

Abstract
The present study provides an experimental model of the apoptotic death of pyramidal neurons in rat olfactory cortex after total bulbectomy. Terminal transferase (TdT)-mediated deoxyuridine triphosphate (d-UTP)-biotin nick end labeling (TUNEL), DNA electrophoresis, and neuronal ultrastructure were used to provide evidence of apoptosis; neurons in olfactory cortex were counted by stereology. Maximal TUNEL staining occurred in the piriform cortex between 18 and 26 hr postbulbectomy. Within the survival times used in the present study (up to 48 hr postlesion), cell death was observed exclusively in the piriform cortex; there was no evidence of cell death in any other areas connected with the olfactory bulb. Neurons undergoing apoptosis were pyramidal cells receiving inputs from, but not projecting to, the olfactory bulb. The apical dendrites of these neurons were contacted by large numbers of degenerating axonal terminals. Gel electrophoresis of DNA purified from lesioned olfactory cortex showed a ladder pattern of fragmentation. Inflammatory cells or phagocytes were absent in the environment of degenerating neurons in the early stages of the apoptotic process. The present model suggests that deafferentation injury in sensory systems can cause apoptosis. In addition, olfactory bulbectomy can be used for investigating molecular mechanisms that underlie apoptosis in mature mammalian cortical neurons and for evaluating strategies to prevent the degeneration of cortical neurons.
Keywords: anterograde degeneration, cell death, DNA fragmentation, neurodegenerative disease, olfactory, TUNEL
Animal models have become essential research tools to study neurodegenerative diseases because they provide relatively simple and reliable paradigms that can be followed over time (i.e., conditions essential to investigate mechanisms of neuronal injury and death but impossible to establish on human autopsy material). Although several injury models of neurological disease that target various populations of neurons have been proposed (Snider et al., 1992), cortical pyramidal neurons are notoriously resistant to simple experimental lesions (i.e., axotomy). The inclusion of a significant portion of the axonal arbor of these neurons within the cortical gray matter in the form of local collaterals protects these cells from white matter lesions designed to avoid direct perikaryal damage. A recent model targeting entorhinal neurons with perforant pathway transections has produced, to date, inconsistent results (Cummings et al., 1992; Koliatsos et al., 1993; Peterson et al., 1994). The rat olfactory cortex is an especially attractive area for investigations of pyramidal neuronal death for two reasons. First, it has been demonstrated in several rodent and primate models that there is remarkable plasticity in central target fields of somatosensory and special sensory systems after manipulations (including ablations) of peripheral inputs (Van der Loos and Woolsey, 1973; Hubel et al., 1977;Kaas et al., 1983; Pons et al., 1991). Second, it has been observed that bulbectomy in adult rats causes argyrophilic degenerative changes and cell death of neurons in piriform cortex ipsilateral to the lesion (Heimer and Kalil, 1978).
In the present study we characterize the type and time course of the degeneration of olfactory pyramidal neurons after unilateral bulbectomy in the rat, and we show that these neurons undergo apoptosis within 1 d postlesion. The critical factor underlying apoptotic death is the removal of afferent input rather than axotomy. Dying neurons are detected by in situ methods [i.e., terminal transferase (TdT)-mediated deoxyuridine triphosphate (d-UTP)-biotin nick end labeling (TUNEL)] that label DNA fragmentation and electron microscopy (EM), and apoptosis is confirmed by agarose electrophoresis. The course of apoptosis and its severity are studied by stereological and semiquantitative methods. We propose that deafferentation injury in the olfactory system leads to apoptotic cell death of cortical neurons. Degeneration of olfactory cortical neurons postbulbectomy can be very useful for investigations of molecular mechanisms that underlie the death of mature cortical neurons and for trials of therapeutic agents that target cortical degeneration.
MATERIALS AND METHODS
Bulbectomy. Adult male Sprague Dawley rats (n = 37; 250–300 gm) were anesthetized with a mixture of enflurane, nitrous oxide, and oxygen (1, 66, and 33%, respectively) and adjusted on a Kopf rat stereotaxic device. The olfactory bulbs were exposed with a midline craniotomy involving the frontal bones. The right olfactory bulb was transected coronally with the aid of a blade mounted on a stereotaxic electrode holder 1 mm anterior to the frontal pole. The blade was kept in place while the bulb stump anterior to it was aspirated to prevent accidental damage to the hemispheres. Control (sham) animals were subjected to craniotomy, followed by a transverse incision of the dura overlying the root of the right olfactory bulb 6.5 mm anterior to bregma. A systematic examination of the lesion site after the brains were removed from the skulls showed that all bulbectomies were performed rostral to the anterior olfactory nucleus, without any direct damage to this structure or to the overlying frontal cortex.
Preparation of tissues—time and localization of cell death.To characterize the time course and anatomical distribution of cell death, we allowed bulbectomized animals to survive for 14, 18, 22, 26, 30, 34, 38, 42, and 48 hr postbulbectomy (n = 2 per time point). Animals were anesthetized deeply with sodium pentobarbital (100 mg/kg) and perfused transcardially through the ascending aorta with a brief flash of PBS, pH 7.4, followed by 4% freshly depolymerized paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, at 4°C for 25 min (∼30 ml of perfusate per minute). Brains were rinsed in 20% PB sucrose overnight, frozen in isopentane, and stored at −80°C until processed. Serial coronal sections were cut through the entire olfactory cortex on a cryostat set at 80 μm. Every sixth section was collected, starting at a random level at the beginning of the olfactory cortex, and sections were processed for TUNEL (n = 12 slides per animal) and counterstained lightly with cresyl violet. This processing reduced section thickness to 30–32 μm, a change likely caused by the digestion of floating sections with proteinase K.
To assess the magnitude of cell loss in the olfactory cortex postbulbectomy, we compared the numbers of neurons in the right piriform cortex of sham-operated animals (n = 5) and bulbectomized rats with a postsurgical survival of 48 hr (n = 5). Bulbectomized and control rats were anesthetized and perfused transcardially, as described above. Brains were paraffin-embedded and cut in 25-μm-thick serial coronal sections starting at a random position at the beginning of the olfactory cortex. Every 15th section was selected in a systematic uniform manner, yielding an average of 14 sections per animal. We deparaffinized and stained sections with cresyl violet to visualize neuronal nuclei. After processing, the mean section thickness was 24.8 μm (SD = 1.2).
EM–morphological features of dying neurons and their neuropil.To study the morphology of degenerating cortical neurons and other types of cells or processes adjacent to degenerating neurons, we prepared cortical tissues from bulbectomized rats 12, 15, 18, and 22 hr postlesion (n = 3 per time point) for EM. Animals were subjected to bulbectomy as described above and were killed with an overdose of sodium pentobarbital, followed by perfusion fixation through the ascending aorta as follows: 30 ml of 0.1 m PB, presaturated with 95% oxygen/5% carbon dioxide and prewarmed at 37°C (for 1 min), and 2% glutaraldehyde and 2% paraformaldehyde in 0.1 m PB at 37°C for 10 min and at 5°C for 20 min. Brains were post-fixed in the glutaraldehyde/paraformaldehyde solution overnight. The olfactory cortex ipsilateral to the lesion at the level of the anterior commissure was subdissected, and blocks were processed for transmission EM by standard methods. Briefly, specimens were treated with 2% osmium tetroxide for 1 hr, rinsed in 0.1 mPB, dehydrated via graded alcohols, defatted in toluene, and embedded in Epon/Araldite. Thin sections were cut on a Sorvall MT2-B ultramicrotome and mounted on 200-mesh uncoated grids. Sections were stained with uranyl acetate and lead citrate before they were viewed with an Hitachi H-600 electron microscope.
Immunocytochemistry (ICC) for the presence of reactive cells near dying neurons. To investigate the occurrence and type of the microglial response in the rat olfactory cortex postbulbectomy, we used ICC for the CR3-like epitope OX-42 and the myelomonocytic epitope ED1 (Graeber et al., 1990; Milligan et al., 1991; Koliatsos et al., 1994). Two rats were bulbectomized and perfused transcardially 24 and 48 hr postlesion, as described above. Brains were frozen in isopentane, and serial coronal sections (40 μm) were cut through a forebrain plane corresponding to the optic chiasm. Sections were stored free-floating in 0.02 m Tris-buffered saline (TBS), pH 7.4, at 4°C until processed for ICC.
For ICC, sections were rinsed in 0.1% Triton X-100 in TBS, blocked with 4% normal goat serum (NGS) for 1 hr, and incubated for 72 hr with the primary antibody diluted 1:50 in 0.1% Triton X-100 and 2% NGS in TBS at 4°C. Then sections were washed in TBS for 30 min and incubated with biotin-conjugated anti-mouse immunoglobulin (Vectastain, Vector Laboratories, Burlingame, CA) and 2% NGS in TBS for 1 hr. Sections were washed extensively in TBS, followed by incubation in an avidin–peroxidase solution (Vectastain, Vector) made in TBS for 1 hr. After a 15 min wash in TBS, sections were developed in 0.05% 3,3′-diaminobenzidine (Sigma, St. Louis, MO) in TBS containing 0.01% hydrogen peroxide (2–5 min). All reactions, except for the incubation in the primary antibody, were performed at room temperature (RT). After ICC, sections were washed with TBS and counterstained with cresyl violet.
Terminal transferase (TdT)-mediated deoxyuridine triphosphate (d-UTP)-biotin nick end labeling (TUNEL). The original TUNEL method for labeling the free 3′-hydroxyl terminals of cleaved DNA (Gavrieli et al., 1992) was modified to accommodate thick sections, necessary for three-dimensional quantitation. Sections were processed free-floating to expose both surfaces and to obtain optimal penetration of reagents. Sections were stored floating in PBS at 4°C until processed.
Section processing began with dehydration in ethanol solutions of increasing concentrations, delipidation in xylene for 15 min, and rehydration via ethanols of decreasing concentrations. Sections subsequently were rinsed in PBS (5 min) and treated with 20 μg/ml proteinase K (Boehringer Mannheim, Indianapolis, IN) in PBS (15 min, RT). Then sections were washed 4× in PBS (2 min per wash) and rinsed in 3% hydrogen peroxide in PBS (5 min) to inactivate endogenous peroxidase. Subsequently, sections were preincubated at RT in TdT buffer containing 30 mm Tris-HCl, pH 7.2, 140 mm sodium cacodylate, and 1 mm cobalt chloride (Boehringer Mannheim) at RT (10 min) and finally incubated in TdT buffer containing 0.5 U of TdT (Boehringer Mannheim) per microliter and 40 μm biotin-16-dUTP (Boehringer Mannheim) in a humid chamber at 37°C (for 2 hr). During the final incubation, sections were placed in 25 μl of the incubation solution on a hydrophobic plastic coverslip to allow the incubation solution to penetrate the section from both sides.
The reaction was stopped with 2× standard saline citrate buffer (300m NaCl and 30 m sodium citrate) for 15 min at RT. After a 5 min wash in PBS, sections were rinsed in 2% bovine serum albumin in PBS to inactivate nonspecific reacting sites (10 min), washed again in PBS (5 min), and then incubated in an avidin–peroxidase solution (Vectastain, Vector) in a humid chamber (1 hr at 37°C). After a 5 min wash in PBS, sections were developed in 0.008% 3,3′-diaminobenzidine (Sigma) in PBS containing 0.001% hydrogen peroxide (10 min, RT). Stained sections were mounted on slides pretreated with Vectabond (Vector) and counterstained lightly with cresyl violet.
TUNEL-stained sections from sham-operated rats (n = 2) were used as negative controls. Positive controls were processed by preincubation in 1 μl/ml DNase I (Boehringer Mannheim) in TdT for 10 min and then in TdT/biotinylated d-UTP. All neuronal nuclei were stained after DNase treatment. TUNEL-stained cells were not seen in sections from unlesioned brains.
Retrograde fluorescent labeling. To visualize neurons in the piriform cortex projecting to the olfactory bulb, we used retrograde tracing with 1% aqueous Fast Blue (FB; Sigma). We have established previously that these concentrations of FB do not cause neurotoxicity in retrogradely labeled neurons (Koliatsos et al., 1988, 1990;Koliatsos and Price, 1993). After surgical exposure of the olfactory bulbs in three rats (see Bulbectomy), we injected a total of 1 μl of the dye in multiple areas of the right olfactory bulb. Animals underwent surgery again 48 hr later, and the injected bulb was removed (see Bulbectomy). Rats were anesthetized deeply with sodium pentobarbital postbulbectomy (24 hr) and killed by transcardial perfusion with 4% paraformaldehyde in PBS. Frozen sections (20 μm) were cut on a cryostat at the coronal plane through the rostral half of piriform cortex, dry-mounted on gelatin-subbed slides, and counterstained with 0.001% aqueous propidium iodide (Molecular Probes, Eugene, OR; used as an in situ DNA marker). Dual labeling with FB and propidium iodide allowed for the identification of neurons projecting to the olfactory bulb and neurons undergoing apoptosis (including potential double-labeled neurons) on the same tissue section.
To demonstrate that FB visualization is possible in the cytoplasm of apoptotic neurons, we prepared a group of control rats (n = 3). These animals were injected in posterior olfactory cortex (0.3 mm posterior to bregma, 4.8 mm lateral to midline, and 1 mm dorsal to the skull base) with 80 nl of 1% FB. The dye was allowed to undergo retrograde transport to pyramidal neurons within anterior areas of the primary olfactory cortex, including planes of the primary olfactory cortex heavily affected by bulbectomy. At 2 d postinjection, animals underwent unilateral bulbectomies on the side of the FB injections, as described above. Animals were processed for fluorescent histology with propidium iodide counterstain as described above.
Gel detection of DNA fragmentation. To detect oligonucleosomal DNA fragmentation as a marker of apoptosis, we used olfactory cortices from control rats and bulbectomized animals with survival times of 22 and 24 hr (n = 2 each). As previously reported in detail (Portera-Cailliau et al., 1995), DNA was extracted from fresh tissue, and samples were labeled by using the Genius 5 oligonucleotide 3′ end-labeling kit (Boehringer Mannheim). DNA was electrophoresed in a 1.5% agarose gel, incubated with a digoxigenin antibody conjugated to alkaline phosphatase, and detected with Lumi Phos 530 (Boehringer Mannheim) after exposure to Kodak X-Omat AR film (Eastman Kodak, Rochester, NY).
Counts of TUNEL-stained neurons. TUNEL-labeled cells were counted on two rats at each of the following time points: 14, 18, 22, 26, 30, 34, 39, 42, and 48 hr postlesion. Systematically sampled serial sections (every sixth) through the entire olfactory cortex, starting at the beginning of the olfactory cortex ipsilateral to the lesion, then were examined at a magnification of 200× with an Olympus BH-2 light microscope. All TUNEL-stained cells located within layer II were counted regardless of morphology. The total number of apoptotic cells in each rat was estimated by multiplying the sum of TUNEL-stained cells in all slides by the sampling interval (equal to six). Thick sections (30 μm on average after processing) were used to minimize edge effects and bias because of cell size.
Stereological estimation of overall cell loss. Five control rats and five lesioned rats killed 48 hr postbulbectomy were used to estimate the total cell loss in the lesioned olfactory cortex. As we have shown previously, the optical dissector combined with the Cavalieri point-counting method (Gundersen and Jensen, 1987) provides an efficient, unbiased estimate of cell number (Mouton et al., 1994). Stereological analyses were performed on sections stained with cresyl violet to visualize neuronal nuclei. Volumes of olfactory cortex were determined by using a point grid with area per point of 0.02244 mm2, yielding an average of 171 points per animal. The optical dissector method (Gundersen, 1986) allows the estimation of neuronal density by unbiased sampling of neuronal nuclei in a known three-dimensional volume. The dissector had a base area of 1161 mm2 and a height of 14 μm (volume = 16,254 mm3). An average of 124 dissectors was analyzed per animal, and the mean number of neurons counted per animal was 301 (SD = 117). Efficient, systematic sampling and estimation strategies produced a mean coefficient of error of 5.3%. Sampling and identification of neuronal nuclei were done at a magnification of 3361× by using an Olympus BH-2 microscope coupled with a stage controller, z-axis microcator, and GRID software (Interactivision, Aarhus, Denmark). Data analysis was performed with JMP statistical software (SAS Institute, Cary, NC). The nonparametric Wilcoxon rank-sum test was used for statistical inferences about group difference.
RESULTS
Bulbectomy causes apoptosis in olfactory cortical neurons
To assess the occurrence and type of death of olfactory cortical neurons postbulbectomy, we processed tissue samples containing the olfactory cortex for in situ labeling of apoptotic neurons, using TUNEL (with aldehyde-fixed sections), and for studies of DNA fragmentation, using agarose gel electrophoresis (with fresh tissue homogenates). The latter method has been used extensively for the biochemical validation of apoptosis (Arends et al., 1990). TUNEL, a more recent method (Gavrieli et al., 1992) with a limited record of applications in the nervous system, has been shown to mark motor neurons undergoing physiological cell death in the rat spinal cord at the E13–E14 stage (Wilcox et al., 1995) as well as neurons undergoing degeneration after surgical lesions (Wilcox et al., 1995), neurotoxic lesions (Portera-Cailliau et al., 1995), and anoxia/ischemia (Portera-Cailliau et al., 1995) and in the course of Huntington’s disease (Portera-Cailliau et al., 1995) and Alzheimer’s disease (Su et al., 1994; Lassmann et al., 1995; Troncoso et al., 1996). In the majority of these settings, TUNEL has been shown to coexist with a DNA fragmentation pattern consistent with apoptosis (i.e., a ladder profile on agarose gel electrophoresis).
In bulbectomized rats (Fig. 1) TUNEL stained degenerating cells in the olfactory cortex ipsilateral to the lesion (Fig. 2A,B), but not in adjacent brain areas or in other regions connected with the olfactory bulb (Fig. 3). Regions from the latter category that were examined include the anterior olfactory nucleus, taenia tecta and induseum griseum, the olfactory tubercle, the basal nucleus complex, the amygdala, the bed nucleus of stria terminalis, the lateral preoptic area, the entorhinal cortex, the locus coeruleus, and the raphe nuclei (Shipley et al., 1995). Minimal labeling was seen in the contralateral olfactory cortex. Almost all TUNEL-positive nuclei belonged to pyramidal neurons in layer IIa (Fig.4A,B). Labeled nuclei displayed several patterns of staining (Figs. 2C,4E,F), the most common of which was confluent round fragments and the rarest of which was a ring of staining located under the nuclear membrane. In preparations counterstained with aniline dyes (cresyl violet), TUNEL-positive neuronal nuclei showed reduced cytoplasm with low basophilia. In these double-stained preparations, as well as in sections stained only with cresyl violet, apoptotic nuclear profiles were visualized as intensely basophilic round bodies, either inside neuronal cell bodies or without any clear cellular affiliations (Fig. 4C–F). Basophilic particles outside the neurons presumably represent end products of the apoptotic process (apoptotic bodies) awaiting phagocytosis (see below). These structures were TUNEL-negative in double-stained preparations and became especially abundant by the second day postbulbectomy. In several cases chromatin fragments inside neurons were double-stained with both TUNEL and cresyl violet (Fig. 4F).
Fig. 1.

Simplified anatomy of the lesion implemented in the present study and its consequences (left) as compared with the normal circuitry (right) between the olfactory bulb (OB) and piriform (olfactory) cortex (PO). Connections are projected against the ventral surface of the rat forebrain. Although the lesion disrupts many connections of the olfactory bulb, anterior olfactory nucleus, and piriform cortex, the critical disconnection leading to apoptosis of cortical neurons is the removal of the mitral cell input to pyramidal cells in layer IIa. The majority of these neurons projects to interneurons and other pyramids within the piriform cortex.
Fig. 2.

Specificity (A, B) and types (C) of TUNEL staining in olfactory cortical neurons postbulbectomy. A, B, Two magnifications of the basal forebrain region (at the level of the decussation of the anterior commissure ipsilateral to the lesion) showing conspicuous TUNEL labeling in olfactory cortex (arrows) against a clear background. C, Apoptotic olfactory neurons show distinct nuclear TUNEL patterns corresponding to chromatin fragmentation in round pieces (1, most common), uniform chromatin condensation (2, common), and chromatin margination against the nuclear membrane (3, occasional). Scale bars: A, 1 mm; B, 0.5 mm;C, 20 μm.
Fig. 3.

Primary olfactory cortex (PO) is the only brain area connected with the olfactory bulb that undergoes apoptosis after bulbectomy. Sections shown in this figure represent two forebrain planes in which olfactory cortex is continuous with other structures interconnected with the bulb [i.e., with the anterior olfactory nucleus (AON; A, B) and the olfactory tubercle (Tu; C, D)]. Sections have been processed for TUNEL and counterstained with cresyl violet.B and D represent magnifications of the areas of transition in A and C that are indicated with arrowheads. A, B, Apoptotic profiles (B, arrows) are contained strictly within the boundaries of PO; none is seen beyond thePO–AON border. A blood vessel (V) is indicated for the purpose of orientation. C, D, Visualization of apoptotic cells (D, arrows) stops abruptly at the border between PO and Tu. Other labeled structures are the endopiriform nucleus (En), fundus striati (FSt), substantia innominata (SI), and magnocellular preoptic nucleus (MPN). Scale bars: A, C, 250 μm;B, D, 60 μm.
Fig. 4.
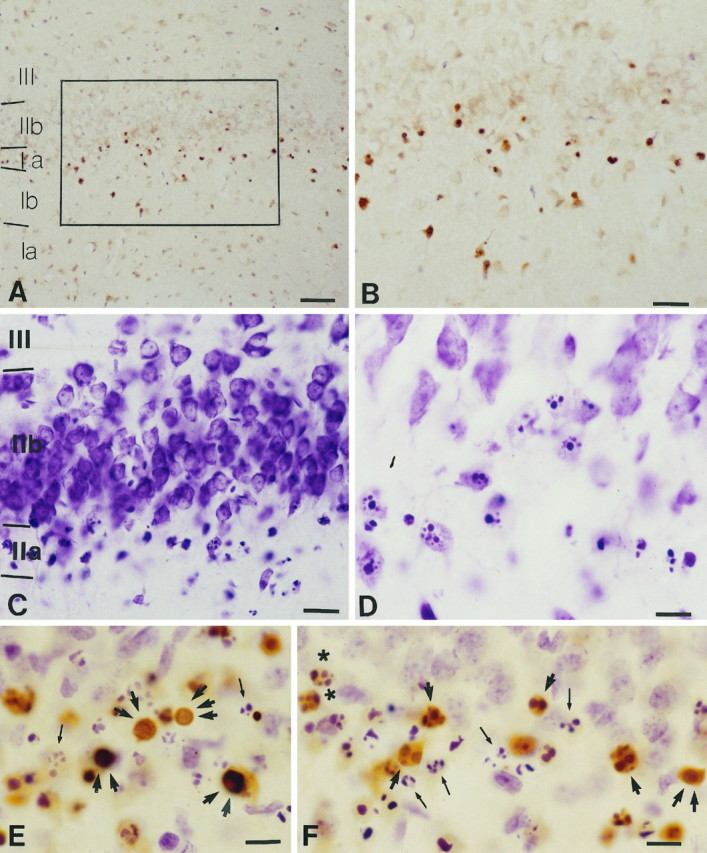
Apoptotic profiles in the rat olfactory cortex 22 hr postbulbectomy as depicted with TUNEL (A, B), an aniline dye (cresyl violet; C, D), and a combination of the two (E, F). A, B, TUNEL-stained nuclei cluster in layer IIa.B, A magnification of the framed area inA. Magnifications: A, 20×;B, 40×. C, D, Apoptotic profiles in layer IIa depicted with cresyl violet. Magnifications:C, 40×; D, 100×. E, F, TUNEL staining followed by cresyl violet counterstain shows similarities and differences of profiles visualized by the two methods. Compare E and F with single-labeledA–D. Note fragmented TUNEL nuclei (single arrow), nuclei diffusely stained with TUNEL (two arrows), and TUNEL-stained nuclei with marginated chromatin (three arrows). Also note apoptotic profiles stained with cresyl violet (thin arrow) and with both TUNEL and cresyl violet (star). In general, cresyl violet preferentially reveals fragmented nuclei and stains small, intensely basophilic particles frequently seen outside the neurons and representing apoptotic bodies (the end products of apoptosis). These bodies are visualized almost exclusively with cresyl violet. Scale bars: A, 80 μm; B, C, 40 μm;D–F, 20 μm.
Ultrastructural analysis of the bulbectomized brain through commissural planes of the olfactory cortex revealed numerous neurons undergoing apoptotic degeneration (Fig.5A). At earlier time points (e.g., 12–15 hr postbulbectomy) we observed some condensation of the cytoplasm; organelles were clearly discernible, whereas blebbing was absent. At 22 hr postbulbectomy, the cytoplasm appeared severely condensed, and its outer portion displayed intense vacuolation (“blebbing”); it was impossible to discern cytoplasmic detail or organelles (Fig. 5A,A′,B). At all stages of degeneration the nucleus appeared to be condensed in one or more large, electron-dense bodies dispersed within the cytoplasm without clear demarcation with a membrane (Fig. 5A′,B). Apoptotic profiles in advanced (i.e., 22 hr postlesion), but not in early (i.e., 12–15 hr postlesion), stages of degeneration were juxtaposed to or completely enclosed in numerous astrocytic processes containing a few mitochondria and clusters of round, electron-dense particles (Fig.5A′,B).
Fig. 5.

Morphological features of degenerating olfactory cortical neurons and the surrounding neuropil 22 hr postbulbectomy. A, A′, Four apoptotic profiles (arrows) are featured in this field. All apoptotic profiles show intense cytoplasmic blebbing. A few myelinated axons are also present (x). A′, A magnification of the rightmost apoptotic neuron and the associated neuropil in A. This dying neuron, like other apoptotic cells in A, is surrounded by newly formed astrocytic processes (a), which contain occasional mitochondria but are devoid of typical filament bundles. Dendrites (d), featured by regularly spaced, densely packed microtubules and the abundance of mitochondria, also are labeled to facilitate orientation in the illustrated field. B, This high-power micrograph reveals the close association (in this case, literal engulfment) of apoptotic neurons with astrocytic processes (a). Compare the morphology of astrocytic processes with that of longitudinally and transversely sectioned dendrites (d). An axonal terminal (t) and a myelinated axon (x) are shown also. Magnifications:A, 3000×; A′, 12,000×;B, 6000×.
We were unable to see phagocytic profiles in juxtaposition to apoptotic neurons up to 22 hr postlesion. The absence of evident phagocytosis in the EM material was consistent with our findings, using ICC for the myelomonocytic epitope ED1. The study of ED-1-stained sections through the olfactory cortex 22 hr postlesion failed to show phagocytic transformation of CNS glia or migration of blood monocytes/macrophages in the vicinity of degenerating neurons (Fig.6A,B).
Fig. 6.

Disposition of phagocytic glial cells toward apoptotic neurons of the olfactory cortex shown in preparations stained for the myelomonocytic epitope ED1. A, B, Cells in the vicinity of degenerating cortical neurons (A, arrowheads) do not express the phagocytic epitope ED1. However, adjacent vascular pericytes are ED1-positive (A,arrows), as are cells in pial vessels in the ventral aspect of the brain underneath the lesioned olfactory cortex (B, arrows). Scale bars:A, 40 μm; B, 20 μm.
Agarose gel electrophoresis of DNA samples purified from the disconnected olfactory cortices 22 and 24 hr postlesion showed oligonucleosomal bands in a “ladder” pattern of migration, whereas samples of olfactory cortex from unlesioned animals (controls) yielded only a short smear (Fig. 7). In most cases samples from the 24 hr postlesion time point showed smaller DNA fragments than samples from 22 hr postlesion. In all cases oligonucleosomal bands coexisted with various degrees of random DNA degradation (smearing). We were unable to discriminate small DNA fragments at time points earlier than 22 hr postlesion, presumably because of a lower concentration of apoptotic nuclei in those samples; at time points later than 24 hr, there was a predominance of DNA smearing.
Fig. 7.

Molecular characterization of the type of cell death in olfactory cortex of bulbectomized rats, based on the “ladder” pattern of DNA fragmentation by gel electrophoresis. Segments of DNA columns showing laddering are delineated (arrows). Note that at later time points (24hvs 22h) ladder bands assume smaller molecular sizes. Examples of oligonucleosomal bands are 360 Da (bottom arrow, 22h), 720 Da (top arrow,22h), and 540 Da (middle arrow,24h). Lane C depicts DNA from control, unlesioned olfactory cortex. Lane M contains molecular weight markers.
Neuronal counts and stereology confirm cell death and establish a brief time course of the apoptotic process
To establish an approximate time course for apoptosis in the olfactory cortex of bulbectomized rats, we performed a semiquantitative analysis of TUNEL-stained neurons in the olfactory cortex at selected time points postlesion (14–48 hr postbulbectomy in 4 hr intervals). At 14 hr postlesion, only a few cortical neurons were stained with TUNEL. At 14–22 hr, the number of TUNEL-positive cells increased markedly and reached a peak at 22–26 hr postlesion. The number of TUNEL-positive neurons declined at 30–42 hr. At 48 hr postlesion, few labeled cells were present in the olfactory cortex (Fig.8A). At this time point, nuclear remnants in the form of confluent, intensely basophilic fragments were identified in sections stained with cresyl violet (similar to particles shown in Fig. 4E,F).
Fig. 8.

Quantitation of TUNEL-stained cells at different times postbulbectomy (A) and at different anteroposterior planes (B). A, This time course illustrates that cell death begins at 14 hr and reaches a peak 22–26 hr postlesion. By 48 hr, cell death has almost ceased. Numbers represent TUNEL profiles throughout the extent of the olfactory cortex. B, Distribution of cell death from the anterior part of the olfactory cortex (closest to the lesion) to the most posterior part at three time points postlesion. Distance (in mm) is measured from the beginning of the olfactory cortex (corresponding to 0.0). A minimal number of cells undergo death 14 hr postlesion. The marked increase in cell death at 18 hr postlesion is especially evident in the anterior olfactory cortex. As the apoptotic process evolves (26 hr), the intensity of cell death migrates caudally.
At earlier time points (14–26 hr), labeled cells were concentrated in the rostral piriform cortex. At later time points (30–42 hr), labeling extended—and eventually predominated—in caudal planes of the olfactory cortex (Fig. 8B).
To assess the neuronal loss in piriform cortex and to confirm that TUNEL has accurately detected the extent of cell death in the bulbectomy model, we estimated the total number of neurons in cortical layers II–III at a time point coinciding with the conclusion of the apoptotic process (48 hr postlesion, as estimated with the time course of TUNEL staining), using unbiased stereology. Animals killed 48 hr postlesion were compared with unlesioned rats (controls). The total number of neurons in the unlesioned olfactory cortex was 297,315 (± 6,789 SEM), whereas the number of olfactory cortical neurons 48 hr postlesion reached a total of 243,887 (± 11,531 SEM); the percentage of difference was 18% (p = 0.02) (Fig.9). The mean difference of 53,428 neurons between control and lesioned olfactory cortices is slightly higher than the average (39,396) of TUNEL-positive cells in lesioned animals from all 4 hr interval counts throughout the 48 hr time course postlesion. Although these two numbers originate from different quantitative methods and are not directly comparable, the difference (14,032) suggests that the duration of TUNEL staining for individual neurons is <4 hr. Based on previous observations of the rapid (i.e., a few hours) progression of apoptosis in vitro and in vivo(Arends et al., 1990; Kerr and Harmon, 1991; Pittman et al., 1993), the <4 hr duration of TUNEL staining of individual cortical neurons postbulbectomy is additional evidence for an apoptotic mechanism of cell death in the bulbectomy model.
Fig. 9.

Neuronal loss in the olfactory cortex 48 hr postbulb-ectomy was measured by stereology. The total number of neurons in layer II of olfactory cortex 48 hr postlesion (n = 5) is 18% less than in unlesioned animals (n = 5). This difference represents an average loss of 53,400 neurons (p = 0.02).
Neurons in olfactory cortex undergo apoptosis as a result of afferent deprivation
Central neurons are especially vulnerable to axonal injury. Depending on the brain area and the distance of the lesion from the cell body, many axotomized neurons of the CNS degenerate and die (Koliatsos et al., 1994), sometimes displaying morphological features consistent with apoptosis (Wilcox et al., 1995). Bulbectomy interrupts many efferent and afferent connections of pyramidal neurons in the piriform cortex and, therefore, is a complex lesion for these neurons. Both anterograde and retrograde signals need to be considered.
To characterize the critical lesion that leads to cell death in the bulbectomy model, first we asked whether apoptotic profiles in the olfactory cortex can be labeled retrogradely with FB injected into the olfactory bulb before bulbectomy (Fig.10A). Under those conditions, labeled (i.e., dying) neurons would be identified as cells projecting to the bulb and, therefore, as cells undergoing axotomy-induced retrograde degeneration. The examination of sections through the piriform cortex revealed a large number of FB-labeled neurons in layer IIb. However, none displayed evidence of apoptosis as labeled with propidium iodide fluorescence. Apoptotic nuclei were observed only in superficially located neurons, as shown in double-illuminated preparations (Fig. 10A).
Fig. 10.

The design (left) and results (right) of the retrograde tracing experiment. FB injections into the olfactory bulb (OB) and piriform cortex (PO) were implemented to define projection targets of dying cortical neurons. Axotomized neurons projecting to the olfactory bulb (top neuron in thediagram) remain intact after the lesion. Nonaxotomized neurons projecting within piriform cortex (bottom neuronin A, corresponding to neurons in A–C) undergo apoptosis. A, Portion of the olfactory cortex of a bulbectomized rat retrogradely labeled with FB injected into the olfactory bulb before the lesion and counterstained with propidium iodide. The animal survived for 24 hr. FB labels neurons in layer IIb and does not colocalize in apoptotic cells (arrowsindicate an apoptotic neuron). B, C, Two apoptotic profiles (arrows) labeled postinjection of FB into the posterior piriform cortex. Fluorescent labeling shows that they are projection (pyramidal) neurons. The fact that these neurons cannot be labeled postinjection into olfactory bulb but become apoptotic postbulbectomy indicates that they are trans-synaptically affected by the lesion. Arrowheads in B show the axon of an apoptotic profile undergoing Wallerian degeneration. Scale bars, 10 μm.
Although apoptotic neurons were not labeled with injections of FB into the olfactory bulb, these cells transported FB retrogradely when the tracer was injected into the posterior subdivisions of piriform cortex before bulbectomy. All of these retrogradely labeled apoptotic neurons were located in layer IIA (Fig. 10B,C). Therefore, we conclude that the subpopulation of piriform neurons undergoing apoptosis postbulbectomy is not neurons projecting to the olfactory bulb.
Direct evidence that apoptotic cortical neurons are cells innervated by the olfactory bulb was obtained in olfactory cortex tissues prepared for electron microscopy 12–15 hr postlesion. At these time points numerous terminals were seen in layer I undergoing electron-dense degeneration (Peters et al., 1991). These terminals contacted abnormal, swollen dendrites with disrupted microtubular structure via asymmetric (gray type 1) densities (Fig.11A); the latter are presumed to be associated with excitatory neurotransmission (Peters et al., 1991). The location and morphology of these structures and the frequency of the terminal–dendrite apposition indicated that degenerating terminals deriving from olfactory bulb axons contact degenerating neurons, the cell bodies of which are located in layer II.
Fig. 11.
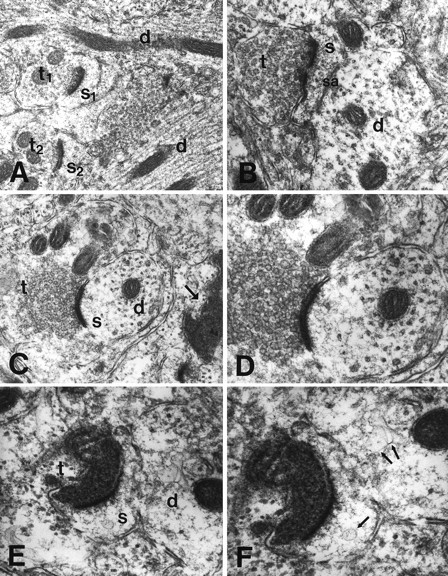
Degenerating neuronal profiles in olfactory cortex after bulbectomy belong to cells receiving inputs from the olfactory bulb, as evidenced by ultrastructural observations on layer I of the olfactory cortex 12–15 hr postlesion. A, A field taken from layer I of control olfactory cortex featuring normal asymmetrical synaptic contacts between two terminals (t1 andt2) and two synaptic spines (s1 ands2). Two adjacent normal dendrites are indicated also (d).B, A normal asymmetrical contact between an axonal terminal (t) and a dendritic spine.s, Spine; d, dendrite; sa, spinal apparatus. C, D, A field from the lesioned olfactory cortex showing a normal asymmetrical axonospinous synapse (t, terminal; s, spine; d, dendrite) beside a densely degenerating terminal (right, arrow). D, A further magnification of C. The uniformly round vesicles in terminals depicted in C and D (as well as in B) are consistent with excitatory neurotransmission. Transversely sectioned microtubules are in evidence in the receiving dendrite in D. E, F, A field from a lesioned olfactory cortex showing in detail a densely degenerating axonal terminal (t) contacting the spine (s) of a dendrite (d) in an asymmetrical manner. There is little residual structure in the receiving spine and dendrite, both of which exhibit abnormal vesicular elements (F, arrows). Magnifications: A, 26,500×; B, 58,500×; C, 41,000×;D, 66,000×; E, 64,500×;F, 86,500×.
DISCUSSION
Cell death of piriform cortical neurons postbulbectomy is apoptotic
Many lines of evidence in the present study indicate that, after their separation from the olfactory bulb, cortical olfactory neurons commit themselves to apoptotic death. At the ultrastructural level these cells show cytoplasmic condensation and blebbing, compaction and fragmentation of nuclear chromatin, and the eventual dissolution of membranous barriers between organelles and cytoplasm; all of these morphological features are associated classically with apoptotic death (Kerr et al., 1972). The rapid progression of degenerating profiles to advanced apoptotic forms demonstrates a rapid dying process, also indicative of apoptosis (Arends et al., 1990; Bursch et al., 1990; Kerr and Harmon, 1991; Pittman et al., 1993). Additional evidence for the rapid progression of death of olfactory neurons (<4 hr) was afforded by comparing counts of surviving neurons, based on Nissl staining at the end of the degenerative process (48 hr postbulbectomy), with cumulative counts of TUNEL-positive neurons in 4 hr intervals in the 14–48 hr postlesion period. The absence of inflammatory cells in the vicinity of dying neurons and especially the lack of prompt recruitment of phagocytic cells in proximity to affected olfactory neurons is also characteristic of apoptotic, but not necrotic, cell death (Kerr and Harmon, 1991). Nissl- and TUNEL-stained chromatin structures in olfactory cortical neurons postbulbectomy have the shape and location of apoptotic profiles as characterized previously (Wilcox et al., 1995) and in the present study with ultrastructural methods (i.e., multiple round basophilic or TUNEL-positive bodies or peripherally translocated chromatin). Finally, DNA electrophoresis of samples of lesioned cortex showed a ladder pattern of DNA migration by the end of the first day postlesion, which was changed into a smearing pattern during the second day postbulbectomy. These electrophoretic patterns indicate early internucleosomal cleavage (Wyllie, 1980) that then progresses to random DNA fragmentation as proteases digest further the histone backbone of single nucleosomes (Portera-Cailliau et al., 1995).
The time course of TUNEL staining of olfactory cortical neurons established a time window of 14–48 hr postbulbectomy within which these neurons undergo apoptotic death. In the area of olfactory cortex that experiences the maximal impact of the lesion (i.e., planes of piriform cortex near the anterior commissure), the previous “apoptotic window” can be narrowed further to 14–24 hr. If the greatest portion of loss of cortical neurons in the anterior piriform cortex occurs within this 10 hr period, then it follows that, in the bulbectomy model, signals pertaining to apoptosis are processed, transported, and transduced in a fairly rapid sequence.
The establishment of a relatively narrow apoptotic window in the bulbectomy model makes this lesion a very efficient assay system for detailed investigations of the expression of mammalian genes associated with cell death (Driscoll, 1996; Johnson et al., 1996). The use of transgenic mice with overexpression/deletion of some of these genes as experimental animals can be particularly useful. The rapid progression of cell death in the bulbectomy model also makes this lesion a very efficient system for testing substances that may prevent or delay cortical apoptosis in vivo.
Deafferentation injury and plasticity in sensory systems
The tract-tracing experiments in the present study have established that apoptotic neurons in the olfactory cortex are projection (pyramidal) neurons projecting in short pathways within the olfactory cortex, but not to the olfactory bulb. Moreover, our ultrastructural observations have demonstrated directly that processes of degenerating neurons are contacted by degenerating terminals 12–15 hr postbulbectomy. The emergence of these degenerating terminals within a short time period after the lesion and before the appearance of the first apoptotic profiles is very significant for understanding the mechanisms of apoptotic death in our model. The previous pattern of terminal degeneration is compatible with the temporal progression of Wallerian degeneration of axons originating in the bulb (from mitral cells), but not with a secondary axonal degeneration originating in the apoptotic cortical neurons themselves or in cortical interneurons. Indeed, the temporal precedence of terminal degeneration to the appearance of apoptotic death in the olfactory cortex suggests that degeneration of olfactory bulb inputs is instrumental in the mediation of apoptosis, i.e., that the latter is a transneuronal effect of the bulbectomy. The ultrastructural appearance of dendrites contacted by degenerating terminals, i.e., the presence of swelling in dendritic spines/shafts with abnormal dilation, and destruction of microtubules is consistent with early evidence of excitotoxic injury (Olney et al., 1979). If this hypothesis is correct, then the excitotoxic provocator is likely to be an excessive amount of glutamate that is released from the degenerating terminals (Choi, 1992). Perhaps the reason that pyramids in layer IIa of the primary olfactory cortex are the only afferent neurons undergoing degeneration after bulbectomy (as compared with, e.g., deafferented neurons in the olfactory tubercle and anterior olfactory nucleus) is that these nerve cells receive exclusive inputs from the bulb, i.e., they are exposed to higher doses of glutamate released from degenerating bulb terminals. This hypothesis would need to be tested experimentally by infusing glutamate antagonists in the vicinity of degenerating neurons and observing them for evidence of delay or abortion of the apoptotic process.
The anterograde effects of neurons on their targets (including other neurons and specialized end organs) have been a subject of investigation for many decades. The majority of the landmark experiments has used lesion approaches, in which inputs to specific target sites in the nervous system were interrupted during development or in adult animals. The dramatic effects observed after these manipulations ranged from extensive reorganization in terminal fields (principally CNS) to degeneration/atrophy of the target structure (mostly PNS). In the CNS, sensory systems have been investigated intensely, and anterograde effects have been demonstrated in auditory and vestibular nuclei (Levi-Montalcini, 1949; Powell and Erulkar, 1962;Schwaber et al., 1993), the visual system (Cook et al., 1951; Kupfer and Palmer, 1964; Hubel et al., 1977; Kaas et al., 1990; Chino et al., 1992), somatosensory cortex (Van der Loos and Woolsey, 1973; Garraghty and Kaas, 1991; Florence et al., 1994; Florence and Kaas, 1995), and the olfactory system (Matthews and Powell, 1962). In the vast majority of the above studies investigators have focused on the reorganization of adult sensory maps in the corresponding modalities, but some of the earlier studies (Cook et al., 1951; Powell and Erulkar, 1962; Kupfer and Palmer, 1964) have reported transneuronal degeneration. In the above studies, which have used both developing (Kupfer and Palmer, 1964) and adult (Cook et al., 1951; Powell and Erulkar, 1962) animals, investigators have found shrinkage and some phenotypic changes in deafferented neurons, but not cell death. An examination of the figures included in these studies does not reveal any evidence of apoptosis. A striking exception to the relative resistance of central neurons to deafferentation is the olfactory cortical neurons in layer IIa. Since the first demonstration of degeneration and death of these neurons after bulbectomy (Heimer and Kalil, 1978), a few more studies have been published on the same phenomenon (Cutler et al., 1983; Friedman and Price, 1986a,b), which have strongly suggested a transneuronal effect, although retrograde factors were not ruled out. In addition, Friedman and Price demonstrated that olfactory cortical neurons of neonates are not vulnerable to the effects of bulbectomy, presumably because of a robust and prompt proliferation of intracortical axons that reinnervate the deafferented dendrites of layer IIa neurons (Friedman and Price, 1986a,b).
In general, the consequences of deafferentation are more severe in the periphery, especially during development. For example, muscle denervation causes a major reduction in actin and myosin as well as muscle fiber atrophy (Tower, 1937; Harris and Ward, 1974; Grinnell and Herrera, 1980). Although most muscle properties that change after denervation can be reproduced by pharmacological paralysis of the muscle (Lomo and Rosenthal, 1972; Purves and Sakmann, 1974; Berg and Hall, 1975), the possibility of the involvement of an “anterograde” trophic molecule remains open. For example, the 100–150 kDa protein agrin, transported by motor axons to synaptic endings in muscle (Ruegg et al., 1992), induces synaptic clustering of several muscle surface proteins essential for differentiation of the developing neuromuscular system (Reist et al., 1992). Peripheral sensory structures (e.g., mammalian taste buds) (Zalewski, 1969) and the Herbst corpuscles present in the bills of some aquatic birds (Saxod, 1978) are also under anterograde trophic control by sensory fibers. In both of these cases end organs cease to develop or degenerate completely after denervation. Effects similar to those exerted by neurons on peripheral end organs have been reported on autonomic, i.e., parasympathetic (McMahan and Kuffler, 1971), and developing sympathetic (Black et al., 1972) ganglia.
On the basis of the above discussion, the present study demonstrates that central targets of a sensory pathway can undergo apoptotic cell death after afferent deprivation. It is unknown at this time whether this phenomenon is exclusive to the olfactory system (olfactory cortex) or also can be found in other sensory systems, provided that specific attention is paid to rapid postlesion developments. If the mechanism of deafferentation-induced death in sensory systems is apoptotic, the rapidly progressing, noninflammatory degeneration typical of this type of death can easily escape observation.
Footnotes
This work was supported by Grants from the U.S. Public Health Service (National Institutes of Health NS 20471 and AG 05146). D.L.P. and V.E.K. are recipients of a Leadership and Excellence in Alzheimer’s Disease (LEAD) Award (NIA AG 07914) and a Javits Neuroscience Investigator Award (National Institutes of Health NS 10580). We gratefully acknowledge the assistance of Mr. Frank Barksdale for EM and photography.
Correspondence should be addressed to Dr. Vassilis E. Koliatsos, Division of Neuropathology, The Johns Hopkins University School of Medicine, 558 Ross Research Building, 720 Rutland Avenue, Baltimore, MD 21205-2196.
REFERENCES
- 1.Arends MJ, Morris RG, Wyllie AH. Apoptosis. The role of the endonuclease. Am J Pathol. 1990;136:593–608. [PMC free article] [PubMed] [Google Scholar]
- 2.Berg DK, Hall ZW. Increased extrajunctional acetylcholine sensitivity produced by chronic post-synaptic neuromuscular blockade. J Physiol (Lond) 1975;244:659–676. doi: 10.1113/jphysiol.1975.sp010818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black IB, Hendry IA, Iversen LL. Effects of surgical decentralization and nerve growth factor on the maturation of adrenergic neurons in a mouse sympathetic ganglion. J Neurochem. 1972;19:1367–1377. doi: 10.1111/j.1471-4159.1972.tb01461.x. [DOI] [PubMed] [Google Scholar]
- 4.Bursch W, Paffe S, Putz B, Barthel G, Schulte-Hermann R. Determination of the length of the histological stages of apoptosis in normal liver and in altered foci of rats. Carcinogenesis. 1990;11:847–853. doi: 10.1093/carcin/11.5.847. [DOI] [PubMed] [Google Scholar]
- 5.Chino YM, Kaas JH, Smith EL, Langston AL, Cheng H. Rapid reorganization of cortical maps in adult cats following restricted deafferentation in retina. Vision Res. 1992;32:789–796. doi: 10.1016/0042-6989(92)90021-a. [DOI] [PubMed] [Google Scholar]
- 6.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 7.Cook WH, Walker JH, Barr ML. A cytological study of transneuronal atrophy in the cat and rabbit. J Comp Neurol. 1951;94:267–291. doi: 10.1002/cne.900940207. [DOI] [PubMed] [Google Scholar]
- 8.Cummings BJ, Yee GJ, Cotman CW. bFGF promotes the survival of entorhinal layer II neurons after perforant path axotomy. Brain Res. 1992;591:271–276. doi: 10.1016/0006-8993(92)91707-l. [DOI] [PubMed] [Google Scholar]
- 9.Cutler JR, Bredesen DE, Edwards R, Simon RP. Failure of naloxone to reverse vascular neurologic deficits. Neurology. 1983;33:1517–1518. doi: 10.1212/wnl.33.11.1517. [DOI] [PubMed] [Google Scholar]
- 10.Driscoll M. Cell death in C. elegans: molecular insights into mechanisms conserved between nematodes and mammals. Brain Pathol. 1996;6:411–425. doi: 10.1111/j.1750-3639.1996.tb00873.x. [DOI] [PubMed] [Google Scholar]
- 11.Florence SL, Kaas JH. Large-scale reorganization at multiple levels of the somatosensory pathway follows therapeutic amputation of the hand in monkeys. J Neurosci. 1995;15:8083–8095. doi: 10.1523/JNEUROSCI.15-12-08083.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Florence SL, Garraghty PE, Wall JT, Kaas JH. Sensory afferent projections and area 3b somatotopy following median nerve cut and repair in macaque monkeys. Cereb Cortex. 1994;4:391–407. doi: 10.1093/cercor/4.4.391. [DOI] [PubMed] [Google Scholar]
- 13.Friedman B, Price JL. Plasticity in the olfactory cortex: age-dependent effects of deafferentation. J Comp Neurol. 1986a;246:1–19. doi: 10.1002/cne.902460102. [DOI] [PubMed] [Google Scholar]
- 14.Friedman B, Price JL. Age-dependent cell death in the olfactory cortex: lack of transneuronal degeneration in neonates. J Comp Neurol. 1986b;246:20–31. doi: 10.1002/cne.902460103. [DOI] [PubMed] [Google Scholar]
- 15.Garraghty PE, Kaas JH. Large-scale functional reorganization in adult monkey cortex after peripheral nerve injury. Proc Natl Acad Sci USA. 1991;88:6976–6980. doi: 10.1073/pnas.88.16.6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graeber MB, Streit WJ, Kiefer R, Schoen SW, Kreutzberg GW. New expression of myelomonocytic antigens by microglia and perivascular cells following lethal motor neuron injury. J Neuroimmunol. 1990;27:121–132. doi: 10.1016/0165-5728(90)90061-q. [DOI] [PubMed] [Google Scholar]
- 18.Grinnell AD, Herrera AA. Physiological regulation of synaptic effectiveness at frog neuromuscular junctions. J Physiol (Lond) 1980;307:301–317. doi: 10.1113/jphysiol.1980.sp013436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gundersen HJG. Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J Microsc. 1986;143:3–45. [PubMed] [Google Scholar]
- 20.Gundersen HJG, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- 21.Harris JB, Ward MR. A comparative study of “denervation” in muscles from mice with inherited neuromuscular disorders. Exp Neurol. 1974;42:169–180. doi: 10.1016/0014-4886(74)90015-6. [DOI] [PubMed] [Google Scholar]
- 22.Heimer L, Kalil R. Rapid transneuronal degeneration and death of cortical neurons following removal of the olfactory bulb in adult rats. J Comp Neurol. 1978;178:559–610. doi: 10.1002/cne.901780310. [DOI] [PubMed] [Google Scholar]
- 23.Hubel DH, Wiesel TN, LeVay S. Plasticity of ocular dominance columns in monkey striate cortex. Philos Trans R Soc Lond [Biol] 1977;278:377–409. doi: 10.1098/rstb.1977.0050. [DOI] [PubMed] [Google Scholar]
- 24.Johnson EM, Jr, Deckwerth TL, Deshmukh M. Neuronal death in developmental models: possible implications in neuropathology. Brain Pathol. 1996;6:397–409. doi: 10.1111/j.1750-3639.1996.tb00872.x. [DOI] [PubMed] [Google Scholar]
- 25.Kaas JH, Merzenich MM, Killackey HP. The reorganization of somatosensory cortex following peripheral nerve damage in adult and developing mammals. Annu Rev Neurosci. 1983;6:325–356. doi: 10.1146/annurev.ne.06.030183.001545. [DOI] [PubMed] [Google Scholar]
- 26.Kaas JH, Krubitzer LA, Chino YM, Langston AL, Polley EH, Blair N. Reorganization of retinotopic cortical maps in adult mammals after lesions of the retina. Science. 1990;248:229–231. doi: 10.1126/science.2326637. [DOI] [PubMed] [Google Scholar]
- 27.Kerr JFR, Harmon BV. Definition and incidence of apoptosis: an historical perspective. In: Tomei LD, Cope FO, editors. Apoptosis. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1991. pp. 5–29. [Google Scholar]
- 28.Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koliatsos VE, Price DL. Nonhuman primate models in trophic factor research. In: Boulton AA, Baker GB, Hefti F, editors. Animal models of neurological disorders. Humana; Clifton, NJ: 1993. pp. 331–370. [Google Scholar]
- 30.Koliatsos VE, Martin LJ, Walker LC, Richardson RT, DeLong MR, Price DL. Topographic, non-collateralized basal forebrain projections to amygdala, hippocampus, and anterior cingulate cortex in the rhesus monkey. Brain Res. 1988;463:133–139. doi: 10.1016/0006-8993(88)90535-5. [DOI] [PubMed] [Google Scholar]
- 31.Koliatsos VE, Martin LJ, Price DL. Efferent organization of the mammalian basal forebrain. In: Steriade M, Biesold D, editors. Brain cholinergic systems. Oxford UP; Oxford: 1990. pp. 120–152. [Google Scholar]
- 32.Koliatsos VE, Price DL, Clatterbuck RE, Markowska AL, Olton DS, Wilcox BJ. Neurotrophic strategies for treating Alzheimer’s disease: lessons from basic neurobiology and animal models. Ann NY Acad Sci. 1993;695:292–299. doi: 10.1111/j.1749-6632.1993.tb23069.x. [DOI] [PubMed] [Google Scholar]
- 33.Koliatsos VE, Price WL, Pardo CA, Price DL. Ventral root avulsion: an experimental model of death of adult motor neurons. J Comp Neurol. 1994;342:35–44. doi: 10.1002/cne.903420105. [DOI] [PubMed] [Google Scholar]
- 34.Kupfer C, Palmer P. Lateral geniculate nucleus: histological and cytochemical changes following afferent denervation and visual deprivation. Exp Neurol. 1964;9:400–409. doi: 10.1016/0014-4886(64)90074-3. [DOI] [PubMed] [Google Scholar]
- 35.Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski HM. Cell death in Alzheimer’s disease evaluated by DNA fragmentation in situ. Acta Neuropathol (Berl) 1995;89:35–41. doi: 10.1007/BF00294257. [DOI] [PubMed] [Google Scholar]
- 36.Levi-Montalcini R. The development of the acoustico-vestibular centers in the chick embryo in the absence of the afferent root fibers and of descending fiber tracts. J Comp Neurol. 1949;91:209–241. doi: 10.1002/cne.900910204. [DOI] [PubMed] [Google Scholar]
- 37.Lomo T, Rosenthal J. Control of ACh sensitivity by muscle activity in the rat. J Physiol (Lond) 1972;221:493–513. doi: 10.1113/jphysiol.1972.sp009764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matthews MR, Powell TPS. Some observations on transneuronal cell degeneration in the olfactory bulb of the rabbit. J Anat. 1962;96:89–102. [PMC free article] [PubMed] [Google Scholar]
- 39.McMahan UJ, Kuffler SW. Visual identification of synaptic boutons on living ganglion cells and of varicosities in postganglionic axons in the heart of the frog. Proc R Soc Lond [Biol] 1971;177:485–508. doi: 10.1098/rspb.1971.0044. [DOI] [PubMed] [Google Scholar]
- 40.Milligan CE, Levitt P, Cunningham TJ. Brain macrophages and microglia respond differently to lesions of the developing and adult visual system. J Comp Neurol. 1991;314:136–146. doi: 10.1002/cne.903140113. [DOI] [PubMed] [Google Scholar]
- 41.Mouton PR, Pakkenberg B, Gundersen HJG, Price DL. Absolute number and size of pigmented locus coeruleus neurons in the brains of young and aged individuals. J Chem Neuroanat. 1994;7:185–190. doi: 10.1016/0891-0618(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 42.Olney JW, Fuller T, deGubareff T. Acute dendrotoxic changes in the hippocampus of kainate-treated rats. Brain Res. 1979;176:91–100. doi: 10.1016/0006-8993(79)90872-2. [DOI] [PubMed] [Google Scholar]
- 43.Peters A, Palay SL, Webster HF. The fine structure of the nervous system. Oxford UP; New York: 1991. [Google Scholar]
- 44.Peterson DA, Lucidi-Phillipi CA, Eagle KL, Gage FH. Perforant path damage results in progressive neuronal death and somal atrophy in layer II of entorhinal cortex and functional impairment with increasing postdamage age. J Neurosci. 1994;14:6872–6885. doi: 10.1523/JNEUROSCI.14-11-06872.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pittman RN, Wang S, DiBenedetto AJ, Mills JC. A system for characterizing cellular and molecular events in programmed neuronal cell death. J Neurosci. 1993;13:3669–3680. doi: 10.1523/JNEUROSCI.13-09-03669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pons TP, Garraghty PE, Ommaya AK, Kaas JH, Taub E, Mishkin M. Massive cortical reorganization after sensory deafferentation in adult macaques. Science. 1991;252:1857–1860. doi: 10.1126/science.1843843. [DOI] [PubMed] [Google Scholar]
- 47.Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powell TPS, Erulkar SD. Transneuronal cell degeneration in the auditory relay nuclei of the cat. J Anat. 1962;96:249–268. [PMC free article] [PubMed] [Google Scholar]
- 49.Purves D, Sakmann B. Membrane properties underlying spontaneous activity of denervated muscle fibres. J Physiol (Lond) 1974;239:125–153. doi: 10.1113/jphysiol.1974.sp010559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reist NE, Werle MJ, McMahan UJ. Agrin released by motor neurons induces the aggregation of acetylcholine receptors at neuromuscular junctions. Neuron. 1992;8:865–868. doi: 10.1016/0896-6273(92)90200-w. [DOI] [PubMed] [Google Scholar]
- 51.Ruegg MA, Tsim KWK, Horton SE. The agrin gene codes for a family of basal lamina proteins that differ in function and distribution. Neuron. 1992;8:691–699. doi: 10.1016/0896-6273(92)90090-z. [DOI] [PubMed] [Google Scholar]
- 52.Saxod R. Development of cutaneous sensory receptors in birds. In: Jacobson M, editor. Development of sensory systems. Springer; Berlin: 1978. pp. 337–417. [Google Scholar]
- 53.Schwaber MK, Garraghty PE, Kaas JH. Neuroplasticity of the adult primate auditory cortex following cochlear hearing loss. Am J Otolaryngol. 1993;14:252–258. [PubMed] [Google Scholar]
- 54.Shipley MT, McLean JH, Ennis M. Olfactory system. In: Paxinos G, editor. The rat nervous system. Academic; San Diego: 1995. pp. 899–926. [Google Scholar]
- 55.Snider WD, Elliott JL, Yan Q. Axotomy-induced neuronal death during development. J Neurobiol. 1992;23:1231–1246. doi: 10.1002/neu.480230913. [DOI] [PubMed] [Google Scholar]
- 56.Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. NeuroReport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 57.Tower SS. Trophic control of non-nervous tissues by the nervous system: a study of muscle and bone innervated from an isolated and quiescent region of spinal cord. J Comp Neurol. 1937;67:241–261. [Google Scholar]
- 58.Troncoso JC, Sukhov RR, Kawas CH, Koliatsos VE. In situ labeling of dying cortical neurons in normal aging and in Alzheimer’s disease: correlations with senile plaques and disease progression. J Neuropathol Exp Neurol. 1996;55:1134–1142. doi: 10.1097/00005072-199611000-00004. [DOI] [PubMed] [Google Scholar]
- 59.Van der Loos H, Woolsey TA. Somatosensory cortex: structural alterations following early injury to sense organs. Science. 1973;179:395–398. doi: 10.1126/science.179.4071.395. [DOI] [PubMed] [Google Scholar]
- 60.Wilcox BJ, Applegate MD, Portera-Cailliau C, Koliatsos VE. Nerve growth factor prevents apoptotic cell death in injured central cholinergic neurons. J Comp Neurol. 1995;359:573–585. doi: 10.1002/cne.903590405. [DOI] [PubMed] [Google Scholar]
- 61.Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 62.Zalewski AA. Combined effects of testosterone and motor, sensory, or gustatory nerve reinnervation on the regeneration of taste buds. Exp Neurol. 1969;24:285–297. doi: 10.1016/0014-4886(69)90022-3. [DOI] [PubMed] [Google Scholar]