

Abstract
Tryptophan hydroxylase (TPH), the initial and rate-limiting enzyme in the biosynthesis of the neurotransmitter serotonin (5-HT), is irreversibly inactivated by nitric oxide (NO). We have expressed brain TPH as a recombinant glutathione-S-transferase fusion protein and delineated the catalytic domain of the enzyme as the region spanning amino acids 99–444. Highly purified TPH catalytic core, like the native enzyme from brain, is inactivated by NO in a concentration-dependent manner. Removal of iron from TPH produces an apoenzyme with low activity that can be reconverted to its highly active holo-form by the addition of ferrous iron. Apo-TPH exposed to NO cannot be reactivated by iron. Treatment of holo-TPH (iron-loaded) with the disulfide 5,5′-dithio-bis (2-nitrobenzoic acid) (DTNB) causes an inactivation of TPH that is readily reversed by dithiothreitol (DTT). DTNB-treated TPH [sulfhydryl (SH)-protected] exposed to NO is returned to full activity by thiol reduction with DTT. The inactivation of native TPH by NO cannot be reversed by either iron or DTT. These data indicate that NO inactivates TPH by selective action on critical SH groups (i.e., cysteine residues) while sparing catalytic iron sites within the enzyme. The results are interpreted with reference to the substituted amphetamines, which are neurotoxic to 5-HT neurons, that inactivate TPH in vivo and are now known to produce NO and other reactive oxygen species in vivo.
Keywords: tryptophan hydroxylase, nitric oxide, sulfhydryls, catalytic iron site, serotonin, neurotoxic amphetamines
Tryptophan hydroxylase [EC1.14.16.4; l-tryptophan, tetrahydropteridine: oxygen oxidoreductase (5-hydroxylating)] (TPH) is the initial and rate-limiting enzyme in the biosynthesis of the neurotransmitter serotonin (5-HT) (Jequier et al., 1967). The synthesis of 5-HT can proceed only through this enzyme-catalyzed step. The activity of TPH controls the amount of 5-HT produced and released from neurons (Gartside et al., 1992; Oluyomi et al., 1994), indicating that this enzyme regulates not just 5-HT synthesis but its function as well. As a neurotransmitter, 5-HT mediates pain, sleep, thermoregulation, and food intake. From a clinical perspective, altered 5-HT function has been implicated in depression, obsessive-compulsive disorder, autism, and impulsive self-destructive behaviors such as aggression, suicide, and drug taking (Schatzberg and Nemeroff, 1995).
Selected amphetamines have profound effects on the 5-HT neuronal system. Methylenedioxymethamphetamine (ecstasy) (MDMA) andp-chloroamphetamine cause extensive destruction of 5-HT neurons. An early manifestation of their effects is a significant inactivation of TPH (for reviews, see Gibb et al., 1993; Steele et al., 1994; Seiden and Sabol, 1996). The mechanisms underlying these effects on TPH are not known, but emerging data implicate drug-induced production of reactive oxygen species (ROS) and nitric oxide (NO). The cellular effects of ROS or NO cannot be predicted a priori: NO can be toxic to some cells (Lipton et al., 1993; Dawson et al., 1994), it is a neurotransmitter–neuromodulator in other cells (Jaffrey and Snyder, 1996), and it can protect still other cells from known toxins (Wink et al., 1995, 1996). The recent demonstration that TPH is inactivated by NO in vitro (Kuhn and Arthur, 1996, 1997) establishes the possibility that this important brain enzyme is susceptible to inactivation by NO in vivo and could form the basis for loss of TPH activity when NO levels are elevated in brain (e.g., amphetamine-induced).
Our initial experiments with NO–TPH interactions focused on crude brain extracts in an attempt to establish that NO can target TPH in a complex protein milieu (Kuhn and Arthur, 1996, 1997). Having done this, we have shifted our attention to determining the molecular mechanisms by which NO and ROS inactivate TPH. These studies are made possible by the cloning and expression of a recombinant TPH as a glutathione-S-transferase (GST) fusion protein. This enzyme can be produced in large quantity in Escherichia coli and purified to homogeneity by a single affinity chromatographic step. Primary targets in proteins for NO attack are iron and SH groups (Stamler et al., 1992; Stamler, 1994). By altering independently the iron or SH status of TPH, we demonstrate that NO inactivates TPH by selective attack on critical SH groups, sparing catalytic iron sites altogether.
MATERIALS AND METHODS
Materials. The following materials were obtained from Sigma (St. Louis, MO): sodium nitroprusside, dithiothreitol (DTT), hemoglobin, o-phenanthroline, ferrous ammonium sulfate, glutathione (GSH), GSH-agarose beads, cysteine, tryptophan, and 5,5′-dithio-bis (2-nitrobenzoic acid) (DTNB). The GST fusion protein vector pGEX4T-2 was purchased from Pharmacia Biotech (Piscataway, NJ). T4 DNA ligase and restriction endonucleases were purchased from New England Biolabs (Beverly, MA). Catalase was a product of Boehringer Mannheim (Indianapolis, IN). Tetrahydrobiopterin was purchased from Dr. B. Schircks Laboratories (Jona, Switzerland). Isopropyl β-d-thiogalactopyranoside (IPTG) was obtained from Gold Biotechnologies (St. Louis, MO). BL-21 E. colicells were purchased from Invitrogen (Carlsbad, CA).
Cloning and expression of TPH. TPH was cloned and expressed as a GST fusion protein as described previously (D’Sa et al., 1996;Kuhn et al., 1997). Both a full-length form of wild-type (WT) TPH (amino acids 1–444; GST–TPH–WT) or a deletional mutant constituting the catalytic core (amino acids 99–444; GST–TPH–CC) were expressed in BL-21 (E. coli) cells. Bacteria transformed with the plasmid bearing the TPH–CC cDNA were induced with 0.1 mmIPTG for 2 hr at 30°C. Bacteria were washed with 10% glycerol and then with 0.1 vol of 50 mm Tris-HCl, pH 7.5. After sonication and centrifugation (40,000 × g) to sediment insoluble material, the supernatants were adsorbed on GSH-agarose for 30 min at 4°C. Affinity beads were washed three times with 50 vol of 50 mm Tris-HCl, pH 7.5, at 4°C and used immediately.
Conversion of TPH between apo- and holoenzyme. TPH–CC was treated with the ferrous iron chelator o-phenanthroline (100–1000 μm) at 4°C for 30 min in the presence of 1 mm DTT. After treatment, GST–TPH–CC beads were washed three times with 50 mm Tris-HCl, pH 7.5, at 4°C to removeo-phenanthroline, and the enzyme was either assayed for residual activity or exposed to NO as described below. Recovery of TPH activity after o-phenanthroline treatment was tested by assaying the enzyme ± 100 μm ferrous iron (ferrous ammonium sulfate).
Treatment of TPH with DTNB. Oxidation of SH groups in proteins by disulfides such as DTNB is specific for cysteine residues and results in the formation of mixed disulfides (Van Iwaarden et al., 1992). GST–TPH–CC was exposed to varying concentrations of DTNB in the absence of DTT at 30°C for 15 min. DTNB was removed by three washes of the affinity beads with 50 mm Tris-HCl, pH 7.5, at 4°C. After the last wash the levels of residual TPH activity were determined. Recovery of TPH activity after treatment with DTNB was tested by exposing treated TPH to varying concentrations of DTT at 4°C for 30 min. The DTT was then removed by washing the affinity beads as described. All assays of TPH pretreated with DTNB–DTT were performed without DTT to allow an assessment of the status of the SH groups before assay. Omission of DTT from the assay does not alter enzyme activity.
Treatment of TPH with NO. GST–TPH–CC was treated with varying concentrations of the NO generator sodium nitroprusside (SNP) (Feelisch and Noack, 1987; Shibuki, 1990; Southam and Garthwaite, 1991). Because the generation of NO from SNP depends on a reductant such as DTT, all experiments with SNP were performed ±1 mmDTT. Controls contained DTT only. Exposure to SNP was at 4°C for 30 min as described previously for brain TPH (Kuhn and Arthur, 1996,1997). In some experiments TPH was treated with eithero-phenanthroline or DTNB as described above before SNP. After these pretreatments, reagents were removed by three washes of the affinity beads before addition of SNP. After exposure to SNP, the beads were washed three more times with 50 mm Tris-HCl, pH 7.5, at 4°C, and residual TPH activity was determined.
Assay of TPH catalytic activity. TPH activity was assayed by measuring the formation of 5-hydroxytryptophan (5-HTP) as described previously (Kuhn et al., 1997). TPH activity was assayed while the protein remained bound to the GSH-affinity beads, and results are expressed as nanomoles of 5-HTP per minute per milligram (D’Sa et al., 1996; Kuhn et al., 1997). Adsorption of the TPH fusion protein to GSH-affinity beads does not interfere with the catalytic function of the enzyme. The levels of protein in all enzyme samples were measured with bead-bound preparations using the method of Bradford (1976). Controls contained the appropriate buffers, solvents, and beads unexposed to protein. The beads did not cause the bovine serum albumin standard curve to depart from linearity.
Statistics. Concentration-effects of iron- or SH-reacting reagents on TPH activity (see Figs. 1, 2, 3) were tested for statistical significance by ANOVA. Student’s t tests were used to compare independent group effects with their controls when reversal of TPH inactivation was attempted (see Figs. 4, 5).
Fig. 1.

Conversion of TPH between apo- and holo-forms witho-phenanthroline. TPH–CC protein was expressed in BL-21 cells and adsorbed to GSH-affinity beads as described in Materials and Methods. The enzyme was exposed to varying concentrations ofo-phenanthroline at 4°C for 30 min in the presence of 1 mm DTT. After this treatment, affinity beads were washed three times with 50 mm Tris-HCl, pH 7.5, at 4°C, and residual TPH activity was determined in the absence or presence of 100 μm ferrous ammonium sulfate. Results are shown as % control TPH activity (57.5 nmol of 5-HTP · min−1 · mg−1) and are the mean ± SEM of four independent experiments run in duplicate. The effect of o-phenanthroline (−iron) was statistically significant by ANOVA; p < 0.001.
Fig. 2.
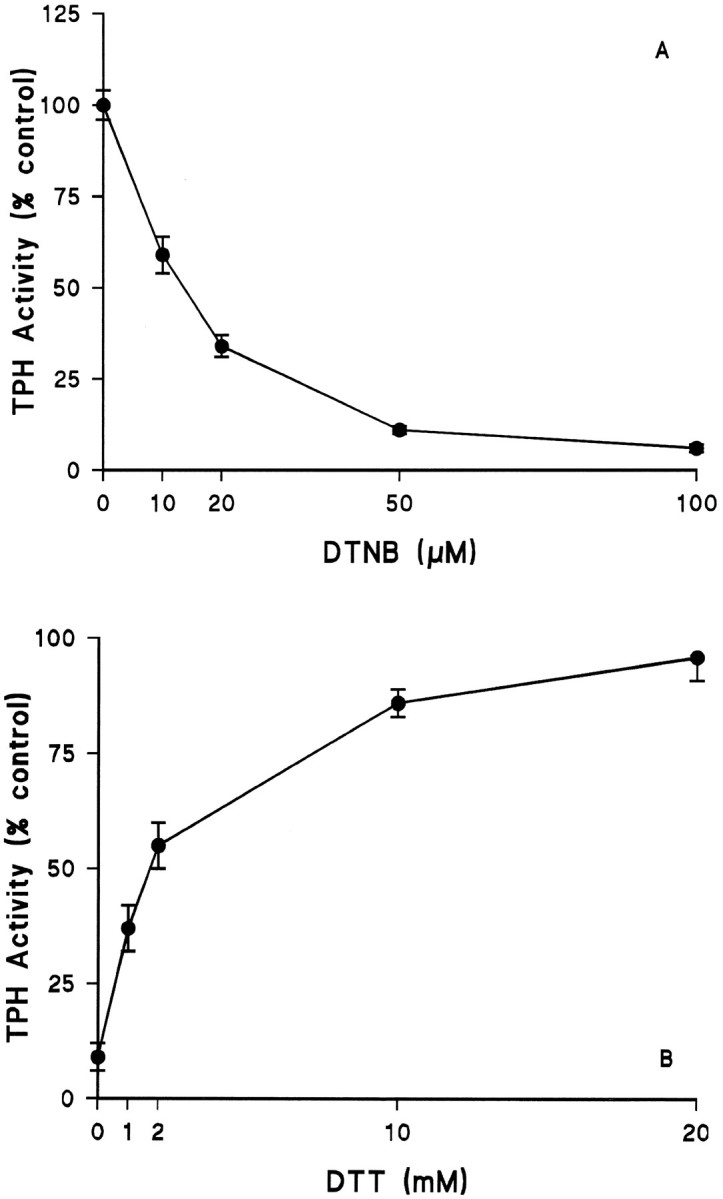
Effects of the disulfide DTNB on TPH. TPH–CC protein was prepared and adsorbed to GSH-affinity beads as described in Materials and Methods. A, The enzyme was exposed to varying concentrations of DTNB (without DTT) at 30°C for 15 min followed by three washes with 50 mm Tris-HCl, pH 7.5, at 4°C. Residual TPH activity was determined in the presence of 100 μm ferrous ammonium sulfate without DTT.B, TPH–CC was first exposed to 50 μm DTNB as described in Figure 2A and then GSH-affinity beads were washed free of DTNB. The enzyme was next exposed to varying concentrations of DTT at 4°C for 30 min. After DTT treatment, beads were washed as before, and residual TPH activity was determined in assays that omitted DTT. The results in both A andB are expressed as % control TPH activity (51.5 nmol of 5-HTP · min−1 · mg−1) and are the mean ± SEM of four independent experiments performed in duplicate. The effects of DTNB in A and that of DTT in B were both statistically significant by ANOVA;p < 0.001 for each.
Fig. 3.

The effects of the NO generator SNP on TPH. Both GST fusion constructs TPH–WT and TPH–CC were prepared and adsorbed to GSH-affinity beads as described and exposed to varying concentrations of SNP ± DTT (1 mm). Enzymes were exposed to SNP at 4°C for 30 min followed by three washes with 50 mmTris-HCl, pH 7.5, to remove the reagent. Residual TPH activity was determined in assays that omitted DTT. The results are presented as % control TPH activity (47.6 and 54.4 nmol · min−1 · mg−1 for WT and CC forms, respectively) and are the mean ± SEM of four independent experiments performed in duplicate. The effects of SNP on both forms of TPH were statistically significant by ANOVA;p < 0.001. The responses of TPH–WT and TPH–CC to SNP did not differ significantly (p > 0.1).
Fig. 4.

The effect of SNP on apo- and holo-TPH. TPH–CC was prepared and adsorbed to GSH-affinity beads as described and exposed to 20 μmo-phenanthroline at 4°C for 30 min to convert TPH to its apo-form (−iron). Controls (holoenzyme) were incubated with buffer alone. Beads were washed three times with 50 mm Tris-HCl, pH 7.5, at 4°C to removeo-phenanthroline. After this initial treatment, holo-TPH and apo-TPH were exposed to 100 μm SNP at 4°C for 30 min, and beads were washed free of SNP. Residual TPH activity was then determined ± ferrous ammonium sulfate (100 μm). The results are expressed as % control TPH activity (49.5 nmol of 5-HTP · min−1 · mg−1) and are the mean ± SEM of four independent experiments performed in duplicate. The effects of SNP and o-phenanthroline were statistically different from control in all cases by Student’st test; *p < 0.01. The effect of iron was significant only in the case of apo-TPH by Student’st test; **p < 0.01.
Fig. 5.

Effect of SNP on SH-protected TPH. TPH–CC was prepared and adsorbed to GSH-affinity beads as described and exposed to 50 μm DTNB at 30°C for 15 min without DTT. Controls were incubated with buffer alone. Beads were washed three times with 50 mm Tris-HCl, pH 7.5, at 4°C to remove DTNB. After this initial treatment, control TPH and DTNB-treated TPH were exposed to 100 μm SNP at 4°C for 30 min, and beads were washed free of SNP. The last step involved exposure of each enzyme preparation to 10 mm DTT at 4°C for 30 min. Controls were incubated with buffer alone. After a final three washes to remove DTT, residual TPH activity was assayed without DTT in the presence of 100 μm iron. The results are presented as % control TPH activity (45.2 nmol of 5-HTP · min−1 · mg−1) and are the mean ± SEM of four independent experiments run in duplicate. The effects of DTNB and SNP were statistically significant from controls by Student’s t test; *p < 0.01. The effect of 10 mm DTT was also significantly different from control after these multiple incubation steps (*p < 0.01). The effect of DTT (10 mm) to reverse DTNB inactivation of TPH was significant for both control and SNP-treated enzyme (**p < 0.01 for each by Student’s t test).
RESULTS
TPH was studied while it remained bound to GSH-affinity beads as a GST fusion protein. The immobilized fusion protein retains all essential catalytic and molecular functions of the native enzyme and offers the distinct advantages of rapid purification and convenient washing before assays of catalytic activity. Because the GST–TPH–CC deletional mutant did not differ significantly from the full-length enzyme in its responses to the treatments used here (e.g., iron, DTT,o-phenanthroline, DTNB) (D’Sa et al., 1996), we concluded that the deleted regulatory domain (amino acids 1–98) does not influence responsiveness of the enzyme catalytic function to iron or disulfides. With the exception of the initial studies with NO-induced inactivation of both the wild-type (full-length) and catalytic core forms of TPH, all remaining experiments used the GST–TPH–CC deletional mutant.
Conversion of TPH between holo- and apo-forms
If TPH-bound iron is a target for NO, it is essential to determine whether TPH could be protected from reaction with NO. We prepared TPH in the presence of the ferrous iron chelatoro-phenanthroline. This reagent has been shown to reduce the iron content of phenylalanine hydroxylase (Shiman et al., 1994), and it significantly inhibits TPH activity (Kuhn et al., 1980). Figure1 shows that o-phenanthroline causes a concentration-dependent reduction of TPH activity. At a concentration of 20 μm, TPH activity was reduced to ∼34% of control when assayed without iron. Inhibition of TPH exceeded 95% at an o-phenanthroline concentration of 100 μm. This effect of o-phenanthroline was statistically significant (p < 0.001; ANOVA). When TPH is then assayed in the presence of 100 μm iron, activity is fully restored to control levels. These experiments establish the importance of iron in TPH function and demonstrate that TPH can be reversibly converted between the apo- and holo-forms.
Modification of SH groups in TPH
TPH was treated with the SH reagent DTNB. The results in Figure2A demonstrate that DTNB causes a concentration-dependent reduction in TPH activity. TPH was quite sensitive to DTNB, and concentrations as low as 50 μm cause a >80% reduction in catalytic activity. The effect of DTNB was statistically significant (p< 0.001; ANOVA). If DTNB-inhibited (50 μm) TPH was subsequently exposed to DTT for 30 min at 4°C, enzyme activity was restored in a concentration-dependent manner. Figure2B shows that a concentration of DTT of 10 mm restores TPH activity to 86% of control, and at 20 mm DTT, TPH activity was 96% of control. The effect of DTT was also statistically significant (p < 0.001). These results establish the essential role played by SH groups in TPH catalytic function and demonstrate that TPH can be cycled reversibly through inactive and active states by oxidation (DTNB) and reduction (DTT) of cysteine residues.
Effect of NO on recombinant TPH
The full-length form (GST–TPH–WT) and the deletional mutant (GST–TPH–CC) of TPH were exposed to increasing concentrations of the NO generator SNP at 4°C for 30 min, and the effects of this treatment are presented in Figure 3. The results demonstrate that SNP causes a concentration-dependent decrease in both forms of TPH in the presence of DTT. This effect of SNP was statistically significant for each construct (p< 0.001; ANOVA), and the two enzymes did not differ in their responsiveness to SNP. A concentration of SNP of 200 μmlowered TPH activity to 36% of control. Generation of NO from SNP is known to depend on DTT or other reductants. In confirmation of this, SNP alone (−DTT) does not significantly alter TPH activity over the same concentration range. The effects of SNP on TPH were not reversed by either iron or DTT treatment (see below). Finally, the effect of SNP was prevented if incubations were performed in the presence of 500 μm hemoglobin (data not shown), as described previously (Kuhn and Arthur, 1996, 1997). Because both TPH–WT and TPH–CC respond to SNP in the same manner, all remaining experiments used the TPH–CC form to rule out alterations that might occur in the regulatory domain of TPH.
Effect of NO on apo-TPH
Chelation and removal of TPH-bound iron witho-phenanthroline causes the enzyme to lose activity (Fig.1). We hypothesized that if NO inactivates TPH by reaction with enzyme-bound iron, then removal of the iron should render TPH unresponsive to NO. The results in Figure4 demonstrate that the holoenzyme is slightly stimulated by iron but the inactivation caused by SNP is not reversed by iron. When holo-TPH was converted to the apo-form byo-phenanthroline treatment and then exposed to SNP, the results were not different. Iron was not capable of restoring activity to the SNP-treated apoenzyme. As shown previously (Fig. 1), the activity of apo-TPH not treated with SNP is readily restored to control levels by iron. These results indicate that the reaction of NO with TPH-bound iron does not appear to be the mechanism of inactivation.
Effect of NO on DTNB-oxidized TPH
Using a similar reasoning and approach as in the previous experiment, we inactivated TPH by DTNB-induced oxidation of SH groups before exposure to NO. We hypothesized that if NO inactivates TPH through attack on SH groups critical for catalytic function, DTNB-oxidized TPH should be unresponsive to NO. The results in Figure5 show that DTNB-induced inactivation can be reversed by treatment with DTT. We also noted that in these experiments with multiple preincubation and washing steps, this high concentration of DTT (10 mm) itself caused ∼50% reductions in control TPH activity. Figure 5 also demonstrates that DTNB itself has no additional influence on the SNP effect; however, DTT restored activity to the enzyme after SNP treatment as long as TPH was SH-protected with DTNB before SNP exposure. These results indicate that preoxidation of SH groups with DTNB prevented the irreversible inactivation of TPH by NO and suggest that NO is attacking critical SH groups to cause the loss of activity in the enzyme.
DISCUSSION
TPH is an important neuronal enzyme that has substantial influence over a number of physiological and clinical processes mediated by the neurotransmitter 5-HT. The understanding of TPH, both in vivo and in vitro, has been hampered by the extreme difficulty in obtaining useful amounts of the enzyme for proper biochemical characterization. This problem can be attributed to the low levels of expression of TPH in brain and to the intrinsic instability of the enzyme (Kuhn et al., 1980). We recently cloned and expressed TPH in recombinant form as a GST fusion protein (D’Sa et al., 1996). This approach offers numerous advantages by providing (1) large-scale production of enzyme in a bacterial host; (2) rapid, single-step purification with GSH-affinity chromatography; (3) expression of a form of TPH retaining high levels of activity, with normal regulatory and molecular properties while it is adsorbed to affinity beads; and 4) a reagent for molecular characterization of TPH not heretofore possible.
These properties of GST–TPH have been exploited here to characterize the mechanisms by which TPH is inactivated by NO. Our studies were pursued with the substituted amphetamines in mind. Some members of this class of drugs are used clinically (e.g., fenfluramine), whereas others have gained notoriety as drugs of abuse (e.g., MDMA). Regardless of where along the use–abuse continuum these drugs fall, they share the ability to inactivate TPH and to cause long-term depletion of 5-HT (Gibb et al., 1993; Steele et al., 1994; Seiden and Sabol, 1996). Numerous mechanisms have been proposed to account for this selectivity, and a fuller understanding of it would lead to more effective treatments for human abusers. Unfortunately, a unifying mechanism has not yet congealed from the available data, leaving open the search for a mechanism of action.
Recent advances have focused attention on NO and ROS as mediators of toxicity to neurons and their constitutive proteins. Many amphetamines are now known to produce various ROS and NO in brain (Abekawa et al., 1995; Bowyer et al., 1995; Cerruti et al., 1995; Colado and Green, 1995; Giovanni et al., 1995; Hirata et al., 1995; Di Monte et al., 1996). If the focus is restricted to NO, a priori conclusions about its effects on cells or proteins cannot be made. For example, NO is a neurotransmitter in some neurons (Jaffrey and Snyder, 1996), it can cause toxicity in others (Lipton et al., 1993; Dawson et al., 1994), and it can protect others from toxicity (Wink et al., 1995, 1996). At the protein level, NO activates guanylate cyclase (Ignarro et al., 1986; Wedel et al., 1995), inactivates NO synthase (Abu-Soud et al., 1995), and has no effect on aconitase (Castro et al., 1994; Hausladen and Fridovich, 1994), which are all heme-iron containing proteins. With these caveats in mind, the amphetamine-induced production of NO does not establish a mechanism of toxicity to TPH or 5-HT neurons. We demonstrated recently that brain TPH is inactivated by NO (Kuhn and Arthur, 1996). The finding that the natural cofactor for TPH, tetrahydrobiopterin, significantly potentiates the effect of NO on the enzyme (Kuhn and Arthur, 1997) could indicate a mechanistic role for the cofactor in the toxic specificity associated with the amphetamines, assuming that the amphetamines produce NO. These results also link TPH inactivation to the amphetamines through NO. Further mechanistic studies on the process have been limited to crude TPH preparations from brain extracts. A fruitful line of investigation to follow in the search for the mechanisms of TPH inactivation by MDMA was established by Stone et al. (1989a,b) when these investigators demonstrated that TPH inactivated by MDMA treatment in vivo could be significantly reversed by treatment of the enzyme in vitrowith strong reducing conditions (iron plus DTT under anaerobic conditions). These findings suggested that a process induced by MDMA and that led to inactivation of TPH causes an oxidation of either iron or SH sites in the enzyme. On the basis of the importance of understanding the mechanisms by which amphetamines exert neuronal toxicity, we pursued more mechanistic studies with a highly active and purified recombinant TPH.
The chemistry of NO and the molecular properties of TPH align these two molecules, making certain predictions possible. NO is known to react with iron and SH groups in biological systems, with deleterious effects on both (Stamler et al., 1992; Stamler, 1994; Cohen, 1994). TPH has a strict requirement for iron and reduced SH groups for optimal catalytic function. Removal of prosthetic iron from TPH by treatment with the ferrous iron chelator o-phenanthroline converts the highly active holoenzyme to the apo-form with low activity. The simple addition of iron to the enzyme restores full activity within minutes. We hypothesized that if NO inactivates TPH via attack on enzyme-bound iron, the removal of iron from TPH would render TPH unresponsive to NO; however, apo-TPH treated with NO could not be reactivated by the addition of iron to the enzyme, indicating that NO–iron interactions could not explain the loss of activity associated with NO.
Next, we hypothesized that if NO inactivates TPH through attack on critical SH groups, blocking (through oxidation) reduced cysteines with DTNB would render TPH less responsive to NO. TPH is extremely sensitive to inactivation by DTNB. This SH oxidant caused a concentration-dependent inactivation of the holoenzyme (iron-loaded). DTNB-treated TPH could be restored to full activity by reduction of the enzyme with DTT. Under similar conditions, NO-treated holo-TPH cannot be recovered by DTT; however, if DTNB-treated TPH was exposed to NO, subsequent thiol reduction with DTT restored activity, as it did in the enzyme exposed to DTNB alone. These data were interpreted in view of the assumption that DTNB is reacting selectively with protein SH groups (Van Iwaarden et al., 1992), but alternative explanations exist. It is possible that DTNB is also protecting iron sites within TPH or that the DTNB-sensitive SH groups are indirectly influencing iron sites so that they are less susceptible to attack by NO.
The present results lead to several interesting conclusions about TPH. First, they reaffirm the importance of iron and SH groups (free cysteines) in TPH catalytic function. Second, they establish the feasibility and value of using highly purified, recombinant TPH to probe the molecular mechanisms regulating this enzyme. Finally, they point to SH groups as targets for NO-induced inactivation of the enzyme. Apo- and holo-TPH are equally reactive with NO, minimizing a role for disruption of the TPH iron site as the mechanism of NO-induced inactivation. The mechanism by which NO modifies SH groups in TPH is not clear. Disulfide exchange, like that caused by DTNB, is probably not occurring, because the effect of NO is irreversible. Perhaps NO oxidizes SH groups that are no longer amenable to reduction to a higher state of oxidation. The NO modification of SH groups could also cause an unfolding of the TPH. Given the often critical role of disulfide bonds (via cysteine residues) in the regulation of protein folding (Chang, 1997), this mechanism could apply as well. It is also possible that NO could modify cysteines in TPH through a nitrosylation and ADP-ribosylation pathway. Precedence for this mechanism has been established for brain glyceraldehyde phosphate dehydrogenase (Mohr et al., 1994, 1996), and ADP-ribosylation has been implicated in NO-stimulated long-term potentiation (Duman et al., 1993) and in the central actions of lithium (Nessler et al., 1995). These possibilities are currently under investigation.
The results of Stone et al. (1989a,b) are important because they provided evidence that MDMA-induced inactivation of TPH in vivo resulted from a drug-induced modification of sulfhydryl sites, and they influenced the direction of the current mechanistic studies. These authors demonstrated that MDMA-inactivated TPH could be reactivated in vitro by treatment of brain extracts with iron and DTT under anaerobic conditions. These same conditions reactivate TPH that has been inactivated by strong SH-oxidizing conditions in vitro (Kuhn et al., 1980). Although the present results are in general agreement with those of Stone et al. (1989a,b), one important distinction remains: the MDMA effect on TPH is partially reversible, whereas the NO-induced inactivation of TPH is not. Before a closer parallel can be drawn between the results of Stone et al. (1989a,b) and the present findings, attempts to reactivate TPH (after NO treatment) with iron and DTT under anaerobic conditions should be made. These studies are under way.
In conclusion, the present results substantiate the possibility that drug-induced damage to the 5-HT neuronal system, including inactivation of TPH, could be mediated by NO or other ROS that are capable of oxidizing SH groups in proteins. Drug-induced production of NO–ROS in brain per se does not establish a basis for cellular toxicity associated with the neurotoxic amphetamines; however, the demonstration that a known target for these drugs, TPH, is directly inactivated by a substance produced by these drugs, NO, strengthens the possibility that this mechanism has in vivo relevance. A distinction must be drawn between amphetamine-induced inactivation of TPH and 5-HT neurotoxicity. Although drugs such as MDMA cause both processes to occur, we are not implying that TPH inactivation itself is the direct cause of 5-HT depletion. These two processes may be linked to the same causal event (e.g., NO or ROS), or they could be independent. In either case, the similarities between NO-inactivated TPH (Kuhn and Arthur, 1996, 1997; present results) and MDMA-inactivated TPH (Stone et al., 1989a,b) are compelling. Furthermore, the potential role of NO in mediating apoptosis (Brune et al., 1996; Jacobson, 1996; Simonian and Coyle, 1996) and the recent claim that MDMA induces apoptosis in a human serotonergic cell line (Simantov and Tauber, 1997) draws another interesting parallel between NO and amphetamine-induced alterations in 5-HT neurons. We are presently developing probes for NO-modified TPH in an attempt to identify amphetamine-modified TPH in vivo.
Footnotes
This research was supported in part by National Institute of Environmental Health Sciences Center Grant (Principal Investigator: Raymond Novak) ES 06639, and by the Joe Young Sr Psychiatric Research Fund of the Department of Psychiatry and Behavioral Neurosciences. We thank Drs. Tom Uhde and Raymond Novak for their support and encouragement throughout these studies.
Correspondence should be addressed to Donald M. Kuhn, Department of Psychiatry and Behavioral Neurosciences, Gordon H. Scott Hall, Room 2125, 540 East Canfield, Detroit, MI 48201.
REFERENCES
- 1.Abekawa T, Ohmori T, Koyama T. Effects of nitric oxide synthesis inhibition on methamphetamine-induced dopaminergic and serotonergic neurotoxicity in the rat brain. J Neural Trans. 1995;103:671–680. doi: 10.1007/BF01271227. [DOI] [PubMed] [Google Scholar]
- 2.Abu-Soud HM, Wang J, Rousseau DL, Fukuto JM, Ignarro LJ, Steuhr DJ. Neuronal nitric oxide synthase self-inactivates by forming a ferrous-nitrosyl complex during aerobic catalysis. J Biol Chem. 1995;270:22997–22306. doi: 10.1074/jbc.270.39.22997. [DOI] [PubMed] [Google Scholar]
- 3.Bowyer JF, Clausing P, Gough B, Slikker W, Jr, Holson RR. Nitric oxide regulation of methamphetamine-induced dopamine release in caudate/putamen. Brain Res. 1995;699:62–70. doi: 10.1016/0006-8993(95)00877-s. [DOI] [PubMed] [Google Scholar]
- 4.Bradford MM. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 5.Brune B, Mohr S, Messmer UK. Protein thiol modification and apoptotic cell death as cGMP-independent nitric oxide (NO) signalling pathways. Rev Physiol Biochem Pharmacol. 1996;127:1–30. doi: 10.1007/BFb0048263. [DOI] [PubMed] [Google Scholar]
- 6.Castro L, Rodriguez M, Radi R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J Biol Chem. 1994;269:29409–29415. [PubMed] [Google Scholar]
- 7.Cerruti C, Sheng P, Ladenheim B, Epstein CJ, Cadet JL. Involvement of oxidative and l-arginine-NO pathways in the neurotoxicity of drugs of abuse in vitro. Clin Exp Pharmacol Physiol. 1995;22:381–382. doi: 10.1111/j.1440-1681.1995.tb02025.x. [DOI] [PubMed] [Google Scholar]
- 8.Chang JY. A two-stage mechanism for the reductive unfolding of disulfide-containing proteins. J Biol Chem. 1997;272:69–75. [PubMed] [Google Scholar]
- 9.Cohen G. Enzymatic/nonenzymatic sources of oxyradicals and regulation of antioxidant defenses. Ann NY Acad Sci. 1994;738:8–14. doi: 10.1111/j.1749-6632.1994.tb21784.x. [DOI] [PubMed] [Google Scholar]
- 10.Colado MI, Green AR. The spin trap reagent alpha-phenyl-N-tert-butyl nitrone prevents “ecstasy”-induced neurodegeneration of 5-hydroxytryptamine neurones. Eur J Pharmacol. 1995;280:343–346. doi: 10.1016/0014-2999(95)00298-y. [DOI] [PubMed] [Google Scholar]
- 11.Dawson TM, Zhang J, Dawson VL, Snyder SH. Nitric oxide: cellular regulation and neuronal injury. Prog Brain Res. 1994;103:365–369. doi: 10.1016/s0079-6123(08)61150-4. [DOI] [PubMed] [Google Scholar]
- 12.Di Monte DA, Royland JE, Jakowec MW, Langston JW. Role of nitric oxide in methamphetamine neurotoxicity: protection by 7-nitroindazole, an inhibitor of neuronal nitric oxide synthase. J Neurochem. 1996;67:2443–2450. doi: 10.1046/j.1471-4159.1996.67062443.x. [DOI] [PubMed] [Google Scholar]
- 13.D’Sa CM, Arthur RE, Jr, Kuhn DM. Expression and deletion mutagenesis of tryptophan hydroxylase fusion proteins: delineation of the enzyme catalytic core. J Neurochem. 1996;67:917–926. doi: 10.1046/j.1471-4159.1996.67030917.x. [DOI] [PubMed] [Google Scholar]
- 14.Duman RS, Terwilliger RZ, Nessler EJ. Alterations in nitric oxide-stimulated endogenous ADP-ribosylation associated with long-term potentiation in rat hippocampus. J Neurochem. 1993;61:1542–1545. doi: 10.1111/j.1471-4159.1993.tb13652.x. [DOI] [PubMed] [Google Scholar]
- 15.Feelisch M, Noack EA. Correlation between nitric oxide formation during degradation of organic nitrates and activation of guanylate cyclase. Eur J Pharmacol. 1987;139:19–39. doi: 10.1016/0014-2999(87)90493-6. [DOI] [PubMed] [Google Scholar]
- 16.Gartside SE, Cowen PJ, Sharp T. Evidence that the large neutral amino acid l-valine decreases electrically-evoked release of 5-HT in rat hippocampus in vivo. Psychopharmacology. 1992;109:251–253. doi: 10.1007/BF02245511. [DOI] [PubMed] [Google Scholar]
- 17.Gibb JW, Johnson M, Stone DM, Hanson GR. Mechanisms mediating biogenic amine deficits induced by amphetamine and its congeners. NIDA Res Monogr. 1993;136:226–236. doi: 10.1037/e495922006-012. [DOI] [PubMed] [Google Scholar]
- 18.Giovanni A, Liang LP, Hastings TG, Zigmond MJ. Estimating hydroxyl radical content in rat brain using systemic and intraventricular salicylate: impact of methamphetamine. J Neurochem. 1995;64:1819–1825. doi: 10.1046/j.1471-4159.1995.64041819.x. [DOI] [PubMed] [Google Scholar]
- 19.Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994;269:29405–29408. [PubMed] [Google Scholar]
- 20.Hirata H, Ladenheim B, Rothman RB, Cadet JL. Methamphet-amine-induced serotonin neurotoxicity is mediated by superoxide radicals. Brain Res. 1995;677:345–347. doi: 10.1016/0006-8993(95)00218-f. [DOI] [PubMed] [Google Scholar]
- 21.Ignarro LJ, Adams JB, Horwitz PM, Wood KS. Activation of soluble guanylate cyclase by NO-hemeproteins involves NO-heme exchange. Comparison of heme-containing and heme-deficient enzyme forms. J Biol Chem. 1986;261:4997–5002. [PubMed] [Google Scholar]
- 22.Jacobson MD. Reactive oxygen species and programmed cell death. Trends Biochem Sci. 1996;21:83–86. [PubMed] [Google Scholar]
- 23.Jaffrey SR, Snyder SH. Nitric oxide: a neural messenger. Annu Rev Cell Dev Biol. 1996;11:417–440. doi: 10.1146/annurev.cb.11.110195.002221. [DOI] [PubMed] [Google Scholar]
- 24.Jequier E, Lovenberg W, Sjoerdsma A. Tryptophan hydroxylase inhibition: the mechanism by which p-chlorophenylalanine depletes brain serotonin. Mol Pharmacol. 1967;3:274–278. [PubMed] [Google Scholar]
- 25.Kuhn DM, Arthur RE., Jr Inactivation of brain tryptophan hydroxylase by nitric oxide. J Neurochem. 1996;67:1072–1077. doi: 10.1046/j.1471-4159.1996.67031072.x. [DOI] [PubMed] [Google Scholar]
- 26.Kuhn DM, Arthur RE., Jr Inactivation of tryptophan hydroxylase by nitric oxide: enhancement by tetrahydrobiopterin. J Neurochem. 1997;68:1495–1502. doi: 10.1046/j.1471-4159.1997.68041495.x. [DOI] [PubMed] [Google Scholar]
- 27.Kuhn DM, Ruskin B, Lovenberg W. Tryptophan hydroxylase: the role of oxygen, iron, and sulfhydryl groups as determinants of stability and catalytic activity. J Biol Chem. 1980;255:4137–4143. [PubMed] [Google Scholar]
- 28.Kuhn DM, Arthur RE, Jr, States JC. Phosphorylation and activation of brain tryptophan hydroxylase: identification of serine-58 as a substrate site for protein kinase A. J Neurochem. 1997;68:2220–2223. doi: 10.1046/j.1471-4159.1997.68052220.x. [DOI] [PubMed] [Google Scholar]
- 29.Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HSV, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;264:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 30.Mohr S, Stamler JS, Brüne B. Mechanism of covalent modification of glyceraldehyde-3-phosphate dehydrogenase at its active site thiol by nitric oxide, peroxynitrite and related nitrosylating agents. FEBS Lett. 1994;348:223–227. doi: 10.1016/0014-5793(94)00596-6. [DOI] [PubMed] [Google Scholar]
- 31.Mohr S, Stamler JS, Brune B. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J Biol Chem. 1996;271:4209–4214. doi: 10.1074/jbc.271.8.4209. [DOI] [PubMed] [Google Scholar]
- 32.Nessler EJ, Terwilliger RZ, Duman RS. Regulation of endogenous ADP-ribosylation by acute and chronic lithium in rat brain. J Neurochem. 1995;64:2319–2324. doi: 10.1046/j.1471-4159.1995.64052319.x. [DOI] [PubMed] [Google Scholar]
- 33.Oluyomi AO, Gibson EL, Barnfield AMC, Curzon G. d-fenfluramine and d-norfenfluramine hypophagias do not require increased hypothalamic 5-hydroxytryptamine release. Eur J Pharmacol. 1994;264:111–115. doi: 10.1016/0014-2999(94)90646-7. [DOI] [PubMed] [Google Scholar]
- 34.Schatzberg AF, Nemeroff CB. Textbook of psychopharmacology. The American Psychiatric Press; Washington, DC: 1995. [Google Scholar]
- 35.Seiden LS, Sabol KE. Methamphetamine and methylenedioxy-methamphetamine neurotoxicity: possible mechanisms of cell destruction. NIDA Res Monogr. 1996;163:251–276. [PubMed] [Google Scholar]
- 36.Shibuki K. An electrochemical microprobe for detecting nitric oxide release in brain tissue. Neurosci Res. 1990;9:69–76. doi: 10.1016/0168-0102(90)90048-j. [DOI] [PubMed] [Google Scholar]
- 37.Shiman R, Grey DW, Hill MA. Regulation of rat liver phenylalanine hydroxylase. I. Kinetic properties of the enzymes reduction site. J Biol Chem. 1994;269:24637–24646. [PubMed] [Google Scholar]
- 38.Simantov R, Tauber M. The abused drug MDMA (ecstasy) induces programmed death of serotonergic cells. FASEB J. 1997;11:141–146. doi: 10.1096/fasebj.11.2.9039956. [DOI] [PubMed] [Google Scholar]
- 39.Simonium NA, Coyle JT. Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 1996;36:83–106. doi: 10.1146/annurev.pa.36.040196.000503. [DOI] [PubMed] [Google Scholar]
- 40.Southam E, Garthwaite J. Comparative effects of some nitric oxide donors on cyclic GMP levels in rat cerebellar slices. Neurosci Lett. 1991;130:107–111. doi: 10.1016/0304-3940(91)90239-p. [DOI] [PubMed] [Google Scholar]
- 41.Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 42.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 43.Steele TD, McCann UD, Ricuarte GA. 3,4-methylenedioxy-methamphetamine (MDMA, “ecstasy”): pharmacology and toxicology in animals and humans. Addiction. 1994;89:539–551. doi: 10.1111/j.1360-0443.1994.tb03330.x. [DOI] [PubMed] [Google Scholar]
- 44.Stone DM, Hanson GR, Gibb JW. In vitro reactivation of rat cortical tryptophan hydroxylase following in vivo inactivation by methylenedioxymethamphetamine. J Neurochem. 1989a;53:572–581. doi: 10.1111/j.1471-4159.1989.tb07372.x. [DOI] [PubMed] [Google Scholar]
- 45.Stone DM, Johnson M, Hanson GR, Gibb JW. Acute inactivation of tryptophan hydroxylase by amphetamine analogs involves the oxidation of sulfhydryl sites. Eur J Pharmacol. 1989b;172:93–97. doi: 10.1016/0922-4106(89)90048-5. [DOI] [PubMed] [Google Scholar]
- 46.Van Iwaarden PR, Driessen AJM, Konings WN. What we can learn from the effects of thiol reagents on transport proteins. Biochim Biophys Acta. 1992;1113:161–170. doi: 10.1016/0304-4157(92)90037-b. [DOI] [PubMed] [Google Scholar]
- 47.Wedel B, Harteneck C, Foerster J, Friebe A, Schultz G, Koesling D. Functional domains of soluble guanylate cyclase. J Biol Chem. 1995;270:24871–24875. doi: 10.1074/jbc.270.42.24871. [DOI] [PubMed] [Google Scholar]
- 48.Wink DA, Cook JA, Krishna MC, Hanbauer I, DeGraff W, Gamson J, Mitchell JB. Nitric oxide protects against alkyl peroxide-mediated cytotoxicity: further insights into the role of nitric oxide plays in oxidative stress. Arch Biochem Biophys. 1995;319:402–407. doi: 10.1006/abbi.1995.1310. [DOI] [PubMed] [Google Scholar]
- 49.Wink DA, Hanbauer I, Grisham MB, Laval F, Nims RW, Laval J, Cook J, Pacelli R, Liebmann J, Krishna M, Ford PC, Mitchell JB. Chemical biology of nitric oxide: regulation and protective and toxic mechanisms. Curr Top Cell Reg. 1996;34:159–187. doi: 10.1016/s0070-2137(96)80006-9. [DOI] [PubMed] [Google Scholar]