

Abstract
Ca2+-induced Ca2+ release (CICR) from intracellular stores amplifies the Ca2+signal that results from depolarization. In neurons, the amplification has been described as a graded process. Here we show that regenerative CICR develops as an all-or-none event in cultured rat dorsal root ganglion neurons in which ryanodine receptors have been sensitized to Ca2+ by caffeine. We used indo-1-based microfluorimetry in combination with whole-cell patch-clamp recording to characterize the relationship between Ca2+ influx and Ca2+ release. Regenerative release of Ca2+ was triggered when action potential-induced Ca2+ influx increased the intracellular Ca2+ concentration ([Ca2+]i) above threshold. The threshold was modulated by caffeine and intraluminal Ca2+. A relative refractory period followed CICR. The pharmacological profile of the response was consistent with Ca2+ influx through voltage-gated Ca2+ channels triggering release from ryanodine-sensitive stores. The activation of a suprathreshold response increased more than fivefold the amplitude and duration of the [Ca2+]i transient. The switch to a suprathreshold response was regulated very precisely in that addition of a single action potential to the stimulus train was sufficient for this transformation. Confocal imaging experiments showed that CICR facilitated propagation of the Ca2+ signal from the plasmalemma to the nucleus. This all-or-none reaction may serve as a switch that determines whether a given electrical signal will be transduced into a local or widespread increase in [Ca2+]i.
Keywords: Ca2+-induced Ca2+release, ryanodine receptors, dorsal root ganglion, intracellular Ca2+, Ca2+ stores, voltage-gated Ca2+ channels, nucleoplasmic Ca2+
Ca2+-induced Ca2+ release (CICR) describes a process in which an elevation in [Ca2+]i mediated by plasmalemmal Ca2+ channels subsequently activates ryanodine receptors to mobilize intracellular Ca2+stores (Endo et al., 1970; Ford and Podolsky, 1970; Fabiato, 1983). Ryanodine receptors are widely distributed in the brain, with the cardiac (type 2) isoform expressed most abundantly (McPherson et al., 1991; Lai et al., 1992; Furuichi et al., 1994). These receptors represent functional Ca2+ stores in neurons as indicated by Ca2+ mobilization evoked by methylxanthines, compounds known to sensitize ryanodine receptors (Smith et al., 1983; Thayer et al., 1988; Seymour-Laurent and Barish, 1995; Usachev and Verkhratsky, 1995). Ryanodine receptors are selectively modulated by and named after a plant alkaloid that blocks the channel at high concentrations (>100 μm) and locks the channel in an open subconductance state at lower concentrations (Fill and Coronado, 1988). On the basis of a sensitivity to ryanodine and other modulators of Ca2+ stores, it has been suggested that CICR plays a role in a number of neuronal processes, including cell excitability (Kuba, 1980; Currie and Scott, 1992), neurotransmission (Peng, 1996), cell development and differentiation (Holliday et al., 1991; Gomez et al., 1995), and synaptic plasticity (Obenaus et al., 1989; Reyes and Stanton, 1996; Wang et al., 1996). Although functional ryanodine-sensitive stores are present in neurons and seem to participate in physiological processes, it is not clear how the stores contribute functionally to Ca2+signaling.
CICR is initiated in heart muscle when an elevation in [Ca2+]i mediated by L-type Ca2+ channels activates ryanodine receptors (Cannell et al., 1995; Lopez-Lopez et al., 1995). Single-channel recordings have shown that ryanodine receptors isolated from brain and incorporated into planar lipid bilayers are also activated by cytosolic Ca2+ (Bezprozvanny et al., 1991; Lai et al., 1992). Depolarization-induced Ca2+ influx will release Ca2+ from ryanodine-sensitive stores in sympathetic (Hua et al., 1993), central (Llano et al., 1994), and sensory neurons (Shmigol et al., 1995). Release of Ca2+ from the store increased in a graded manner with increasing stimulus strength, suggesting that CICR in neurons was a simple amplifier of Ca2+ influx (Hua et al., 1993; Kostyuk and Verkhratsky, 1994; Berridge et al., 1995). In the presence of caffeine, however, [Ca2+]i was induced to oscillate, indicating that under suitable conditions Ca2+ release could be regenerative (Lipscombe et al., 1988; Kostyuk et al., 1991; Friel and Tsien, 1992). Such regenerative responses support the concept of a positive feedback loop previously postulated for muscle, in which released Ca2+ triggers additional Ca2+release by interacting with neighboring ryanodine receptors (Endo et al., 1970; Ford and Podolsky, 1970). This feed-forward amplification would be predicted to produce responses that were all-or-none events. Indeed, all-or-none increases in [Ca2+]i have been described for snail neurons (Kostyuk et al., 1989; Mironov and Usachev, 1990).
In this report we show that all-or-none CICR can be evoked in mammalian sensory neurons by short bursts of action potentials. We used indo-1-based microfluorimetry in combination with whole-cell patch-clamp recording to characterize the relationship between Ca2+ influx and Ca2+ release. CICR displayed a discrete threshold for activation that was subject to modulation. Recruitment of regenerative CICR increased severalfold the amplitude and duration of the [Ca2+]itransient, and confocal imaging experiments showed that it facilitated inward propagation of the [Ca2+]i rise throughout the soma. Ryanodine-sensitive stores can function in neurons as a switch that regulates coupling between the plasma membrane and intracellular signaling. Selective modulation of the threshold provides a precise tuning of this coupling.
A preliminary report of this work has been published previously (Usachev and Thayer, 1996).
MATERIALS AND METHODS
Cell culture. Rat dorsal root ganglion (DRG) neurons were grown in primary culture as described previously (Thayer and Miller, 1990). In brief, DRG neurons were dissected from the thoracic and lumbar regions of 1- to 3-d-old Sprague Dawley rats. Ganglia were then incubated at 37°C in collagenase–dispase (0.8 and 6.4 U/ml, respectively) for 45 min. Ganglia were dissociated by trituration through a flame-constricted pipette and then plated onto laminin-coated (50 μg/ml) glass coverslips (25 mm diameter). Cells were grown in Ham’s F12 media supplemented with 5% heat-inactivated horse serum, 50 ng/ml NGF, 4.4 mm glucose, 2 mml-glutamine, modified Eagle’s medium vitamins, and penicillin–streptomycin (100 U/ml and 100 μg/ml, respectively). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2. Cells were used on the second and third day after plating.
Simultaneous whole-cell patch-clamp and microfluorimetric recording. Electrical measurements and [Ca2+]i were recorded from cultured DRG neurons by using the whole-cell patch-clamp technique (Hamill et al., 1981) in combination with indo-1-based microfluorimetry (Grynkiewicz et al., 1985). The instrumentation has been described previously in detail (Werth et al., 1996). Cells were placed in a flow-through chamber (Thayer and Miller, 1990) (10 sec solution exchange) that was mounted on the stage of an inverted epi-fluorescence microscope (Leitz, Wetzlar, Germany) equipped with a 70× objective [numerical aperture (NA) = 1.15]. Indo-1 was loaded into the cells via the patch pipette. The dye was excited at 350 (10) nm, and emission was detected at 405 (20) and 490 (20) nm. Fluorescence was monitored by a pair of photomultiplier tubes (Thorn, EMI, Fairfield, NJ) operating in photon-counting mode. The 5 V output signals were then integrated by 8-pole Bessel filters and digitized, along with patch-clamp data (PC501 amplifier, Warner Instrument, Hamden, CT), at 1 kHz with an analog-to-digital converter (Indec Systems, Sunnyvale, CA). Data were stored and analyzed on an IBM-compatible computer. Patch pipettes were pulled from borosilicate glass (2–4 MΩ; Narishige, Tokyo, Japan) on a Sutter Instrument (Novato, CA) P-87 micropipette puller and filled with the following solution (in mm): potassium gluconate 125, KCl 10, Mg-ATP 3, MgCl2 1, HEPES 10, indo-1 0.1, pH 7.25 with KOH, 290 mOsm/kg with sucrose. Extracellular recording solution contained (in mm): NaCl 140, KCl 5, CaCl2 2, MgCl2 1, HEPES 10, glucose 10, pH 7.35 with NaOH, 310 mOsm/kg with sucrose. To isolate Ca2+currents from other currents, Cs+ was substituted for K+ in the pipette solution and extracellular Na+ and K+ were replaced with TEA+.
Fluorescence changes were converted to [Ca2+]i by using the formulaKdβ(R −Rmin)/(Rmax −R), where R is 405/490 nm fluorescent ratio (Grynkiewicz et al., 1985). The dissociation constant used for indo-1 was 250 nm, and β was the ratio of fluorescence emitted at 490 nm and measured in the absence and presence of Ca2+. Rmin,Rmax, and β were determined in intact cells by applying 10 μm ionomycin in Ca2+-free buffer (1 mm EGTA) and saturating Ca2+ (5 mmCa2+). Values forRmin, Rmax, and β ranged from 0.26 to 0.28, 2.0 to 2.4, and 2.7 to 3.0, respectively. Background light levels were collected in the cell-attached configuration.
Data are presented as mean ± SEM.
Confocal imaging of intracellular Ca2+.[Ca2+]i imaging was performed on an upright Olympus AX70 microscope (Olympus Optical, Tokyo, Japan) equipped with the Bio-Rad MRC 1024 laser-scanning confocal imaging system (Bio-Rad, Hercules, CA) and an Olympus 60× water-immersion objective (NA = 0.90). Cells were loaded with indicator by incubation in media containing 5 μm calcium green-1 AM and 2% (w/w) pluronic F-127 for 30 min at room temperature. The dye was excited with a krypton–argon laser at 488 nm and detected at 522 (32) nm. The iris was set to 2 mm, providing an estimatedz-axis resolution of <1 μm. Images of 128 × 128 pixels were collected at 2 Hz. Each pixel corresponded to ∼0.28 μm. To decrease noise, each experiment was repeated twice, and the corresponding images were averaged. Images were then spatially filtered using a 3 × 3 median filter (MetaMorph 2.5 software). [Ca2+] was presented as the intensity of calcium green-1 fluorescence normalized to that in a resting cell (F/F0). All measurements were corrected for background fluorescence. Transient elevations in [Ca2+] were evoked by extracellular field stimulation. Field potentials were generated by passing current between two platinum electrodes via a Grass S44 stimulator and a stimulus isolation unit (Quincy, MA) as described previously (Werth et al., 1996).
Reagents. Indo-1, calcium green-1 AM, and pluronic F-127 were obtained from Molecular Probes (Eugene, OR). Cyclic ADP-ribose (cADPr) was a gift from Dr. T. F. Walseth (University of Minnesota). All other reagents were purchased from Sigma (St. Louis, MO).
RESULTS
Elevation of [Ca2+]i above threshold triggers regenerative CICR
[Ca2+]i transients were recorded from large (28–34 μm diameter) DRG neurons with indo-1-based microfluorimetry. Resting membrane potential measured at the beginning of each experiment was −56.4 ± 0.2 mV (n = 141). Cells were voltage-clamped to −56 mV using the whole-cell configuration of the patch-clamp technique. Recordings were performed in 5 mm caffeine, which readily crosses the plasmalemma, to increase the sensitivity of ryanodine receptors to cytosolic Ca2+ (Usachev et al., 1993). Initial application of caffeine produced a transient increase in [Ca2+]i in most cells that completely recovered to basal levels in 3–6 min. In 14% of neurons (n = 141) no significant change in [Ca2+]i was detected. Recording was started 10 min after the addition of caffeine. Depolarization to −20 mV activated voltage-gated Ca2+ influx, resulting in an increase in [Ca2+]i that rose linearly with the duration of the depolarizing step (Fig.1). Repolarization terminated the increase and [Ca2+]i recovered to baseline (Fig. 1, trace 1). When the duration of the depolarizing pulse was sufficient to elevate [Ca2+]i above 124 ± 5 nm (n = 11) (Fig. 1, horizontal arrow), termination of the stimulus did not stop the [Ca2+]i increase, which continued to rise for 5–10 sec to peak at 376 ± 12 nm(n = 11) (Fig. 1, trace 2). This poststimulus [Ca2+]i rise was regenerative and independent of Ca2+ influx, because Ca2+ channels were deactivated by repolarization. A further increase in the duration of the pulse did not change the amplitude of the [Ca2+]i transient (Fig. 1, compare traces 2 and 3). Thus, [Ca2+]i increased in proportion to the length of the depolarizing pulse until threshold was reached, at which point a maximal [Ca2+]i response was triggered. [Ca2+]i transients of intermediate amplitude, with the peaks falling between 104 ± 4 nm (subthreshold response) and 376 ± 12 nm (n = 11; maximal response), could not be elicited under these conditions.
Fig. 1.

Elevating [Ca2+]iabove threshold triggers a regenerative [Ca2+]i response. A large DRG neuron was voltage-clamped at −56 mV in the presence of 5 mmcaffeine. The membrane was depolarized to −20 mV for 36 sec (trace 1), 39 sec (trace 2), or 68 sec (trace 3). If the [Ca2+]i rose above threshold (horizontal arrow), a regenerative [Ca2+]i response developed (traces 2 and 3, but not1). The dotted lines indicate repolarization. The inset provides an example of the all-or-none generation of action potentials. Depolarizing current pulses (700 pA) were applied to a DRG neuron held in whole-cell current clamp. The dotted lines indicate termination of the pulses. The threshold for triggering an action potential is indicated by the horizontal arrow.
This all-or-none [Ca2+]i transient resembles the generation of the action potential. For example, injection of current into DRG neurons produced a graded increase in membrane potential (Fig. 1, inset). When the duration of the current injection was sufficient to increase membrane potential above threshold (Fig. 1, horizontal arrow in inset), an action potential was generated. The amplitude of the action potential was constant and did not depend on the duration of a suprathreshold current injection.
We found that like the action potential, regenerative [Ca2+]i transients displayed a discrete threshold for activation and that once elicited, a full-size response developed. To understand the mechanism controlling this regenerative increase in [Ca2+]i, we studied the relationship between Ca2+ influx and corresponding changes in [Ca2+]i. We simultaneously measured Ca2+ currents (ICa) and [Ca2+]i, which allowed us to control the duration and magnitude of Ca2+ influx. [Ca2+]i transients were elicited by step depolarizations of various duration to +10 mV from a holding potential of −60 mV (Fig. 2). The increase in [Ca2+]i was plotted as a function of electrical charge transferred by Ca2+(∫ICadt). In the absence of caffeine, [Ca2+]i rose in proportion to Ca2+ influx (Fig. 2A,D, open circles), suggesting that voltage-gated Ca2+channels mediated the [Ca2+]iincrease. In the presence of 5 mm caffeine (Fig.2B,D, solid squares), the [Ca2+]i elevation was proportional to Ca2+ influx for stimuli shorter than 30 msec. For test pulses longer than 30 msec (Ca2+ load ≥ 35 ± 3 pC; n = 7), the amplitude of the response jumped approximately fivefold and then showed a modest dependence on further increases in Ca2+ influx. For these full-size responses, [Ca2+]i rose regeneratively for 5–10 sec after termination of the Ca2+ current (Fig. 2C). Increasing [Ca2+]i over this range corresponds to a steep increase in the open probability of cerebellar ryanodine-sensitive Ca2+ release channels (Bezprozvanny et al., 1991) and a strong Ca2+-dependent facilitation of ryanodine binding to the receptor in sympathetic neurons (Hernandez-Cruz et al., 1995). The discontinuity in the relationship between Ca2+influx and changes in [Ca2+]isuggested that the switch to the full-size response resulted from recruitment of CICR from intracellular stores. Once triggered by elevation of [Ca2+]i above threshold (Fig. 2C, horizontal arrow) the release became regenerative and developed independent of Ca2+ influx.
Fig. 2.

Combined [Ca2+]iand ICa recordings indicate activation of regenerative CICR. A, B, [Ca2+]i transients andICa were elicited by step depolarizations of various duration from −60 to +10 mV in the absence (A) or presence (B) of 5 mm caffeine. The duration of the depolarization is indicated above the current traces. Pulses were applied every 2 min. The traces are presented in order of increasing pulse duration, although long and short stimuli were intermixed in the actual experiment. The insets show correspondingICa on an expanded time scale. Thevertical and horizontal bars in theinsets correspond to 1 nA and 100 msec, respectively.C, The traces produced by a 30 msec depolarization inB are displayed on an expanded time scale. Note that the rapid Ca2+ influx phase during the test pulse is followed by the regenerative phase that develops after termination of the stimulus. The horizontal arrow indicates the threshold [Ca2+]i for activation of a regenerative [Ca2+]i rise. Thedotted line indicates cessation ofICa. D, The dependence of the amplitude of the [Ca2+]i transient on Ca2+ influx is plotted for the same cell in the absence (open circles) and presence (solid squares) of 5 mm caffeine. Ca2+influx was quantified as electric charge transferred by Ca2+ during the stimulus (∫ICadt). This plot is representative of seven experiments. The increases in [Ca2+]i produced by influx and release do not sum, because the two processes peak at different times. Depolarization-induced [Ca2+]itransients peaked 0.4–0.8 sec after the stimulus, whereas CICR peaked 5–10 sec after activation.
Regenerative CICR increases the size and duration of [Ca2+]i transients evoked by trains of action potentials
We next tested whether all-or-none CICR could be evoked by a series of action potentials. [Ca2+]itransients were evoked by trains of action potentials (2 Hz, 0.5–10 sec) in the presence of 5 mm caffeine (Fig.3). The number of action potentials was used as an index of Ca2+ influx. Similar to the experiments described in Figure 2, the increase in [Ca2+]i for subthreshold stimuli was proportional to the number of action potentials (Fig. 3A, transients elicited by 2, 4, and 6action potentials). On reaching threshold (Fig. 3A, horizontal dashed line), the [Ca2+]i was amplified significantly (compare transient elicited by 6 to that resulting from 7 action potentials) and then remained constant (compare transients elicited by 7, 10, 15, and20 action potentials). The threshold, defined as the [Ca2+]i at the end of the minimal stimulus that elicited a full-size response, was 127 ± 5 nm (n = 26). This trigger was exquisitely sensitive in that adding a single action potential to a 2 Hz burst was sufficient to evoke regenerative CICR. Regenerative [Ca2+]i responses were observed in 92% of large DRG neurons tested (n = 153). Only 1 of 12 small diameter (17–22 μm) DRG neurons that were examined demonstrated regenerative CICR. The action potential was more broad and elicited changes in [Ca2+]i that were significantly greater in small (40 ± 9 nm;n = 8) relative to large DRG neurons (10 ± 1 nm; n = 19). This suggests that Ca2+ signaling in small cells with a large surface-to-volume ratio is predominantly mediated by Ca2+ influx and in large cells with a small surface-to-volume ratio, Ca2+ stores transmit the Ca2+ signal. Activation of CICR resulted in significant changes in the size, shape, and duration of [Ca2+] transients. These characteristics are summarized in Figure 3. The amplitude of [Ca2+]i transients (Δ[Ca2+]i) increased from 61 ± 4 to 306 ± 14 nm (n = 26) (Fig. 3C). The time (Δ t) from termination of the stimulus to the peak of the [Ca2+]i transient extended from 1.2 ± 0.1 sec in the absence to 16.1 ± 1.8 sec on activation of CICR (n = 26) (Fig. 3D). Recovery of [Ca2+]i transients elicited by Ca2+ influx alone were fitted well by a single exponential with a half-recovery time (t1/2) of 9.8 ± 2 sec (n = 26). In contrast, after the recruitment of CICR, the recovery process had more complex kinetics and was slower, with at1/2 of 64.2 ± 6.3 sec (n= 26) (Fig. 3E). These data show that once activated, CICR became the predominant factor determining the size, shape, and duration of [Ca2+]i transients elicited by bursts of action potentials.
Fig. 3.

Recruitment of regenerative CICR significantly modifies action potential-elicited [Ca2+]i responses. A, Regenerative [Ca2+]i transients were elicited by 2 Hz trains of action potentials (APs) in the presence of 5 mm caffeine. Action potentials were evoked in current-clamp, and the number of action potentials in each stimulus train is indicated above the voltage trace. Thehorizontal dashed line indicates the threshold [Ca2+]i for triggering regenerative CICR. Trains of action potentials were evoked every 3 min. The traces are presented in order of increasing number of action potentials, although long and short stimulus trains were intermixed in the actual experiment. B, An action potential-induced [Ca2+]i transient is displayed on an expanded time scale. [Ca2+]itransients for sub- and suprathreshold stimuli were compared for changes in amplitude (Δ[Ca2+]i), the time between termination of the stimulus and the peak [Ca2+]i (Δt), and the time required for [Ca2+]i to recover from the peak to half of its amplitude (t1/2). C–E, Comparison of Δ[Ca2+]i (C), Δt (D), andt1/2 (E) for responses that were below (sub.) or above (CICR) the threshold for regenerative CICR. All three parameters increased significantly on activation of CICR (n = 26; p< 0.001; paired Student’s t test).
Pharmacology of the regenerative [Ca2+]i response
If the switch from a subthreshold to a full-size [Ca2+]i response is caused by activation of CICR, this process should be sensitive to ryanodine and antagonists of voltage-gated Ca2+ channels. We characterized the pharmacology of the all-or-none [Ca2+]i response. Trains of action potentials in the presence of 5 mm caffeine were used to elicit regenerative changes in [Ca2+]ibefore and after the addition of drugs (Fig.4). A suprathreshold stimulus was chosen at the beginning of each recording. For the experiment shown in Figure4A, a train of 10 action potentials applied at 2 Hz was found to be above threshold. When applied at 4 min intervals, this stimulus elicited reproducible regenerative responses (484 and 496 nm at the peaks). The first stimulus applied in the presence of 10 μm ryanodine elicited a [Ca2+]i rise comparable to control, which is consistent with the use-dependence of ryanodine effects (Thayer et al., 1988). The rate of recovery slowed, suggesting that ryanodine impaired the ability of the stores to sequester Ca2+. A subsequent stimulus in the presence of drug produced a [Ca2+]i transient that peaked at 112 nm, which although significantly smaller was above threshold for eliciting CICR in this cell. Increasing the number of action potentials in the stimulus train to 30 increased the amplitude of [Ca2+]i response, although CICR was not recruited. In the presence of ryanodine, [Ca2+]i always began to return to basal levels within a second after termination of the stimulus, indicating a failure to trigger a regenerative [Ca2+]i rise (n = 6). The effect of ryanodine did not reverse during a 30 min wash period.
Fig. 4.

Ryanodine and Cd2+ prevent triggering of a regenerative [Ca2+]iresponse. A, Ryanodine irreversibly blocks triggering of a regenerative [Ca2+]i response. [Ca2+]i transients were elicited by action potentials (APs) applied at 2 Hz. The number of action potentials is indicated above the voltage trace. Treatment with 10 μm ryanodine is indicated by thehorizontal bar. This experiment is representative of six replicates in which ryanodine completely blocked CICR.B, Cd2+ blocks action potential-induced CICR. Cd2+ (200 μm) was applied at the time indicated by the horizontal bar. [Ca2+]i transients were elicited by action potentials applied at 1 Hz, except for the train of 50 action potentials that was elicited at 5 Hz. The number of action potentials is indicated above the voltage trace.
If CICR were triggered by depolarization-evoked Ca2+influx, it would be predicted to be inhibited by blockers of voltage-gated Ca2+ channels. In Figure4B, reproducible full-size [Ca2+]i responses were elicited by 1 Hz trains of 10 action potentials. Cd2+ (200 μm) blocked this response, indicating that Ca2+ influx through voltage-gated Ca2+ channels was required to trigger CICR. DRG neurons express multiple types of voltage-gated Ca2+channels (Nowycky et al., 1985; Scroggs and Fox, 1992). We tried to identify more specifically a particular role for Ca2+ channel subtypes in triggering regenerative CICR. In cardiac muscle, L-type channels were found to couple specifically with ryanodine receptors to activate CICR (Cannell et al., 1995; Lopez-Lopez et al., 1995). Although neurons predominantly express the cardiac isoform of ryanodine receptor, we did not find a significant effect of the L-type channel antagonist nimodipine (2 μm) on CICR (n = 4). Activation of regenerative CICR was blocked by ω-conotoxin GVIA (1 μm; n = 4; data not shown), implying participation of N-type channels. The response, however, was not linked exclusively to N-type channels, because the block produced by ω-conotoxin could be overcome by increasing the stimulus strength. Thus, a preferential source of Ca2+ for triggering CICR was not identified. These findings are consistent with observations that L-type channels are weakly expressed in large DRG neurons (Scroggs and Fox, 1992) and not essential in mediating action potential-induced [Ca2+]i increases in these cells (Piser et al., 1994). The threshold [Ca2+]i required for eliciting CICR was not affected by inhibition of Ca2+ channels.
Modulation of the threshold for eliciting regenerative CICR by caffeine
Caffeine increases the sensitivity of ryanodine receptors to Ca2+ (Rousseau and Meissner, 1989; Sitsapesan and Williams, 1990), and regenerative CICR results from the activity of multiple ryanodine receptors releasing Ca2+ in synchrony. Thus, sensitization of single releasing channels to Ca2+ by caffeine is predicted to decrease the threshold [Ca2+]i required to trigger regenerative CICR. We tested this idea by studying the effect of various caffeine concentrations on the threshold at which CICR was triggered. The threshold was determined by applying short (1–2 sec) trains of action potentials at a frequency of 2 Hz separated by 2.5 sec rest periods (Fig. 5A). [Ca2+]i began to decline within 1 sec after the end of each train unless the threshold [Ca2+]i concentration was reached. Thus, the [Ca2+]i at the end of the train that was followed by a regenerative rise in [Ca2+]i was defined as threshold (indicated by horizontal arrows in Fig. 5A). Successive [Ca2+]i transients were separated by 3 min intervals to ensure replenishment of Ca2+ stores. As shown in Figure 5, increasing the caffeine concentration from 2.5 to 5 mm decreased the threshold from 150 ± 11 to 112 ± 9 nm(n = 8; p < 0.05; paired Student’st test). The dependence of the threshold on caffeine concentration is summarized in Figure 5B. For concentrations ≤2.5 mm, the initial application of caffeine did not increase [Ca2+]i, although CICR was evoked by depolarization in these cells. Only 36% of cells (n = 14) that exhibited regenerative CICR in 5 mm caffeine also responded in 1 mm caffeine. These data show that the threshold for eliciting CICR is susceptible to modulation, identifying a novel mechanism by which the transduction of electrical stimuli into a second messenger response can be regulated.
Fig. 5.

Caffeine decreases the threshold for eliciting a regenerative response. A, [Ca2+]i transients were elicited in the presence of 2.5 or 5.0 mm caffeine as indicatedabove the traces. Trains (2 Hz) of four action potentials were delivered every 2.5 sec until a regenerative rise in [Ca2+]i had started. Thehorizontal arrows indicate the threshold [Ca2+]i for triggering CICR. Thevertical dotted lines mark termination of the stimulus.B, Dependence of the threshold on caffeine concentration obtained for 5–14 cells. Data points are mean ± SEM for responding cells.
We explored the possibility that endogenous modulators of the receptor may adjust the threshold for CICR, similar to the actions of caffeine. A likely candidate for such a modulator is cADPr, a metabolite of nicotinamide-adenine dinucleotide that regulates the release of Ca2+ from ryanodine-sensitive Ca2+ stores in sea urchin eggs (Lee et al., 1989;Galione et al., 1991) and other tissues (Lee et al., 1994b). We were not able to evoke regenerative CICR when cADPr was applied through the patch pipette at concentrations of 1 or 10 μm(n = 9). We cannot rule out a role for cADPr in regulating all-or-none CICR in DRG neurons and continue to explore recording conditions that might reveal an effect. Alternatively, cADPr might not act as a modulator of ryanodine receptor isoform 2 in situ. Single-channel studies on cardiac ryanodine receptors found that cADPr competed with ATP for the same binding site on the receptor and thus was ineffective at physiological levels of ATP (Sitsapesan et al., 1995).
Activation of CICR facilitates spread of the [Ca2+]i elevation throughout the soma
The regenerative nature of the CICR described here suggests that like the action potential, it too may facilitate the spread of information. The spatial distribution of the [Ca2+]i signal depends on the source of Ca2+. Depolarization-induced Ca2+ influx results in an elevation in [Ca2+]i localized initially under the plasma membrane (Hernandez-Cruz et al., 1990; Blumenfeld et al., 1992), followed by diffusion toward the center of the soma. Powerful buffering, sequestration, and efflux processes (Miller, 1991; Werth et al., 1996) severely restrict the distance that Ca2+signals will travel. Therefore, depolarization-induced increases in [Ca2+]i diminish as they spread deeper into the soma. Ryanodine receptors are distributed throughout the soma in central and peripheral neurons (Marrion and Adams, 1992;Seymour-Laurent and Barish, 1995; our unpublished observations). We explored the possibility that recruitment of regenerative CICR would transform passive diffusion into active propagation of the Ca2+ signal throughout the soma, providing a widespread rather than localized elevation in [Ca2+]i.
We used confocal microscopy to image changes in [Ca2+]i in DRG somata produced by action potentials elicited by extracellular field stimulation (Werth et al., 1996). In the absence of caffeine (control), brief trains of action potentials evoked an increase in [Ca2+]i that was ∼40% lower near the center of the soma relative to the rim (Fig.6A,B,F). The nucleoplasmic Ca2+ concentration was comparable to [Ca2+]i in adjacent cytosolic regions (Fig. 6F), consistent with the idea that Ca2+ freely diffuses through the pores of the nuclear envelope (Al-Mohanna et al., 1994; O’Malley, 1994). A similar spatial distribution was observed when a subthreshold stimulus was applied in the presence of 5 mm caffeine (data not shown). When the stimulus was increased above threshold, Ca2+ influx triggered CICR (Fig. 6C,D). The [Ca2+]i gradient was detected in the image that was captured 0.5 sec after the stimulus, but subsequent frames described a regenerative rise in [Ca2+]i that developed homogeneously throughout the soma (Fig. 6C,D). The increases in [Ca2+]i at the center of the cell and in the nucleus became much greater and were indistinguishable from Ca2+ levels at the rim. As summarized in Figure6F, the ratio of the [Ca2+]i at the center of the soma and in the nucleus relative to the rim were 0.56 ± 0.09 and 0.59 ± 0.13, respectively, in control conditions and increased to 0.96 ± 0.05 and 0.90 ± 0.05 (n = 6) when regenerative CICR was activated. These observations demonstrate that activation of regenerative CICR enhances propagation of Ca2+signals initiated at the plasmalemma throughout the soma and into the nucleus.
Fig. 6.

Regenerative CICR facilitates propagation of the Ca2+ signal from the plasmalemma to the center of the soma and into the nucleus. A, C, Calcium green-1-based confocal images show the distribution of [Ca2+] during electrical stimulation for the same cell in the absence (A) and presence (C) of 5 mm caffeine. Fluorescent intensity was normalized to that at rest (F/F0) and used as an index of [Ca2+]. The horizontal color bar indicates relative fluorescence intensity. Elevations in [Ca2+] were elicited by extracellular field stimulation applied at 5 Hz for 0.9 sec (A) or 0.4 sec (C). The stimulus strength was adjusted to produce comparable peak [Ca2+] levels. Times at which the images were captured are indicated under the corresponding images. The resting images were taken immediately before stimulation. B, D, The time course of the changes in [Ca2+] is plotted in the absence (B) and presence (D) of 5 mm caffeine. [Ca2+] was measured at the rim (red), center (green), and nucleus (black) of the cell as shown inE. The duration of the stimulus is indicatedunder each plot. The vertical arrowsindicate the times at which the images presented in Aand C were captured. E, Raw calcium green-1 fluorescence for the cell described in A–D.Rectangles indicate areas for which changes in [Ca2+]i are plotted inB and D. The intensity of fluorescence in a resting cell is much higher in the nucleus (O’Malley, 1994), enabling its unambiguous identification. F, Changes in [Ca2+] in the center of the soma and in the nucleus are compared with those at the rim when CICR was (open bar) and was not (solid bar) recruited (n = 6). *p < 0.05; **p < 0.01; paired Student’s ttest.
Intraluminal Ca2+ modulates the threshold for eliciting regenerative CICR
Similar to the action potential, a refractory period followed generation of all-or-none CICR. This was demonstrated using a paired-pulse protocol (Fig.7A). The first (control) stimulus activated regenerative CICR-depleting ryanodine-sensitive Ca2+ stores. After a delay, a second (test) stimulus of the same intensity was applied. If the time between responses was insufficient to replenish the stores (<30 sec for this cell), a regenerative [Ca2+]i response did not develop (Fig. 7B). One explanation for this phenomenon is that the threshold depends on intraluminal calcium such that the threshold increases for depleted stores, requiring a stronger stimulus to elicit CICR. We tested this possibility by measuring the threshold for activation of CICR for depleted and completely replenished stores. As described above (Fig. 5), the threshold was defined as the [Ca2+]i at the end of the stimulus that evoked a regenerative rise in [Ca2+]i. The degree to which Ca2+ stores were filled was varied by changing the delay between stimuli; 3 min provided complete replenishment of the stores. For full stores, regenerative release was induced by a series of action potential trains stimulated at 1 Hz and had a threshold of 121 ± 8 nm (n = 7) (Fig. 7C, horizontal arrows). To elicit a regenerative response immediately after recovery of the previous [Ca2+]itransient, the frequency of stimulation was increased to 5 Hz. Despite incomplete refilling of the stores, release was still activated, but the threshold increased significantly to 213 ± 16 nm(n = 7) (Fig. 7C,D). An alternative means to reduce the amount of Ca2+ accumulated in the stores is to treat with cyclopiazonic acid (CPA), a specific inhibitor of endoplasmic reticulum Ca2+-ATPases (Thomas and Hanley, 1994). We compared the threshold after a 180 sec delay in the absence or presence of CPA (Fig. 7E,F). After treatment with 1 μm CPA, the threshold for eliciting CICR increased significantly from 122 ± 14 to 174 ± 9 nm (n = 6; p < 0.001; paired Student’s t test). A regenerative [Ca2+]i response could not be evoked if neurons were incubated in CPA at concentrations of 5 μm or higher, presumably because the store could not refill at all. Thus, the threshold is subject to modulation by intraluminal Ca2+. A relative refractory period after all-or-none CICR seems to be a consequence of an increase in the threshold [Ca2+]i when the Ca2+ level inside the stores is low.
Fig. 7.
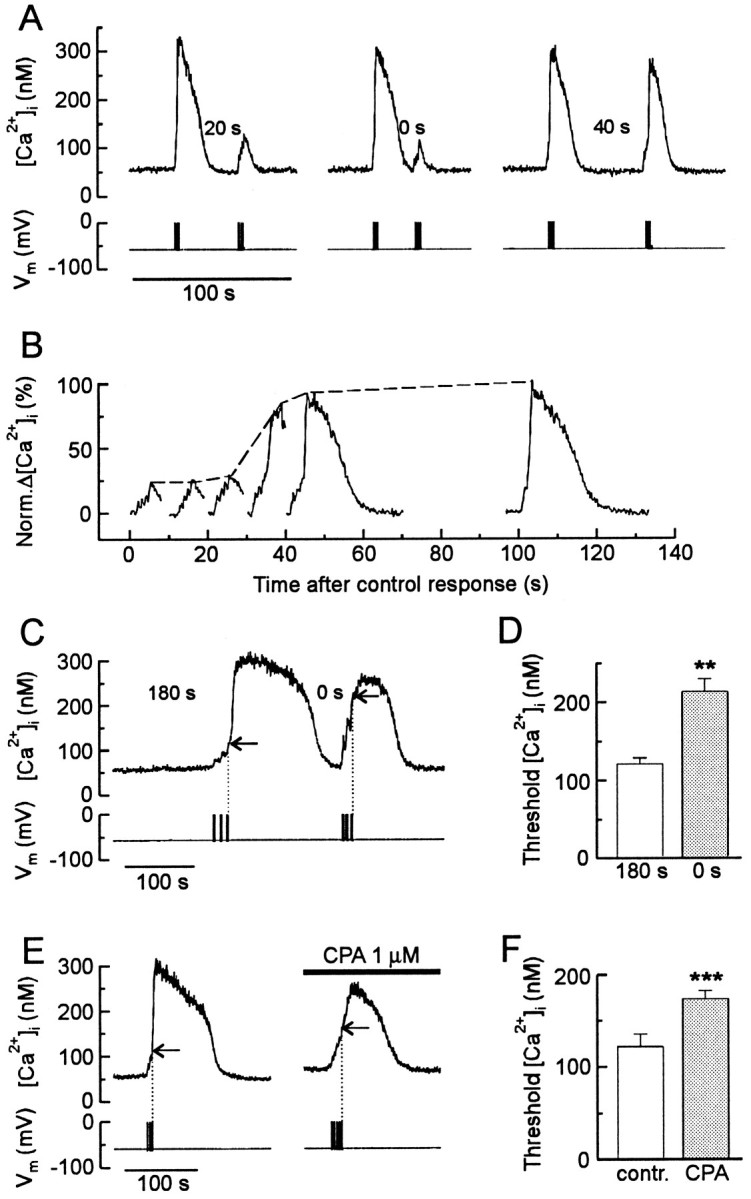
CICR is refractory after depletion of the Ca2+ store. A, A paired-pulse protocol was used to study the effects of refilling the Ca2+ stores on CICR. In the presence of 5 mm caffeine, a train of action potentials (1 Hz, 4 sec) elicited reproducible regenerative [Ca2+]i transients when the [Ca2+]i was returned to basal levels for 40 sec. Applying a test stimulus 0 or 20 sec after recovery of the control response failed to elicit a regenerative response.B, A series of test responses elicited at various times after recovery of the control response are shown on the same time scale. Graded responses were not observed. C, The threshold for regenerative CICR was elevated in cells with depleted Ca2+ stores. Representative traceshows initial control response (180 sec recovery time) followed by response evoked with no time at rest (0 sec recovery time).Horizontal arrows indicate threshold in the cell when stores are full (stimulus = 3 bursts of 3 sec at 1 Hz) and depleted (stimulus = 3 bursts of 2.5 sec at 5 Hz).D, Histogram displays average threshold [Ca2+]i from seven experiments such as the one described in C. **p < 0.01; paired Student’s t test. E, The threshold for regenerative CICR was elevated when refilling of intracellular Ca2+ stores was slowed by treatment with cyclopiazonic acid (CPA). Representative traces show test responses rested for 180 sec in the absence (left) or presence (right) of 1 μm CPA. Horizontal arrows indicate threshold for CICR in the absence (stimulus = 3 bursts of 1.5 sec at 2 Hz) and presence (stimulus = 5 bursts of 1.5 sec at 2 Hz) of CPA. F, Histogram displays average threshold [Ca2+]i from six experiments such as the one described in E. ***p < 0.001; paired Student’s t test.
DISCUSSION
Recruitment of all-or-none CICR transforms the size and shape of [Ca2+]i transients
In this report we describe a form of CICR in neurons that develops as an all-or-none response. On activation of CICR, the stores contributed 80% of the total [Ca2+]ielevation evoked by membrane depolarization, similar to that seen in cardiac myocytes (Wier, 1990; Lipp et al., 1992). For subthreshold responses, [Ca2+]i began a monoexponential recovery to basal levels within 1 sec of termination of the stimulus, presumably caused by Ca2+ efflux via the plasma membrane Ca2+ ATPase (Werth et al., 1996). For suprathreshold stimuli, however, the response was regenerative, as indicated by a continued increase in [Ca2+]i for several seconds after Ca2+ influx had ceased. This provided an easily discerned temporal separation of the portion of the increase in [Ca2+]i produced by influx from that contributed by release (Fig. 2C). A brief poststimulus increase in the average [Ca2+]i might result from local saturation of the indicator followed by the redistribution of Ca2+. For the size of cells used in this study, however, the increase would not be predicted to exceed 500 msec (Hernandez-Cruz et al., 1990; Blumenfeld et al., 1992). Furthermore, the regenerative phase of the [Ca2+]i transient was blocked by ryanodine. After the rising phase, [Ca2+]i declined slowly, possibly because of continued release countered by Ca2+efflux. Then, after ∼60 sec, the rate of recovery increased, producing a rapid falling phase to terminate the response. This final recovery seems to result in part from reuptake of Ca2+ into the store, as indicated by the marked slowing of this phase produced by ryanodine (Fig. 4A) or CPA (Fig. 7E). The marked changes in the size and shape of the [Ca2+]i transient that result from the activation of regenerative CICR modifies the relationship between electrical activity and the resulting Ca2+signal.
Induction of regenerative CICR
Previous studies found that depolarization-induced CICR in neurons developed in a graded manner that depended on Ca2+influx, suggesting that CICR acted as a simple amplifier of Ca2+ influx (Hua et al., 1993; Kostyuk and Verkhratsky, 1994; Berridge et al., 1996). We have described a regenerative, all-or-none form of CICR that when activated becomes the predominant factor in shaping the [Ca2+]i transient. Whether a given CICR response becomes regenerative is determined in part by the degree to which Ca2+ released from individual ryanodine receptors activates neighboring channels. Individual channels display stochastic opening events, producing small transient increases in [Ca2+]i that are temporally and spatially isolated (Ca2+ sparks) (Cheng et al., 1993). The frequency of sparks in cardiac muscle is increased by activation of local L-type Ca2+ channels (Cannell et al., 1995; Lopez-Lopez et al., 1995). This so-called “Ca2+ synapse” (Stern, 1992) provides a mechanism of local control in which an increase in Ca2+ influx increases the number of independent release events that sum to produce graded CICR (Wier et al., 1994). Regenerative responses result when release sites are sufficiently coupled to enable the Ca2+ elevation to spread from one release unit to another, overcoming fast buffering processes (Lipp et al., 1992; Han et al., 1994). Thus, factors such as the density of ryanodine receptors, the level of Ca2+ within the stores, and the sensitivity of ryanodine receptors to cytosolic Ca2+ determine whether stimulus-evoked sparks will sum to a regenerative response. Indeed, we found that sensitization of ryanodine receptors with caffeine and replenishment of intracellular stores with Ca2+ after their release were important to obtain regenerative CICR in DRG neurons.
All-or-none CICR exhibited a discrete threshold for activation
We found that all-or-none CICR was triggered when an elevation in [Ca2+]i reached a threshold. This was the critical point at which more Ca2+ was released than was buffered, presumably coupling release units. Threshold was experimentally defined as the minimum [Ca2+]i at which a regenerative response was initiated. The presence of a discrete threshold for activation differs from a study by Hernandez-Cruz et al. (1997) in which caffeine-induced CICR was dependent on the rate of rise in [Ca2+]i. Presumably a discrete threshold was resolved in our study because CICR was triggered by the influx of Ca2+ from an infinite source, minimizing the effects of buffering processes on triggering the regenerative response. This paradigm also kept the caffeine concentration, and thus the sensitivity of ryanodine receptors to Ca2+, constant. The CICR responses described here were smaller than those observed in sympathetic neurons (Hernandez-Cruz et al., 1997), in which peak [Ca2+]i values of 500–1000 nm might produce complex kinetics for activation attributable to the binding of Ca2+ to a low-affinity inactivation site on the ryanodine receptor (Bezprozvanny et al., 1991; Hernandez-Cruz et al., 1995). In the presence of caffeine, the threshold was poised to discriminate bursts of action potentials from weaker signals. Caffeine decreased the threshold in a concentration-dependent manner, consistent with its ability to sensitize ryanodine receptors to Ca2+ (Sitsapesan and Williams, 1990). Thus, caffeine concentration determined the intensity of electrical stimulation required to evoke regenerative CICR. These observations suggest that in this system, ryanodine receptors act as a coincidence detector requiring both an elevation in [Ca2+]i, which is a function of time-averaged electrical activity, and the presence of a sensitizing agent such as caffeine to trigger all-or-none CICR.
That ryanodine receptors might lie at the junction of multiple signaling pathways is consistent with the large number of regulatory sites on these receptors and their high susceptibility to modulation (Sorrentino, 1996; Sutko and Airey, 1996). We speculate that endogenous modulators of the receptor may adjust the threshold for CICR, similar to the actions of caffeine. A candidate for such a modulator is cADPr, although we have not found it to be effective in our recording conditions. Other modulators of the receptor that may play a role in modulating the threshold for activation of all-or-none CICR are calmodulin (Lee et al., 1994a), protein kinases (Sorrentino, 1996), and immunophillins (Brillantes et al., 1994).
Discharge of the store was followed by a relative refractory period during which the threshold for activation was raised. The threshold decreased in parallel with the time course for refilling the stores, and inhibition of the Ca2+ ATPases that sequester Ca2+ into the stores increased the duration of the refractory period. Loading the store may determine the strength of the coupling between release sites by increasing the driving force for Ca2+ through the open channel (Han et al., 1994). This explanation posits that the sensitivity of the ryanodine receptor to Ca2+ does not change and that the decreased coupling can be compensated by elevated [Ca2+]i. Alternatively, there may be a threshold for luminal Ca2+ (Nelson and Nelson, 1990) mediated by Ca2+ binding proteins within the store interacting with ryanodine receptors (Diaz-Munoz et al., 1990;Gilchrist et al., 1992). We did not observe the initiation of regenerative responses on the falling phase of stimulus-induced [Ca2+]i transients, suggesting that the luminal Ca2+ level modulates but does not trigger CICR. The refractory period places important constraints on the generation of all-or-none CICR by limiting the frequency of regenerative [Ca2+]i transients.
Regenerative CICR alters the spatial distribution of the Ca2+ signal
Ca2+ influx that failed to evoke regenerative CICR produced an elevation in [Ca2+]ithat was restricted to regions near the plasma membrane (Fig.6A,B), similar to previous observations (Hernandez-Cruz et al., 1990; Blumenfeld et al., 1992; O’Malley, 1994). These localized increases in [Ca2+]i are in good agreement with a high degree of cytoplasmic Ca2+ buffering in neurons (Miller, 1991). Induction of regenerative CICR facilitated the inward spread of the Ca2+ signal, so that the increase in [Ca2+]i was homogeneous (Fig.6C,D). In contrast to the passive diffusion of Ca2+ after subthreshold stimulation, all-or-none CICR responses were maintained by the regenerative activity of ryanodine receptors as if moving through an excitable medium (Lechleiter et al., 1991). The described enhancement of the inward propagation of the [Ca2+]i wave may be important for the selective regulation of Ca2+targets distant from the plasma membrane (Kennedy, 1989; Ghosh and Greenberg, 1995; Ginty, 1997). We noted in particular that recruitment of CICR promoted elevation of the Ca2+ concentration in the nucleus. Nucleoplasmic Ca2+ regulates transcription (Ghosh and Greenberg, 1995) mediated by Ca2+–calmodulin-dependent protein kinases and the cyclic-AMP response element-binding protein (Sheng et al., 1991;Hardingham et al., 1997). Thus, all-or-none CICR may be a key component in a mechanism that controls excitation–transcription coupling.
The implications of regenerative CICR to neuronal Ca2+ signaling
[Ca2+]i controls diverse functions in neurons (Kennedy, 1989; Ghosh and Greenberg, 1995). Cellular processes activated by an elevation in [Ca2+]i depend on the size, duration, and spatial distribution of the Ca2+ signal. Activation of all-or-none CICR changed considerably all of these parameters; however, regenerative CICR was only evoked by the coincident presence of a sensitizing agent and a suprathreshold electrical stimulus. Thus, this all-or-none response may be important for Ca2+-regulated processes that display a threshold for activation, such as exocytosis (Peng and Zucker, 1993;Seward et al., 1995; Huang and Neher, 1996), synaptic plasticity (Neveu and Zucker, 1996), and gene expression (Hardingham et al., 1997). Indeed many of these responses depend on the operation of ryanodine-sensitive stores (Kostyuk and Verkhratsky, 1994; Simpson et al., 1995). The regenerative CICR described here was an all-or-none response, but the threshold for its activation was subject to modulation. Factors that adjust threshold have the potential to significantly alter the transduction of electrical activity into cellular responses.
Footnotes
This work was supported by the National Science Foundation (IBN9412654, IBN9723796) and National Institutes of Health (DA07304, DA09293). Y.M.U. was supported by National Institute on Drug Abuse training Grant T32DA07234. We thank Drs. Martha Nowycky and David Friel for their comments on an earlier version of this manuscript, Kyle T. Baron for excellent technical assistance, and Gerald Sedgewick for help with image processing.
Correspondence should be addressed to Dr. S. A. Thayer, Department of Pharmacology, University of Minnesota Medical School, 3-249 Millard Hall, 435 Delaware Street SE, Minneapolis, MN 55455.
REFERENCES
- 1.Al-Mohanna FA, Caddy KWT, Bolsover SR. The nucleus is insulated from large cytosolic calcium ion changes. Nature. 1994;367:745–750. doi: 10.1038/367745a0. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ, Cheek TR, Bennett DL, Bootman MD. Ryanodine receptors and intracellular calcium signaling. In: Sorrentino V, editor. Ryanodine receptors. CRC; Boca Raton, FL: 1996. pp. 119–153. [Google Scholar]
- 3.Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calciumresponse curve of Ins(1,4,5,)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 4.Blumenfeld H, Zablow L, Sabatini B. Evaluation of cellular mechanisms for modulation of calcium transients using a mathematical model of Fura-2 Ca-2+ imaging in Aplysia sensory neurons. Biophys J. 1992;63:1146–1164. doi: 10.1016/S0006-3495(92)81670-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 6.Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- 7.Cheng H, Lederer W, Cannell M. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 8.Currie KPM, Scott RH. Calcium-activated currents in cultured neurones from rat dorsal root ganglia. Br J Pharmacol. 1992;106:593–602. doi: 10.1111/j.1476-5381.1992.tb14381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diaz-Munoz M, Hamilton SL, Kaetzel MA, Hazarika P, Dedman JR. Modulation of Ca2+ release channel activity from sarcoplasmic reticulum by annexin VI (67 kDa calcimedin). J Biol Chem. 1990;265:15894–15899. [PubMed] [Google Scholar]
- 10.Endo M, Tanaka M, Ogawa Y. Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature. 1970;228:34–36. doi: 10.1038/228034a0. [DOI] [PubMed] [Google Scholar]
- 11.Fabiato A. Calcium-induced calcium release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 12.Fill M, Coronado R. Ryanodine receptor channel of sarcoplasmic reticulum. Trends Neurosci. 1988;11:453–457. doi: 10.1016/0166-2236(88)90198-1. [DOI] [PubMed] [Google Scholar]
- 13.Ford LE, Podolsky RJ. Regenerative calcium release within muscle cells. Science. 1970;167:58–59. doi: 10.1126/science.167.3914.58. [DOI] [PubMed] [Google Scholar]
- 14.Friel DD, Tsien RW. Phase-dependent contributions from Ca2+ entry and Ca2+ release to caffeine-induced [Ca2+]i oscillations in bullfrog sympathetic neurons. Neuron. 1992;8:1109–1125. doi: 10.1016/0896-6273(92)90132-w. [DOI] [PubMed] [Google Scholar]
- 15.Furuichi T, Furutama D, Hakamata Y, Nakai J, Takeshima H, Mikoshiba K. Multiple types of ryanodine receptor/Ca2+ release channels are differentially expressed in rabbit brain. J Neurosci. 1994;14:4794–805. doi: 10.1523/JNEUROSCI.14-08-04794.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galione A, Lee HC, Busa WB. Ca2+-induced Ca2+ release in sea urchin egg homogenates: modulation by cyclic ADP-ribose. Science. 1991;253:1143–1146. doi: 10.1126/science.1909457. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 18.Gilchrist JSC, Belcastro AN, Katz S. Intraluminal Ca2+ dependence of Ca2+ and ryanodine-mediated regulation of skeletal muscle sarcoplasmic reticulum Ca2+ release. J Biol Chem. 1992;267:20850–20856. [PubMed] [Google Scholar]
- 19.Ginty DD. Calcium regulation of gene expression: isn’t that spatial? Neuron. 1997;18:183–186. doi: 10.1016/s0896-6273(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 20.Gomez TM, Snow DM, Letourneau PC. Characterization of spontaneous calcium transients in nerve growth cones and their effect on growth cone migration. Neuron. 1995;14:1233–1246. doi: 10.1016/0896-6273(95)90270-8. [DOI] [PubMed] [Google Scholar]
- 21.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 22.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 23.Han S, Schiefer A, Isenberg G. Ca2+ load of guinea-pig ventricular myocytes determines efficacy of brief Ca2+ currents as trigger for Ca2+ release. J Physiol (Lond) 1994;480:411–421. doi: 10.1113/jphysiol.1994.sp020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- 25.Hernandez-Cruz A, Sala F, Adams PR. Subcellular calcium transients visualized by confocal microscopy in a voltage-clamped vertebrate neuron. Science. 1990;247:858–862. doi: 10.1126/science.2154851. [DOI] [PubMed] [Google Scholar]
- 26.Hernandez-Cruz A, Diaz-Munoz M, Gomez-Chavarin M, Canedo-Merino R, Protti DA, Escobar AL, Sierralta J, Suarez-Isla BA. Properties of the ryanodine-sensitive release channels that underlie caffeine-induced Ca2+ mobilization from intracellular stores in mammalian sympathetic neurons. Eur J Neurosci. 1995;7:1684–1699. doi: 10.1111/j.1460-9568.1995.tb00690.x. [DOI] [PubMed] [Google Scholar]
- 27.Hernandez-Cruz A, Escobar AL, Jimenez N. Ca2+-induced Ca2+ release phenomena in mammalian sympathetic neurons are critically dependent on the rate of rise of trigger Ca2+. J Gen Physiol. 1997;109:147–167. doi: 10.1085/jgp.109.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holliday J, Adams RJ, Sejnowski TJ, Spitzer NC. Calcium-induced release of calcium regulates differentiation of cultured spinal neurons. Neuron. 1991;7:787–796. doi: 10.1016/0896-6273(91)90281-4. [DOI] [PubMed] [Google Scholar]
- 29.Hua SY, Nohmi M, Kuba K. Characteristics of Ca2+ release induced by Ca2+ influx in cultured bullfrog sympathetic neurones. J Physiol (Lond) 1993;464:245–272. doi: 10.1113/jphysiol.1993.sp019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang L, Neher E. Ca2+-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron. 1996;17:135–145. doi: 10.1016/s0896-6273(00)80287-1. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy MB. Regulation of neuronal function by calcium. Trends Neurosci. 1989;12:417–420. doi: 10.1016/0166-2236(89)90089-1. [DOI] [PubMed] [Google Scholar]
- 32.Kostyuk P, Verkhratsky A. Calcium stores in neurons and glia. Neuroscience. 1994;63:381–404. doi: 10.1016/0306-4522(94)90537-1. [DOI] [PubMed] [Google Scholar]
- 33.Kostyuk PG, Mironov SL, Tepikin AV, Belan PV. Cytoplasmic free Ca in isolated snail neurons as revealed by fluorescent probe fura-2: mechanisms of Ca recovery after Ca load and Ca release from intracellular stores. J Membr Biol. 1989;110:11–18. [Google Scholar]
- 34.Kostyuk PG, Belan PV, Tepikin AV. Free calcium transients and oscillations in nerve cells. Exp Brain Res. 1991;83:459–464. doi: 10.1007/BF00231173. [DOI] [PubMed] [Google Scholar]
- 35.Kuba K. Release of calcium ions linked to the activation of potassium conductance in a caffeine-treated sympathetic neurone. J Physiol (Lond) 1980;298:251–269. doi: 10.1113/jphysiol.1980.sp013079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai AF, Dent M, Wickenden C, Xu L, Kumari G, Misra M, Lee HB, Sar M, Meissner G. Expression of a cardiac Ca2+-release channel isoform in mammalian brain. Biochem J. 1992;288:553–564. doi: 10.1042/bj2880553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lechleiter J, Girard S, Peralta E, Clapham D. Spiral calcium wave propagation and annihilation in Xenopus laevis oocytes. Science. 1991;252:123–126. doi: 10.1126/science.2011747. [DOI] [PubMed] [Google Scholar]
- 38.Lee HC, Walseth TF, Bratt GT, Hayes RN, Clapper DL. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J Biol Chem. 1989;264:1608–1615. [PubMed] [Google Scholar]
- 39.Lee HC, Aarhus R, Graeff R, Gurnack ME, Walseth TF. Cyclic ADP ribose activation of the ryanodine receptor is mediated by calmodulin. Nature. 1994a;370:307–309. doi: 10.1038/370307a0. [DOI] [PubMed] [Google Scholar]
- 40.Lee HC, Galione A, Walseth TF. Cyclic ADP-ribose: metabolism and calcium mobilizing function. Vitam Horm. 1994b;48:199–257. doi: 10.1016/s0083-6729(08)60499-9. [DOI] [PubMed] [Google Scholar]
- 41.Lipp P, Pott L, Callewaert G, Carmeliet E. Calcium transients caused by calcium entry are influenced by the sarcoplasmic reticulum in guinea-pig atrial myocytes. J Physiol (Lond) 1992;454:321–338. doi: 10.1113/jphysiol.1992.sp019266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lipscombe D, Madison DV, Poenie M, Reuter H, Tsien RW, Tsien RY. Imaging of cytosolic Ca2+ transients arising from Ca2+ stores and Ca2+ channels in sympathetic neurons. Neuron. 1988;1:355–365. doi: 10.1016/0896-6273(88)90185-7. [DOI] [PubMed] [Google Scholar]
- 43.Llano I, DiPolo R, Marty A. Calcium-induced calcium release in cerebellar Purkinje cells. Neuron. 1994;12:663–673. doi: 10.1016/0896-6273(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 44.Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- 45.Marrion NV, Adams PR. Release of intracellular calcium and modulation of membrane currents by caffeine in bull-frog sympathetic neurones. J Physiol (Lond) 1992;445:515–535. doi: 10.1113/jphysiol.1992.sp018937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McPherson PS, Kim Y-K, Valdivia H, Knudson CM, Takekura H, Franzini-Armstrong C, Coronado R, Campbell KP. The brain ryanodine receptor: a caffeine-sensitive calcium release channel. Neuron. 1991;7:17–25. doi: 10.1016/0896-6273(91)90070-g. [DOI] [PubMed] [Google Scholar]
- 47.Miller RJ. The control of neuronal Ca2+ homeostasis. Prog Neurobiol. 1991;37:255–285. doi: 10.1016/0301-0082(91)90028-y. [DOI] [PubMed] [Google Scholar]
- 48.Mironov SL, Usachev YM. Sr and Ba transients in isolated snail neurones studied with fura-2. The recovery from depolarization induced load and modulation of Ca release from intracellular stores. Neurosci Lett. 1990;112:184–189. doi: 10.1016/0304-3940(90)90200-s. [DOI] [PubMed] [Google Scholar]
- 49.Nelson TE, Nelson KE. Intra- and extraluminal sarcoplasmic reticulum membrane regulatory sites for Ca2+-induced Ca2+ release. FEBS Lett. 1990;263:292–294. doi: 10.1016/0014-5793(90)81396-6. [DOI] [PubMed] [Google Scholar]
- 50.Neveu D, Zucker RS. Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron. 1996;16:619–629. doi: 10.1016/s0896-6273(00)80081-1. [DOI] [PubMed] [Google Scholar]
- 51.Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channels with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- 52.Obenaus A, Mody I, Baimbridge KG. Dantrolene-Na (dantrium) blocks induction of long-term potentiation in hippocampal slices. Neurosci Lett. 1989;98:172–178. doi: 10.1016/0304-3940(89)90505-3. [DOI] [PubMed] [Google Scholar]
- 53.O’Malley DM. Calcium permeability of the neuronal nuclear envelope: evaluation using confocal volumes and intracellular perfusion. J Neurosci. 1994;14:5741–58. doi: 10.1523/JNEUROSCI.14-10-05741.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peng YY. Ryanodine-sensitive component of calcium transients evoked by nerve firing at presynaptic nerve terminals. J Neurosci. 1996;16:6703–6712. doi: 10.1523/JNEUROSCI.16-21-06703.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng YY, Zucker RS. Release of LHRH is linearly related to the time integral of presynaptic Ca2+ elevation above a threshold level in bullfrog sympathetic ganglia. Neuron. 1993;10:465–473. doi: 10.1016/0896-6273(93)90334-n. [DOI] [PubMed] [Google Scholar]
- 56.Piser TM, Lampe RA, Keith RA, Thayer SA. ω-Grammotoxin blocks action-potential-induced Ca2+ influx and whole-cell Ca2+ current in rat dorsal root ganglion neurons. Pflügers Arch. 1994;426:214–220. doi: 10.1007/BF00374774. [DOI] [PubMed] [Google Scholar]
- 57.Reyes M, Stanton PK. Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci. 1996;16:5951–5960. doi: 10.1523/JNEUROSCI.16-19-05951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rousseau E, Meissner G. Single cardiac sarcoplasmic reticulum Ca2+-release channel: activation by caffeine. Am J Physiol. 1989;256:H328–H333. doi: 10.1152/ajpheart.1989.256.2.H328. [DOI] [PubMed] [Google Scholar]
- 59.Scroggs RS, Fox AP. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J Physiol (Lond) 1992;445:639–658. doi: 10.1113/jphysiol.1992.sp018944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seward EP, Chernevskaya NI, Nowycky MC. Exocytosis in peptidergic nerve terminals exhibits two calcium-sensitive phases during pulsatile calcium entry. J Neurosci. 1995;15:3390–3399. doi: 10.1523/JNEUROSCI.15-05-03390.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seymour-Laurent KJ, Barish M. Inositol 1,4,5-trisphosphate and ryanodine receptor distributions and patterns of acetylcholine- and caffeine-induced calcium release in cultured mouse hippocampal neurons. J Neurosci. 1995;15:2592–2608. doi: 10.1523/JNEUROSCI.15-04-02592.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheng M, Thompson AM, Greenberg EM. CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 63.Shmigol A, Verkhratsky A, Isenberg G. Calcium-induced calcium release in rat sensory neurons. J Physiol (Lond) 1995;489:627–636. doi: 10.1113/jphysiol.1995.sp021078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simpson PB, Challiss RAJ, Nahorski SR. Neuronal Ca2+ stores: activation and function. Trends Neurosci. 1995;18:299–306. doi: 10.1016/0166-2236(95)93919-o. [DOI] [PubMed] [Google Scholar]
- 65.Sitsapesan R, Williams AJ. Mechanisms of caffeine activation of single calcium-release channels of sheep cardiac sarcoplasmic reticulum. J Physiol (Lond) 1990;423:425–439. doi: 10.1113/jphysiol.1990.sp018031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sitsapesan R, McGarry SJ, Williams AJ. Cyclic ADP-ribose, the ryanodine receptor and Ca2+ release. Trends Pharmacol Sci. 1995;16:386–391. doi: 10.1016/s0165-6147(00)89080-x. [DOI] [PubMed] [Google Scholar]
- 67.Smith SJ, MacDermott AB, Weight FF. Detection of intracellular Ca2+ transients in sympathetic neurones using Arsenazo III. Nature. 1983;304:350–352. doi: 10.1038/304350a0. [DOI] [PubMed] [Google Scholar]
- 68.Sorrentino V. Molecular biology of ryanodine receptors. In: Sorrentino V, editor. Ryanodine receptors. CRC; Boca Raton, FL: 1996. pp. 85–100. [Google Scholar]
- 69.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sutko JL, Airey JA. Ryanodine receptor Ca2+ release channels: does diversity in form equal diversity in function? Physiol Rev. 1996;76:1027–1071. doi: 10.1152/physrev.1996.76.4.1027. [DOI] [PubMed] [Google Scholar]
- 71.Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J Physiol (Lond) 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thayer SA, Hirning LD, Miller RJ. The role of caffeine sensitive calcium stores in the regulation of free intracellular calcium in rat sympathetic neurons in vitro. Mol Pharmacol. 1988;34:664–673. [PubMed] [Google Scholar]
- 73.Thomas D, Hanley MR. Pharmacological tools for perturbing intracellular calcium storage. Methods Cell Biol. 1994;40:65–89. doi: 10.1016/s0091-679x(08)61110-3. [DOI] [PubMed] [Google Scholar]
- 74.Usachev Y, Shmigol A, Pronchuk N, Kostyuk P, Verkhratsky A. Caffeine-induced calcium release from internal stores in cultured rat sensory neurons. Neuroscience. 1993;57:845–859. doi: 10.1016/0306-4522(93)90029-f. [DOI] [PubMed] [Google Scholar]
- 75.Usachev Y, Verkhratsky A. IBMX induces calcium release from intracellular stores in rat sensory neurones. Cell Calcium. 1995;17:197–206. doi: 10.1016/0143-4160(95)90034-9. [DOI] [PubMed] [Google Scholar]
- 76.Usachev YM, Thayer SA. All-or-none Ca2+ release from intracellular stores triggered by Ca2+ influx through voltage-gated Ca2+ channels in rat DRG neurons. Soc Neurosci Abstr. 1996;22:340. doi: 10.1523/JNEUROSCI.17-19-07404.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Y, Wu J, Rowan MJ, Anwyl R. Ryanodine produces a low frequency stimulation-induced NMDA receptor-independent long-term potentiation in the rat dentate gyrus in vitro. J Physiol (Lond) 1996;495:755–767. doi: 10.1113/jphysiol.1996.sp021631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Werth JL, Usachev YM, Thayer SA. Modulation of calcium efflux from cultured rat dorsal root ganglion neurons. J Neurosci. 1996;16:1008–1015. doi: 10.1523/JNEUROSCI.16-03-01008.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wier WG. Cytoplasmic [Ca2+]i in mammalian ventricle: dynamic control by cellular processes. Annu Rev Physiol. 1990;52:467–488. doi: 10.1146/annurev.ph.52.030190.002343. [DOI] [PubMed] [Google Scholar]
- 80.Wier WG, Egan TM, Lopez-Lopez JR, Balke CW. Local control of excitation-contraction coupling in rat heart cells. J Physiol (Lond) 1994;474:463–471. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]