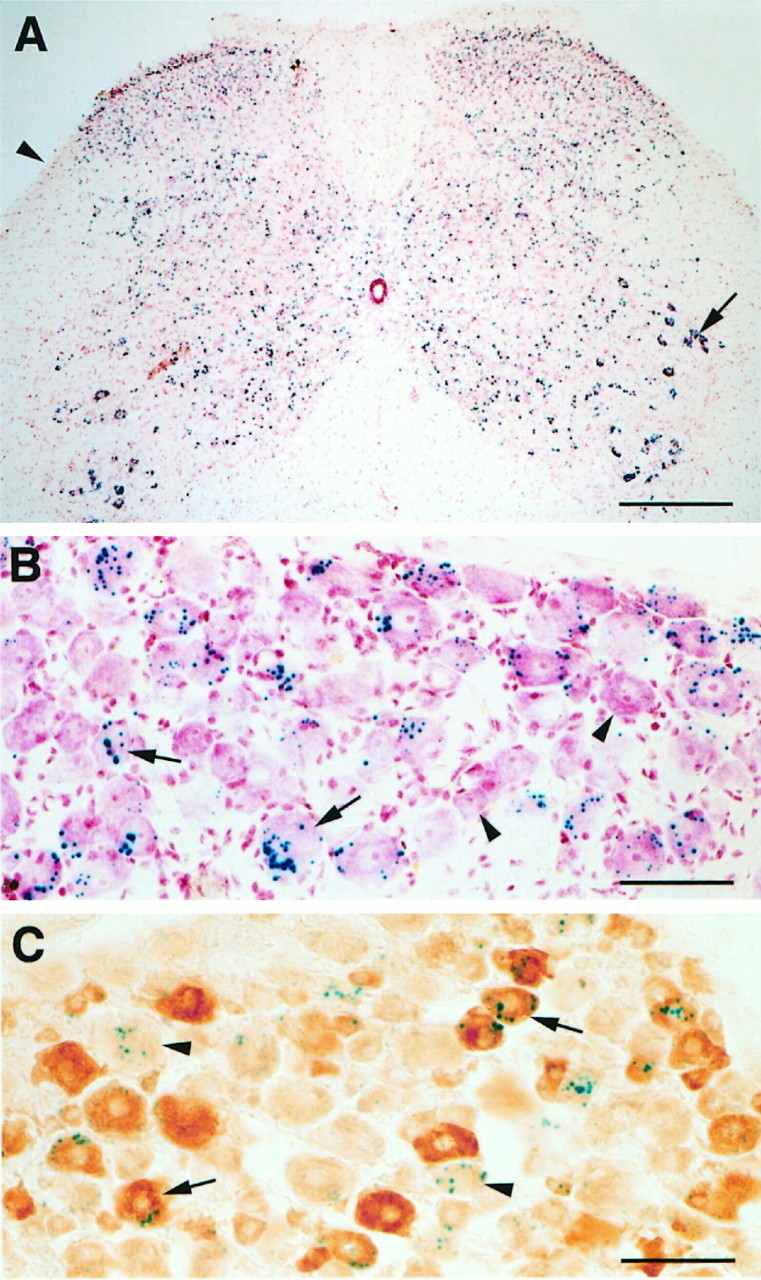
RIβlacZ expression in DRG cells revealed by β-gal staining. A, β-Gal activity (blue dots) was observed in all laminae of the spinal cord but not in the white matter (arrowhead). The arrowpoints to heavily labeled motoneurons. The sections were counterstained with neutral red. Scale bar, 320 μm. B, In the DRG, β-gal activity was found in both small (arrows) and large cells. Some DRG cells did not stain for β-gal (arrowheads). Scale bar, 100 μm. C, β-Gal activity was observed in both trkA-positive (arrows) and trkA-negative (arrowheads) DRG neurons. Other DRG cells showed neither trkA immunoreactivity nor β-gal activity. Scale bar, 100 μm.
Complement of primary afferent axons in the dorsal root
To rule out the possibility that the reduced inflammation and nociceptive behavior observed in the mutant mice resulted from a developmental abnormality, specifically a loss of primary afferent fibers, we used electron microscopy to compare the number of axons in the L5 dorsal roots of two wild-type and two mutant mice. Neither the morphology nor the numbers of myelinated or unmyelinated axons differed between mutant and wild-type mice. Specifically, the number of myelinated fibers was 2246 and 2655 in the two wild-type mice and 2276 and 2383 in the two mutant mice. The number of unmyelinated fibers was 4836 and 5651 in the wild-type mice and 5033 and 5214 in the mutant mice.
DISCUSSION
In the present study, we used mutant mice that lack the neuronal-specific RIβ subtype of PKA to evaluate the contribution of this regulatory subunit to the behavioral and anatomical consequences of tissue and nerve injury and to identify its neuronal site of action. Our results suggest that RIβ PKA is necessary for the full expression of neurogenic inflammation and for the paw edema and persistent pain produced by tissue injury. We found no evidence that the RIβ subunit of PKA contributes to the development of neuropathic pain behavior produced by peripheral nerve injury.
Mice that lack the RIβ of PKA did not differ from wild-type mice in tests of acute pain. In the absence of injury, the mice showed comparable mechanical and thermal nociceptive thresholds. Furthermore, the phase I response in the formalin test, a measure of acute pain, was not altered. These results suggest that activation of C-fibers per se, which involves thermal, mechanical, or chemical transduction mechanisms, does not require RIβ. In contrast, in the setting of persistent tissue injury, we found profound deficits of nociceptive processing in the mutant mice, including a significant decrease in the magnitude of the second-phase behavior of the formalin test. This phase of pain behavior results from both inflammation-evoked primary afferent activity and from sensitization of dorsal horn neurons secondary to the afferent activity generated during phase I (Dickenson and Sullivan, 1990; Puig and Sorkin, 1996). Because we found that formalin-evoked paw swelling of the hindpaw was significantly decreased in the mutant mice, we believe that the major defect in these animals resulted from a decreased inflammatory response. The magnitude of firing of primary afferent C-fibers would thus be reduced, and this would result in a reduction of second-phase pain behavior. The decrease in the activity of primary afferent nociceptors would also explain the reduced Fos immunoreactivity in the spinal cord dorsal horn.
Although a reduction of inflammation and consequent pain could arise from changes in the central and peripheral nervous systems, the fact that capsaicin-evoked plasma extravasation was reduced in the RIβ mutant mice indicates that there is a significant peripheral defect in these animals. Capsaicin selectively activates small-diameter, unmyelinated, neuropeptide-containing primary afferent fibers to produce neurogenic inflammation (see Bevan et al., 1987; Holzer, 1991). Although activity in the CNS can regulate this response, it is not required. Furthermore, although increased activity in sympathetic efferents contributes to the neurogenic inflammatory response (Coderre et al., 1989), we found no evidence of expression of RIβlac in sympathetic preganglionic neurons. In fact, because we found high levels of expression of PKA RIβlac in DRG cells, including many that coexpressed trkA, a marker of nociceptive afferents (Averill et al., 1995), notably those that coexpress calcitonin gene-related peptide, we suggest that a defect in the peptide-containing primary afferent nociceptor is critical to the phenotype that was revealed.
Because cAMP analogs and activators of PKA enhance peptide release from cultured sensory neurons (Hintgen et al., 1995; Supowit et al., 1995), we hypothesize that the reduced capsaicin-evoked neurogenic inflammation and the reduced inflammation and second-phase behavioral response in the formalin test in the mutant mice results from a defect in PKA-mediated regulation of neurotransmitter release from the peripheral terminals of primary afferent C-fibers. One possibility is that cAMP-dependent phosphorylation of a Ca2+channel (Hell et al., 1995), or of proteins required for vesicle docking and transmitter release from primary afferent C fibers, is abnormal in the mutant mice. Because PGE2 increases the release of peptide neurotransmitters from cultured DRG cells (Nicol et al., 1992), it is likely that comparable changes in the phosphorylation state of the same proteins located on the central terminals of C-fibers underlie the reduced hyperalgesia that we observed after intrathecal injection of PGE2 in the mutant mice.
Other mechanisms, however, must be be identified to account for the entire “pain” phenotype of the mutant mice. Specifically, the hyperalgesia produced by intraplantar injection of PGE2cannot be attributed to changes in the release of neuropeptides from the primary afferent terminal. Rather, PGE2-evoked hyperalgesia in this model results from a sensitization of the C-fiber terminal, which does not require Ca2+-dependent neurotransmitter release. Sensitization is manifest as a lowered threshold for firing of the terminal; this results in a behavioral allodynia in which non-noxious stimuli can evoke withdrawal reflexes and pain behavior (Martin et al., 1986; Schaible and Schmidt, 1988).
Previous studies demonstrated that the lowering of the pain threshold produced by PGE2 can be reduced with nonselective inhibitors of PKA (Taiwo et al., 1989; Taiwo and Levine, 1991). Our results are consistent with those observations and indicate that the RIβ subunit of PKA, although not required for activation of the primary afferent terminal, contributes to changes in specific ionic conductance produced by sensitizing agents such as PGE2. Because PGE2 increases the conductance of a TTX-resistant Na+ channel that is highly expressed in capsaicin-sensitive, small-diameter DRG cells, and because cAMP mimics these effects (England et al., 1996; Gold et al., 1996), it is possible that phosphorylation of this channel via PKA requires the RIβ subunit. The fact that this channel is expressed in small-diameter peripheral afferents, as well as in DRG cells (Jeftinija, 1994; Pearce and Duchen, 1994; Arbuckle and Docherty, 1995), suggests that the RIβ subunit is, in fact, involved at the peripheral terminal. It is also possible that the membrane proteins that transduce thermal and mechanical noxious stimuli are phosphorylated by PKA when PGE2 is released in the setting of injury, resulting in a lowered threshold for activation of the primary afferent by these stimuli.
Although we observed profound changes in tissue injury models of pain, nerve injury-evoked thermal and mechanical allodynia did not differ in mutant and wild-type mice. There are many features that distinguish the persistent pain evoked by tissue and nerve injury. Importantly, only the former is associated with significant inflammation. By contrast, injured nerve fibers develop bursting, spontaneous activity and become very sensitive to locally applied and circulating noradrenaline (Wall and Gutnick, 1974). Ectopic discharges also develop in DRG cells that give rise to the injured afferents (Wall and Devor, 1983); sprouting of sympathetic postganglionic axons into the DRG in the setting of nerve injury (McLachlan et al., 1993) may contribute to these changes. Our results suggest that PKA RIβ does not contribute to any of these manifestations of nerve injury.
An alternative explanation for the lack of defect in the neuropathic pain state in the RIβ PKA mutant mice is that the nerve injury produces a stimulus that results in higher cAMP concentrations, which in turn stimulate other PKA subtypes. For example, in the wild-type mouse, a threefold to sevenfold higher concentration of cAMP is required to activate the RIα subunit compared with the RIβ subunit (Cadd et al., 1990). However, because there is a significant upregulation of the RIα subunit in the RIβ mutant mice (Brandon et al., 1995), RIα may substitute for RIβ in the mutant mice. This could preserve the nerve injury-evoked pain phenotype in these animals. Upregulation of the RIα subunit or other transduction cascade, such as PKC, may also be responsible for a portion of the residual responses that we observed in the tissue injury and inflammatory models of pain and after intrathecal injection of PGE2.
In summary, our studies suggest that a specific regulatory subunit of PKA, RIβ, is critical for the full expression of inflammatory and pain responses produced by tissue injury, including the neurogenic component of inflammation and sensitization of primary afferent nociceptors. We suggest that the latter features of inflammation respectively involve PKA phosphorylation of proteins involved in transmitter release and/or of ion channels or receptor proteins located in small-diameter primary afferent nociceptors. The reduction of thermal allodynia after intrathecal injection of PGE2injection indicates that the RIβ subunit also influences nociceptive processing at the level of the spinal cord. By contrast, we found no evidence of a contribution of PKA RIβ to nerve injury-evoked pain. Our results are consistent with the fact that cyclooxygenase inhibitors, which block the synthesis of prostaglandins and reduce inflammation and tissue injury-evoked pain, are of little value in the treatment of neuropathic pain conditions.
Footnotes
These studies were supported by National Institutes of Health Grants NS 14627 and 21445 (A.I.B.) and GM 32875 (G.S.M.), the Swedish Cancer Foundation (A.B.M.) and the Pharmaceutical Research and Manufacturers of America Foundation (A.B.M.). We thank Dr. Helen Wang for help with electron microscopic analysis of the dorsal roots.
Correspondence should be addressed to Annika B. Malmberg, Department of Anatomy 0452, University of California, San Francisco, San Francisco, CA 94143-0452.
REFERENCES
- 1.Abbadie C, Brown JL, Mantyh PW, Basbaum AI. Spinal cord substance P receptor immunoreactivity increases in both inflammatory and nerve injury models of persistent pain. Neuroscience. 1996;70:201–209. doi: 10.1016/0306-4522(95)00343-h. [DOI] [PubMed] [Google Scholar]
- 2.Arbuckle JB, Docherty RJ. Expression of tetrodotoxin-resistant channels in capsaicin-sensitive dorsal root ganglion neurons of adult rats. Neurosci Lett. 1995;185:70–73. doi: 10.1016/0304-3940(94)11227-a. [DOI] [PubMed] [Google Scholar]
- 3.Averill S, McMahon SB, Lary DO, Reichardt LF, Priestley JV. Immunocytochemical localization of trkA receptors in chemically identified subgroups of adult rat sensory neurons. Eur J Neurosci. 1995;7:1484–1494. doi: 10.1111/j.1460-9568.1995.tb01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bevan SJ, James IF, Rang HP, Winter J, Wood JN. The mechanism of action of capsaicin—a sensory neurotoxin. In: Jenner P, editor. Neurotoxins and their pharmacological implications. Raven; New York: 1987. pp. 261–277. [Google Scholar]
- 5.Brandon EP, Zhuo M, Huang YY, Qi M, Gerhold KA, Burton KA, Kandel ER, McKnight GS, Idzerda RL. Hippocampal long-term depression and depotentiation are defective in mice carrying a targeted disruption of the gene encoding the RI beta subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1995;92:8851–8855. doi: 10.1073/pnas.92.19.8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cadd G, McKnight GS. Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron. 1989;3:71–79. doi: 10.1016/0896-6273(89)90116-5. [DOI] [PubMed] [Google Scholar]
- 7.Cadd GG, Uhler MD, McKnight GS. Holoenzymes of cAMP-dependent protein kinase containing the neural form of type I regulatory subunit have an increased sensitivity to cyclic nucleotides. J Biol Chem. 1990;265:19502–19506. [PubMed] [Google Scholar]
- 8.Cerne R, Jiang MC, Randic M. Cyclic adenosine 3′5′-monophosphate potentiates excitatory amino acid and synaptic responses of rat spinal dorsal horn neurons. Brain Res. 1992;596:111–123. doi: 10.1016/0006-8993(92)91538-p. [DOI] [PubMed] [Google Scholar]
- 9.Cerne R, Rusin KI, Randic M. Enhancement of the N-methyl-d-aspartate response in spinal dorsal horn neurons by cAMP-dependent protein kinase. Neurosci Lett. 1993;161:124–128. doi: 10.1016/0304-3940(93)90275-p. [DOI] [PubMed] [Google Scholar]
- 10.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Res. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 11.Coderre TJ, Basbaum AI, Levine JD. Neural control of vascular permeability: interactions between primary afferents, mast cells, and sympathetic efferents. J Neurophysiol. 1989;62:48–58. doi: 10.1152/jn.1989.62.1.48. [DOI] [PubMed] [Google Scholar]
- 12.Dickenson AH, Sullivan AF. Differential effects of excitatory amino-acid antagonists on dorsal horn nociceptive neurons in the rat. Brain Res. 1990;506:31–39. doi: 10.1016/0006-8993(90)91195-m. [DOI] [PubMed] [Google Scholar]
- 13.Dubner R, Basbaum AI. Spinal dorsal horn plasticity following tissue or nerve injury. In: Wall PD, Melzack R, editors. The textbook of pain. Churchill–Livingstone; London: 1994. pp. 225–241. [Google Scholar]
- 14.England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase cascade. J Physiol (Lond) 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferreira SH, Nakamura M. Prostaglandin hyperalgesia, a cAMP/Ca2+ dependent process. Prostaglandins. 1979;18:179–190. doi: 10.1016/0090-6980(79)90103-5. [DOI] [PubMed] [Google Scholar]
- 16.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 17.Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:141–167. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 19.Hell JW, Yokoyama CT, Breeze LJ, Chavkin C, Catterall WA. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO J. 1995;14:3036–3044. doi: 10.1002/j.1460-2075.1995.tb07306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′,5′-cyclic monophosphate transduction cascade. J Neurosci. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hökfelt T, Zhang X, Wiesenfeld-Hallin Z. Messenger plasticity in primary sensory neurons following axotomy and its functional implications. Trends Neurosci. 1994;17:22–30. doi: 10.1016/0166-2236(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 22.Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev. 1991;43:144–201. [PubMed] [Google Scholar]
- 23.Hsu S, Raine L, Fanger H. A comparative study of the antiperoxidase method and an avidin-biotin complex method for studying polypeptide hormones with radioimmunoassay antibodies. Am J Clin Pathol. 1981;75:734–738. doi: 10.1093/ajcp/75.5.734. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y-Y, Kandel ER, Varshavsky L, Brandon EP, Qi M, Idzerda RL, McKnight GS, Bourtchouladze R. A genetic test of the effect of mutations in PKA on mossy fiber LTP and its relation to spatial and contextual learning. Cell. 1995;83:1211–1222. doi: 10.1016/0092-8674(95)90146-9. [DOI] [PubMed] [Google Scholar]
- 25.Hylden JLK, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 26.Jeftinija S. The role of tetrodotoxin-resistant sodium channels of small primary afferent fibers. Brain Res. 1994;639:125–134. doi: 10.1016/0006-8993(94)91772-8. [DOI] [PubMed] [Google Scholar]
- 27.Kress M, Rödl J, Reeh PW. Stable analogues of cyclic AMP but not cyclic GMP sensitize unmyelinated primary afferents in rat skin to heat stimulation but not to inflammatory mediators, in vitro. Neuroscience. 1996;74:609–617. doi: 10.1016/0306-4522(96)00181-9. [DOI] [PubMed] [Google Scholar]
- 28.Langford LA, Coggeshall RE. Branching of sensory axons in the dorsal root and evidence for the absence of dorsal root efferent fibers. J Comp Neurol. 1979;184:193–204. doi: 10.1002/cne.901840111. [DOI] [PubMed] [Google Scholar]
- 29.Lembeck F, Holzer P. Substance P as neurogenic mediator of antidromic vasodilatation and neurogenic plasma extravasation. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:175–183. doi: 10.1007/BF00500282. [DOI] [PubMed] [Google Scholar]
- 30.Lin Q, Peng YB, Willis WD. Possible role of protein kinase C in the sensitization of primate spinothalamic tract neurons. J Neurosci. 1996;16:3026–3034. doi: 10.1523/JNEUROSCI.16-09-03026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lozier AP, Kendig JJ. Long-term potentiation in an isolated peripheral nerve-spinal cord preparation. J Neurophysiol. 1995;74:1001–1009. doi: 10.1152/jn.1995.74.3.1001. [DOI] [PubMed] [Google Scholar]
- 32.Malmberg AB, Hamberger A, Hedner T. Effects of prostaglandin E2 and capsaicin on behavior and cerebrospinal fluid amino acid concentrations of unanesthetized rats: a microdialysis study. J Neurochem. 1995;65:2185–2193. doi: 10.1046/j.1471-4159.1995.65052185.x. [DOI] [PubMed] [Google Scholar]
- 33.Mao J, Mayer DJ, Hayes RL, Price DD. Spatial patterns of increased spinal cord membrane-bound protein kinase C and their relation to increases in 14C-2-deoxyglucose metabolic activity in rats with painful peripheral mononeuropathy. J Neurophysiol. 1993;70:70–81. doi: 10.1152/jn.1993.70.2.470. [DOI] [PubMed] [Google Scholar]
- 34.Martin HA, Basbaum AI, Kwiat GC, Goetzl EJ, Levine JD. Leukotriene and prostaglandin sensitization of cutaneous high-threshold C- and A-delta mechanonociceptors in the hairy skin of rat hindlimbs. Neuroscience. 1986;22:651–659. doi: 10.1016/0306-4522(87)90360-5. [DOI] [PubMed] [Google Scholar]
- 35.McLachlan EM, Janig W, Devor M, Michaelis M. Nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature. 1993;363:543–545. doi: 10.1038/363543a0. [DOI] [PubMed] [Google Scholar]
- 36.Menétrey D, Giesler GJ, Besson JM. An analysis of response properties of spinal cord dorsal horn neurons to non-noxious and noxious stimuli in the spinal rat. Exp Brain Res. 1977;27:15–33. doi: 10.1007/BF00234822. [DOI] [PubMed] [Google Scholar]
- 37.Minami T, Uda R, Horiguchi S, Ito S, Hyodo M, Hayaishi O. Allodynia evoked by intrathecal administration of prostaglandin E2 to conscious mice. Pain. 1994a;57:217–223. doi: 10.1016/0304-3959(94)90226-7. [DOI] [PubMed] [Google Scholar]
- 38.Minami T, Nishihara I, Uda R, Ito S, Hyodo M, Hayaishi O. Involvement of glutamate receptors in allodynia induced by prostaglandins E2 and F2α injected into conscious mice. Pain. 1994b;57:225–231. doi: 10.1016/0304-3959(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 39.Nicol GD, Klingberg DK, Vasko MR. Prostaglandin E2 increases calcium conductance and stimulates release of substance P in Avian sensory neurons. J Neurosci. 1992;12:1917–1927. doi: 10.1523/JNEUROSCI.12-05-01917.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palecek J, Paleckova V, Dougherty PM, Willis WD. The effect of phorbol esters on the responses of primate spinothalamic neurons to mechanical and thermal stimuli. J Neurophysiol. 1994;71:529–537. doi: 10.1152/jn.1994.71.2.529. [DOI] [PubMed] [Google Scholar]
- 41.Pearce RJ, Duchen MR. Differential expression of membrane currents in dissociated mouse sensory neurons. Neuroscience. 1994;63:1041–1056. doi: 10.1016/0306-4522(94)90571-1. [DOI] [PubMed] [Google Scholar]
- 42.Puig S, Sorkin LS. Formalin-evoked activity in identified primary afferent fibers: systemic lidocaine suppresses phase-2 activity. Pain. 1996;64:345–355. doi: 10.1016/0304-3959(95)00121-2. [DOI] [PubMed] [Google Scholar]
- 43.Randic M, Jiang MC, Cerne R. Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. J Neurosci. 1993;13:5228–5241. doi: 10.1523/JNEUROSCI.13-12-05228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogers KV, Boring LF, McKnight GS, Clegg CH. Promotor for the regulatory type Iβ subunit of the 3′,5′-cyclic adenosine monophosphate-dependent protein kinase directs transgene expression in the central nervous system. Mol Endocrinol. 1992;6:1756–1765. doi: 10.1210/mend.6.10.1448119. [DOI] [PubMed] [Google Scholar]
- 45.Schaible HG, Schmidt RF. Excitation and sensitization of fine articular afferents from cat’s knee joint by prostaglandin E2. J Physiol (Lond) 1988;403:91–104. doi: 10.1113/jphysiol.1988.sp017240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- 47.Supowit SC, Christensen MD, Westlund KN, Hallman DM, DiPette DJ. Dexamethasone and activators of the protein kinase A and C signal transduction pathway regulate neuronal calcitonin gene-related peptide expression and release. Brain Res. 1995;686:77–86. doi: 10.1016/0006-8993(95)00461-x. [DOI] [PubMed] [Google Scholar]
- 48.Taiwo Y, Levine JD. Indomethacin blocks central nociceptive effects of PGF2a. Brain Res. 1986;373:81–84. doi: 10.1016/0006-8993(86)90317-3. [DOI] [PubMed] [Google Scholar]
- 49.Taiwo Y, Levine JD. Prostaglandins inhibit endogenous control mechanisms by blocking transmission at spinal noradrenergic synapses. J Neurosci. 1988;8:1346–1349. doi: 10.1523/JNEUROSCI.08-04-01346.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taiwo YO, Levine JD. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience. 1991;44:131–135. doi: 10.1016/0306-4522(91)90255-m. [DOI] [PubMed] [Google Scholar]
- 51.Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD. Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience. 1989;32:577–580. doi: 10.1016/0306-4522(89)90280-7. [DOI] [PubMed] [Google Scholar]
- 52.Uda R, Horiguchi S, Ito S, Hyodo M, Hayaishi O. Nociceptive effects by intrathecal administration of prostaglandin D2, E2 or F2α to conscious mice. Brain Res. 1990;510:26–32. doi: 10.1016/0006-8993(90)90723-o. [DOI] [PubMed] [Google Scholar]
- 53.Wall PD, Devor M. Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain. 1983;17:321–339. doi: 10.1016/0304-3959(83)90164-1. [DOI] [PubMed] [Google Scholar]
- 54.Wall PD, Gutnick M. Ongoing activity in peripheral nerves: the physiology and pharmacology of impulses originating from a neuroma. Exp Neurol. 1974;43:580–593. doi: 10.1016/0014-4886(74)90197-6. [DOI] [PubMed] [Google Scholar]
- 55.Woolf CJ, Thompson S. The induction and maintenance of central sensitization is dependent on N-methyl-d-aspartic acid receptor activation: implications for the treatment of post-injury pain hypersensitivity states. Pain. 1991;44:293–299. doi: 10.1016/0304-3959(91)90100-C. [DOI] [PubMed] [Google Scholar]