

Abstract
Previous work indicated that the temperature-dependent block of synaptic transmission in cysteine string protein (csp) mutants ofDrosophila was attributable to a failure of nerve impulses to trigger transmitter release. The current investigations were undertaken to resolve in more detail the mechanism of this transmission deficit. Our studies reveal that the spider venom toxin α-latrotoxin can trigger a sustained discharge of quanta at neuromuscular junctions of csp mutant larvae at nonpermissive temperature. The same is true of the calcium ionophore ionomycin. However, solutions with an elevated concentration of K or Ca ions fail to circumvent the block of quantal secretion in these mutants. Likewise, 4-aminopyridine, which augments transmitter release at permissive temperature in csp mutants, fails to reverse the inhibition of impulse-evoked transmitter release at elevated temperature. These data are consistent with the hypothesis that there is a deficit either in Ca ion entry or in the ability of Ca ions to trigger exocytosis in csp mutants at nonpermissive temperatures. In part, because of previous work showing that csps are important for the functional expression of N-type Ca channels in frog oocytes, we favor the idea that csps participate in a regulatory interaction involving presynaptic Ca channels.
Keywords: cysteine string proteins, synaptic transmission, α-latrotoxin, presynaptic calcium channels, Drosophila, shibire
Cysteine string proteins (csps) (Zinsmaier et al., 1990) are highly conserved proteins associated with synaptic vesicles and other secretory organelles (Mastrogiacomo et al., 1994; Braun and Scheller, 1995; Mastrogiacomo and Gundersen, 1995;Chamberlain et al., 1996; Buchner and Gundersen, 1997). Immunohistochemistry reveals that csps are distributed ubiquitously at nerve endings in the central and peripheral nervous systems of both invertebrates and vertebrates (Zinsmaier et al., 1994; Kohan et al., 1995). However, the precise role of csps in presynaptic events remains uncertain. Early investigations using csp antisense RNA suggested that csps were important for the function of presynaptic, N-type Ca channels (Gundersen and Umbach, 1992). Subsequently, csp mutant alleles of Drosophila were shown to display aberrant electroretinograms, temperature-sensitive (ts) paralysis, and premature death (Zinsmaier et al., 1994). The cellular basis of the ts paralysis of csp mutants was shown to reflect the failure of nerve impulses to trigger neurotransmitter release at nonpermissive temperatures (Umbach et al., 1994). Interestingly, spontaneous transmitter release persisted in these mutants (Umbach et al., 1994). Because of the retention of this form of quantal secretion, we hypothesized that csp mutant Drosophila were defective at a step in the process that couples nerve excitation to secretion (Umbach et al., 1994).
A recent review (Sudhof, 1995) contained an alternative hypothesis for csp function. This hypothesis was based on two observations. First, adult csp mutants display a paucity of synaptic vesicles at central synapses (Zinsmaier et al., 1994). Second, csps harbor in their primary sequence a J domain, a putative site for interaction in vivo with heat shock protein 70 (Hsp70) molecular chaperones (Caplan et al., 1993; Silver and Way, 1993). Because at least one member of the large family of Hsp70 chaperones is involved in the uncoating of clathrin-coated vesicles (Chappell et al., 1986), Sudhof (1995) postulated that csps might participate in this process of clathrin coat removal. By inference, the diminished number of synaptic vesicles at nerve endings in the brain of adult csp mutants might therefore arise from a defect in synaptic vesicle recycling (Sudhof, 1995).
In light of these widely divergent hypotheses concerning csp function, we undertook investigations to determine whether a defect in synaptic vesicle recycling could explain the ts failure of synaptic transmission in csp mutant larvae. Our studies reveal that even when nerve impulses fail to trigger the release of a single quantum of transmitter, nerve endings of csp mutantDrosophila retain a large store of readily releasable quanta. These quanta can be released in response to the spider venom toxin α-latrotoxin (α-Ltx) and the calcium ionophore ionomycin. However, various strategies that require the opening of presynaptic Ca channels fail to elicit vesicular exocytosis. Thus, instead of a role in synaptic vesicle recycling, our data, together with previous results (Gundersen and Umbach, 1992), suggest that csps participate in a novel modulatory interaction involving presynaptic Ca channels.
MATERIALS AND METHODS
Electrophysiology.Electrophysiological recordings were made of spontaneous miniature excitatory junctional potentials (mejps) and nerve impulse-evoked excitatory junctional potentials (ejps), events using dorsal longitudinal muscle fibers 6 and 7 of wild-type (Canton S) or cspX1 (Zinsmaier et al., 1994) larval Drosophila exactly as described by Umbach et al. (1994). We emphasize that for all experiments using thiscsp mutant strain, a preliminary screening for temperature sensitivity was conducted as discussed by Umbach et al. (1994). Experiments using shibire mutant Drosophila used the shits1 strain (Grigliatti et al., 1973). The standard recording solution contained (in mm): 140 NaCl, 2 KCl, 1 CaCl2, 5 MgCl2, 5 glucose, and 5 HEPES with pH adjusted to 7.0 using NaOH.
Reagents and alternative recording conditions.α-Ltx, a 125 kDa component of black widow spider venom, was purified as described byGrasso and Mastrogiacomo (1992). This toxin preparation was recently shown to be active in promoting high-frequency mejp discharges in wild-type Drosophila (J. Umbach, unpublished observations) and was bath-applied to achieve a final concentration of 5–10 nm.
To assess the effects of solutions with elevated Ca or K ion composition, we augmented the normal recording solution with CaCl2 to 5 mm (high-Ca solution) or raised the KCl concentration to 60 mm and lowered NaCl to 82 mm (high-K solution). Analysis of mejp frequency in experiments using high-K solution was done with the muscle fiber resting potential held at −60 mV by the injection of continuous negative DC current. To deplete shibire mutants of releasable quanta, the high-K solution had 3 mmCaCl2 and 3 mm MgCl2. For experiments using the calcium ionophore ionomycin (Calbiochem, La Jolla, CA), a stock of ionomycin was prepared in dimethylsulfoxide (Sigma, St. Louis, MO) and diluted to the working concentration (10 μm ionomycin) in recording solution under conditions in which the final concentration of solvent was <0.1%. Finally, 4-aminopyridine (4-AP; Aldrich, Milwaukee, WI) was used at 1 mm in the recording solution.
RESULTS
To examine the possibility that transmitter release fails incsp mutant preparations because of a lack of recycled synaptic vesicles, we set out to investigate whether releasable quanta persist in csp mutant nerve endings at nonpermissive temperature. Moreover, because ts shibire mutantDrosophila are known to be defective in the recycling of synaptic vesicles (Poodry and Edgar, 1979; Kosaka and Ikeda, 1983), we were interested in comparing the behavior of csp andshibire mutant organisms under similar experimental conditions. As shown in Figure1A, both cspand shibire mutant Drosophila display a complete loss of nerve impulse-evoked transmitter release at nonpermissive temperature. For csp mutants, as reported previously (Umbach et al., 1994), this transmission block develops slowly (over 10–20 min) and occurs independently of any efforts to evoke quantal transmitter secretion. However, to achieve the ts block of transmitter release in shibire mutants (as illustrated in Fig.1A), it is necessary first to deplete the nerve endings of their preformed store of quanta by enhancing quantal transmitter release (which was done in this experiment using elevated K solution; Fig. 1A). Clearly, this conditional block of transmitter release (which requires enhanced activity inshibire) is one phenotypic difference between cspand shibire mutant Drosophila. Once depleted of releasable quanta by the high-K solution, no further evoked response can be recorded in shibire mutants in normal recording solution at nonpermissive temperature (Fig. 1A). Nevertheless, both shibire and csp mutantDrosophila recover evoked responses after being returned to permissive temperature (21°C, Fig. 1A). Thus, in both of these mutants the ts block of transmitter release is reversible with cooling.
Fig. 1.

Response of wild-type and mutant preparations to nerve stimulation and α-Ltx at permissive and nonpermissive temperatures. A, Nerve impulse-evoked ejps in the standard recording solution in wild-type (WT) andcsp (CSP) and shibire(Shi) mutant preparations at permissive (21°C) and after 15 min at nonpermissive (32°C) temperature. To obtain this ts blockade of ejps in shibire mutants it was necessary to pretreat the preparation in high-K solution until mejps ceased at 32°C. This was done by warming the preparation to 32°C and incubating it for 15 min in high-K solution (see Materials and Methods), followed by washing with normal recording solution. No such treatment was needed for csp mutants, which were fully blocked after 10–15 min at 32°C. Shibire andcsp mutants recover ejps after 15 min at 21°C.B, Effect of α-Ltx on mejp discharges in wild-type,csp, and shibire mutant preparations at permissive (21°C) and nonpermissive temperatures (32°C). Toxin (5 nm) was added at time 0, and representative mejp traces are presented at selected intervals thereafter. Note that prolonged exposure of wild-type larvae to 32°C often results in spontaneous ejps seen as spikes in the bottom traces. This spiking is not seen in csp or depleted shibirepreparations. Again, as in A, the shibiremutant preparation was depleted of releasable quanta at 32°C by exposure to high-K solution. Note that a similar pretreatment of control or csp mutant preparations with high-K solution did not alter the α-Ltx effect (data not shown). Data are representative of results from a minimum of three experiments for each condition.
Next, we evaluated the effect of α-Ltx on quantal transmitter release in wild-type and mutant preparations at permissive and nonpermissive temperatures. As indicated in Figure 1B, 5 nm α-Ltx triggers a sustained increase of mejp frequency in wild-type preparations. This mejp cascade begins 3–5 min after bath application of the toxin and lasts for at least 40 min (Fig.1B). During the period of high mejp frequency, the muscle fiber depolarizes, and individual mejps cannot be resolved (Fig.1B). Because of this, we did not attempt to quantitate mejp frequency during this quantal cascade. Instead, we noted that with 5 nm α-Ltx, both the time of onset (3–5 min) and duration (40–50 min) of this period of enhanced mejp activity were very consistent among wild-type preparations.
When α-Ltx is applied to a csp mutant preparation at 21°C, it induces a prompt (within 5 min) and sustained (>30 min) increase in mejp frequency (Fig. 1B). Interestingly, α-Ltx triggers a similar response in csp mutants at 32°C (Fig. 1B). In other words, at a time when nerve impulses fail to elicit the discharge of a single quantum of transmitter (Fig. 1A), α-Ltx induces a massive burst of mejps (Fig. 1B). The onset and duration of this increased mejp activity are indistinguishable from those of wild-type controls (Fig. 1B). These results indicate that csp mutant Drosophila retain a sizable pool of quanta that can be released by α-Ltx but not by nerve impulses.
Application of α-Ltx to shibire mutant preparations also triggers a massive outpouring of quanta at 21°C (Fig.1B). However, this situation changes inshibire mutant preparations at nonpermissive temperature (32°C). In shibire mutant preparations that have been depleted of vesicles at 32°C, α-Ltx fails to trigger any further detectable mejps (Fig. 1B). Indeed, in contrast tocsp mutant preparations that still show spontaneous mejps at nonpermissive temperature (Fig. 1B; Umbach et al., 1994), one seldom (less than one per minute) detects mejps in these depleted shibire preparations (Fig. 1B). Although these latter results conform to expectations based on the known depletion of synaptic vesicles in shibire mutants (Poodry and Edgar, 1979), they also highlight the prominent difference in the effect of α-Ltx on a preparation with a known membrane-recycling defect (the shibire mutants) and thecsp mutants. From these results, we conclude that the ts block of transmitter release in csp mutants cannot be attributed to a failure of synaptic vesicle recycling.
Based on the fact that quantal transmitter release can be elicited by α-Ltx in csp mutant preparations, it is necessary to consider other mechanisms to explain the failure of nerve impulses to evoke transmitter release at restrictive temperature. The mechanisms that are consistent with the available data (see Discussion) are either that presynaptic Ca channels fail to open or that Ca influx or action is altered in some manner that precludes effective triggering of transmitter secretion. To distinguish among these possibilities, we conducted several additional experiments.
To minimize problems in obtaining stable recordings, we have generally used bath solutions with a high ratio of Mg ions to Ca ions (Fig. 1;Umbach et al., 1994). Although this reduces muscle twitching in response to nerve impulses (by keeping the quantal content low), it may also accentuate problems with Ca ion triggering of transmitter release in csp mutants at elevated temperature. Thus, our first experiment was to raise the concentration of Ca ions in the bath solution to determine whether this overcomes the blockade of synaptic transmission in csp mutants. We found that increasing bath Ca from 1 to 5 or 10 mm does not restore evoked responses (data not shown). Thus, this expedient of increasing the electrochemical gradient for Ca ion entry does not overcome the block of transmitter release in csp mutants.
Our second strategy was to use solutions with an elevated K ion concentration. As illustrated in Figure2, in a csp mutant preparation that fails to release any transmitter in response to nerve stimulation, high-K solution is also without any detectable effect on quantal transmitter secretion. In n = 3 experiments, mejp frequency at restrictive temperature was 1.7 ± 0.2/sec (mean ± SD) before high K and 1.6 ± 0.1/sec (mean ± SD) during high K. Thus, there is no significant change of mejp frequency incsp mutants at elevated temperatures. However, when the preparation of Figure 2 is cooled back to room temperature, high K has its characteristic effect (reported originally by Liley, 1956) of dramatically enhancing mejp frequency. Indeed, mejp frequency rises to a level that precludes accurate determination of the frequency. From these results, we conclude that the transmitter release apparatus ofcsp mutants is immune to the release triggering action of high K solution at restrictive temperature.
Fig. 2.
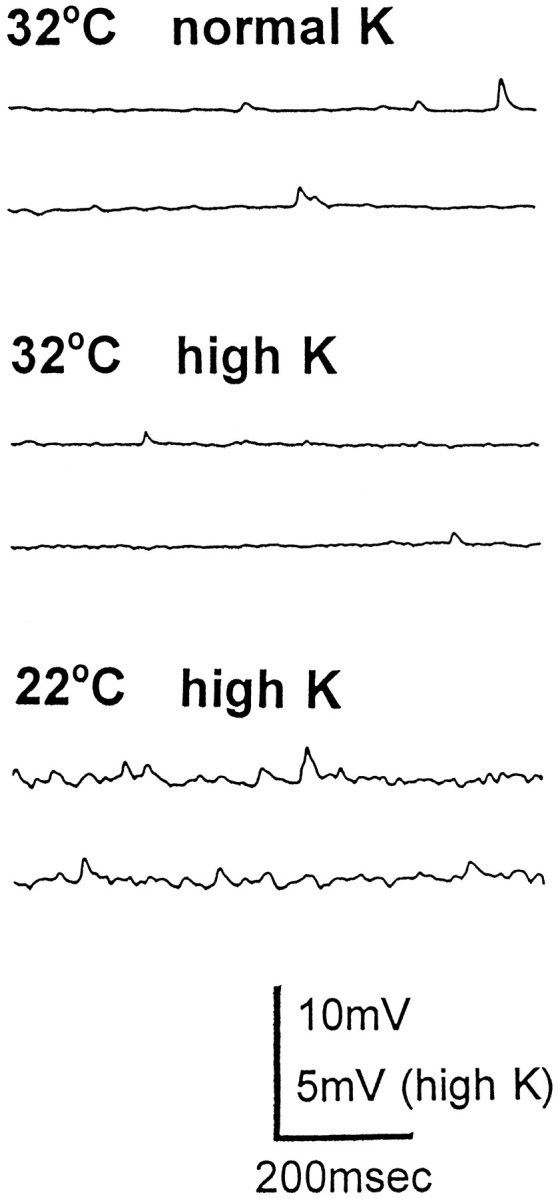
Effect of high-K solution on quantal transmitter release in a csp mutant preparation. Acsp mutant preparation was warmed to 32°C until nerve impulse-evoked responses failed completely (as in Fig.1A). The initial record (32°C normal K) shows mejps that persist under these circumstances. The bath solution was then replaced with high-K solution (32°C high K), and mejp frequency was not detectably altered. (However, the membrane potential dropped from −45 to −28 mV.) When this preparation was cooled back to 22°C for 15 min, one sees a large increase in mejp frequency (22°C high K), although the membrane potential of the muscle fiber remains depolarized (at −27 mV).
Another approach that has been very effective at potentiating neurotransmitter release is to use 4-AP (e.g., see Heuser et al., 1979;Katz and Miledi, 1979). Again, we were interested in whether 4-AP would overcome the blockade of stimulus-evoked transmitter release incsp mutants at 32°C. The results in Figure3 indicate that 4-AP is ineffectual in this context. In a preparation that fails to respond to nerve impulses at 32°C, we see no restoration of quantal secretion in the presence of 4-AP (1 mm). Interestingly, even after removing the 4-AP from the bath solution and cooling this preparation back to room temperature, we routinely detected a large and protracted ejp in response to nerve stimulation (compare ejps at 21°C in Fig. 3,top and bottom). Note that we also tested 4-AP with elevated Ca (5 mm) solution and saw no relief of the transmission block in csp mutants (data not shown). These results indicate that 4-AP potentiates transmitter secretion incsp mutants at permissive temperature but not at nonpermissive temperature.
Fig. 3.

Effect of 4-AP on quantal transmitter release in acsp mutant preparation. A ts block of evoked transmitter release was obtained in a csp mutant preparation (as in Fig.1A). 4-AP was added after verifying the block of evoked release at 32°C (note the absence of evoked response at 32°C; − 4AP). No recovery of evoked responses was seen with 4-AP (+ 4AP) at 32°C. However, if one cools the 4-AP-treated preparation to 21°C, one detects the protracted ejp associated with the action of this K channel blocker. Comparable results were obtained in three experiments.
Elevated Ca, high-K solution, and 4-AP are all treatments that should potentiate transmitter release only if presynaptic Ca channels are capable of opening and conducting Ca ions. This is illustrated in Figure 4, which shows that Cd ions, which are nonselective blockers of all known voltage-gated Ca channels (Tsien et al., 1988), completely antagonize the release-enhancing effect of elevated Ca, high-K solution, or 4-AP on quantal transmitter release in wild-type controls at 21–22°C. (Note that these data also illustrate the release-enhancing effect of these treatments in control preparations.) Because of these results, it appeared plausible that the inability of these treatments to overcome the failure of quantal secretion in csp mutants might owe to an inhibition in these organisms of presynaptic Ca channels. If this were the case, we postulated that it should be possible to circumvent these channels and to trigger a flurry of mejps using the Ca ionophore ionomycin.
Fig. 4.

Cadmium ion antagonism of the release-enhancing effects of high-Ca solution, high-K solution, or 4-AP in wild-type preparations at 21–22°C. In these experiments, the initial record inA–C is the control situation before addition of: A, high-Ca solution; B, high-K solution; and C, 4-AP. The second record in each series shows the release-enhancing effect of each of these treatments. The third record indicates what happens when 1 mm Cd is added to the bath solution. In A and C, we measured evoked responses, whereas in high K (B), only spontaneous events were recorded. In our experience, 1 mm Cd not only abolished the stimulus-evoked responses in A and C, but it also considerably reduced mejp frequency in this preparation, as indicated in B.
In our initial experiments using ionomycin, we encountered the unexpected difficulty that this ionophore efficiently raised mejp frequency at 22°C, but it became appreciably less effective in raising mejp frequency at temperatures >30°C. This effect was obtained in six different wild-type preparations. However, we also noticed that this temperature effect was rapidly reversible, and as ionomycin-treated preparations were cooled, mejp frequency increased dramatically. Thus, it appears that the Ca-transporting activity of ionomycin may be altered in Drosophila membranes at >30°C. [We drew this conclusion because normal, nerve impulse-evoked responses could still be obtained at 30°C in wild-type, ionophore-treated larvae, which indicates that the transmitter release apparatus is not compromised (data not shown).] Because of this behavior of the ionophore, our studies usingcsp mutants were designed to assess the action of ionophore during a brief period of cooling from 32°C. As indicated (Fig.5), heating to 32°C produces the typical block of evoked responses. In Figure 5, third panel, ionomycin was added, and the preparation was cooled to 29°C. With this cooling, mejp frequency increases to a level that precludes accurate counting. What is important is that there is no recovery of nerve impulse-evoked responses at the same time that ionomycin is promoting this mejp discharge (Fig. 5) [Interestingly, we used this same paradigm using high-K solution and saw no increase in mejp frequency within 5 min of cooling at 29°C (data not shown).] Under normal circumstances, when we cool a csp mutant preparation from 30–32°C back to 21–22°C, it takes at least 5 min for a full recovery of evoked responses (Umbach et al., 1994). This means that when cooling to 29°C, we have a several minute window before evoked responses show any recovery, and this allows us to gauge the effect of ionomycin on mejp frequency before ejps begin to reappear. Finally, this release-enhancing effect of Ca ionophore is not seen if Ca in the bath solution is replaced with Mg (data not shown). Thus, Ca ionophore does circumvent the block of transmitter release in ts cspmutants, and it does so by facilitating Ca entry across the plasma membrane.
Fig. 5.

Action of ionomycin on a csp mutant preparation. In the top (21°C), mejps (first two traces) and the ejp are shown in acsp mutant preparation at permissive temperature. The characteristic failure of the ejp, but not the mejps, is illustrated in the center (32°C). Finally, the effect of ionomycin to increase dramatically the frequency of mejps is shown in the bottom (29°C). At this time, the evoked response remains inhibited (final record), indicating that ionomycin promotes quantal secretion (mejps) at a time when evoked responses (ejps) are blocked. This result is representative of data obtained in three separate experiments.
DISCUSSION
Two important conclusions emerge from this investigation. First, our data show that the failure of synaptic transmission incsp mutant larvae is not caused by a defect in synaptic vesicle recycling. Second, our results support the conclusion that csps regulate an early step in the Ca-dependent triggering of quantal transmitter secretion. We begin by reviewing evidence germane to the recycling issue and follow this with arguments in favor of the hypothesis that this “early step” in the secretory cascade involves a regulatory interaction between csps and presynaptic Ca channels.
The idea that csps are involved in synaptic vesicle recycling (Sudhof, 1995) fails to account for the inhibition of transmitter secretion incsp mutants at restrictive temperature. In contrast toshibire mutant Drosophila, which are defective in dynamin, a protein that is important for clathrin-dependent membrane recycling (Poodry and Edgar, 1979; Kosaka and Ikeda, 1983; Chen et al., 1991; van der Bliek and Meyerowitz, 1991), csp mutants retain abundant releasable quanta at a time when nerve impulses fail to elicit any quantal transmitter release. This is one of many pointed contrasts between these two mutant strains. For instance, we have shown that transmitter release is eliminated in csp mutants without any prerequisite for a period of enhanced quantal secretion. The situation is quite different in shibire larvae, where a period of quantal release activity is necessary to produce the loss of releasable quanta and morphologically identifiable synaptic vesicles (Poodry and Edgar, 1979; Kosaka and Ikeda, 1983). Thus, in the current experiments, we used solutions with elevated K ions to depleteshibire nerve terminals of preformed vesicles. Subsequently, these shibire mutants showed almost no detectable spontaneous mejp activity at restrictive temperature, whereascsp mutant larvae continued to display mejps. These phenotypic traits distinguish these two mutant strains and force us to consider other hypotheses (than a loss of synaptic vesicles) to explain the inhibition of synaptic transmission in csp mutants at 32°C.
By a process of elimination, we can infer from the available data that Ca influx via presynaptic Ca channels is attenuated in cspmutants at nonpermissive temperatures. This reasoning begins with the α-Ltx and ionomycin results. Thus, even when nerve impulses fail to trigger the release of a single quantum of transmitter, both of these reagents elicit a sustained discharge of mejps at csp mutant nerve terminals. With α-Ltx, we followed this cascade of mejps for >30 min. These results unambiguously demonstrate that there is no insurmountable barrier to the ongoing recruitment of synaptic vesicles to participate in the release process in these mutants. [As an interesting counterpoint, Broadie et al. (1995) showed thatDrosophila strains that are defective in syntaxin or synaptobrevin are resistant to the release-promoting effects of an extract of black widow spider venom glands.] Instead, our data argue that there is a disruption of the normal process that couples nerve impulses to transmitter release in csp mutants.
Physiologically, nerve impulses evoke the release of neurotransmitter by the depolarization-dependent opening of presynaptic Ca channels (Katz, 1969; Augustine et al., 1987). Ca ions entering via these channels bind to “Ca sensors” (presumed to include members of the synaptotagmin family; see Sudhof, 1995; Augustine et al., 1996) to trigger the final sequence of steps leading to the exocytotic discharge of transmitter. Because action potential conduction into the terminal is unaffected in csp mutants (Umbach et al., 1994), the ts failure of secretion must reflect a defect at some later stage of the release-coupling process. There are four general explanations that could account for this failure of excitation–secretion coupling. First, if Ca influx is attenuated, then the defect lies in the operation of the Ca channels that normally (i.e., at permissive temperature) participate in the triggering of transmitter release. However, if the pathways for Ca entry remain unaltered (relative to what occurs at permissive temperature), then transmitter release may fail for one of the following three reasons: (1) Ca buffering (or extrusion) is so efficient that it precludes triggering of exocytosis; (2) release sites become physically dissociated from sites of Ca entry to the extent that release coupling fails; and (3) the Ca sensors for exocytosis are defective. Although the available data do not permit us to distinguish unequivocally among these possibilities, we review evidence that favors the hypothesis that Ca channel function is altered in ts csp mutants.
A facile interpretation of the ionomycin data is that this ionophore bypasses dysfunctional Ca channels in the csp mutants and provides an alternative pathway for Ca entry that is adequate to trigger high-frequency mejp discharges. The fact that ionomycin promotes quantal transmitter release, whereas solutions with high K, high Ca, or 4-AP do not, is also consistent with this interpretation. These latter three treatments all demand that presynaptic Ca channels be capable of opening and conducting Ca ions as a prelude to the release-triggering process. The finding that none of these treatments elicit any detectable secretion at nonpermissive temperature incsp mutants is consistent with the hypothesis that the target Ca channels are dysfunctional.
A more systematic approach to interpreting the release data forcsp mutants is to assume that the presynaptic Ca channels remain functional and to ask whether any of the three alternative explanations (1–3, above) can account for the observed results. First, is it plausible that enhanced buffering or extrusion of Ca ions would interfere with release evoked by nerve impulses or high K without also blunting secretion triggered by ionomycin [or α-Ltx, which appears to act at least in part by raising cytosolic ionized Ca (Meldolesi et al., 1986; Surkova, 1994)]? If the buffering and extrusion hypothesis were valid, there should have been a demonstrable attenuation of the release-triggering effect of ionomycin or α-Ltx. [Supporting this assumption are preliminary results showing that the action of α-Ltx to enhance quantal secretion is diminished in Drosophilalarvae pretreated with the permeant Ca chelator BAPTA-AM (J. Umbach, unpublished observation).] Because ionomycin and α-Ltx displayed similar efficacy as secretogogues in wild-type and cspmutant preparations, this makes it unlikely that Ca buffering or extrusion changes in the mutants.
Second, we can inquire whether a physical separation of release sites from the sites of Ca entry underlies the ts block of release incsp mutants. If this situation existed, one might predict that ionomycin [and α-Ltx, if it can act as a mobile Ca ionophore (see Meldolesi et al., 1986; Surkova, 1994)] should provide sufficiently diffuse Ca ion entry to trigger release in these organisms. However, if this were the case, it is a challenge to explain why α-Ltx and ionomycin promote such a dramatic increase in mejp frequency, whereas sustained depolarization by high K and nerve stimulation in 4-AP are completely ineffectual at triggering secretion. Thus, although our data do not favor this explanation, we cannot completely exclude it at this time.
The third alternative to explain the ts failure of secretion incsp mutants is that the Ca-sensing machinery fails to trigger exocytosis. Here again, we regard it as somewhat arbitrary to assume that ionomycin (or α-Ltx) can overcome a defect at this level, whereas nerve impulses or high K cannot. Thus, it remains that the most parsimonious explanation of our data is that presynaptic Ca channels fail in a temperature-dependent manner in csp mutant larvae.
The last argument in favor of a csp–Ca channel link stems from physiological results obtained during the initial cloning of a vertebrate csp cDNA (Gundersen and Umbach, 1992). In these studies, csp antisense RNA selectively reduced (or abolished) N-type Ca channel currents recorded in frog oocytes injected with Torpedoelectric lobe mRNA. Conversely, csp sense RNA augmented N-type Ca channel currents. Although the molecular basis of these observations has not been established, we now know that csps are synaptic vesicle proteins that are tethered to the vesicle surface in an orientation that allows them to interact directly or indirectly with presynaptic Ca channels (Mastrogiacomo et al., 1994; Umbach et al., 1995). This precedent for a csp–Ca channel link supports our conclusion that the remnants of this regulatory interaction fail in csp mutantDrosophila, leading to the ts block of secretion.
Footnotes
This work was supported in part by National Institutes of Health Grant NS31934 (J.A.U.). We thank Dr. A. Grasso and Dr. A. Mastrogiacomo for the α-latrotoxin and Theresa Sama for preparing this manuscript.
Correspondence should be addressed to Dr. Joy Umbach, Department of Molecular and Medical Pharmacology, University of California at Los Angeles School of Medicine, Los Angeles, CA 90095-1735.
REFERENCES
- 1.Augustine GJ, Charlton MP, Smith SJ. Calcium action in synaptic transmitter release. Annu Rev Neurosci. 1987;10:633–693. doi: 10.1146/annurev.ne.10.030187.003221. [DOI] [PubMed] [Google Scholar]
- 2.Augustine GJ, Burns ME, DeBello WM, Pettit DL, Schweizer FE. Exocytosis: proteins and perturbations. Annu Rev Pharmacol Toxicol. 1996;36:659–701. doi: 10.1146/annurev.pa.36.040196.003303. [DOI] [PubMed] [Google Scholar]
- 3.Braun JEA, Scheller RE. Cysteine string protein, a DnaJ family member, is present on diverse secretory vesicles. Neuropharmacology. 1995;34:1361–1369. doi: 10.1016/0028-3908(95)00114-l. [DOI] [PubMed] [Google Scholar]
- 4.Broadie K, Prokop A, Bellen HJ, O’Kane CJ, Schulze KL, Sweeney ST. Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron. 1995;15:663–673. doi: 10.1016/0896-6273(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 5.Buchner E, Gundersen CB. The DnaJ like cysteine string protein and neurotransmitter release. Trends Neurosci. 1997;20:223–227. doi: 10.1016/s0166-2236(96)10082-5. [DOI] [PubMed] [Google Scholar]
- 6.Caplan AJ, Cyr DM, Douglas MG. Eukaryotic homologs of Escherichia coli DnaJ: a diverse protein family that functions with Hsp70 stress proteins. Mol Biol Cell. 1993;4:555–563. doi: 10.1091/mbc.4.6.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlain LH, Henry J, Burgoyne RD. Cysteine string proteins are associated with chromaffin granules. J Biol Chem. 1996;271:19514–19517. doi: 10.1074/jbc.271.32.19514. [DOI] [PubMed] [Google Scholar]
- 8.Chappell TG, Welch WJ, Schlossman DM, Palter KB, Schlesinger MJ, Rothman JE. Uncoating ATPase is a member of the 70 kilodalton family of stress proteins. Cell. 1986;45:3–13. doi: 10.1016/0092-8674(86)90532-5. [DOI] [PubMed] [Google Scholar]
- 9.Chen MS, Obar RA, Schroeder CC, Austin TW, Poodry CA, Wadsworth SC, Vallee RB. Multiple forms of dynamin are encoded by shibire, a Drosophila gene involved in endocytosis. Nature. 1991;351:583–586. doi: 10.1038/351583a0. [DOI] [PubMed] [Google Scholar]
- 10.Grasso A, Mastrogiacomo A. α-Latrotoxin: preparation and effects on calcium fluxes. FEMS Microbiol Immunol. 1992;105:131–138. doi: 10.1111/j.1574-6968.1992.tb05895.x. [DOI] [PubMed] [Google Scholar]
- 11.Grigliatti TA, Hall L, Rosenbluth R, Suzuki DT. Temperature-sensitive mutants in Drosophila melanogaster. XIV. A selection of immobile adults. Mol Gen Genet. 1973;120:107–114. doi: 10.1007/BF00267238. [DOI] [PubMed] [Google Scholar]
- 12.Gundersen CB, Umbach JA. Suppression cloning of the cDNA encoding a candidate presynaptic calcium channel subunit of Torpedo. Neuron. 1992;9:527–537. doi: 10.1016/0896-6273(92)90190-o. [DOI] [PubMed] [Google Scholar]
- 13.Heuser JE, Reese TS, Dennis MJ, Jan Y, Jan L, Evans L. Exocytosis of synaptic vesicles captured by quick freezing and correlated with quantal transmitter release. J Cell Biol. 1979;81:275–300. doi: 10.1083/jcb.81.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katz B. The release of neural transmitter substances. Liverpool University; Liverpool, UK: 1969. [Google Scholar]
- 15.Katz B, Miledi R. Estimates of quantal content during chemical potentiation of transmitter release. Proc R Soc Lond [Biol] 1979;205:369–378. doi: 10.1098/rspb.1979.0070. [DOI] [PubMed] [Google Scholar]
- 16.Kohan SA, Pescatori M, Brecha NC, Mastrogiacomo A, Umbach JA, Gundersen CB. Cysteine string protein immunoreactivity in the nervous system and adrenal gland of the rat. J Neurosci. 1995;15:6230–6238. doi: 10.1523/JNEUROSCI.15-09-06230.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kosaka T, Ikeda K. Possible temperature-dependent blockage of synaptic vesicle recycling induced by a single gene mutation in Drosophila. J Neurobiol. 1983;14:207–225. doi: 10.1002/neu.480140305. [DOI] [PubMed] [Google Scholar]
- 18.Liley AW. The effects of presynaptic polarization on the spontaneous activity at the mammalian neuromuscular junction. J Physiol (Lond) 1956;134:427–443. doi: 10.1113/jphysiol.1956.sp005655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mastrogiacomo A, Gundersen CB. The nucleotide and deduced amino acid sequence of a rat cysteine string protein. Mol Brain Res. 1995;28:12–18. doi: 10.1016/0169-328x(94)00172-b. [DOI] [PubMed] [Google Scholar]
- 20.Mastrogiacomo A, Parsons SM, Zampighi GA, Jenden DJ, Umbach JA, Gundersen CB. Cysteine string proteins: a potential link between synaptic vesicles and presynaptic calcium channels. Science. 1994;263:981–982. doi: 10.1126/science.7906056. [DOI] [PubMed] [Google Scholar]
- 21.Meldolesi J, Scheer H, Madeddu L, Wanke E. Mechanism of action of α-latrotoxin: the presynaptic stimulatory toxin of black widow spider venom. Trends Pharmacol Sci. 1986;7:151–155. [Google Scholar]
- 22.Poodry CA, Edgar L. Reversible alterations in the neuromuscular junction of Drosophila melanogaster bearing a temperature-sensitive mutation, shibire. J Cell Biol. 1979;81:520–527. doi: 10.1083/jcb.81.3.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silver PA, Way JC. Eukaryotic DnaJ homologs and the specificity of Hsp70 activity. Cell. 1993;74:5–6. doi: 10.1016/0092-8674(93)90287-z. [DOI] [PubMed] [Google Scholar]
- 24.Sudhof T. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 25.Surkova I. Can exocytosis induced by α-latrotoxin be explained solely by its channel-forming activity? Ann NY Acad Sci. 1994;710:48–64. doi: 10.1111/j.1749-6632.1994.tb26613.x. [DOI] [PubMed] [Google Scholar]
- 26.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 27.Umbach JA, Zinsmaier KE, Eberle KK, Buchner E, Benzer S, Gundersen CB. Presynaptic dysfunction in Drosophila csp mutants. Neuron. 1994;13:899–908. doi: 10.1016/0896-6273(94)90255-0. [DOI] [PubMed] [Google Scholar]
- 28.Umbach JA, Mastrogiacomo A, Gundersen CB. Cysteine string proteins and presynaptic function. J Physiol (Paris) 1995;89:95–101. doi: 10.1016/0928-4257(96)80556-0. [DOI] [PubMed] [Google Scholar]
- 29.van der Bliek A, Meyerowitz EM. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature. 1991;351:411–414. doi: 10.1038/351411a0. [DOI] [PubMed] [Google Scholar]
- 30.Zinsmaier KE, Hofbauer A, Heimbeck G, Pflugfelder GO, Buchner S, Buchner E. A cysteine-string protein is expressed in retina and brain of Drosophila. J Neurogenet. 1990;7:15–29. doi: 10.3109/01677069009084150. [DOI] [PubMed] [Google Scholar]
- 31.Zinsmaier KE, Eberle KK, Buchner E, Walter K, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]