

Abstract
Botulinum (BoNT/A–G) and tetanus toxins (TeNT) are zinc endopeptidases that cleave proteins associated with presynaptic terminals (SNAP-25, syntaxin, or VAMP/synaptobrevin) and block neurotransmitter release. Treatment of hippocampal slice cultures with BoNT/A, BoNT/C, BoNT/E, or TeNT prevented the occurrence of spontaneous or miniature EPSCs (sEPSCs or mEPSCs) as well as the [Ca2+]o-independent increase in their frequency induced by phorbol ester, 0.5 nm α-latrotoxin, or sucrose. [Ca2+]o-independent and -dependent release thus requires that the target proteins of clostridial neurotoxins be uncleaved. In contrast, significant increases in mEPSC frequency were produced in BoNT-treated, but not TeNT-treated, cultures by application of the Ca2+ionophore ionomycin in the presence of 10 mm[Ca2+]o. The frequency of sEPSCs was increased in BoNT-treated, but not TeNT-treated, cultures by increasing [Ca2+]o from 2.8 to 5–10 mm or by applying 5 mm Sr2+. Large Ca2+ and Sr2+ influxes thus can rescue release after BoNT treatment, albeit less than in control cultures. The nature of the toxin-induced modification of Ca2+-dependent release was assessed by recordings from monosynaptically coupled CA3 cell pairs. The paired-pulse ratio of unitary EPSCs evoked by two presynaptic action potentials in close succession was 0.5 in control cultures, but it was 1.4 and 1.2 in BoNT/A- or BoNT/C-treated cultures when recorded in 10 mm[Ca2+]o. Log–log plots of unitary EPSC amplitude versus [Ca2+]o were shifted toward higher [Ca2+]o in BoNT/A- or BoNT/C-treated cultures, but their slope was unchanged and the maximal EPSC amplitudes were reduced. We conclude that BoNTs reduce the Ca2+ sensitivity of the exocytotic machinery and the number of quanta released.
Keywords: Ca2+, clostridial neurotoxins, exocytosis, α-latrotoxin, protein kinase, transmitter release
Synaptic vesicle exocytosis is accomplished by a series of protein interactions in the axon terminal (for review, see Schweizer et al., 1995; Südhof, 1995). The so-called synaptosomal-associated receptor (SNARE) proteins include the vesicle-associated membrane proteins (v-SNAREs), vesicle-associated membrane protein (VAMP)/synaptobrevin and synaptotagmin, and the plasma-associated target proteins (t-SNAREs), synaptosomal-associated protein (SNAP-25) and syntaxin. VAMP/synaptobrevin, syntaxin, and SNAP-25 form a tight heterotrimeric complex in vitro that has been proposed to be essential for exocytosis (Söllner et al., 1993a,b). Evidence that the SNARE proteins are required for neurotransmitter release comes from studies showing that tetanus toxin (TeNT) and the seven serotypes of botulinum neurotoxin (BoNT) block transmitter release by acting as zinc-dependent endopeptidases that proteolyse several SNARE proteins (for review, see Niemann et al., 1994; Schiavo et al., 1995). For example, TeNT cleaves the v-SNARE VAMP/synaptobrevin (Link et al., 1992; Schiavo et al., 1992), BoNT/A and BoNT/E cleave the t-SNARE SNAP-25 (Blasi et al., 1993a; Schiavo et al., 1993), and BoNT/C cleaves the t-SNARE syntaxin alone (Blasi et al., 1993b) or together with SNAP-25 (Foran et al., 1996; Osen-Sand et al., 1996; Williamson et al., 1996).
Clostridial neurotoxins are powerful inhibitors of exocytosis from various non-neuronal cells and nerve terminals (for review, see Dolly et al., 1994; Poulain et al., 1995). How the cleavage of SNARE proteins by clostridial toxins causes this inhibition of release is not known. Large numbers of synaptic vesicles remain associated with presynaptic release sites after clostridial toxin treatment (Harris and Miledi, 1971; Hunt et al., 1994; Broadie et al., 1995). In addition, SNARE proteins cleaved by clostridial toxins remain capable of forming heterotrimeric complexes, and these can be dissociated by ATP, much like complexes formed by intact proteins (Hayashi et al., 1994;Pellegrini et al., 1995). If the docking of vesicles to the plasma membrane and the assembly of fusion complexes are not inhibited, some other physiological mechanism(s) must underlie the actions of clostridial toxins.
The fusion of neurotransmitter-containing vesicles with the plasma membrane can occur spontaneously or in response to action potential-induced Ca2+ influx (Katz, 1969). Spontaneous exocytosis occurs at a low rate in the absence of Ca2+ influx, and this rate is increased by elevations of [Ca2+]i (Kita and Van der Kloot, 1974). A number of agents promote exocytosis by mechanisms that may not involve an increase in [Ca2+]i, however, including phorbol ester activators of protein kinase C (PKC) (Malenka et al., 1987; Gillis et al., 1996); the black widow spider venom component, α-latrotoxin (α-LTx) (Longenecker et al., 1970) (for review, seeRosenthal and Meldolesi, 1989); and hyperosmotic, sucrose-containing saline (Fatt and Katz, 1952) (for review, see Van der Kloot and Molgó, 1994). At the Drosophila neuromuscular junction, syntaxin and VAMP/synaptobrevin must be present and intact to support α-LTx-induced, but not sucrose-induced, release (Broadie et al., 1995; Sweeney et al., 1995). In contrast, α-LTx may remain effective at clostridial toxin-treated mammalian neuromuscular junctions, but not sucrose-containing saline (Dreyer et al., 1987;Gansel et al., 1987). Unfortunately, the actions of secretagogues have not been studied extensively at toxin-treated vertebrate CNS synapses.
To address these issues, we incubated hippocampal slice cultures with clostridial neurotoxins and then examined the effects of this treatment on spontaneous glutamate release, on [Ca2+]o-dependent-evoked release, and on [Ca2+]o-independent release elicited by phorbol ester, sucrose, or α-LTx.
MATERIALS AND METHODS
Slice cultures, electrophysiology, and drug application.Organotypic hippocampal slice cultures were prepared from 6-d-old rat pups as described in detail elsewhere (Gähwiler, 1981). After 2–4 weeks in vitro, cultures were placed in a recording chamber on an inverted microscope and superfused with control saline containing (in mm): 137 NaCl, 2.7 KCl, 2.8 CaCl2, 2.5 MgCl2, 11.6 NaHCO3, 0.4 NaH2PO4, and 5.6 glucose, pH 7.4, at 24°C. SrCl2 was applied in Ca2+-free saline. CA3 pyramidal cells were whole-cell voltage-clamped to −70 mV, using pipettes containing (in mm): 140 K-gluconate, 10 KCl, 5 HEPES, 2 MgCl2, and 1.1 EGTA, pH 7.3. Cell capacitance and series resistance (10.0 ± 0.5 MΩ; n = 50, randomly selected) were monitored routinely. Miniature EPSCs (mEPSCs) and spontaneous EPSCs (sEPSCs) were acquired and analyzed as described previously (Capogna et al., 1996). Unitary EPSCs between pairs of monosynaptically connected CA3 pyramidal neurons were recorded as described previously (Debanne et al., 1996). The presynaptic cell was recorded in current-clamp mode with a 1 mKMeSO4-filled sharp microelectrode, and action potentials were elicited with 20 msec depolarizing current pulses (0.1–0.2 Hz). The postsynaptic cell was whole-cell voltage-clamped to −70 mV. All statistical comparisons were performed with two-tailed paired or two sample t tests; numerical values are given as mean ± SEM.
Serum-free medium containing TeNT or BoNTs was applied in the test tube containing a slice culture under sterile conditions after >14 din vitro. Then the culture was returned to the incubator for ∼48 hr at 36°C before immunocytochemistry, immunoblotting, or electrophysiological recording was performed. α-LTx (Alomone Labs, Jerusalem, Israel) was applied focally by drop application (20 nl, 0.5 or 3 nm) after saline perfusion was stopped, as described elsewhere (Capogna et al., 1996). Tetrodotoxin (TTX, Sankyo, Tokyo, Japan), bicuculline methochloride (Fluka AG, Buchs, Switzerland), AMPA (Tocris Cookson, Bristol, UK), and phorbol 12,13-diacetate (PDAc) and ionomycin (LC Laboratories Europe, Läufelfingen, Switzerland) were bath-applied. BoNT/C was purchased from WAKO Chemicals (Neuss, Germany).
Immunocytochemistry. Hippocampal cultures were fixed overnight with 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, washed in incubation medium (PB, 1.5% horse serum, 1.5% goat serum, and bovine serum albumin), and then left overnight in incubation medium containing Triton X-100 (0.4% v/v) at 4°C. A mouse anti-synaptophysin monoclonal antibody (SY38, 1:20 dilution; Boehringer Mannheim, Mannheim, Germany) was applied (84 hr at 4°C) in incubation medium containing 0.4% Triton X-100, together with anti-VAMP/synaptobrevin-2 (1:100 dilution), anti-SNAP-25 (1:200 dilution), or anti-syntaxin (1:200 dilution) affinity-purified antibodies from rabbit [provided by C. Montecucco, University of Padua, Italy; for detailed description of the epitopes recognized by these antibodies, see Rossetto et al. (1996) and Williamson et al. (1996)]. Cultures were washed in incubation medium, followed by application of a biotinylated anti-rabbit antibody (1:200 dilution; 3 hr at 22°C), and then washed again before using an anti-mouse antibody (1:150 dilution; 20 min at 22°C) directly conjugated to FITC to reveal the anti-synaptophysin antibodies. Cultures were washed again before avidin–Texas Red was applied to reveal the anti-SNARE antibodies. Cultures were mounted in Slow Fade (Molecular Probes, Portland, OR) and imaged on a laser scanning confocal microscope (Zeiss LSM-410, Oberkochen, Germany), using lasers tuned to 488 nm (FITC) and 543 nm (Texas Red) and a 100× objective (1.4 numerical aperture). Ten optical sections were collected from area CA3 at 0.2-μm-depth intervals, and three-dimensional reconstruction was made with IMARIS software (Bitplane AG, Zurich, Switzerland). Immunocytochemistry was performed “blind,” and treated and untreated cultures were processed simultaneously. The mean pixel intensity and SD for each of the SNARE proteins were calculated from the original unprocessed optical section by Voxelshop software (Bitplane AG).
Immunoblotting. Four control or clostridial neurotoxin-treated cultures were pooled and washed three times in PBS containing a protease inhibitor cocktail (Complete, Boehringer Mannheim). The samples were solubilized in the same buffer containing Triton X-100 (1% v/v) for 1 hr before centrifugation at 80,000 ×g for 1 hr. Equal volumes of the supernatants were diluted into SDS-PAGE sample buffer, boiled for 5 min at 95°C, and analyzed on 12.5% polyacrylamide gels. After blotting, a protein stain of the nitrocellulose revealed no obvious differences in the amount or pattern of proteins loaded. The blots were probed by incubation with antibodies against synaptophysin (SY38, Boehringer Mannheim), SNAP-25 (SMI 81, Affiniti Research Products, Nottingham, UK), syntaxin (HPC1, Sigma, Deisenhofen, Germany), and VAMP/synaptobrevin-2 and synaptotagmin (kindly provided by M. Takahashi, Mitsubishi Kasei Institute of Life Sciences, Tokyo, Japan; for details, see Oho et al., 1995). Note that these antibodies recognize different epitopes of the SNARE proteins from those used for immunocytochemistry. Bound immunoreactivity was detected by ECL (Amersham, Little Chalfont, UK), as described previously (Pellegrini et al., 1995). The analysis was performed blind on three separate sets of cultures.
RESULTS
Clostridial neurotoxins cleave SNARE proteins in hippocampal slice cultures
Rat hippocampal slice cultures were incubated with TeNT (50–100 ng/ml, 45 ± 4 hr, n = 23), BoNT/A (50–100 ng/ml, 55 ± 2 hr, n = 44), BoNT/C (50–100 ng/ml, 50 ± 2 hr, n = 34), or BoNT/E (1 μg/ml, 59 ± 5 hr, n = 6). The effectiveness of the neurotoxin treatment was examined with immunocytochemistry and with Western blots (Fig. 1). VAMP/synaptobrevin-2 immunoreactivity was virtually absent in TeNT-treated cultures. In BoNT/A-treated cultures SNAP-25 was immunocytochemically undetectable with an antibody directed against the C-terminal portion of the molecule (Williamson et al., 1996), and Western blot analysis revealed a decrease in the molecular weight of essentially all SNAP-25. BoNT/C treatment abolished SNAP-25 and syntaxin immunocytochemical staining, strongly reduced the amount of uncleaved syntaxin in the immunoblots, and decreased the molecular weight of the majority of SNAP-25, as detected by using another antibody that recognizes BoNT-cleaved SNAP-25. In contrast, in toxin-treated cultures, synaptic terminals remained densely stained with the anti-synaptophysin antibody, suggesting that synaptic vesicles were retained in the terminals of treated cultures. The amount of synaptophysin immunoreactivity appeared to be reduced slightly in TeNT-treated cultures; however, none of the neurotoxins affected the amount of the synaptic vesicle protein synaptotagmin in immunoblots (data not shown). TeNT thus cleaved VAMP/synaptobrevin-2, BoNT/A cleaved SNAP-25, and BoNT/C cleaved syntaxin and SNAP-25 in hippocampal slice cultures, consistent with previous reports in isolated synaptic vesicles, synaptosomes, and cultured cells (Foran et al., 1996; Osen-Sand et al., 1996; Williamson et al., 1996) (for review, see Schiavo et al., 1995).
Fig. 1.
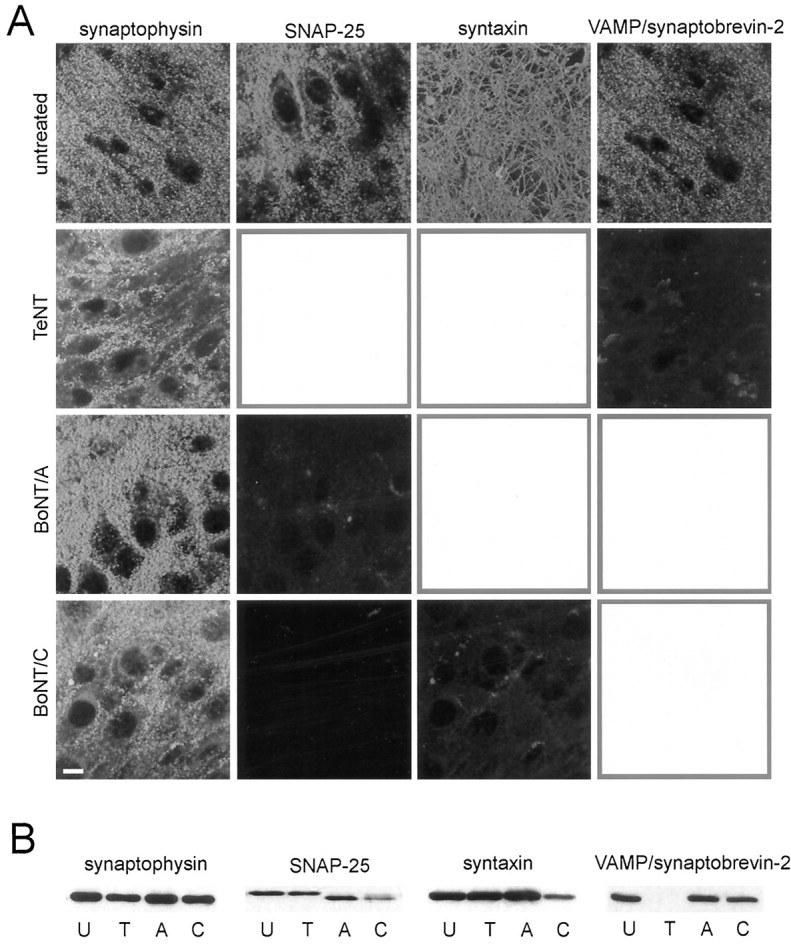
Clostridial neurotoxins effectively cleave SNAREs. Control and clostridial toxin-treated hippocampal slice cultures were labeled with antibodies directed against SNAREs and the synaptic vesicle marker synaptophysin, as indicated, or subjected to immunoblot analysis. A, Immunocytochemical staining for SNAREs was abolished in large part in toxin-treated cultures, whereas staining for the synaptic vesicle-associated protein synaptophysin remained. Cytotoxic effects of toxin treatment were not apparent (cf. Osen-Sand et al., 1996). Scale bar, 10 μm. Autofluorescence of macrophages can be seen in toxin-treated cultures stained for SNAREs. Mean pixel intensity and SD were calculated for each of the SNARE proteins from the unprocessed original data sets. The mean pixel intensities for both the treated and untreated cultures for the different SNARE proteins are as follows: SNAP-25—untreated, 59.23 ± 34.33; BoNT/A, 18.86 ± 11.68; BoNT/C, 11.83 ± 6.65;syntaxin—untreated, 60.09 ± 29.03; BoNT/C, 19.53 ± 13.81; VAMP/synaptobrevin-2—untreated, 56.19 ± 29.79; TeNT, 16.66 ± 12.39. B, VAMP/synaptobrevin-2 and syntaxin immunoreactivity was absent or strongly reduced in Western blots of TeNT toxin-treated (T) and BoNT/C-treated (C) cultures, respectively, as compared with untreated control cultures (U), whereas synaptophysin immunoreactivity was present. BoNT/A (A) and BoNT/C treatment reduced the molecular weight of SNAP-25.
Clostridial neurotoxins block basal and [Ca2+]o-independent release
Incubation of cultures with clostridial neurotoxins invariably blocked glutamate release in control saline. The frequency of mEPSCs recorded from CA3 pyramidal cells in the presence of 0.5 μm TTX and 40 μm bicuculline methochloride (to block action potential-dependent release and GABAAreceptors, respectively) was reduced significantly by all toxins relative to control cultures (p < 0.001) (Table1; compare Fig.2A vs Figs.3 and 5A–C). Furthermore, the frequency of action potential-dependent sEPSCs recorded in control saline (without TTX) also was reduced significantly by all neurotoxins (p < 0.001) (Table 1; compare Fig.2B–D vs Figs. 4 and5D–F).
Table 1.
Effects of clostridial neurotoxins on hippocampal synaptic transmission
| Control | TeNT | BoNT/A | BoNT/C | |||||
|---|---|---|---|---|---|---|---|---|
| Before | After | Before | After | Before | After | Before | After | |
| mEPSC frequency (Hz) | ||||||||
| Control | 0.92 ± 0.10 (45)° | – | 0.11 ± 0.02 (14)1-159 | – | 0.07 ± 0.01 (24)1-159 | – | 0.04 ± 0.01 (15)∥ | – |
| PDAc | 1.50 ± 0.22 | 7.40 ± 0.90 (8)‡ | 0.11 ± 0.03 | 0.18 ± 0.06 (9)* | 0.05 ± 0.01 | 0.06 ± 0.02 (7)* | 0.07 ± 0.04 | 0.08 ± 0.04 (6)* |
| α-LTx (0.5 nm) | 0.79 ± 0.14 | 11.16 ± 1.67 (26)‡ | 0.13 ± 0.04 | 0.11 ± 0.05 (6)* | 0.10 ± 0.04 | 0.11 ± 0.05 (7)* | 0.05 ± 0.02 | 0.07 ± 0.02 (8)* |
| α-LTx (3 nm/10 mmCa) | – | – | 0.15 ± 0.09 | 0.14 ± 0.10 (3)* | 0.06 ± 0.02 | 6.70 ± 1.20 (4)§§ | 0.09 ± 0.03 | 8.07 ± 3.50 (3)* |
| Ionomycin (2.8 mm Ca) | 1.07 ± 0.15 | 7.03 ± 1.96 (15)§§ | 0.13 ± 0.05 | 0.16 ± 0.08 (6)* | 0.07 ± 0.03 | 0.14 ± 0.10 (5)* | 0.09 ± 0.02 | 0.18 ± 0.06 (5)* |
| Ionomycin (10 mmCa) | – | – | 0.11 ± 0.04 | 0.64 ± 0.33 (4)* | 0.05 ± 0.02 | 3.11 ± 0.79 (8)§§ | 0.09 ± 0.02 | 2.65 ± 0.74 (5)§ |
| sEPSC frequency (Hz) | ||||||||
| Control | 1.68 ± 0.25 (18)° | – | 0.09 ± 0.01 (32)1-159 | – | 0.09 ± 0.01 (41)1-159 | – | 0.09 ± 0.02 (22)∥ | – |
| 5 mm Ca/0.5 mm Mg | 1.17 ± 0.17 | 4.71 ± 0.84 (7)† | 0.10 ± 0.02 | 0.10 ± 0.04 (9)* | 0.09 ± 0.02 | 0.91 ± 0.23 (15)† | 0.09 ± 0.03 | 1.25 ± 0.32 (9)§§ |
| 10 mm Ca/0.5 mm Mg | 2.12 ± 0.33 | 19.12 ± 4.03 (4)§ | 0.10 ± 0.02 | 0.69 ± 0.29 (8)* | 0.11 ± 0.02 | 4.24 ± 1.36 (10)§§ | 0.08 ± 0.02 | 2.54 ± 0.37 (10)‡ |
| 2.8 mm Sr/2.5 mm Mg | 1.45 ± 0.25 | 3.45 ± 0.72 (6)§ | 0.10 ± 0.03 | 0.05 ± 0.01 (3)* | 0.09 ± 0.05 | 0.62 ± 0.12 (7)§§ | 0.14 ± 0.06 | 1.04 ± 0.28 (5)§ |
| 5 mm Sr/0.5 mmMg | 1.45 ± 0.25 | 7.30 ± 1.70 (6)† | 0.09 ± 0.02 | 0.05 ± 0.01 (6)* | 0.09 ± 0.04 | 1.13 ± 0.33 (5)§ | 0.16 ± 0.06 | 1.83 ± 0.58 (6)§ |
| 7.5 mm Sr/0.5 mmMg | – | – | 0.12 ± 0.03 | 0.15 ± 0.01 (4)* | – | – | – | – |
| Sucrose | 1.80 ± 0.20 | 11.07 ± 1.07 (3)§§ | 0.07 ± 0.03 | 0.05 ± 0.01 (5)* | 0.08 ± 0.01 | 0.09 ± 0.06 (3)* | 0.03 ± 0.01 | 0.03 ± 0.02 (3)* |
Significance assessed relative to values in same cells before treatment:
not significantly different (p > 0.05);§p < 0.05;§§p < 0.01;
p < 0.005;
p < 0.001; –, not determined.
Significance of sEPSC vs mEPSC frequency:
F1-159: not significantly different (p > 0.1);∥p < 0.04; °p > 0.001.
The number of observations is given in parentheses. The effects of α-LTx and ionomycin on mEPSC frequency in control cultures include data from Capogna et al. (1996).
Fig. 2.

In untreated cultures the frequency of mEPSCs is enhanced by phorbol ester, and the frequency of sEPSCs is increased by saline containing 5 or 10 mm Ca2+/0.5 mm Mg2+, 2.8 or 5 mmSr2+/0.5 mm Mg2+, or sucrose. Each sweep shows a continuous recording before and after application of 3 μm PDAc (A,phorbol ester), 5 or 10 mmCa2+/0.5 mm Mg2+, 2.8 or 5 mm Sr2+/0.5 mmMg2+, or sucrose (100 mm).A, PDAc increased mEPSC frequency in a [Ca2+]o-independent manner in untreated cultures (2.2 Hz before PDAc, 8.1 Hz after PDAc, and 8.5 Hz after subsequent application of Ca2+-free/1 mm EGTA-containing saline). B,C, Both 5 or 10 mmCa2+/0.5 mm Mg2+ and 2.8 or 5 mm Sr2+/0.5 mmMg2+ increased sEPSC frequency in untreated cultures (1.6 Hz before, and 8.7 and 21.2 Hz after 5 and 10 mmCa2+, respectively; 1.8 Hz before, and 2.6 and 7.9 Hz after 2.8 and 5 mm Sr2+, respectively). D, Application of 100 mmsucrose enhanced sEPSC frequency in control cultures (1.6 Hz before and 13.2 Hz after sucrose). Data on the increases in mEPSC frequency induced by ionomycin and α-LTx in control cultures are reported inCapogna et al. (1996).
Fig. 3.
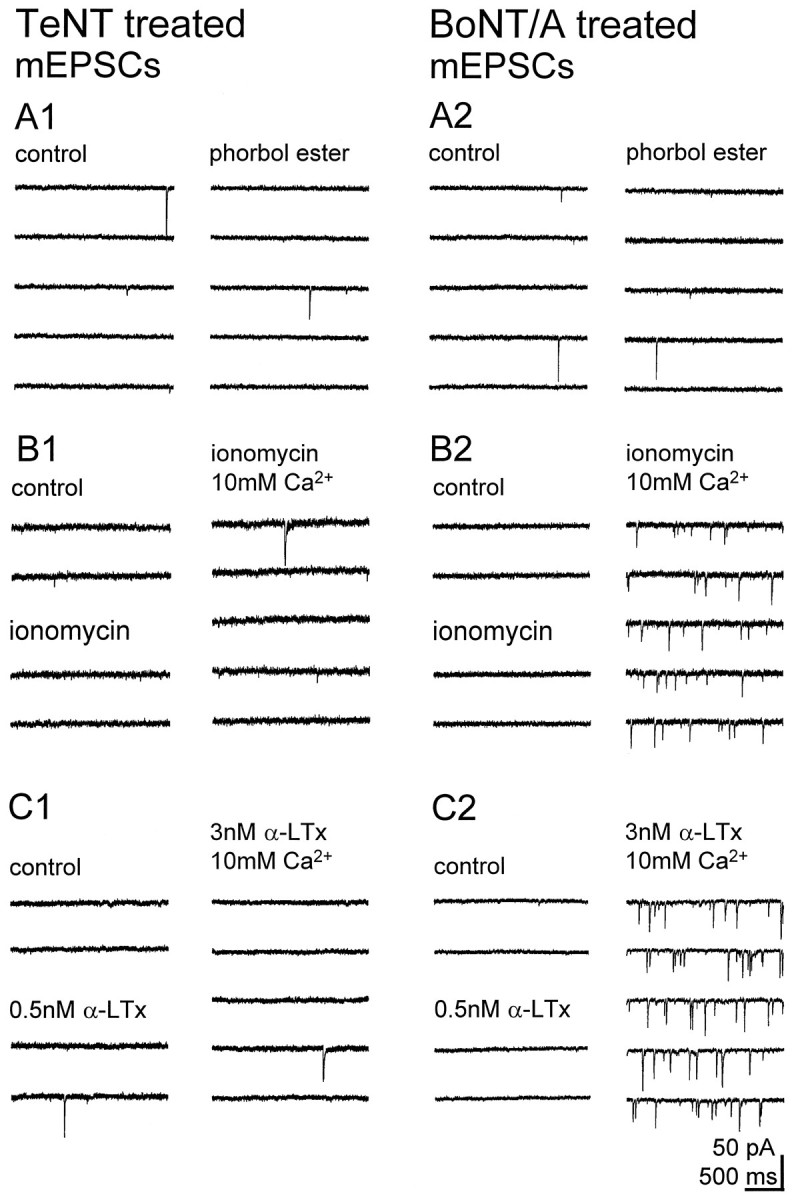
The frequency of mEPSCs is very low in clostridial toxin-treated cultures but is increased by large Ca2+ influx into the terminal in BoNT/A-treated, but not TeNT-treated, cultures. Each sweep shows a continuous recording before and after application of 3 μm PDAc (A, phorbol ester), the Ca2+ ionophore ionomycin (B), and α-LTx (C). A, PDAc did not increase mEPSC frequency in either TeNT- or BoNT/A-treated cultures (0.1 Hz before and 5 min after PDAc; 0.07 Hz before and 0.06 Hz 5 min after PDAc, respectively). B1, C1, Neither 2.5 μm ionomycin applied in control or 10 mmCa2+ containing saline nor 0.5 nmα-LTx applied in control saline or 3 nm α-LTx applied in 10 mm Ca2+ increased mEPSC frequency in TeNT-treated cultures (0.06 Hz in control, 0.07 Hz 5 min after ionomycin in 2.8 mm Ca2+, and 0.11 Hz 5 min after ionomycin in 10 mm Ca2+; 0.06 Hz in control, 0.05 Hz 5 min after 0.5 nm α-LTx in 2.8 mm Ca2+, and 0.05 Hz 5 min after 3 nm α-LTx in 10 mm Ca2+).B2, C2, Ionomycin and α-LTx increased mEPSC frequency only after [Ca2+]o was raised from 2.8 (control) to 10 mm in BoNT/A-treated cultures (0.04 Hz in control, 0.07 Hz 5 min after ionomycin in 2.8 mmCa2+, and 3.6 Hz 5 min after ionomycin in 10 mm Ca2+; 0.06 Hz in control, 0.05 Hz 5 min after 0.5 nm α-LTx in 2.8 mmCa2+, and 5.2 Hz 5 min after 3 nmα-LTx in 10 mm Ca2+).
Fig. 5.

In BoNT/C-treated cultures the low frequency of sEPSCs or mEPSCs is increased only by large Ca2+ or Sr2+ influx into the axon terminal.A, PDAc (3 μm) did not increase mEPSC frequency in BoNT/C-treated cultures (0.05 Hz before and 0.03 Hz 5 min after PDAc). B, C, Ionomycin and α-LTx each increased mEPSC frequency only after [Ca2+]o was raised from 2.8 (control) to 10 mm in BoNT/C-treated cultures (0.13 Hz in control, 0.36 Hz 5 min after ionomycin in 2.8 mmCa2+, and 5.3 Hz 5 min after ionomycin in 10 mm Ca2+; 0.08 Hz in control, 0.05 Hz 5 min after 0.5 nm α-LTx in 2.8 mmCa2+, and 3.3 Hz 5 min after 3 nmα-LTx in 10 mm Ca2+).D, Raising [Ca2+]o from 2.8 mm (control) to 5 or 10 mm and decreasing [Mg2+]o from 2.5 mm to 0.5 mm increased sEPSC frequency in BoNT/C-treated cultures (0.05 Hz control, 0.5 Hz 5 min after 5 mmCa2+, and 3.9 Hz 5 min after 10 mmCa2+). E, Application of either 2.8 mm Sr2+/2.5 mmMg2+ or 5 mm Sr2+/0.5 mm Mg2+ containing saline increased sEPSC frequency in BoNT/C-treated cultures (0.04 Hz control, 0.85 Hz 5 min after 2.8 mm Sr2+, and 1.73 Hz 5 min after 5 mm Sr2+). F, Application of 100 mm sucrose did not increase sEPSC frequency in BoNT/C-treated cultures (0.06 Hz before and 5 min after sucrose).
Fig. 4.

The frequency of sEPSCs is very low in clostridial neurotoxin-treated cultures and is increased by large Ca2+ or Sr2+ influxes in BoNT/A-treated, but not TeNT-treated, cultures. Each sweep shows a continuous recording before and after application of saline containing 10 mm Ca2+/0.5 mmMg2+ (A, high [Ca2+]o), saline containing 5 mm Sr2+/0.5 mmMg2+ (B, high [Sr2+]o), or sucrose (100 mm). A1, B1, Neither high [Ca2+]o nor high [Sr2+]o increased sEPSC frequency in TeNT-treated cultures (0.09 Hz before and 5 min after high [Ca2+]o; 0.14 Hz before and 0.09 Hz 5 min after high [Sr2+]o). A2,B2, Both high [Ca2+]oand high [Sr2+]o increased sEPSC frequency in BoNT/A-treated cultures (0.12 Hz before and 4.6 Hz 5 min after high [Ca2+]o; 0.04 Hz before and 2.4 Hz 5 min after high [Sr2+]o). C1,C2, Application of 100 mm sucrose did not increase sEPSC frequency in TeNT- or BoNT/A-treated cultures (0.06 Hz before and 5 min after sucrose, and 0.05 Hz before and 5 min after sucrose, respectively).
The toxins had no effect on postsynaptic glutamate sensitivity, as indicated by their lack of effect on mean mEPSC amplitudes (control, 13.3 ± 0.7 pA, n = 32; TeNT, 16 ± 1.3 pA,n = 12; BoNT/A, 14 ± 1.2 pA, n = 24; BoNT/C, 12.6 ± 1.3 pA, n = 14;p > 0.05 vs control for all toxins), or on the amplitude of inward currents elicited by application of AMPA (30 sec, 1 μm) (control, 147 ± 12 pA, n = 5; TeNT, 167 ± 31 pA, n = 5; p > 0.05). In untreated cultures the mean amplitude of sEPSCs was greater than the mean amplitude of mEPSCs (sEPSCs 18.6 ± 1.8 pA,n = 16; mEPSCs 13.3 ± 0.7 pA, n = 32; p < 0.001), consistent with a significant contribution of action potential-induced multiquantal events in the absence of TTX. In contrast, there was no significant difference in the amplitude of spontaneous events before and after application of TTX to clostridial neurotoxin-treated cultures, consistent with an inhibition of action potential-evoked transmitter release (TeNT—sEPSCs 17.7 ± 1.1 pA, n = 32; mEPSCs 16.0 ± 1.3 pA,n = 12; p > 0.1; BoNT/A—sEPSCs 16.4 ± 1.0 pA, n = 41; mEPSCs 14.0 ± 1.2 pA, n = 24; p > 0. 1; BoNT/C—sEPSCs 13.9 ± 1.5 pA, n = 22; mEPSCs 12.6 ± 1.3 pA, n = 14; p > 0.5). Further support for this conclusion is that the frequency of spontaneous synaptic currents in toxin-treated cultures in large part was unaffected by TTX (Table 1).
α-LTx was found previously to be able to trigger glutamate release from hippocampal slice cultures in a [Ca2+]o-independent manner when applied at <1 nm (Capogna et al., 1996). In addition, PDAc (3 μm) produced an increase in mEPSC frequency in untreated cultures that also was unaffected by removal of Ca2+ from the extracellular saline (1.7 ± 0.4 Hz before and 9.2 ± 0.9 Hz 5 min after application of 3 μm PDAc, and 8.9 ± 1.3 Hz 3 min after subsequent application of Ca2+-free/1 mmEGTA-containing saline, n = 3; Fig.2A). After treatment with BoNT/A, BoNT/C, or TeNT, however, the frequency of mEPSCs was not increased by application of either 3 μm PDAc (Figs. 3A, 5A) or 0.5 nm α-LTx (Figs. 3C, 5C).
The effects of clostridial toxins on neurotransmitter release were dependent on the concentration and time of toxin exposure. In cultures treated with 50 ng/ml BoNT/C for only 18–21 hr, for example, the frequency of mEPSCs was 0.6 ± 0.25 Hz, and subsequent application of 3 μm PDAc increased the frequency of mEPSCs to 3.43 ± 0.29 Hz (n = 3; compare Table 1). Positive correlations between the extent of reduction of the frequency of sEPSCs or mEPSCs and the amount of immunocytochemically detectable uncleaved SNAREs also were found (data not shown).
TeNT, BoNT/A, or BoNT/C thus were equally effective in blocking glutamate release as well as in the ability of phorbol esters and α-LTx to induce [Ca2+]o-independent exocytosis in hippocampal cultures. All of these forms of exocytosis thus depend on the integrity of VAMP/synaptobrevin, SNAP-25, and syntaxin. Finally, these results provided a positive control that the toxins had impaired release from virtually all of the excitatory synaptic terminals within the treated cultures.
Large influxes of Ca2+ or Sr2+ rescues transmitter release in BoNT/A- or BoNT/C-treated cultures
The block of transmitter release in control saline could be overcome by inducing large increases in intraterminal [Ca2+] via several means in BoNT/A- or BoNT/C-treated, but not in TeNT-treated, cultures (Table 1).
Application of the Ca2+ ionophore ionomycin (2.5 μm) in control saline increased mEPSC frequency in untreated (Capogna et al., 1996), but not in clostridial neurotoxin-treated, cultures. If the concentration of Ca2+ in the extracellular saline was increased from 2.8 to 10 mm during ionomycin application, however, then significant increases in mEPSC frequency were produced in BoNT/A- or BoNT/C-treated cultures, but not in TeNT-treated cultures (Figs.3B, 5B). The frequency of mEPSCs under these conditions nevertheless was lower than the frequency of mEPSCs in control cultures after application of ionomycin in control saline. In saline containing 10 mm Ca2+, increases in mEPSC frequency also could be produced in BoNT/A- or BoNT/C-treated, but not TeNT-treated, cultures by applying a high concentration of α-LTx (3 nm, Figs. 3C, 5C), which triggers release from hippocampal cultures in a [Ca2+]o-dependent manner (Capogna et al., 1996).
We next examined whether increased Ca2+ influx through voltage-dependent Ca2+ channels also could restore action potential-mediated sEPSCs in the absence of TTX. In control cultures a significant elevation of the sEPSC frequency (Table1; Fig. 2B) was produced by increasing release probability with saline containing 5 or 10 mmCa2+/0.5 mm Mg2+. In BoNT/A- or BoNT/C-treated cultures, sEPSC frequency also was increased by saline containing 5 or 10 mm Ca2+/0.5 mm Mg2+ (Table 1; Figs.4A2, 5D). In TeNT-treated cultures, in contrast, no significant increases in sEPSC frequency were observed after raising [Ca2+]o (Table 1; Fig.4A1). We conclude that the rescue of release can occur regardless of the means of Ca2+ entry.
Removal of extracellular Ca2+ and application of saline containing 5 mm Sr2+/0.5 mm Mg2+ induce desynchronized release (Table 1; Fig. 2C), perhaps by activating a distinct Ca2+ sensor protein (Goda and Stevens, 1994; Li et al., 1995). We therefore tested the ability of Sr2+to rescue release in toxin-treated cultures. Indeed, replacement of extracellular Ca2+ with Sr2+resulted in an increase in sEPSC frequency in BoNT/A- or BoNT/C-treated cultures, but not in TeNT-treated cultures (Table 1; Figs.4B1,B2, 5E). Both Ca2+and Sr2+ thus were able to rescue release after cleavage of SNAP-25 alone or together with SNAP-25.
Increasing the osmolarity of the extracellular saline by adding 100 mm sucrose also increased sEPSC frequency in control cultures (Table 1; Fig. 2D). This effect was independent of [Ca]o, because the frequency of sEPSCs in 50 mm sucrose was not reduced by applying Ca2+-free/1 mm EGTA-containing saline (5.7 ± 0.9 Hz before and 5.4 ± 1.4 Hz after;n = 3), in agreement with Rosenmund and Stevens (1996). No increase in sEPSC frequency was produced by 100 mmsucrose in cultures treated with any clostridial toxin (Table 1; Figs.4C, 5F).
Effects of BoNT/E treatment
BoNT/E cleaves more of SNAP-25 than BoNT/A (26 vs 9 amino acids) (Binz et al., 1994). We therefore asked whether rescue of release could be obtained after BoNT/E treatment. Cleavage of SNAP-25 with BoNT/E resulted in the same electrophysiological phenotype as BoNT/A or BoNT/C treatment. In BoNT/E-treated cultures the mean frequency of mEPSCs was very low (0.07 ± 0.05 Hz,n = 6) and was not increased by application of phorbol ester (0.08 ± 0.06 Hz and 0.07 ± 0.03 Hz, n= 5; p > 0.05; before and after 3 μmPDAc). In contrast, significant increases in mEPSC frequency were produced by ionomycin when it was applied in saline containing 10 mm Ca2+ (0.07 ± 0.06 Hz and 1.2 ± 0.06 Hz, n = 4; p < 0.01; before and after ionomycin). Similarly, the mean frequency of sEPSCs was very low in control saline (0.06 ± 0.02 Hz, n= 12) and was significantly increased by 10 mmCa2+/0.5 mmMg2+-containing saline (0.1 ± 0.02 Hz and 1.2 ± 0.3 Hz, n = 6; p < 0.05; before and after 10 mm Ca2+/0.5 mm Mg2+). Enhanced influx of Ca2+ also was able to rescue release after a large portion had been cleaved from the C terminus of SNAP-25.
Actions of BoNT/A and BoNT/C on evoked unitary EPSCs
When two action potentials are elicited in close succession in a single cell, the second action potential triggers the release of more vesicles than the first, because the release probability remains transiently elevated because of the residual Ca2+that remains in the nerve terminal after the first action potential (Katz and Miledi, 1968). We therefore examined the effects of BoNT/A and BoNT/C treatment on unitary EPSCs elicited by two action potentials at 50–80 msec intervals with dual recordings of monosynaptically coupled CA3 pyramidal cells. The paired-pulse ratio (PPR = amplitude of all EPSC2s/amplitude of all EPSC1s) of unitary EPSCs was inversely dependent on release probability (Fig.6), as previously reported (Debanne et al., 1996). In untreated cultures in control saline (Fig. 6) (n = 5), the amplitude of EPSC1 was 41 ± 14 pA, the probability that the first presynaptic action potential failed to trigger an EPSC was 0.05 ± 0.03, and the mean PPR was 1.4 ± 0.5. Application of saline containing 10 mmCa2+/0.5 mm Mg2+increased the mean amplitude of EPSC1 to 63 ± 5 pA, decreased the failure probability to 0, and decreased the PPR to 0.5 ± 0.1 (n = 3), consistent with an increase in release probability. Lowering release probability with saline containing 1 mm Ca2+/2.5 mmMg2+ saline produced opposite changes (n = 3; data not shown). In BoNT/A-treated (n = 5) or BoNT/C-treated (n = 3) cultures (Fig. 6) in the presence of 10 mmCa2+/0.5 mm Mg2+saline, in contrast, the mean amplitude of EPSC1 was very small (BoNT/A, 7 ± 3 pA; BoNT/C, 13 ± 6 pA pA), the failure probability was very high (BoNT/A, 0.56 ± 0.15; BoNT/C, 0.38 ± 0.24), and the PPR was much greater than in control cultures (BoNT/A, 1.4 ± 0.2; BoNT/C, 1.2 ± 0.2). We conclude that the basal release probability in BoNT/A- or BoNT/C-treated cultures was much lower than in control cultures but that it could be increased moderately by elevating [Ca2+]o. Cleavage of either SNAP-25 with BoNT/A or SNAP-25 and syntaxin with BoNT/C thus resulted in a decrease in the ability of Ca2+ influx through voltage-dependent channels to trigger synaptic vesicle exocytosis.
Fig. 6.

Unlike in control cultures, paired-pulse facilitation (PPF) occurs in both control and 10 mmCa2+/0.5 mm Mg2+containing saline after BoNT/A or BoNT/C treatment. A, Dual recordings of monosynaptically connected CA3 pyramidal neurons recorded in a control (left) and a BoNT/A-treated culture (right). Top traces are pairs of presynaptic action potentials with an interval of 50 msec.Bottom traces are representative single sweeps of postsynaptic unitary currents elicited either in control saline containing 2.8 mm Ca2+/2.5 mm Mg2+ or in saline containing 10 mm Ca2+/0.5 mmMg2+. In the control cell pair, application of 10 mm Ca2+/0.5 mmMg2+ produced an increase in release probability, and paired-pulse depression was observed in 97% of the individual trials. In contrast, in the cell pair from a BoNT/A-treated culture, PPF was observed in 64% of the individual trials in the presence of 10 mm Ca2+/0.5 mmMg2+ containing saline, and failures of EPSC1 occurred in 93% of trials in control saline. B, In control cultures the mean paired-pulse ratio between two CA3 pyramidal cells was >1 in control saline but <1 after the release probability was increased by raising the Ca2+/Mg2+ ratio. In BoNT/A- and BoNT/C-treated cultures, the paired-pulse ratio was >1 in saline containing 10 mm Ca2+/0.5 mmMg2+. Failures of transmission were included in the calculation of the paired-pulse ratio.
Does BoNT/A or BoNT/C treatment impair the cooperative action of multiple Ca2+ ions, as previously suggested for BoNT/A treatment at the neuromuscular junction (Cull-Candy et al., 1976)? In control cultures log–log plots of mean unitary EPSC amplitude as a function of [Ca2+]owere fit with a straight line for [Ca2+]o ≤1 mm. The slope of this line, an index of Ca2+ cooperativity, was 2.0 ± 0.1 (n = 3) (Fig.7), consistent with observations at the neuromuscular junction for this range of [Ca2+]o (Dodge and Rahamimoff, 1967). In BoNT/A-treated (n = 3) or BoNT/C-treated (n = 2) cultures, the curves were shifted toward higher [Ca2+]o, but the slope of the initial portion of the relationship was not affected (BoNT/A, 2.2 ± 0.6; BoNT/C, 2.3 ± 0.2) (Fig. 7). We conclude that the cooperative action of multiple Ca2+ ions in initiating vesicle fusion is unaffected by cleavage of SNAP-25 or both SNAP-25 and syntaxin.
Fig. 7.

BoNT/A or BoNT/C treatment decreases the maximum number of quanta released and the Ca2+sensitivity, but not cooperativity, of the exocytotic machinery.A, Action potentials elicited in a presynaptic CA3 pyramidal cell and unitary EPSCs recorded in a postsynaptic CA3 pyramidal cell in control (left traces) and BoNT/A-treated cultures (right traces). Each trace is a representative example from 60 trials for each [Ca2+]o, which was decreased from 2.8 to 0.5 mm. [Mg2+]o was kept constant throughout the experiment at 2.5 mm. B, Log–log plot of mean unitary EPSC amplitudes, evoked by single action potentials, as a function of [Ca2+]o in individual cell pairs in control, BoNT/A-treated, and BoNT/C-treated cultures. The control and BoNT/A data are from the cell pairs illustrated inA. Note that EPSC amplitudes approach a maximum, presumably corresponding to successful release at all synapses. In BoNT/A- or BoNT/C-treated cultures, the relation was shifted to the right, indicating a decrease in Ca2+ sensitivity, and the maximum EPSC amplitude was decreased, indicating a reduction in the maximum number of quanta released. The slope of this relation (for [Ca2+]o = 0.25–1 mm in the control culture; [Ca2+]o = 0.5–2.8 mm in the BoNT/A- or BoNT/C-treated culture) was not affected by toxin treatment, indicating that Ca2+ cooperativity (Dodge and Rahamimoff, 1967) was not changed. Failures of transmission were included in the calculation of mean EPSC amplitude.
DISCUSSION
Both spontaneous and evoked release require SNARE proteins in CNS
We observed that VAMP/synaptobrevin-2 was cleaved after TeNT treatment, that syntaxin and SNAP-25 were cleaved after BoNT/C treatment, and that SNAP-25 was cleaved after BoNT/A treatment, as assessed with both immunohistochemistry and Western blot analysis in hippocampal slice cultures, consistent with previous studies (Foran et al., 1996; Osen-Sand et al., 1996; Williamson et al., 1996) (for review, see Schiavo et al., 1995). In contrast, synaptic vesicles remained in toxin-treated cultures, as indicated by the presence of synaptophysin and synaptotagmin immunoreactivity.
The probability of evoking neurotransmitter release was greatly reduced in toxin-treated cultures, as indicated by the small amplitude of unitary EPSCs between pairs of CA3 pyramidal cells and paired-pulse facilitation in saline containing 10 mmCa2+/0.5 mm Mg2+. In addition, BoNTs and TeNT decreased the frequency of both TTX-sensitive sEPSCs and TTX-insensitive mEPSCs to the same low level of ∼0.1 Hz. We do not know what accounts for those synaptic events that are resistant to the action of the toxin. It is possible that they are released from a subpopulation of vesicles containing SNARE proteins not cleaved by BoNTs or TeNT, perhaps because they were present in heterotrimeric complexes (Hayashi et al., 1994; Pellegrini et al., 1995). This seems unlikely, however. First, the SNARE complexes probably would have turned over many times during the 48 hr of toxin treatment. Second, phorbol esters and α-LTx at concentrations <1 nm were ineffective in the toxin-treated cultures (see below). Alternatively, cleavage of SNAREs may very strongly reduce the likelihood of spontaneous vesicle fusion but not fully block it.
These data are in agreement with the bulk of previous electrophysiological work with clostridial neurotoxins at central synapses (Bergey et al., 1987; Finch et al., 1990) and at the neuromuscular junction (Harris and Miledi, 1971; Cull-Candy et al., 1976; Dreyer and Schmitt, 1983; Dreyer et al., 1983, 1987; Gansel et al., 1987; Molgó et al., 1989). Our results are also consistent with the abolition of both constitutive and evoked glutamate release at the neuromuscular junction of Drosophila embryos lacking syntaxin (Broadie et al., 1995; Schulze et al., 1995). In transgenicDrosophila embryos expressing TeNT, evoked release is abolished, but the frequency of spontaneous glutamate release is reduced by only 50%, in contrast to our results (Broadie et al., 1995). We conclude that both constitutive and evoked release of glutamate in the mammalian CNS share a similar molecular mechanism requiring that the presynaptic proteins VAMP/synaptobrevin, SNAP-25, and syntaxin be intact.
Release induced by Ca2+-independent secretagogues is blocked by treatment with clostridial neurotoxins
Exocytosis can be stimulated in the hippocampus by several agents in a manner that is independent from either [Ca2+]o or [Ca2+]i or both, including activators of protein kinases A or Ca (Malenka et al., 1987; Finch and Jackson, 1990; Chavez-Noriega and Stevens, 1994; Capogna et al., 1995; Trudeau et al., 1996b), α-LTx at concentrations <1 nm (Capogna et al., 1996), sucrose (Rosenmund and Stevens, 1996), ruthenium red (Trudeau et al., 1996a), and nitric oxide (Meffert et al., 1996). The effects of 0.5 nm α-LTx, phorbol ester, and sucroseb were prevented by all clostridial toxins. Similarly, the ability of ruthenium red (Trudeau et al., 1996b) or α-LTx at concentrations <1 nm (Capogna et al., 1996) to induce [Ca2+]o-independent release in hippocampus was shown previously to be prevented by TeNT or BoNT/F treatment. Finally, [Ca2+]o-independent nitric oxide-stimulated secretion is impaired by cleavage of any of the SNARE proteins (Meffert et al., 1996). At the mammalian neuromuscular junction, clostridial neurotoxin treatment also blocks sucrose-stimulated release fully (Dreyer et al., 1987). α-LTX, in contrast, is weakly active after TeNT treatment but unaffected by BoNT/A treatment (Dreyer et al., 1987). We thus conclude that, like normal synaptic transmission, these [Ca2+]o-independent forms of release depend on the integrity of VAMP/synaptobrevin, SNAP-25, and syntaxin in the mammalian CNS.
A better description of the molecular mechanism(s) by which these secretagogues act clearly is required before a mechanistic hypothesis for inhibition by clostridial toxins is suggested. Nevertheless, these results strengthen the suggestion that these [Ca2+]o-independent secretagogues trigger release via direct changes in the functional state of the exocytotic machinery, particularly in the final steps of exocytosis. For example, phorbol ester increases the size of the readily releasable pool of secretory granules in chromaffin cells (Gillis et al., 1996), whereas forskolin increases release probability without changing the number of vesicles available for release at hippocampal synapses (Trudeau et al., 1996b). Phosphorylation of SNAP-25 by PKC has been shown to contribute to the stimulation of release from PC12 cells (Shimazaki et al., 1996). We suggest that cleavage of SNAP-25 by clostridial toxins prevents this phosphorylation and thus accounts for the inability of phorbol esters to promote release.
Ca2+-dependent evoked release can be rescued after cleavage of SNAP-25 and syntaxin
The mechanism by which cleavage of SNARE proteins inhibits transmitter release is not known. The SNARE complex assembly and disassembly cycle can still occur after cleavage of SNAREs (Hayashi et al., 1994; Pellegrini et al., 1995). In addition, ultrastructural analysis of presynaptic release sites has shown that synaptic vesicles still dock, at the morphological level, in the absence of either syntaxin or intact VAMP/synaptobrevin (Harris and Miledi, 1971; Hunt et al., 1994; Broadie et al., 1995), although this may be accounted for by the binding of the vesicle protein synaptotagmin to SNAP-25 (Schiavo et al., 1997).
We found that glutamate release could be triggered by large Ca2+ or Sr2+ influxes into axon terminals after cleavage of either SNAP-25 alone or both SNAP-25 and syntaxin, but not after cleavage of VAMP/synaptobrevin-2. It is important to note that the release induced by these manipulations in toxin-treated cultures did not originate from a subpopulation of excitatory terminals not affected by the toxins, because phorbol ester, sucrose, or ionomycin and α-LTx when applied in normal saline failed to induce any release in the same BoNT-treated cultures. Moreover, it is unlikely that the toxin-resistant release came from ectopic or abnormal release sites, because miniature and unitary postsynaptic currents in BoNT-treated cultures appeared indistinguishable in kinetics and latency from those seen in control cultures.
Enhanced Ca2+ influx has been shown previously to rescue acetylcholine release at the neuromuscular junction after treatment with BoNT/A or BoNT/E (Cull-Candy et al., 1976; Dreyer and Schmitt, 1983; Gansel et al., 1987; Molgó et al., 1989) (for review, see Molgó et al., 1990; Poulain et al., 1995). Consistent with our results, partial rescue of secretion from chromaffin cells also has been observed after treatment with BoNT/A, but not with TeNT (Lawrence et al., 1994, 1996; Banerjee et al., 1996). In contrast to our results, however, Ca2+-mediated rescue from these cells was not obtained after BoNT/E or BoNT/C treatment (Banerjee et al., 1996; Foran et al., 1996).
In addition, we found that the mean amplitude of evoked unitary EPSCs in BoNT-treated cultures in the presence of 10 mmCa2+/0.5 mm Mg2+ was less than the mean amplitude of unitary EPSCs in control cultures in saline containing 2.8 mm Ca2+/2.5 mm Mg2+. It therefore would appear that BoNT treatment decreased the maximum number of quanta that could be released. Assuming that the number of docked vesicles is not decreased after toxin treatment (see Harris and Miledi, 1971; Hunt et al., 1994;Broadie et al., 1995), this observation suggests that cleavage of syntaxin and SNAP-25 may impair release at some point subsequent to the action of Ca2+ in triggering release.
In BoNT-treated cultures, plots of unitary EPSC amplitudes as a function of [Ca2+]o yielded a slope similar to that seen in untreated cultures, but the relationship was shifted toward higher [Ca2+]o. This observation indicates that cleavage of SNAP-25 alone or together with syntaxin also decreases release by decreasing the apparent sensitivity of the release machinery for Ca2+. Although the inhibition of Ca2+-dependent evoked release by clostridial neurotoxins could result from an inhibition of voltage-dependent Ca2+ channels or an alteration in intraterminal Ca2+ dynamics, these possibilities have been ruled out previously (Dreyer et al., 1983; Mallart et al., 1989; Mochida et al., 1995; Stanley and Mirotznik, 1997).
There are insufficient biochemical data at present that can account for these effects of BoNT or TeNT treatment. Clostridial neurotoxins change the energetics of the assembly and disassembly of SNARE complexes, as revealed by their ability to withstand denaturation by SDS (Pellegrini et al., 1995). The SDS resistance of the SNARE complex is lower after TeNT than after BoNT/A treatment (Pellegrini et al., 1995). It is thus noteworthy that TeNT caused an inhibition of release that we could not counteract with large Ca2+ or Sr2+ influxes. One possibility is that BoNT treatment modifies the interactions between the heterotrimeric fusion complex and voltage-dependent Ca2+ channels (for review, see Zucker, 1996). A simple “uncoupling” of Ca2+ channels from fusion-competent vesicles cannot account for all of the effects of the toxins, however, because we observed that release also was inhibited by BoNT/A and BoNT/C when Ca2+ was transported directly into the terminal by ionomycin.
In conclusion, the rescue of release by large Ca2+or Sr2+ influx after cleavage of SNAP-25 alone or together with syntaxin adds functional evidence for an essential postdocking role of these proteins in neurotransmitter exocytosis.
Footnotes
This work was supported by the Dr. Eric Slack-Gyr Foundation and the Swiss National Science Foundation (31-42174.94). We thank L. Heeb, R. Kägi, H. Kasper, L. Rietschin, and R. Schöb for technical assistance; Dr. P. Vincent for the software for analyzing synaptic currents; Drs. H. Bigalke, C. Montecucco, and O. Rossetto for the gift of TeNT, BoNTs, and anti-SNARE antibodies; C. Heuss and Drs. F. Benfenati, H. Betz, and J.-C. Poncer for their comments.
Correspondence should be addressed to Dr. Marco Capogna, Brain Research Institute, University of Zurich, August Forel-Strasse 1, CH-8029 Zurich, Switzerland.
aParfitt and Madison (1993) found that the enhancement of mEPSC frequency elicited by phorbol ester was partially attenuated by either Cd2+ or L-type Ca2+ channel antagonists. Differences in experimental conditions (area CA1 in acute slices vs area CA3 in slice cultures, 10 vs 3 μm PDAc; measurement of mEPSC frequency >15 vs 5 min after PDAc application) may account for the differences between their conclusion and ours.
bIn Drosophila, hyperosmotic saline can induce some release after cleavage of VAMP/synaptobrevin by TeNT or in the absence of syntaxin (Broadie et al., 1995). We were unable to test the high concentrations of sucrose used in that study, however.
REFERENCES
- 1.Banerjee A, Kowalchyk JA, DasGupta BR, Martin TFJ. SNAP-25 is required for a late postdocking step in Ca2+-dependent exocytosis. J Biol Chem. 1996;271:20227–20230. doi: 10.1074/jbc.271.34.20227. [DOI] [PubMed] [Google Scholar]
- 2.Bergey GK, Bigalke H, Nelson PG. Differential effects of tetanus toxin on inhibitory and excitatory synaptic transmission in mammalian spinal cord neurons in culture: a presynaptic locus of action for tetanus toxin. J Neurophysiol. 1987;57:121–131. doi: 10.1152/jn.1987.57.1.121. [DOI] [PubMed] [Google Scholar]
- 3.Binz T, Blasi J, Yamasaki S, Baumeister A, Link E, Südhof TC, Jahn R, Niemann H. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J Biol Chem. 1994;269:1617–1620. [PubMed] [Google Scholar]
- 4.Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, De Camilli P, Südhof TC, Niemann H, Jahn R. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 1993a;365:160–163. doi: 10.1038/365160a0. [DOI] [PubMed] [Google Scholar]
- 5.Blasi J, Chapman ER, Yamasaki S, Binz T, Niemann H, Jahn R. Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J. 1993b;12:4821–4828. doi: 10.1002/j.1460-2075.1993.tb06171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broadie K, Prokop A, Bellen HJ, O’Kane CJ, Schulze KL, Sweeney ST. Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron. 1995;15:663–673. doi: 10.1016/0896-6273(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 7.Capogna M, Gähwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. J Neurosci. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capogna M, Gähwiler BH, Thompson SM. Calcium-independent actions of α-latrotoxin on spontaneous and evoked synaptic transmission in the hippocampus. J Neurophysiol. 1996;76:3149–3158. doi: 10.1152/jn.1996.76.5.3149. [DOI] [PubMed] [Google Scholar]
- 9.Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cull-Candy SG, Lundh H, Thesleff SJ. Effects of botulinum toxin on neuromuscular transmission in the rat. J Physiol (Lond) 1976;260:177–203. doi: 10.1113/jphysiol.1976.sp011510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debanne D, Gähwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol (Lond) 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodge FA, Rahamimoff R. Co-operative action of calcium ions in transmitter release at the neuromuscular junction. J Physiol (Lond) 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolly JO, de Paiva A, Foran P, Lawrence G, Daniels-Holgate P, Ashton AC. Probing the process of transmitter release with botulinum and tetanus neurotoxins. Semin Neurosci. 1994;6:149–158. [Google Scholar]
- 14.Dreyer F, Schmitt A. Transmitter release in tetanus and botulinum A toxin-poisoned mammalian motor endplates and its dependence on nerve stimulation and temperature. Pflügers Arch. 1983;399:228–234. doi: 10.1007/BF00656720. [DOI] [PubMed] [Google Scholar]
- 15.Dreyer F, Mallart A, Brigant JL. Botulinum A toxin and tetanus toxin do not affect presynaptic membrane currents in mammalian motor nerve endings. Brain Res. 1983;270:373–375. doi: 10.1016/0006-8993(83)90617-0. [DOI] [PubMed] [Google Scholar]
- 16.Dreyer F, Rosenberg F, Becker C, Bigalke H, Penner R. Differential effects of various secretagogues on quantal transmitter release from mouse motor nerve terminals treated with botulinum A and tetanus toxin. Naunyn Schmiedebergs Arch Pharmacol. 1987;335:1–7. doi: 10.1007/BF00165027. [DOI] [PubMed] [Google Scholar]
- 17.Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. J Physiol (Lond) 1952;117:109–128. [PMC free article] [PubMed] [Google Scholar]
- 18.Finch DM, Jackson MB. Presynaptic enhancement of synaptic transmission in hippocampal cell cultures by phorbol esters. Brain Res. 1990;518:269–273. doi: 10.1016/0006-8993(90)90979-l. [DOI] [PubMed] [Google Scholar]
- 19.Finch DM, Fisher RS, Jackson MB. Miniature excitatory synaptic currents in cultured hippocampal neurons. Brain Res. 1990;518:257–268. doi: 10.1016/0006-8993(90)90978-k. [DOI] [PubMed] [Google Scholar]
- 20.Foran P, Lawrence GW, Shone CC, Foster KA, Dolly JO. Botulinum neurotoxin C1 cleaves both syntaxin and SNAP-25 in intact and permeabilized chromaffin cells: correlation with its blockade of catecholamine release. Biochemistry. 1996;35:2630–2636. doi: 10.1021/bi9519009. [DOI] [PubMed] [Google Scholar]
- 21.Gähwiler BH. Organotypic monolayer cultures of nervous tissue. J Neurosci Methods. 1981;4:329–342. doi: 10.1016/0165-0270(81)90003-0. [DOI] [PubMed] [Google Scholar]
- 22.Gansel M, Penner R, Dreyer F. Distinct sites of action of clostridial neurotoxins revealed by double-poisoning of mouse motor nerve terminal. Pflügers Arch. 1987;409:533–539. doi: 10.1007/BF00583812. [DOI] [PubMed] [Google Scholar]
- 23.Gillis KD, Mössner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 24.Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci USA. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harris AJ, Miledi R. The effect of type D botulinum toxin on frog neuromuscular junctions. J Physiol (Lond) 1971;217:497–515. doi: 10.1113/jphysiol.1971.sp009582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Südhof TC, Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunt JM, Bommert K, Charlton MP, Kistner A, Habermann E, Augustine GJ, Betz H. A post-docking role for synaptobrevin in synaptic vesicle fusion. Neuron. 1994;12:1269–1279. doi: 10.1016/0896-6273(94)90443-x. [DOI] [PubMed] [Google Scholar]
- 28.Katz B. The release of neural transmitter substances. Liverpool UP; Liverpool, UK: 1969. [Google Scholar]
- 29.Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kita H, Van der Kloot W. Calcium ionophore X-537A increases spontaneous and phasic quantal release of acetylcholine at frog neuromuscular junction. Nature. 1974;250:658–660. doi: 10.1038/250658a0. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence GW, Weller U, Dolly JO. Botulinum A and the light chain of tetanus toxins inhibit distinct stages of Mg 249 ATP-dependent catecholamine exocytosis from permeabilised chromaffin cells. Eur J Biochem. 1994;222:325–333. doi: 10.1111/j.1432-1033.1994.tb18871.x. [DOI] [PubMed] [Google Scholar]
- 32.Lawrence GW, Foran JO, Dolly JO. Distinct exocytotic responses of intact and permeabilised chromaffin cells after cleavage of the 25 kDa synaptosomal-associated protein (SNAP-25) or synaptobrevin by botulinum toxin A or B. Eur J Biochem. 1996;236:877–886. doi: 10.1111/j.1432-1033.1996.00877.x. [DOI] [PubMed] [Google Scholar]
- 33.Li C, Davletov A, Südhof TC. Distinct Ca2+ and Sr2+ binding properties of synaptotagmins. J Biol Chem. 1995;270:24898–24902. doi: 10.1074/jbc.270.42.24898. [DOI] [PubMed] [Google Scholar]
- 34.Link E, Edelmann L, Chou JH, Binz T, Yamasaki S, Eisel U, Baumert M, Südhof TC, Niemann H, Jahn R. Tetanus toxin action: inhibition of neurotransmitter release linked to synaptobrevin proteolysis. Biochem Biophys Res Commun. 1992;189:1017–1023. doi: 10.1016/0006-291x(92)92305-h. [DOI] [PubMed] [Google Scholar]
- 35.Longenecker HE, Hurlbut WP, Mauro A, Clark AW. Effects of black widow spider venom on the frog neuromuscular junction. Nature. 1970;225:701–703. doi: 10.1038/225701a0. [DOI] [PubMed] [Google Scholar]
- 36.Malenka RC, Ayoub GS, Nicoll RA. Phorbol esters enhance transmitter release in rat hippocampal slices. Brain Res. 1987;403:198–203. doi: 10.1016/0006-8993(87)90145-4. [DOI] [PubMed] [Google Scholar]
- 37.Mallart A, Molgó J, Angaut-Petit D, Thesleff S. Is the internal calcium regulation altered in type A botulinum toxin-poisoned motor endings? Brain Res. 1989;479:167–171. doi: 10.1016/0006-8993(89)91348-6. [DOI] [PubMed] [Google Scholar]
- 38.Meffert MK, Calakos NC, Scheller RH, Schulman H. Nitric oxide modulates synaptic vesicle docking/fusion reactions. Neuron. 1996;16:1229–1236. doi: 10.1016/s0896-6273(00)80149-x. [DOI] [PubMed] [Google Scholar]
- 39.Mochida S, Saisu H, Kobayashi H, Abe T. Impairment of syntaxin by botulinum neurotoxin C1 or antibodies inhibits acetylcholine release but not Ca2+ channel activity. Neuroscience. 1995;65:905–915. doi: 10.1016/0306-4522(94)00508-3. [DOI] [PubMed] [Google Scholar]
- 40.Molgó J, DasGupta BR, Thesleff S. Characterization of the actions of botulinum neurotoxin type E at the rat neuromuscular junction. Acta Physiol Scand. 1989;137:497–501. doi: 10.1111/j.1748-1716.1989.tb08786.x. [DOI] [PubMed] [Google Scholar]
- 41.Molgó J, Comella JX, Angaut-Petit D, Pecot-Dechavassine M, Tabti N, Faille L, Mallart A, Thesleff S. Presynaptic actions of botulinal neurotoxins at vertebrate neuromuscular junctions. J Physiol (Paris) 1990;84:152–166. [PubMed] [Google Scholar]
- 42.Niemann H, Blasi J, Jahn R. Clostridial neurotoxins: new tools for dissecting exocytosis. Trends Cell Biol. 1994;4:179–185. doi: 10.1016/0962-8924(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 43.Oho C, Seino S, Takahashi M. Expression and complex formation of soluble N-ethyl-maleimide-sensitive factor attachment protein (SNAP) receptors in clonal rat endocrine cells. Neurosci Lett. 1995;186:208–210. doi: 10.1016/0304-3940(95)11317-p. [DOI] [PubMed] [Google Scholar]
- 44.Osen-Sand A, Staple JK, Naldi E, Schiavo G, Rossetto O, Petitpierre S, Malgaroli A, Montecucco C, Catsicas S. Common and distinct fusion proteins in axonal growth and transmitter release. J Comp Neurol. 1996;367:222–234. doi: 10.1002/(SICI)1096-9861(19960401)367:2<222::AID-CNE5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 45.Parfitt KD, Madison DV. Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. J Physiol (Lond) 1993;471:245–268. doi: 10.1113/jphysiol.1993.sp019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pellegrini LL, O’Connor V, Lottspeich F, Betz H. Clostridial neurotoxins compromise the stability of a low energy SNARE complex mediating NSF activation of synaptic vesicle fusion. EMBO J. 1995;14:4705–4713. doi: 10.1002/j.1460-2075.1995.tb00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poulain B, Molgó J, Thesleff S. Quantal neurotransmitter release and the clostridial neurotoxins’ target. In: Montecucco C, editor. Clostridial neurotoxins. Springer; Berlin: 1995. pp. 243–255. [DOI] [PubMed] [Google Scholar]
- 48.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 49.Rosenthal L, Meldolesi J. α-Latrotoxin and related toxins. Pharmacol Ther. 1989;42:115–134. doi: 10.1016/0163-7258(89)90024-7. [DOI] [PubMed] [Google Scholar]
- 50.Rossetto O, Gorza L, Schiavo G, Schiavo N, Scheller RH, Montecucco C. VAMP/synaptobrevin isoforms 1 and 2 are widely and differentially expressed in nonneuronal tissues. J Cell Biol. 1996;132:167–179. doi: 10.1083/jcb.132.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 52.Schiavo G, Rossetto O, Catsicas S, Polverino de Laureto P, DasGupta BR, Benfenati F, Montecucco C. Identification of the nerve terminal targets of botulinum neurotoxin serotypes A, D, and E. J Biol Chem. 1993;268:23784–23787. [PubMed] [Google Scholar]
- 53.Schiavo G, Rossetto O, Tonello F, Montecucco C. Intracellular targets and metalloprotease activity of tetanus and botulism neurotoxins. In: Montecucco C, editor. Clostridial neurotoxins. Springer; Berlin: 1995. pp. 257–274. [DOI] [PubMed] [Google Scholar]
- 54.Schiavo G, Stenbeck G, Rothman JE, Söllner TH. Binding of the synaptic vesicle v-SNARE, synaptotagmin, to the plasma membrane t-SNARE, SNAP-25, can explain docked vesicles at neurotoxin-treated synapses. Proc Natl Acad Sci USA. 1997;94:997–1001. doi: 10.1073/pnas.94.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schulze KL, Broadie K, Perin MS, Bellen HJ. Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
- 56.Schweizer FE, Betz H, Augustine GJ. From vesicle docking to endocytosis: intermediate reactions of exocytosis. Neuron. 1995;14:689–696. doi: 10.1016/0896-6273(95)90213-9. [DOI] [PubMed] [Google Scholar]
- 57.Shimazaki Y, Nishiki T, Omori A, Sekiguchi M, Kamata Y, Kozaki S, Takahashi M. Phosphorylation of 25 kDa synaptosome-associated protein. J Biol Chem. 1996;271:14548–14553. doi: 10.1074/jbc.271.24.14548. [DOI] [PubMed] [Google Scholar]
- 58.Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly–disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993a;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 59.Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993b;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 60.Stanley EF, Mirotznik RR. Cleavage of syntaxin prevents G-protein regulation of presynaptic calcium channels. Nature. 1997;385:340–343. doi: 10.1038/385340a0. [DOI] [PubMed] [Google Scholar]
- 61.Südhof TC. The synaptic vesicle cycle: a cascade of protein–protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 62.Sweeney ST, Broadie K, Keane J, Niemann H, O’Kane CJ. Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron. 1995;14:341–351. doi: 10.1016/0896-6273(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 63.Trudeau L-E, Doyle RT, Emery DG, Haydon PG. Calcium-independent activation of the secretory apparatus by ruthenium red in hippocampal neurons: a new tool to assess modulation of presynaptic function. J Neurosci. 1996a;16:46–54. doi: 10.1523/JNEUROSCI.16-01-00046.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trudeau L-E, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996b;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- 65.Van der Kloot W, Molgó J. Quantal acetylcholine release at the vertebrate neuromuscular junction. Physiol Rev. 1994;74:899–991. doi: 10.1152/physrev.1994.74.4.899. [DOI] [PubMed] [Google Scholar]
- 66.Williamson LC, Halpern JL, Montecucco C, Brown JE, Neale EA. Clostridial neurotoxins and substrate proteolysis in intact neurons. J Biol Chem. 1996;271:7694–7699. doi: 10.1074/jbc.271.13.7694. [DOI] [PubMed] [Google Scholar]
- 67.Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. [DOI] [PubMed] [Google Scholar]