

Abstract
Administration of nerve growth factor (NGF) to aged or lesioned animals has been shown to reverse the atrophy of basal forebrain cholinergic neurons and ameliorate behavioral deficits. To examine the importance of endogenous NGF in the survival of basal forebrain cholinergic cells and in spatial memory, mice bearing a disruption mutation in one allele of the NGF gene were studied. Heterozygous mutant mice (ngf+/−) have reduced levels of NGF mRNA and protein within the hippocampus and were found to display significant deficits in memory acquisition and retention in the Morris water maze. The behavioral deficits observed in NGF-deficient mice were accompanied by both shrinkage and loss of septal cells expressing cholinergic markers and by a decrease in cholinergic innervation of the hippocampus. Infusions of NGF into the lateral ventricle of adultngf+/− mice abolished the deficits on the water maze task. Prolonged exposure to NGF may be required to induce cognitive effects, because reversal of the acquisition deficit was seen after long (5 weeks) but not short (3 d) infusion. Although NGF administration did not result in any improvement in the number of septal cells labeled for choline acetyltransferase, this treatment did effectively correct the deficits in both size of cholinergic neurons and density of cholinergic innervation of the hippocampus. These findings demonstrate the importance of endogenous NGF for survival and function of basal forebrain cholinergic neurons and reveal that partial depletion of this trophic factor is associated with measurable deficits in learning and memory.
Keywords: NGF, neurotrophin, cholinergic, learning, memory, cell death, gene deletion
The basal forebrain cholinergic system has been shown to play a critical role in learning and memory function. Lesions of basal forebrain cholinergic neurons or their projections produce severe memory deficits (for review, see Olton and Wenk, 1987). Nerve growth factor (NGF) is known to promote the survival, sprouting, and phenotypic expression of responsive neuronal populations to various degrees in developing and mature organisms (Mobley et al., 1986; Levi-Montalcini, 1987; Thoenen et al., 1987;Whittemore and Seiger, 1987). The profound response of basal forebrain cholinergic cells to NGF in various experimental paradigms has suggested that NGF may serve as a target-derived trophic factor to regulate survival or function of these neurons (Hefti et al., 1989). Intraventricular infusions of NGF can rescue axotomized cholinergic cells and reverse associated memory deficits (Will and Hefti, 1985;Hefti, 1986; Williams et al., 1986; Kromer, 1987; Gage et al., 1988;Hagg et al., 1988; Koliatsis et al., 1990; Dekker et al., 1992). NGF infusions can ameliorate age-related memory deficits and reverse degeneration of basal forebrain cholinergic cells (Fischer et al., 1987, 1991). These findings on the actions of exogenous NGF have given rise to the hypothesis that NGF serves as a target-derived survival factor for developing basal forebrain cholinergic neurons and suggest a role for NGF in the function of these neurons in the adult brain.
Studies using immunoneutralization and gene deletion have unambiguously demonstrated the importance of NGF for survival of sympathetic and small sensory neurons (Levi-Montalcini and Booker, 1960; Gorin and Johnson, 1979; Johnson et al., 1980; Aloe et al., 1981; Ruit et al., 1992; Crowley et al., 1994). Although numerous studies have indicated that NGF is capable of exerting potent influences on basal forebrain cholinergic neurons, the actions of endogenous NGF on these neurons has not been rigorously examined. Studies using immunoneutralization have been hampered by the limited diffusion of antibodies in the CNS, the limited developmental time window and doses examined, and the potential cross-reactivity of NGF antibodies with other members of the neurotrophin family (Vantini et al., 1989; Nabeshima et al., 1991; Li et al., 1995; Van der Zee et al., 1995). Although the gene deletion approach circumvents these problems, the poor viability of animals homozygous for NGF gene disruption precludes analysis of the role of NGF in the survival or function of cholinergic neurons past the neonatal period (Crowley et al., 1994).
In the present study, heterozygous NGF-deficient mutant mice (ngf+/−) were examined as a model of partial NGF deprivation. The mice were examined for brain content of NGF mRNA and protein, tested for spatial learning and memory function, and examined for morphological alterations in cholinergic neurons projecting from the septum to the hippocampus. To distinguish aspects of the phenotype of adult ngf+/− mice arising from acute insufficiency of NGF in the adult brain from those that may result from NGF deprivation during a critical developmental time window, the ability of NGF treatment in adult life to reverse various phenotypic characteristics of ngf+/− mice was examined.
MATERIALS AND METHODS
Mice. All mice used for study were F1 hybrids obtained by mating wild-type C57BL/6 females and chimeric males carrying an ngf+/− allele on a 129 background. Thus, the genetic background of all mice studied was identical, with the exception of the presence or absence of one disrupted ngfallele. All behavioral testing and data collection were conducted by an investigator blinded to the genotype of the mice.
NGF two-site ELISA. A subgroup of animals (eightngf+/− and eight ngf+/+ mice) was decapitated, and the brains were removed. The hippocampus and entorhinal cortex were dissected and homogenized in a sample/conjugate buffer containing Tris-HCL, NaCl, CaCl2, Triton X-100, and BSA at 4°C. An NGF two-site ELISA was performed in 96-well plates according to a modification of the procedure described by Korsching and Thoenen (1983), using human recombinant NGF as a standard.
Ribonuclease protection assay. The hippocampi from another subgroup of animals (nine ngf+/− and nine ngf+/+ mice) were dissected out, and the RNAzol B method was used to extract total RNA (Tel-Test, Inc.). Tissue was homogenized in 2.7× vol of RNAzol B using a Polytron 3000 homogenizer and PT-DA 3007/2 generator. Total RNA was extracted from each sample, and NGF mRNA levels were determined using the Ambion RPA II Ribonuclease Protection Assay Kit.33P-UTP-labeled RNA probes for mouse NGF and mouse cyclophilin were generated using the Promega Riboprobe Gemini System. The NGF probe was made to the region of the mouse NGF gene that spans nucleotides 230–454. This probe spans the region of the NGF gene deleted in the targeting construct, allowing the probe to specifically recognize only wild-type NGF mRNA in the RNase Protection assay. Quantitation of NGF mRNA was determined from standard curves generated by synthetic sense strand NGF mRNA. The cyclophilin probe was made to span the region of this gene from 36 to 166 bases.
Choline acetyltransferase (ChAT) radioenzyme assay. The hippocampus and entorhinal cortex were dissected out from another subgroup of animals (12 ngf+/− and 14 ngf+/+ mice) and homogenized in a sodium phosphate buffer, pH 7.4, containing Triton X-100. ChAT enzyme levels were then determined using the method described by Fonnum (1974).
Learning and memory testing. Two studies were conducted. In the first, ngf+/− (n = 22) andngf+/+ (n = 34) male and female mice were tested in a Morris water maze. In the second study, ngf+/− (n = 25) and ngf+/+ (n = 25) male and female mice were tested. All mice were F1 hybrids of 129 × C57BL/6 matings and were between 5 and 6 months of age at the start of behavioral testing. The experimenter was blinded to the genotype of the mice during all testing and data collection. The water maze consisted of a circular pool 48 inches in diameter that was painted black and filled with room temperature water. The mice were given four consecutive trials per day starting from four different pseudo-randomized start locations, with a 10 sec intertrial interval and a 90 sec maximum swim latency per trial. Data collection was automated by a computerized video tracking system (San Diego Instruments). The total swim distance, the percentage of swim distance spent in the platform quadrant, and the latency to find the platform were analyzed. The correlation between each of these variables was >90%, so only the swim latency results have been presented. Mice in the first study were tested on the hidden platform task for 8 d (days 1–8). After a 2 week period, all the mice were tested for retention of location of the hidden platform (day 24). The location of the platform was held constant throughout these days of testing. The hidden platform was then moved to a new location on days 30 and 31. After a 6 week delay period, all the mice were tested for retention of this new platform location (day 72).
In the second study, which assessed the effects of exogenous NGF administration, mice were tested on the water maze task using a procedure that was identical to that described above except that the delay interval for retention of the second (“new”) platform location was 4 weeks instead of 6 weeks, and mice were tested for a period of 5 d instead of 8 d in the acquisition of the initial platform location. Additionally, testing on the visible platform task was performed before testing on the hidden platform task. After this pretesting, the ngf+/− mice were equally divided into a group that received chronic intracerebroventricular infusions of NGF via osmotic minipumps or a group that received infusions of vehicle alone. The ngf+/+ mice were divided into a group that received infusions of vehicle or a group that received no infusion. Three days after implantation of the pumps, all the mice were trained on a third hidden platform location (different from the two locations used on testing before the infusions) to assess the effects of the infusions on both acquisition and retention of a new platform location. After a 4 week period, the mice were tested for retention of this hidden platform location. Immediately after that retention testing, all the mice were tested for the acquisition of another new platform location. All animals included in both studies had to swim quickly and accurately to a visible platform to screen out animals that had visual, attentional, motoric, or other noncognitive impairments. All the mice in both studies were able to perform the visible platform task, and thus no mice were excluded from either study.
Pump implantation and NGF infusion. The mice were deeply anesthetized with a combination of acepromazine, rompum, and ketamine, and a 28 gauge stainless steel cannula (Alzet brain infusion kit; Alza Corp., Palo Alto, CA), embedded in a dental acrylic stabilization platform, was lowered into the right lateral ventricle (coordinates: anteroposterior = 0.2 mm, mediolateral = −1.2 mm, and dorsoventral = −2.3 mm relative to Bregma). The stabilization platform was secured to the skull by cyanoacrylate. An Alzet Model 1007D osmotic minipump (Alza), half of which was coated in wax to reduce the flow rate to 0.25 μl/hr, was connected to the cannula with flexible vinyl tubing. The minipump was placed subcutaneously in the neck/shoulder area of the animal and changed after 2 weeks. The scalp was then closed, and the mouse was returned to its home cage. During the surgery to change the minipumps, the animals were anesthetized, and an incision was made in the neck/shoulder area adjacent to the minipump. The minipump was then removed and replaced with a new minipump. The infusion vehicle was a phosphate-buffered artificial cerebrospinal fluid containing 150 mm NaCl, 1.8 mm CaCl2, 1.2 mmMgSO4, 2.0 mmK2HPO4, 10.0 mm glucose, and 0.1% autologous mouse serum, pH 7.4. The concentration of human recombinant NGF used was 50 μg/ml. NGF-infused mice received ∼300 ng/d for a total of 8.4 μg of NGF over the 4 week infusion period.
Activity level testing. After testing on the water maze, a subgroup of mice from the first study (n = 12ngf+/− and n = 12 ngf+/+ mice) was tested on a habituation test. In this task, mice were placed individually into a chamber that automatically measured their locomotor activity (San Diego Instruments) for a period of 20 min, and then they were returned to their home cages.
Hot plate and tail flick testing. All mice included in the first study were tested on the hot plate and the tail flick tasks. Hot plate testing consisted of placing the mouse on a 48°C hot plate and measuring latency to paw lick or shake. For tail flick testing, mice were gently manually restrained, the distal centimeter of their tail was immersed in a beaker of 50°C water, and the latency to tail flick was recorded.
Histology. A subgroup of animals from each behavioral study was perfused with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The brains were removed, post-fixed overnight in 4% paraformaldehyde, and cryoprotected in 30% phosphate-buffered sucrose for 3 d at 4°C. Forty-micrometer-thick coronal sections were cut on a freezing microtome. A series of sections was processed histochemically for acetylcholinesterase (AChE) (Hedreen et al., 1985) to visualize the density of cholinergic fiber innervation in the hippocampus. A second series of sections was stained with a polyclonal antibody (made in goat) specific for ChAT (Chemicon International, Temecula, CA) [for a more complete description of the immunohistochemical staining procedure, see Chen and Gage (1995)]. A series of sections from animals in the first study was also stained with an antibody specific for the p75 receptor (generous gift from Louis Reichardt, University of California, San Francisco).
Morphometric analysis. Cells in the medial septum labeled by antibody to ChAT were counted and sized on a computerized image analysis system, the MCID Image Analysis System (Imaging Research), using a magnification of 33× (10× objective with a 3.3× photo objective). For each animal, a series of three sections, spaced at intervals of 240 μm from each other, was analyzed. Sections from each animal were matched for their anteroposterior level using the decussation of the corpus callosum and the crossing of the anterior commissure as landmarks. The ventral border of the medial septum was defined in each of these sections by the anterior commissure. Cells were defined as stained objects between 70 and 300 μm2 in area. The number of cells in each of the three sections was then summed to obtain a total number of cells for each animal. These numbers were then corrected using the Abercrombie method (Abercrombie, 1946). The mean cell size was also obtained by measuring the cross-sectional area of all the cells that were counted.
Cell counts for p75-labeled cells were performed by microscopic observation by an investigator blinded to the genotype of the specimens. Cells were counted as defined by labeled profiles with an identifiable nucleus.
The extent of fiber innervation into the hippocampus was determined on the AChE-stained sections. The MCID Image Analysis System (Imaging Research), using a magnification of 33× (10× objective with a 3.3× photo objective), was used to quantify fiber density. Fiber density was determined in three AChE-stained sections, spaced at 240 μm intervals, starting with the first section with the entire dorsal and ventral blades of the dentate gyrus visible. To reduce the variability of the AChE staining, all of the AChE-stained sections were stained in two batches. Variability of the AChE staining was assessed by examining the level of background staining within the corpus callosum and the intensity of staining within the thalamus. On the basis of visual inspection, a small number of sections (<5%) were excluded from analysis because of abnormally light staining. For both the CA1 (stratum lacunosum moleculare) and alveus, 20 fields were imaged from both left and right hippocampi of each section. For the dentate gyrus, 30 fields were sampled in the ventral and dorsal blades. An optical density measurement (percentage of total area occupied by stained fibers) was obtained for each field. A mean value for all fields that were quantitated per region per animal was then calculated.
RESULTS
Mice heterozygous for NGF gene deletion (ngf+/−) did not exhibit any gross morphological or behavioral abnormalities and could not be distinguished from wild-type (ngf+/+) littermates by visual inspection. There was a significant reduction in NGF protein levels in the ngf+/− animals as measured by a two-site ELISA (Table 1). NGF levels in the hippocampus of the ngf+/− mice constitute ∼25% of the levels in wild-type mice. Ribonuclease protection assay revealed that NGF mRNA levels were also reduced in the hippocampus of thengf+/− mice (Table 1). No significant differences were observed in hippocampal wet weight, total RNA levels, or cyclophilin mRNA between ngf+/+ and ngf+/− mice.
Table 1.
NGF protein and mRNA levels in hippocampus
| Groups | NGF protein (ng/gm wet wt) | NGF mRNA (fg NGF mRNA/μg cyclophilin mRNA) |
|---|---|---|
| ngf+/− | 0.085 ± 0.023* (n = 8) | 7.60 ± 0.86* (n = 9) |
| ngf+/+ | 0.321 ± 0.024 (n = 8) | 11.25 ± 1.23 (n = 9) |
Significant differences between groups (p < 0.05).
Two separate behavioral and morphological studies were performed. In the first study, a group of ngf+/− mice (n= 24) and their ngf+/+ littermates (n = 34) were tested on the water maze task (Fig.1) beginning at 5 months of age. Results obtained using measures of swim distance were similar to those obtained using latency, thus only the latency data has been presented. Analysis of the results obtained from male and female mice showed no significant differences and were consequently pooled for analysis. Importantly, all animals were able to learn a cued or visible platform task, and there were no significant differences in swim speed (not shown) or latency to find the visible platform between the NGF-deficient and wild-type groups of mice (Fig. 1A).
Fig. 1.

A, Mean latency to find the hidden platform on the water maze task on each day of testing. Eachpoint represents the mean performance of a group of mice on the four trials per day. Days 1–8 = performance during the acquisition of the initial location of the hidden platform; day 24 = performance on testing for retention of the initial platform location after a 16 d delay; days30 and 31 = performance during the acquisition of a new platform location; day72 = performance on testing for retention of the new platform location after a 6 week delay. The pointlabeled Visible represents performance on the visible platform task. B, Mean latency to find the hidden platform on each of the individual four trials on Day 1, and (C) on Day 30.D, Mean latency to find the hidden platform on the first trial of Day 72 (*p < 0.05, **p < 0.01, *** p < 0.0005; one-factor ANOVA with post hoc Fisher PLSD).
In the hidden-platform task of the water maze paradigm, bothngf+/+ and ngf+/− groups exhibited identical latencies to locate the platform on the first trial of testing (Fig.1B) (F(1, 54) = 0.037;p > 0.50). On the second and third trials, as the mice began to learn the location of the hidden platform, there was a trend toward better performance in the ngf+/+ mice compared with the ngf+/− mice. This difference, however, did not achieve statistical significance. By the fourth trial, both groups of mice were not significantly different from each other. After 8 d of testing, the ngf+/+ and ngf+/− mice had both learned to find the location of the platform quickly and accurately, and the performance of the two groups of mice did not differ (Fig.1A, day 8). After a 2 week retention period, all animals were retested on this same hidden platform location, revealing no significant difference between the performance of the two groups (Fig. 1A, day24).
After this initial testing, animals were then analyzed in a more demanding acquisition and retention paradigm. Acquisition was made more difficult by requiring the mice to learn a novel platform location (reversal task), whereas retention testing differed from the initial paradigm by both decreasing the number of days of acquisition testing (from 8 to 2 d) and increasing the retention interval (from 2 to 6 weeks). On day 30 of the experiment, all mice were tested for acquisition of the second platform location (Fig. 1C). Consistent with the lack of a memory component on the first trial of this task, the NGF-deficient and wild-type mice performed identically; however, the wild-type group acquired the new platform location more quickly than the ngf+/− group, performing significantly better on the second (F(1, 54) = 14.77;p < 0.0005) and third trials (F(1, 54) = 5.65; p < 0.05). By the fourth trial, both groups had learned the new platform location and were not significantly different from each other (p > 0.10). When the performance of each group was averaged across all four trials on the first day of the acquisition reversal task (day 30), thengf+/− mice were significantly impaired compared withngf+/+ littermates (F(1, 54) = 6.87;p < 0.01). The two groups did not differ on the next day of testing, and consequently testing on the second platform location was stopped to prevent overtraining. After a retention interval of 6 weeks, the mice were retested on day 72 for retention of the new platform location, and the ngf+/− mice were found to be significantly impaired compared with the ngf +/+ mice (Fig. 1A,D). By the second day of retention testing, day 73, the two groups were no longer significantly different (not shown).
The ngf+/− mice and their ngf+/+ littermates were also tested for locomotor behavior in activity chambers (Table2). There is no difference between the activity levels of the two groups of mice, indicating that thengf+/− mice were neither hyperactive nor hypoactive. Both groups of mice were tested on the tail flick and hot plate test, tasks that assess responsiveness to thermal pain (Table 2). Although a trend toward reduced responsivity of the ngf+/− mice was observed, there was no significant difference between thengf+/+ and the ngf+/− mice on either task (p > 0.10). These results support the conclusion that the deficits exhibited by the ngf+/− mice on the water maze task are not attributable to general sensory, motor, or arousal deficits.
Table 2.
Open field activity and nociceptive function
| Groups | Latency on the tail flick test (sec) | Latency on the hot plate test (sec) | Activity chamber (counts) |
|---|---|---|---|
| ngf+/− | 3.1 ± 0.6 | 31.8 ± 2.4 | 423 ± 47 |
| ngf+/+ | 2.8 ± 0.7 | 28.8 ± 1.5 | 389 ± 41 |
In the second study, which assessed the effects of chronic exogenous NGF administration, another group of adult ngf+/− mice (n = 24) and their ngf+/+ littermates (n = 30) was pretested on the water maze task in a manner that was similar to that described above (see Materials and Methods) (Fig. 2A). The results obtained during this pretesting confirmed those observed in the first study: the ngf+/− mice were significantly impaired on both acquisition and retention of the reversal task (not shown). After pretesting, ngf+/− mice were implanted with mini-osmotic pumps that delivered NGF or vehicle into the lateral ventricle. Controlngf+/+ mice were either untreated or infused with vehicle. Because the performance of the ngf+/+ mice receiving vehicle did not differ from unoperated ngf+/+ mice, results are shown only for the vehicle-infused group. Three days after the start of infusion (day 68), both groups of ngf+/− mice, NGF-infused and vehicle-infused, were still significantly impaired compared with wild-type mice on the acquisition of a new platform location (Fig.2B) (p < 0.05). Four weeks later, ngf+/− mice that received infusions of NGF were significantly improved on the retention of this platform as compared with vehicle-infused ngf+/− mice (F(1, 25) = 7.41; p < 0.05) (Fig. 2C). In fact, the performance of the NGF-infused ngf+/− was not significantly different from the performance of the wild-type mice on this retention task.
Fig. 2.

A, Testing paradigm of the second study, which assessed the effects of exogenous administration of NGF.B, Mean latency to find the hidden platform on the water maze task on the first day of acquisition after intraventricular infusions (Day 68). * denotes a significant difference between the two ngf+/− groups and ngf+/+ group; p < 0.05. C, Mean latency to find the hidden platform on the first trial of the first day of retention of this new platform location (Day 100).D, Mean latency to find the hidden platform during an acquisition task conducted on Day 103, 5 weeks after the onset of NGF infusion. * denotes a difference between NGF-infused and vehicle-infused ngf+/− mice (one-factor ANOVA withpost hoc Fisher PLSD).
Because the mice had received NGF infusions for a period of only 3 d before acquisition testing, the lack of effect observed on acquisition may be attributable to the short duration of NGF treatment rather than to the inability of NGF to ameliorate the acquisition deficit observed in the ngf+/− mice. Therefore, all of the mice were tested for acquisition of another novel platform location. On trials 2 and 3 of the acquisition testing (day 103), conducted 5 weeks after the onset of NGF infusion, the ngf+/− mice that received NGF performed significantly better than ngf+/− mice that received vehicle alone (trial 2: F(1, 25) = 6.53, p < 0.05; trial 3:F(1, 25) = 5.44, p < 0.05) and in fact were no longer impaired as compared with wild-type controls (Fig. 2D).
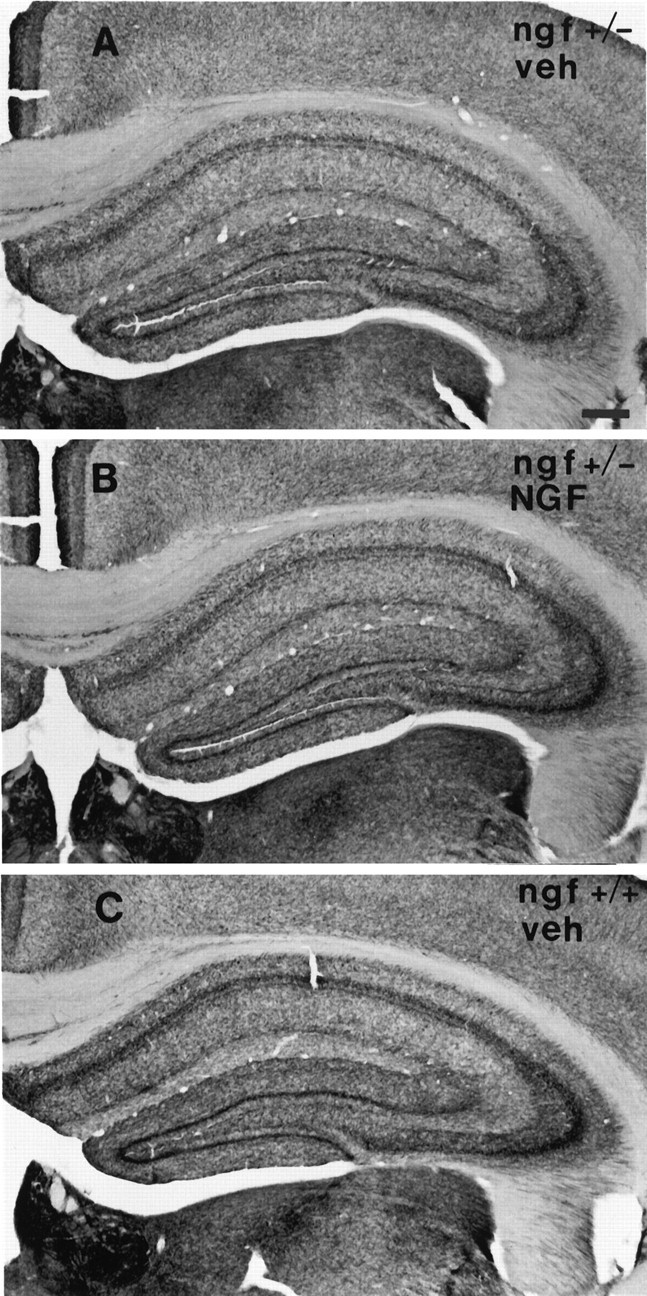
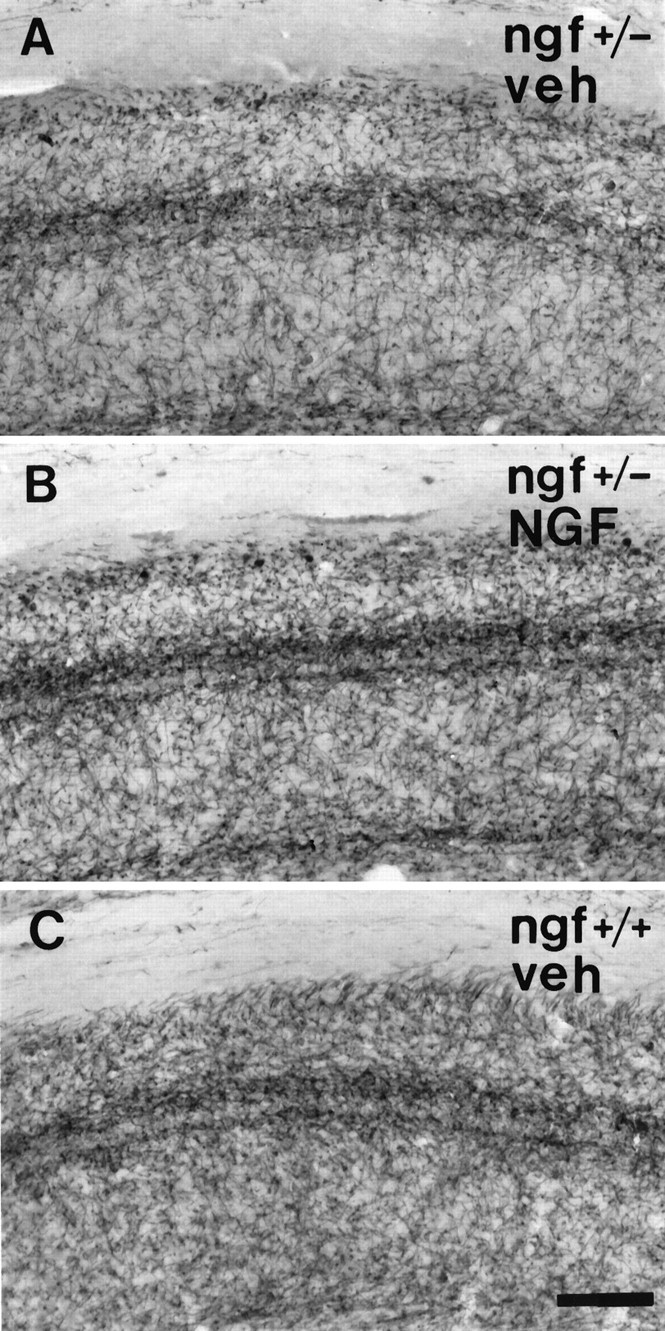
One-half of the mice were perfused with fixative after behavioral testing, and their brains were processed for analysis of basal forebrain cholinergic neurons. A series of sections was stained for ChAT immunocytochemistry, and ChAT-immunoreactive neurons within the medial septal nucleus were sized and counted. ngf+/− mice had significantly fewer ChAT-positive medial septal neurons than theirngf+/+ littermates (Figs. 3,4). Remaining ChAT-positive cells in thengf+/− animals were significantly shrunken compared withngf+/+ controls (Figs. 3, 4). A second series of sections from the same animals was stained with an antibody raised against the p75 receptor, the low-affinity NGF receptor. The cell counts obtained in the p75-stained sections confirmed the loss of cholinergic cells that was observed in the ChAT-stained sections (ngf+/− = 152 ± 14 vs ngf+/+ = 228 ± 13;F(1, 24) = 15.63; p < 0.01). A series of sections through the hippocampal formation was stained for AChE histochemistry to assess the density of cholinergic projections to this region. There was a significant reduction in AChE-positive fibers in CA1, CA3, and dentate gyrus regions of the hippocampus in thengf+/− mice compared with ngf+/+ littermates (Figs. 5,6, 7).
Fig. 3.

Photomicrograph of the medial septal region of (A) a ngf+/− mouse that had received vehicle, (B) a ngf+/− mouse that had received NGF, and (C) angf+/+ mouse that had received vehicle stained with antibodies to ChAT; insets show a higher magnification of stained cells. Scale bar, 100 μm.
Fig. 4.
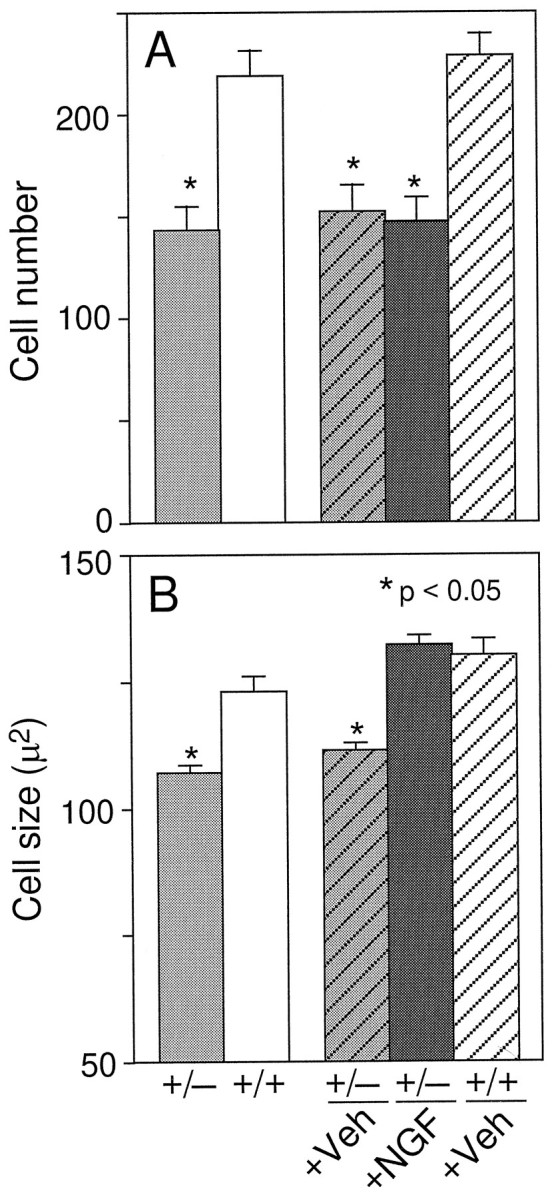
Number (A) and size (B) of ChAT-positive cells in the medial septal region [* denotes a significant difference from either untreated or vehicle-treated wild-type mice (p < 0.05; one-factor ANOVA with post hoc Fisher PLSD)].
Fig. 5.
Photomicrograph of the CA1 region of the hippocampus of (A) a ngf+/− mouse that had received vehicle, (B) angf+/− mouse that had received NGF, and (C) a ngf+/+ mouse that had received vehicle stained with AChE. Scale bar, 100 μm.
Fig. 6.
Photomicrograph of the hippocampus of (A) a ngf+/− mouse that had received vehicle, (B) a ngf+/− mouse that had received NGF, and (C) angf+/+ mouse that had received vehicle stained with AChE. Scale bar, 100 μm.
Fig. 7.
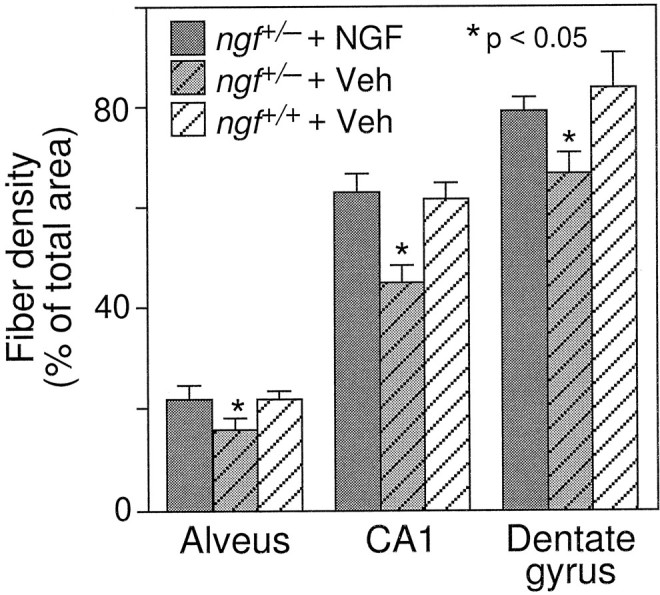
Density of AChE-positive fibers in the alveus, CA1, and dentate gyrus regions in the hippocampus [* denotes a significant difference between vehicle-treated ngf+/− and either ngf+/+ or NGF-treated ngf+/− mice (p < 0.05; one-factor ANOVA withpost hoc Fisher PLSD].
Measurement of ChAT activity revealed a significant reduction in ChAT levels in the hippocampus of the ngf+/− mice compared withngf+/+ littermates (Table 3). ChAT levels in the entorhinal cortex were not significantly reduced in the ngf+/− mice compared with their littermates. The smaller reduction in ChAT levels in the cortex may reflect the presence of intrinsic ChAT-positive cells in the cortex that are not affected by decreased NGF levels.
Table 3.
ChAT levels
| Groups | Hippocampus (×105cpm · min−1 · mg−1 wet wt) | Entorhinal cortex (×105cpm · min−1 · mg−1 wet wt) |
|---|---|---|
| ngf+/− | 3.39 ± 0.203-150 | 5.24 ± 0.27 |
| ngf+/+ | 4.59 ± 0.26 | 5.72 ± 0.27 |
F3-150: Significant differences between groups (p < 0.05).
Chronic intraventricular NGF infusions were able to reverse the shrinkage of cholinergic neurons that is observed in the heterozygotes, but they did not reverse the observed cell loss (Figs. 3, 4). NGF administration was also able to increase AChE-positive fiber innervation of the hippocampus in the ngf+/− mice (Figs.5, 6, 7).
DISCUSSION
In the present study, mice that are heterozygous for NGF gene disruption were found to have decreased levels of NGF mRNA and protein within the hippocampus, a prominent target of basal forebrain cholinergic neurons. ngf+/− mice exhibited significant acquisition and retention deficits in the water maze and displayed morphological deficits in septal cholinergic neurons. Morphological deficits included a loss of approximately one-third of septal cells labeled by ChAT or p75, a reduction in the size of remaining septal cholinergic cell bodies, and loss of both ChAT activity and AChE-positive fiber density within the hippocampal formation. Direct administration of NGF to the brain of adult ngf+/− mice was able to ameliorate the retention and acquisition deficits as well as increase the size of basal forebrain cholinergic neurons and cholinergic fiber density within the hippocampus. This treatment, however, did not produce any increase in the number of ChAT-positive septal neurons, suggesting that the loss of ChAT-positive cells in thengf+/− mice reflects cell death caused by NGF deprivation during a critical developmental time window.
Previous observations in mice homozygous for NGF gene disruption indicate that in the absence of NGF, septal cholinergic neurons differentiate and express their normal complement of phenotypic markers (Crowley et al., 1994). Because of the poor survival ofngf−/− mice, however, it could not be determined whether septal cholinergic neurons acquire dependence on NGF for survival after these cells have established their mature pattern of connections with the target area. The present findings provide evidence that basal forebrain cholinergic neurons do exhibit a dependence on endogenous NGF for survival. Here, we observe that adult mice with a partial deficiency in NGF exhibit evidence for loss of septal neurons labeling with either ChAT or p75. The failure to regain any ChAT-positive cells after an NGF treatment regimen that restores the size (and projections) of remaining cells points strongly to cell loss rather than downregulation of phenotypic markers. Thus, the availability of endogenous NGF during development appears to be limiting for survival of septal cholinergic neurons. Whether the reported lack of cell loss in ngf−/− mice reflects the immature developmental stage at which these mice were studied or whether it indicates that exposure to NGF is necessary to induce dependence of cholinergic neurons for survival remains to be established.
In addition to the reduction in number of septal cholinergic neurons, adult ngf+/− mice exhibited significant shrinkage of remaining ChAT-positive cells and a loss in density of cholinesterase-positive fibers within the hippocampus. The presence of morphological deficits in the cholinergic neurons remaining inngf+/− mice, and the ability of NGF infusion into adult brain to reverse them, suggests that in addition to its limiting role in development, NGF plays a critical role in the adult brain to maintain proper function of basal forebrain cholinergic neurons. These observations are consistent with previous findings indicating that NGF administration to aged rats is capable of reversing both memory deficits and shrinkage of cholinergic cells (Fischer et al., 1987), and with observations that NGF can induce sprouting of mature cholinergic neurons (Kawaja and Gage, 1991; Van der Zee et al., 1992). Thus, although the dependence of cholinergic neurons on NGF for cell survival may be restricted to a critical developmental time window, cholinergic cells also appear to require optimal levels of NGF during adult life for maintenance of an optimally functional phenotype.
In addition to the morphological effects observed on basal forebrain cholinergic neurons, ngf+/− mice exhibited significant deficits in performance in the water maze task. Our results confirm and extend previous findings using immunodeprivation (Nabeshima et al., 1991; Van der Zee et al., 1995) by indicating that even a partial reduction in NGF availability leads to measurable effects on spatial learning and memory. Lack of a more robust behavioral deficit may be the result of compensatory changes induced by decreased levels of NGF, and presumably reduced numbers and sizes of basal forebrain cholinergic cells, throughout development. The memory systems in thesengf+/− mice may have compensated for this chronic hypofunction of the cholinergic system, and thus the resulting deficit may not be as great as the deficit that would result from a comparable decrease of NGF protein occurring in adulthood. Although observations of behavioral deficits in genetically manipulated mice are frequently confounded by the genetic background of the mice investigated (Gerlai, 1996), all of the mice in the current study were F1 hybrids between two inbred strains and consequently are genetically identical, with the exception of the presence or absence of one mutated allele for NGF. In contrast to the CNS abnormalities reported in the 129 strain (Lipp et al., 1995), F1 hybrid mice of C57 and 129 strains perform well in the Morris water maze and exhibit robust long-term potentiation. Thus, the behavioral deficits reported here are attributable to the disruption of a single allele of the NGF gene and not to abnormalities arising from the genetic background of the mice.
Our observations indicate that partial depletion of NGF is associated with deficits in both acquisition and retention of a spatial learning task. The behavioral effects of NGF depletion appear to be specific to learning and memory function, because the performance ofngf+/− mice on the visible platform task did not differ from that of wild-type littermates. This observation argues strongly that the deficits in performance reflect poor acquisition and retention of spatial memory rather than alterations in sensory, motor, or motivational status of the ngf+/− mice. Consistent with this view, the performance of ngf+/− mice was identical to that of wild-type mice in hidden platform trials that did not involve a memory component, i.e., the first trial of a given platform location. Furthermore, swim speeds of ngf+/− mice were indistinguishable from those of wild-type mice, and in an open field test no differences in locomotor activity were observed. Additional evidence that the poor performance in the water maze task reflects a CNS-mediated deficit comes from the ability of direct infusions of NGF into the brain of ngf+/− mice to reverse the deficit.
The ability of NGF infusions to reverse the deficits in performance on the water maze point to a deficiency of NGF within the adult brain as the source of the behavioral abnormality. The deficit seen in thengf+/− mice on acquisition in the water maze task, however, does not appear to result from acute deficiency in NGF at the actual time of cognitive testing, because it can be reversed only by chronic (5 week) and not acute (3 day) infusion of NGF. This finding suggests that the behavioral improvements seen may be mediated via morphological alterations in basal forebrain cholinergic neurons rather than via acute actions of NGF on neuronal activity. As has been seen in aged rodents (Fischer et al., 1992; Chen and Gage, 1995), NGF administration to ngf+/− mice effectively ameliorates learning and memory deficits despite an irreversible loss of cholinergic neurons. These behavioral effects are likely to be mediated via the hypertrophy and sprouting of remaining cholinergic cells and may involve changes in synaptic density similar to those reported with NGF treatment in aged rats (Chen et al., 1995).
Our results indicate that inactivation of a single allele of the NGF gene results in the loss of approximately one-third of septal cholinergic cells, with an equivalent reduction in hippocampal ChAT activity. These changes in the cholinergic system were accompanied by subtle but significant and reproducible deficits in performance on a task of learning and memory. Although the extent of cell loss (35%) matches the relative reduction in hippocampal content of NGF mRNA quite well (33%), it is less than might be predicted from the large reduction in NGF protein content within the hippocampus (75%). The differences between the ratios of mRNA to protein seen in thengf+/− versus ngf+/+ mice may reflect differences in the efficiency of translation, secretion, transport, or utilization of NGF protein. One caveat in the interpretation of the gene dosage effect is that NGF mRNA and protein determinations were performed on samples from adult animals, and as yet the time period of dependence of cholinergic neurons on NGF for survival is unknown. Thus, at present, the quantitative relationship between NGF availability and cholinergic cell survival and function cannot be deduced. Further studies in which both NGF mRNA and protein levels are determined at various points in development in both the basal forebrain and in target areas are needed.
The present findings provide direct evidence for the role of NGF as a survival factor for basal forebrain cholinergic neurons during development and further suggest that NGF plays a role in the mature nervous system to regulate the function of these cells. These observations underscore the importance of endogenous NGF to CNS function by indicating that even partial reduction in levels of this protein results in cell loss and measurable effects on learning and memory.
Footnotes
Received February 3, 1997; revised June 11, 1997; accepted July 18, 1997.
We thank Lanway Ling for assistance on the hot plate and tail flick tests, David Merrill for assistance on the water maze task, David Shelton for instruction in the ChAT assay, and Teddi Colbert and Hank La for animal care.
Correspondence should be addressed to Dr. Heidi S. Phillips, Genentech, Inc., 460 Point San Bruno Boulevard, South San Francisco, CA 94080.
REFERENCES
- 1.Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- 2.Aloe L, Cozzari C, Calissano P, Levi-Montalcini R. Somatic and behavioral postnatal effects of fetal injections of nerve growth factor antibodies in the rat. Nature. 1981;291:413–415. doi: 10.1038/291413a0. [DOI] [PubMed] [Google Scholar]
- 3.Chen KS, Gage FH. Somatic gene transfer of NGF to the aged brain: behavioral and morphological amelioration. J Neurosci. 1995;15:2819–2825. doi: 10.1523/JNEUROSCI.15-04-02819.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen KS, Masliah E, Mallory M, Terry RD, Gage FH. Synaptic loss in cognitively impaired aged rats is ameliorated by chronic human nerve growth factor infusion. Neuroscience. 1995;68:19–27. doi: 10.1016/0306-4522(95)00099-5. [DOI] [PubMed] [Google Scholar]
- 5.Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD, Phillips HS. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- 6.Dekker AJ, Gage FH, Thal LJ. Delayed treatment with nerve growth factor improves acquisition of a spatial task in rats with lesions of the nucleus basalis magnocellularis: evaluation of the involvement of different neurotransmitter systems. Neuroscience. 1992;48:111–119. doi: 10.1016/0306-4522(92)90342-y. [DOI] [PubMed] [Google Scholar]
- 7.Fischer W, Wictorin K, Bjorklund A, Williams LR, Varon S, Gage FH. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329:65–68. doi: 10.1038/329065a0. [DOI] [PubMed] [Google Scholar]
- 8.Fischer W, Bjorklund A, Chen KS, Gage FH. NGF improves spatial memory in aged rodents as a function of age. J Neurosci. 1991;11:1889–1906. doi: 10.1523/JNEUROSCI.11-07-01889.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer W, Chen KS, Gage FH, Bjorklund A. Progressive decline in spatial learning and integrity of forebrain cholinergic neurons in rats during aging. Neurobiol Aging. 1992;13:9–23. doi: 10.1016/0197-4580(92)90003-g. [DOI] [PubMed] [Google Scholar]
- 10.Fonnum F. A rapid radiochemical method for the determination of choline acetyltransferase. J Neurochem. 1974;24:407–409. doi: 10.1111/j.1471-4159.1975.tb11895.x. [DOI] [PubMed] [Google Scholar]
- 11.Gage FH, Armstrong DM, Williams LR, Varon S. Morphologic response of axotomized septal neurons to nerve growth factor. J Comp Neurol. 1988;269:147–155. doi: 10.1002/cne.902690112. [DOI] [PubMed] [Google Scholar]
- 12.Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 13.Gorin P, Johnson EM. Experimental autoimmune model of nerve growth factor deprivation: effects on developing peripheral sympathetic and sensory neurons. Proc Natl Acad Sci USA. 1979;76:5382–5386. doi: 10.1073/pnas.76.10.5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagg T, Manthorpe M, Vahlsing HL, Varon S. Delayed treatment with nerve growth factor reverses the apparent loss of cholinergic neurons after acute brain damage. Exp Neurol. 1988;101:303–312. doi: 10.1016/0014-4886(88)90013-1. [DOI] [PubMed] [Google Scholar]
- 15.Hedreen JC, Bacon JC, Price DL. A modified histochemical method to visualize acetylcholinesterase-containing axons. J Histochem Cytochem. 1985;33:134–140. doi: 10.1177/33.2.2578498. [DOI] [PubMed] [Google Scholar]
- 16.Hefti F. Nerve growth factor (NGF) promotes survival of septal cholinergic neurons after fimbrial transection. J Neurosci. 1986;6:2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hefti F, Hartikka J, Knusel B. Function of neurotrophic factors in the adult and aging brain and their possible use in the treatment of neurodegenerative diseases. Neurobiol Aging. 1989;10:515–533. doi: 10.1016/0197-4580(89)90118-8. [DOI] [PubMed] [Google Scholar]
- 18.Johnson EM, Jr, Gorin PD, Brandeis LD, Pearson J. Dorsal root ganglion neurons are destroyed by exposure in utero to maternal antibody to nerve growth factor. Science. 1980;210:916–918. doi: 10.1126/science.7192014. [DOI] [PubMed] [Google Scholar]
- 19.Kawaja MD, Gage FH. Reactive astrocytes are substrates for the growth of adult CNS axons in the presence of elevated levels of nerve growth factor. Neuron. 1991;7:1019–1030. doi: 10.1016/0896-6273(91)90346-2. [DOI] [PubMed] [Google Scholar]
- 20.Koliatsos VE, Nauta HJW, Clatterbuck RE, Holtzman DM, Mobley WC, Price DL. Mouse nerve growth factor prevents degeneration of axotomized basal forebrain cholinergic neurons in monkeys. J Neurosci. 1990;10:3810–3813. doi: 10.1523/JNEUROSCI.10-12-03801.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korsching S, Thoenen H. Nerve growth factor in sympathetic ganglia and corresponding target organs of the rat: correlation with density of sympathetic innervation. Proc Natl Acad Sci USA. 1983;64:3513–3516. doi: 10.1073/pnas.80.11.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kromer LF. Nerve growth factor treatment after brain injury prevents neuronal death. Science. 1987;235:214–216. doi: 10.1126/science.3798108. [DOI] [PubMed] [Google Scholar]
- 23.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 24.Levi-Montalcini R, Angeletti PU. Nerve growth factor. Physiol Rev. 1968;48:534–569. doi: 10.1152/physrev.1968.48.3.534. [DOI] [PubMed] [Google Scholar]
- 25.Levi-Montalcini R, Booker B. Destruction of the sympathetic ganglia in mammals by an antiserum to a nerve-growth protein. Proc Natl Acad Sci USA. 1960;46:384–391. doi: 10.1073/pnas.46.3.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Holtzman DM, Kromer LF, Kaplan DR, Chua-Couzens J, Clary DO, Knüsel B, Mobley WC. Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci. 1995;15:2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipp H-P, Stagliar-Bozizevic M, Wolfer DP. Behavior in large numbers of knockout mice: results, caveats and perspectives. Behav Genet. 1995;25:275. [Google Scholar]
- 28.Mobley WC, Rutkowski JL, Tennekoon GI, Gemski J, Buchanan K, Johnston MV. Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Mol Brain Res. 1986;1:53–62. doi: 10.1016/0169-328x(86)90020-3. [DOI] [PubMed] [Google Scholar]
- 29.Nabeshima T, Ogawa S, Ishimaru H, Kameyama T, Fukuta T, Takeuchi R, Hayashi K. Memory impairment and morphological changes in rats induced by active fragment of anti-nerve growth factor-antibody. Biochem Biophys Res Commun. 1991;175:215–219. doi: 10.1016/s0006-291x(05)81222-7. [DOI] [PubMed] [Google Scholar]
- 30.Olton DS, Wenk GL. Dementia: animal models of the cognitive impairments produced by degeneration of the basal forebrain cholinergic system. In: Meltzer HY, editor. Psychopharmacology: the third generation of progress. Raven; New York: 1987. pp. 941–953. [Google Scholar]
- 31.Ruit KG, Elliott JL, Osborne PA, Yan Q, Snider WD. Selective dependence of mammalian dorsal root ganglion neurons on nerve growth factor during embryonic development. Neuron. 1992;8:1–20. doi: 10.1016/0896-6273(92)90284-k. [DOI] [PubMed] [Google Scholar]
- 32.Thoenen H, Bandtlow C, Heumann R. The physiological significance of nerve growth factor in the central nervous system: comparison with the periphery. Rev Physiol Biochem Pharmacol. 1987;109:145–178. doi: 10.1007/BFb0031026. [DOI] [PubMed] [Google Scholar]
- 33.Van der Zee CE, Fawcett J, Diamond J. Antibody to NGF inhibits collateral sprouting of septohippocampal fibers following entorhinal cortex lesion in adult rats. J Comp Neurol. 1992;326:91–100. doi: 10.1002/cne.903260108. [DOI] [PubMed] [Google Scholar]
- 34.Van der Zee CE, Lourenssen S, Stanisz J, Diamond J. NGF deprivation of adult rat brain results in cholinergic hypofunction and selective impairments in spatial learning. Eur J Neurosci. 1995;7:160–168. doi: 10.1111/j.1460-9568.1995.tb01030.x. [DOI] [PubMed] [Google Scholar]
- 35.Vantini G, Schiavo N, Di Martino A, Polato P, Triban C, Callegaro L, Toffano G, Leon A. Evidence for a physiological role of nerve growth factor in the central nervous system of neonatal rats. Neuron. 1989;3:267–273. doi: 10.1016/0896-6273(89)90251-1. [DOI] [PubMed] [Google Scholar]
- 36.Whittemore SR, Seiger A. The expression, localization, and functional significance of β-nerve growth factor in the central nervous system. Brain Res Rev. 1987;12:439–464. doi: 10.1016/0165-0173(87)90008-7. [DOI] [PubMed] [Google Scholar]
- 37.Will B, Hefti F. Behavioral and neurochemical effects of chronic intraventricular injections of nerve growth factor in adult rats with fimbria lesions. Behav Brain Res. 1985;17:17–24. doi: 10.1016/0166-4328(85)90004-x. [DOI] [PubMed] [Google Scholar]
- 38.Williams LR, Varon S, Peterson GM, Wictorin K, Fisher W, Bjorklund A, Gage FH. Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria-fornix transection. Proc Natl Acad Sci USA. 1986;83:9231–9235. doi: 10.1073/pnas.83.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]