

Abstract
To investigate the molecular mechanisms of cholinergic sprouting in the hippocampus after removal of entorhinal cortical inputs, we evaluated trophic factor gene expression in the denervated hippocampus. Despite the proposed role for nerve growth factor (NGF) in this sprouting, we observed no change in NGF mRNA or protein at several postlesion time points. In contrast, FGF-2 mRNA was increased within 16 hr. FGF-2 immunoreactivity was localized within GFAP-positive hypertrophic astrocytes distributed specifically within the denervated outer molecular layer after the lesion. To address the functional significance of this increase in FGF-2, we assessed the magnitude of cholinergic sprouting in animals receiving chronic intracerebroventricular infusions of neutralizing antibodies specific for FGF-2 and compared it with that observed in lesioned animals receiving infusate controls. Animals given FGF-2 antibodies displayed a marked reduction in cholinergic sprouting as compared with controls. In fact, many of these animals exhibited virtually no sprouting at all despite histological verification of complete lesions. These results suggest that endogenous FGF-2 promotes cholinergic axonal sprouting in the injured adult brain. Furthermore, immunocytochemical localization of receptors for FGF-2 (i.e., FGFR1) on projecting basal forebrain cholinergic neurons suggests that FGF-2 acts directly on these neurons to induce the lesion-induced sprouting response.
Keywords: entorhinal cortex lesion, FGF-2, hippocampus, NGF, neurotrophin, sprouting
Numerous research efforts have sought to elucidate the function of trophic factors in neural development, maintenance of connections in the mature brain, and reparative processes after injury. Neural tissue contains a variety of trophic molecules and their functional receptors, the expression patterns of which differ during development (Maisonpierre et al., 1990; Ernfors and Persson, 1991;Friedman et al., 1991; Schnurch and Risau, 1991; Stockli et al., 1991) and in response to injury (Ernfors et al., 1991; Woodroofe et al., 1991; Lindholm et al., 1992; Lindvall et al., 1992; Rocamora et al., 1992; Takeda et al., 1993). It is, therefore, likely that multiple growth factors act coordinately to elicit the structural and functional changes that take place during dynamic growth processes.
The majority of studies has investigated the function of nerve growth factor (NGF), the best-characterized member of the neurotrophin family of trophic molecules (Korsching, 1993). Although much evidence supports the role of endogenous NGF in promoting neuronal survival in vivo (Barde, 1989), less is known about its ability to influence axonal growth. NGF does not seem to be involved in the initiation or guidance of axons to their targets during development (Davies et al., 1987; Buchman and Davies, 1993); however, it may be involved in directing certain types of axonal growth in the adult. Uninjured sympathetic (Isaacson et al., 1992) and basal forebrain cholinergic neurons (Kawaja and Gage, 1991; Heisenberg et al., 1994) seem to sprout in response to local delivery of exogenous NGF, demonstrating that axon branching can be directed by NGF in vivo. Interestingly, when branching of uninjured septal cholinergic fibers is induced by NGF, the branching may be facilitated by astrocytes (Kawaja and Gage, 1991; Heisenberg et al., 1994). Antibody delivery studies have shown that collateral sprouting of subpopulations of undamaged neurons is neurotrophin-dependent (Diamond et al., 1987, 1992b; Gloster and Diamond, 1992; Van der Zee et al., 1992). Two studies in the CNS suggest that NGF also can promote the regeneration of axotomized septal cholinergic fibers via nerve grafts (Hagg et al., 1990) or fibroblast grafts (Kawaja et al., 1992) into the hippocampus. The role of NGF in the damaged peripheral nervous system (PNS) may differ from its actions on basal forebrain cholinergic neurons. In the PNS, studies suggest that regeneration of damaged neurons is neurotrophin-independent (Rich et al., 1984; Diamond et al., 1987, 1992a; Gloster and Diamond, 1992).
Our interest in mechanisms of neural injury and repair prompted us to investigate the role of trophic factor interactions in a well characterized model of collateral sprouting in the adult rat hippocampus. Removal of efferent projections from the entorhinal cortex to the hippocampus results in the reactive sprouting of intact fiber systems within the dentate gyrus, including those from basal forebrain cholinergic neurons (Lynch et al., 1972; Storm-Mathisen, 1974;Stanfield and Cowan, 1982), the commissural/associational projection (Lynch et al., 1974; Amaral et al., 1980), and the crossed temporodentate pathway (Steward et al., 1974; Steward, 1976; Deller et al., 1996). We, and others, have proposed that this synaptic reorganization is mediated by the action(s) of neurotrophic molecule(s) (Gage et al., 1988; Fagan and Gage, 1990; Van der Zee et al., 1992;Conner et al., 1994; Guthrie et al., 1995). Reports of changes in the level and/or distribution of a variety of trophic molecules (or their bioactivity) in the denervated hippocampus has been inconsistent (Crutcher and Collins, 1986; Whittemore et al., 1987; Lapchak et al., 1993; Conner et al., 1994; Gwag et al., 1994), likely caused by the various time points analyzed and the different methods used to quantify changes. Van der Zee and colleagues (1992) provided the first demonstration of a functional relevance of a trophic factor in this model by showing that daily intracerebroventricular injections of antibodies to NGF resulted in attenuation of cholinergic sprouting. The particular neurotrophin blocked by the antibody could not be confirmed, however, because their antibody recognized brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) in addition to NGF. To investigate definitively the potential role of different trophic factors in this sprouting model, we assessed the expression of several trophic molecules in the hippocampus after denervation. We found that after hippocampal denervation there was an increase, not in neurotrophin expression but rather in fibroblast growth factor-2 (FGF-2) gene expression, and that infusions of an anti-FGF-2 antibodyin vivo attenuates the cholinergic sprouting that takes place in this model system.
MATERIALS AND METHODS
Reverse transcription PCR (RT-PCR)
Surgery. Adult female Sprague Dawley rats (n = 24) were anesthetized with a mixture of ketamine (75 mg/kg), xylazine (4 mg/kg), and acepromazine (5.6 mg/kg) and placed in a Kopf stereotaxic apparatus. Each rat received a unilateral aspirative lesion of the retrosplenial cortex, which resulted in transection of the perforant path (PP) as described previously (Gage et al., 1988; Fagan and Gage, 1990, 1994). Animals (n = 4 at each time point) were decapitated rapidly 4, 8, or 16 hr after surgery. For each animal, tissue of the dorsal hippocampus ipsilateral to the lesion cavity was microdissected rapidly and quick-frozen on dry ice. Care was taken to avoid tissue immediately adjacent to the lesion cavity. As control tissue, four hippocampi were dissected from additional animals that had been anesthetized as described, but who received no lesion (0 hr).
RT-PCR. Total RNA was extracted by guanidinium thiocyanate (GTC)-cesium chloride gradient as described (Sambrook et al., 1989). Resulting RNA was quantified by optical density and stored in water at −70°C. Messenger RNA (100 ng) was reverse-transcribed in 1× PCR buffer (10 mm Tris-Cl, pH 8.3, and 50 mm KCl), 2.5 mm MgCl2, 1 mm dNTP (Boehringer Mannheim, Indianapolis, IN), 100 pm random hexamers (Boehringer Mannheim), 20 U RNAsin (Promega, Madison, WI), and 12.5 U AMV reverse transcriptase (Promega) in a 20 μl final volume for 75 min at 42°C, followed by 10 min at 95°C. PCR amplification was performed by adding premixed 1× PCR buffer in water, additional MgCl2 (1.75 mm final concentration), 2.5 UTaq polymerase (Perkin-Elmer Cetus, Oak Brook, IL), 0.5 μg of each primer, and 2 μCi of 32[P]dCTP per reaction. Eighty microliters of this mixture were added to each reaction for a total of 100 μl. Samples were overlaid with mineral oil and amplified in a PEC thermocycler 480 as follows: 2 min at 95°C, 2 min at 60°C, and 2 min at 72°C, for 20 cycles. Primer sequences and PCR product lengths are described in Table 1. Amplified samples were analyzed by 6.5% polyacrylamide gel electrophoresis, exposed to PhosphorImager screens (Molecular Dynamics, Sunnyvale, CA), and analyzed with ImageQuant software. Final values for each mRNA species were normalized to values of RPL27A internal control bands within each sample. The RPL27A transcript is a ubiquitous RNA encoding the rat large subunit ribosomal protein 27 (Wool et al., 1990). Normalized values for each mRNA species were computed for each lesioned animal and compared with those from unoperated controls. Data are reported as the mean percentage of control, thus providing an index of the magnitude of the lesion-induced change in the abundance of the particular mRNA. In addition, the following control experiments were performed: (1) determination of the linear amplification range of rat brain mRNA species examined, (2) determination of the specificity of amplification for RNA by exclusion of reverse transcriptase or RNA from samples for amplification, and (3) independent determination of product size and specificity of all primers in separate and mixed primer reactions.
Table 1.
Sequences of primers used in RT-PCR analysis
| mRNA species | Pd lg | Primer sequence | Intron |
|---|---|---|---|
| Trophic factors: | |||
| BDNF | 106 | 5′GGAGCTGAGCGTGTGTGACA | No |
| 5′CGGGACTTTCTCCAGGACT | |||
| FGF-2 | 280 | 5′GCGTGGACGGCGTCCGGGAG | Yes |
| 5′GGCCCCGTTTTGGATCCGAG | |||
| NGF–prelim | 115 | 5′GCAGTGCCCCTGCTGAACCA | No |
| 5′AAACAGCACGCGGGGTGAAC | |||
| NGF–final | 148 | 5′GACCACAGCCACGGACATC | No |
| 5′CTTGGAGTCAATGCCCCGGC | |||
| NT-3 | 91 | 5′TGCCATGGTTACTTCTGCCACG | No |
| 5′GGATGCCACGGAGATAAGCAAG | |||
| Receptors: | |||
| CNTFR | 383 | 5′CGCCTTGGGTCACAACAC | Yes |
| 5′GCTTCAGTGGTGAGATGC | |||
| FGFR1 | 110 | 5′CATTATCTACTGCACCGGGG | Yes |
| 5′CAGCCATCTGGCTATGGAAG | |||
| GLUR5 and R1a | 400 | 5′GATAAGATCTGTGTGCAGTG | Yes |
| 5′AAGATAGCAGTAAATCTGCT | |||
| 5′CAGGTAGCAGGATGTGGTAG | |||
| TRK A | 368 | 5′CACTGGGTGGCAGTTCTCTT | Yes |
| 5′CATGTACTCGAAGACCATGA | |||
| TRK B | 597 | 5′GTTGCTGACCAAACCAATCG | Yes |
| 5′CATGTACTCAAAGACCATGA | |||
| TRK C | 190 | 5′CTACAATCTAAGCCCCACCA | Yes |
| 5′CATGTACTCAAAGACCATGA | |||
| Structural/other: | |||
| GAD | 260/346 | 5′AAGGTTTTGACTTCCACCAC | Yes |
| 5′CATAAGAACAAAGACGGGTGC | |||
| GFAP | 140 | 5′ACCTCGGCACCCTGAAGAAG | Yes |
| 5′CCAGCGACTCAACCTTCCTC | |||
| Nf 60 kDa | 154 | 5′GCCGCCTACTCAAGGCTAAG | Yes |
| 5′GCTTCGCAGCTCATTCTCCAG | |||
| Nf 200 kDa | 300 | 5′GAGGAGATAACTGAGTACCG | Yes |
| 5′CCAAAGCCAATCCGACACTC | |||
| RPL27A | 235 | 5′ATCGGTAAGCACCGCAAGCA | No |
| 5′GGGAGCAACTCCATTCTTGT |
The far left column denotes the mRNA species examined. The left center column is the predicted RT-PCR product length (Pd lg) in nucleotide pairs. The right center column shows the sequence of the oligonucleotide primer pairs in 5′-3′ orientation: the upper primer is sense, and the lower primer is the antisense partner. The far right column indicates whether the primer set is predicted to span intron sequences in the rat genome, thereby precluding amplification of a PCR product from any contaminating genomic DNA. NGF mRNA was examined with two different primer sets (prelim and final), resulting in two different-sized PCR products in different experiments. The GAD primer set recognizes both the adult and embryonic splice variants of the GAD mRNA. All primer pairs were proven functional for amplification of their specific RNA species in independent experiments in our laboratory (data not shown).
Targeted differential display. We used the highly sensitive RT-PCR technique to quantify mRNA levels, using what could be described as a “targeted differential display” approach by which multiple PCR primers, each specific for individual mRNA species, are amplified within a given RNA sample. Because of the large number of primer sets included in a single reaction and the format for examining the products, the end result shares many characteristics of differential display PCR. Each of the primer sets was tested under a variety of experimental conditions and proved to be both sensitive and specific in the amplification of their target mRNAs. Using this multiprimer approach, we were able to obtain information about many molecules from a single RT-PCR reaction. Although this approach did not permit valid conclusions to be drawn regarding differences between the relative levels of the different mRNAs within a sample, it did allow for comparisons of the relative abundance of given mRNAs between lesioned and control tissue samples. RT-PCR was performed on individual RNA samples (n = 4 each) from lesioned and unoperated hippocampal tissue, and reaction products were loaded side by side on a large sequencing-sized polyacrylamide gel (6.5%). The use of a large format gel allowed for maximum separation and definition of our multiple PCR products. In this initial survey, the four adjacent bands from each experimental condition could be quantitated rapidly as a single composite band. To provide an estimation of the magnitude of the difference between mRNA levels in the control and lesioned samples, we quantified these composite bands with the PhosphorImager (Molecular Dynamics). Raw values then were normalized to the RPL27A internal control band within each composite sample, and a ratio was computed corresponding to the percentage of difference in mRNA abundance in the denervated hippocampi as compared with unoperated controls.
Enzyme-linked immunosorbent assay (ELISA) for NGF
Adult female Sprague Dawley rats (n = 5) were decapitated at 7 d after unilateral PP transection, and tissue of the dorsal hippocampus rostral to the lesion cavity was microdissected rapidly and quick-frozen on dry ice. Care was taken to avoid tissue immediately adjacent to the lesion cavity. Hippocampal tissue from an additional set of animals (n = 4) killed 7 d after unilateral fimbria–fornix (FFX) transection was included in the analysis for comparative purposes. Tissue of the dorsal hippocampi of unoperated rats (n = 2) served as normal controls.
Tissue was homogenized by sonication in 0.5 ml of extraction buffer containing EDTA (4 mm) and the protease inhibitors phenylmethylsulfonyl fluoride (PMSF, 1 mm; Sigma, St. Louis, MO) and aprotinin (7 mg/ml; Sigma). Homogenates were ultracentrifuged (100,000 × g) for 10 min at 4°C, and NGF protein levels were measured by two-site ELISA (Boehringer Mannheim) as described previously (Yoshida and Gage, 1991). Purified mouse NGF served as the standard. This assay is specific for NGF, with a detection limit of 5–10 pg NGF/ml, and does not detect BDNF or NT-3 (data not shown). Statistical evaluation was made with a one-way ANOVA with post hoc Fisher comparisons to assess individual group differences. Statistical significance was defined at thep < 0.05 level.
Immunocytochemistry (ICC)
FGF-2 and GFAP. Coronal tissue sections (40 μm) through the level of the dorsal hippocampus from lesioned animals killed at D7 (n = 6) were processed for combined ICC for FGF-2 (rabbit anti-FGF-2, 1:500; gift from Dr. A. Baird, Prism, La Jolla, CA) and glial fibrillary acidic protein (GFAP) (mouse anti-GFAP, 1:1000; Amersham, Arlington Heights, IL) to identify astrocytes. General procedures for double immunolabeling were as described previously (Batchelor et al., 1989).
FGFR1 and ChAT. Horizontal tissue sections (50 μm) through the level of the medial septum were processed for combined fluorescent ICC for the FGF-2 receptor (FGFR1) (Lee et al., 1989; Dionne et al., 1990) (mouse anti-FGFR1, 1:25; gift from Dr. Baird) and choline acetyltransferase (ChAT) (goat anti-ChAT, 1:250, Chemicon, Temecula, CA) as described previously (Peterson et al., 1996). Briefly, sections were incubated for 72 hr in a mixture of the primary antibodies, followed by a 1 hr incubation in Cy5-conjugated donkey anti-goat antibodies to visualize ChAT (1:167, Jackson ImmunoResearch, West Grove, PA) and biotinylated donkey anti-mouse antibodies (1:83) to localize FGFR1. Then FGFR1 immunoreactivity was amplified and visualized after a 2 hr incubation in streptavidin conjugated to FITC (1:250, Jackson Immunoresearch). Finally, stained sections were reacted with propidium iodide (0.1% v/v) for 15 min to label all cells. Fluorescent sections were imaged with a Bio-Rad MRC1024 UV confocal microscope (Richmond, CA), and images were processed as described previously (Peterson et al., 1996).
Chronic intracerebroventricular antibody infusions into PP-lesioned animals
Surgery. Osmotic mini-pumps (Alzet) and attached cannulae (Small Parts, Miami Lakes, FL) were filled with (1) rabbit anti-FGF-2 antibody (1.0 mg/ml; n = 7), (2) normal rabbit serum (RS; 1.0 mg/ml; n = 4), or (3) artificial cerebrospinal fluid (CSF) [(in mm) Na 150, K 3.0, Ca 1.4, Mg 0.8, P 1.0, and Cl 155 (n = 3)]. All antibodies were diluted in artificial CSF. The anti-FGF-2 antibody has been shown to be neutralizing and highly specific for FGF-2 (Baird and Ling, 1987; A. Baird, unpublished observations). Adult female Sprague Dawley rats (n = 14) received a unilateral PP transection, followed immediately by intracerebroventricular (ICV) cannula implantation ipsilateral to the lesion and mini-pump implantation as described previously (Williams et al., 1987). Solutions were infused continuously at a rate of 0.5 μl/hr for 2 weeks (D14), at which time the animals were killed.
Histology. Rats were perfused transcardially with 0.9% saline, followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Brains were post-fixed overnight and cryoprotected in 30% sucrose. Coronal sections were cut at 40 μm on a freezing sliding microtome and stored in cryoprotectant until processing. Adjacent sections through the level of the dorsal hippocampus were processed with the following: (1) thionine to assess lesion and cannula placement; (2) Gallyas silver stain for degenerating axon terminals (Gallyas et al., 1980) to verify lesion completeness; (3) peroxidase ICC, using biotinylated goat anti-rabbit antibodies to assess tissue distribution of anti-FGF-2 and RS to establish proper intraventricular infusate delivery; and (4) acetylcholinesterase (AChE) histochemistry (Hedreen et al., 1985) to assess the density of cholinergic fibers in the outer molecular layer (ML) of the dentate gyrus (DG).
Analysis and quantification. Only those rats exhibiting accurate ICV cannula placement, infusate penetration, and complete and discrete PP lesions were chosen for quantitative analysis (n = 12). A complete and discrete lesion was defined by complete interruption of the perforant path (via ablation of the retrosplenial cortex as assessed by thionine staining), resulting in the presence of degenerating terminals restricted to the outer ML of the DG ipsilateral to the lesion (as assessed by Gallyas staining). Analyses of AChE fiber density were performed on four representative sections (40× objective) through the dorsal hippocampus for each animal in a blinded manner. Both the denervated and contralateral hippocampi were evaluated. The relative density of AChE-positive fibers in the outer ML of the DG (dorsal blade) was assessed with the National Institutes of Health Image analysis system (version 1.57) linked to a Nikon Microphot-FXL microscope as previously described (Holtzman and Lowenstein, 1995). In each of the four tissue sections analyzed per animal, six random fields (130 μm × 91 μm) within the outer ML were sampled (3 in the denervated hippocampus and 3 on the contralateral side), and the mean number of pixels covered by AChE-positive fibers was determined. Sections were analyzed on-line by a macro sequence in the Image program to ensure that a constant density threshold value was applied to each section. Ratios of lesion/contralateral values (×100) were computed to determine the mean percentage of increase in fiber density in the outer ML on the lesioned side, thus providing an index of the magnitude of the lesion-induced cholinergic sprouting response in each animal.
The mean width of the ML in each DG was calculated also to assess gross lesion-induced tissue shrinkage that artifactually could contribute to increases in fiber density values on the lesioned side. The width of the ML was defined as the absolute distance from the hippocampal commissure to the dentate granule cell layer in the dorsal blade of the DG. Values were computed from the sampled regions described above (20× objective). This distance was partitioned further into inner and outer ML regions, defined by the boundary between the AChE fiber-rich outer ML and the AChE fiber-poor inner ML. Expansion of the inner ML on the lesioned side can be considered a gross estimation of the magnitude of the sprouting of the commissural/associational projection that also takes place in this model system (Lynch et al., 1974). Statistical evaluations were made with Student’s t tests to assess group differences (CSF vs serum control; controls vs antibody treatments). Statistical significance was defined at thep < 0.05 level.
RESULTS
RT-PCR analysis of mRNA levels in the control hippocampus and 16 hr after PP transection
In our attempt to elucidate the molecular mechanisms underlying the cholinergic sprouting that takes place in the hippocampus after PP transection, we sought to identify molecules, the mRNA levels of which changed in the denervated hippocampus. To do this, we used a highly sensitive reverse transcription PCR (RT-PCR) technique to quantify mRNA levels, using what could be described as a “targeted differential display” approach by which the PCR primers used are specific for individual mRNA species. Because of the large number of primer sets included in a single reaction and the format for examining the products, the end result shares many characteristics of differential display PCR. We specifically chose to investigate a 16 hr postlesion time point, a time before the initiation of synaptic rearrangements (beginning ∼2–4 d after the lesion) but subsequent to possible activity-dependent changes in mRNAs induced by the trauma itself (within the initial hour after the lesion). We reasoned that by investigating this intermediate time point any molecular changes observed, by their very timing, would make them good candidates for being involved in the initiation of the subsequent structural changes. We investigated a number of trophic molecules (and their receptors) known to be present in the normal hippocampus and hypothesized to be involved in neuronal plasticity. For comparative purposes, we also analyzed two neuronal structural proteins, neurofilament (60 and 200 kDa) and the astrocyte-specific glial fibrillary acidic protein (GFAP), as well as a few molecules hypothesized not to change as a result of the lesion.
This targeted differential display approach was used to quantify multiple RT-PCR products from single hippocampal RNA samples simultaneously (Fig. 1). In general, the intensity of a given PCR band among the four animals in each group (lesioned or control) was relatively invariant. Because of this, the four adjacent bands could be quantitated rapidly as a single composite band in this initial survey. With this preliminary analysis, we observed no significant change in mRNA levels for ciliary neurotrophic factor receptor (CNTFR), glutamate receptors 1 and 5 (GluR1/5), neurofilament 60 and 200 kDa (NF60 and NF200), or the signaling receptors for NGF and NT-3 (trkA and trkC, respectively) 16 hr after PP transection. In contrast, GFAP mRNA was observed to increase, as reported previously (Steward et al., 1990; Poirier et al., 1991), and NT-3 transcript levels decreased. PCR products for the low-affinity neurotrophin receptor (p75NGFR), glial derived neurotrophic factor (GDNF), ciliary neurotrophic factor (CNTF), tyrosine hydroxylase (TH), choline acetyltransferase (ChAT), and interleukin 1α and β (IL-1α and IL-1β) were below the level of detection in hippocampal tissue under the present conditions. These products, however, were detected easily in tissue from other regions, demonstrating the efficacy of each of the primer sets (data not shown).
Fig. 1.
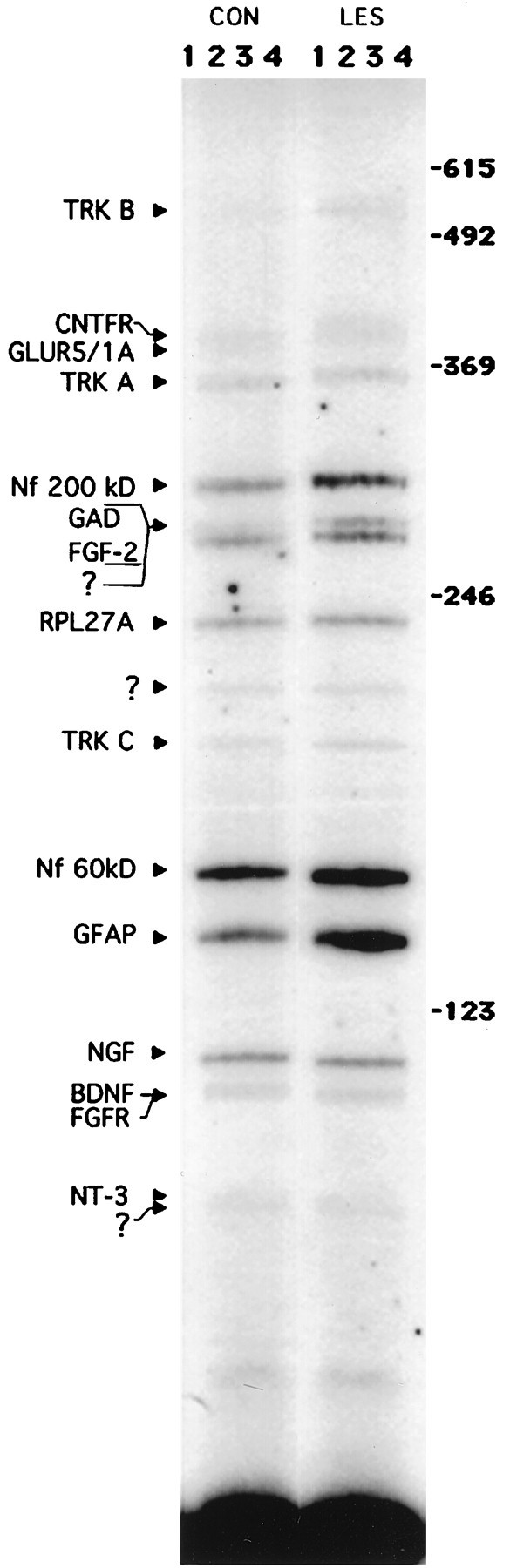
Autoradiogram of RT-PCR products from control and lesioned (16 hr) rat hippocampus. At the top,CON and LES indicate control and lesioned RNA samples, respectively. The numbers at thetop (1–4) indicate the position of the lane for each of the four samples in both groups.Numbers down the right side indicate the position of 123 base pair markers on the autoradiogram.Names and arrows on theleft denote the PCR products predicted to correspond to the individual bands. Question marks indicate PCR products not expected from the original pool of primers used, presumably resulting from chance nonspecific priming of unrelated mRNAs present in the tissue extracts. Because several of the resulting PCR products are very similar in size and overlap on the gel, more than one individual set of primers may contribute to band intensity (e.g., GAD and FGF-2 products are close enough to cause uncertainty in conclusively predicting the identity of clustered bands). In these cases, follow-up analysis (see Fig. 2 and Results) was necessary to confirm the identity and intensity of the PCR products.
We were unable definitively to quantify some of the PCR products with this multiprimer approach, either because of close spacing of two bands (e.g., BDNF/FGFR1; FGF-2/GAD), an aberrant problem with a negative control condition (e.g., NGF), or the large size of the PCR product itself (e.g., trkB). Therefore, any primers that gave PCR product profiles in the denervated hippocampus beyond one SD from control values or that were ambiguous (as described above) were run again in separate reactions, using only two sets of primers per sample. Then these reactions were run on separate gels (Fig. 2), and PCR product bands were quantified individually. This strategy allowed us to determine unambiguously the relative mRNA levels for these molecules in lesioned and control tissue.
Fig. 2.

Autoradiograms of RT-PCR products from a second round of analysis of control and lesioned (16 hr) hippocampal RNA. Primer sets for species of interest or those giving products of uncertain origin from the first examination of control and lesioned hippocampal RNAs were combined in a pair-wise manner and reexamined in individual gels. CON and LES indicate mRNAs from control and lesioned animals, respectively. Mindicates the 123 base pair marker lane, and 1–4indicate the four different mRNA samples in each group.Numbers on the left side show base pair markers. Names and arrows at theright indicate the name and location of the various PCR products. All reactions were run with the RPL27A (L27) internal control primer set. Top left, BDNF and NT-3 analysis; top right, FGF-2 and FGFR1 analysis; middle left,trkB and GAD analysis; middle right, GFAP analysis; bottom center, NGF and additional FGF-2 analysis.
Values for the 16 hr lesioned hippocampus are presented in Table2 as the mean percentage of unoperated control values (after normalizing to the RPL27A internal control bands within each sample). Values for each mRNA species obtained from control tissue were set at 100%. For some mRNA species we observed clear increases or decreases, whereas other mRNA species did not change at this time point. In particular, we detected significant lesion-induced decreases in BDNF and NT-3 mRNA expression, increases in GFAP and FGF-2 mRNA, consistent with previous observations of later time points (Steward et al., 1990; Poirier et al., 1991; Guthrie et al., 1995), and no change in fibroblast growth factor receptor-1 (FGFR1), glutamic acid decarboxylase (GAD; neither embryonic nor adult forms), or the BDNF signaling receptor, trkB. We also observed no change in NGF mRNA levels at this 16 hr time point. For all PCR products, each control condition yielded the appropriate results (data not shown).
Table 2.
Quantification of RT-PCR products in hippocampal tissue obtained from animals killed 0 and 16 hr after unilateral PP transection
| mRNA | 0 hr (%) | 16 hr (%) |
|---|---|---|
| BDNF | 100 ± 12 | *39 ± 1.6 |
| FGF-2 | 100 ± 6.4 | *175 ± 20 |
| FGFR 1 | 100 ± 14 | 74 ± 5.7 |
| GAD | 100 ± 2.5 | 92 ± 7.3 |
| GFAP | 100 ± 7.5 | *243 ± 65 |
| NGF | 100 ± 7.0 | 108 ± 1.0 |
| NT-3 | 100 ± 39 | *35 ± 0.09 |
| TRK B | 100 ± 24 | 107 ± 16 |
Values for each mRNA species obtained from control tissue (0 hr) (n = 4) were set at 100%. Values for the 16 hr lesioned hippocampi (n = 4) are presented as the mean percentage (±SEM) of unoperated control values (after normalizing to the RPL27A internal control bands within each sample). Asterisk indicates difference from control values, p < 0.05.
Changes in NGF transcription and translation after PP transection
The proposed role of NGF in eliciting the cholinergic sprouting that takes place in this model system has prompted many groups to investigate changes in levels of NGF mRNA or protein within the denervated hippocampus. The conclusions drawn from these studies are mixed. Northern blot analysis of hippocampal tissue taken from neonates 10 d after entorhinal cortex (EC) lesion demonstrated no lesion-induced increase in NGF mRNA (Whittemore et al., 1987). In contrast, another group using in situ hybridization analysis demonstrated increased NGF hybridization in the granule cell layer 4 hr after PP transection, but not at 1, 24, or 60 hr postlesion (Gwag et al., 1994). Such a transitory increase, however, may reflect activity-dependent changes associated with the lesion itself (Springer et al., 1994). To address the possibility that we missed an earlier change in NGF transcription, we performed an early time course study. Additional animals were given unilateral PP transections and were killed 4, 8, or 16 hr later (n = 4 each). RT-PCR for NGF was performed on hippocampal RNA obtained from animals at each time point, quantified as described above, and compared with the profiles of GFAP, a molecule that previously showed a significant increase in lesion-induced transcription at 16 hr. Whereas an increase in GFAP mRNA was observed as early as 8 hr after PP transection, reaching statistical significance by 16 hr (p < 0.05), at no time did we observe an increase in NGF mRNA when compared with unoperated control levels (Fig. 3), even at the early 4 hr time point previously reported by Gwag and colleagues (1994). This discrepancy may reflect differences in the lesions used to denervate the hippocampus or the different methods used to quantify mRNA levels. NGF mRNA levels also seemed not to increase significantly at later time points (i.e., D1, D4), although there was a trend toward increased levels by D7 (S. Suhr, unpublished observations).
Fig. 3.

Graph showing quantification ofGFAP and NGF mRNA levels in hippocampal tissue obtained from animals killed at various early time points after unilateral PP transection. RT-PCR was performed on hippocampal tissue from n = 4 animals at each postlesion time point. Values are presented as the mean percentage of unoperated control values (0 hr, after normalizing to the RPL27A internal control bands within each sample). Error bars correspond to ± SEM. Asterisk indicates difference from unoperated control value (0 hr); p < 0.05.
Others have investigated changes in NGF protein levels in this model system but have failed to detect significant increases in response to EC lesioning (Whittemore et al., 1987; Conner et al., 1994) despite an increase in NGF bioactivity (Crutcher and Collins, 1986). Using a sensitive two-site ELISA for NGF, we too observed no significant increase in hippocampal NGF protein 7 d after our PP lesion, despite a significant increase at the same time after FFX transection (CTL, 49.7 ± 5.6 pg NGF/100 mg of tissue; PP, 58.2 ± 6.1 pg NGF/100 mg of tissue; FFX, 73.0 ± 4.1 pg/100 mg of tissue), in agreement with other reports (Gasser et al., 1986; Weskamp et al., 1986; Whittemore et al., 1987). Thus, NGF may be involved in the sprouting response that takes place after PP transection, but it is likely to act via a mechanism independent of increases in transcription or translation, as has been proposed by others (Conner et al., 1994).
Cellular source of increased FGF-2 expression
Our demonstration of an increase in FGF-2 mRNA in the denervated hippocampus as early as 8 hr postlesion and remaining elevated through D7 (the latest time point investigated; data not shown) is consistent with observations of Guthrie and colleagues (1995) using in situ hybridization analysis. To identify the cellular source of FGF-2 in this model, we performed double-labeling immunocytochemistry on hippocampal tissue from animals 7 d after PP transection with antibodies to FGF-2 and GFAP. We observed colocalization of FGF-2 and GFAP immunoreactivity within cells scattered throughout the normal hippocampus, indicating that hippocampal astrocytes contain FGF-2. We also observed FGF-2-positive/GFAP-negative profiles within the CA2 (Fig. 4) and subicular regions. The size and location of these cells suggest that they are neurons, although double labeling with neuron-specific markers and anti-FGF-2 was not performed. This pattern of FGF-2 immunolabeling in the normal hippocampus has been reported by others (Gomez-Pinella et al., 1992; Woodward et al., 1992). Seven days after PP transection, we observed a hypertrophy and distribution of FGF-2-positive/GFAP-positive astrocytes predominantly within the denervated ML (Fig. 4), similar to what has been shown after electrolytic lesioning of the EC (Gomez-Pinella et al., 1992). Although peroxidase immunocytochemistry by the avidin–biotin method does not permit quantitative statements to be made regarding protein levels within individual cells, these hypertrophic astrocytes are likely to be the source of the increased FGF mRNA levels we observed with RT-PCR analysis.
Fig. 4.
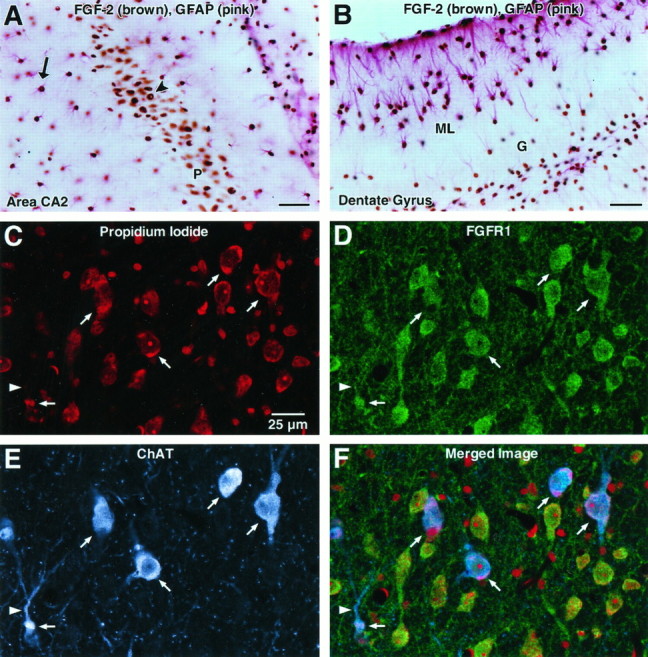
Color photomicrographs of double-labeling immunocytochemistry showing distribution of FGF-2 (brown) and GFAP (pink) immunoreactivity (IR) within hippocampal area CA2 (A) and dentate gyrus (B) and immunofluorescence confocal images localizing FGF receptor-1 (FGFR1) and choline acetyltransferase (ChAT) within the medial septal nucleus (C–F) of adult rats. A, Colocalization of FGF-2-IR (brown) and GFAP-IR (pink) within area CA2 demonstrates that both the pyramidal cells in CA2 (P,arrowhead) and adjacent astrocytes (arrow) express FGF-2. Scale bar, 50 μ.B. Seven days after perforant pathway lesion,FGF-2 (brown) is expressed by GFAP-IRastrocytes (pink) both around the granule cell layer (G) of the dentate gyrus and in the dentate molecular layer (ML). Hypertrophic astrocytes expressing FGF-2 are especially prominent in the outer ML, the region in which the terminals of the lesioned perforant pathway (PP) were located. Scale bar, 50 μ. C, The fluorescent nuclear counterstain propidium iodide stains neurons, glia, and endothelial cells in this field of view in the medial septum. Thearrows and arrowhead are in registration and show the same location within the field of view forC–F. Scale bar, 25 μ for C–F.D, FGFR1 (green) immunofluorescence is localized to a number of cells showing a neuronal morphology and fine processes. E, ChATimmunofluorescence (blue) shows a number of positive neurons (arrows) that are a subset of the number of neurons indicated in C and D. A primary dendrite of a ChAT-IR neuron is indicated by thearrowhead. F, A merge ofC–F demonstrates colocalization of FGFR1 on ChAT-IR cell bodies (arrows) and processes (arrowhead), indicating that cholinergic neurons in the medial septum of the adult rat express FGFR1. In addition, many noncholinergic (ChAT-negative) neurons express FGFR1.
Functional role of FGF-2 in cholinergic sprouting after PP transection
Because we observed an increase in gene expression for FGF-2 and a distribution of FGF-2-positive astrocytes in the denervated ML, we next asked whether endogenous FGF-2 plays a role in cholinergic sprouting after PP transection. Lesioned animals received chronic ICV infusions of either (1) antibodies to FGF-2, (2) artificial CSF, or (3) normal rabbit serum for 14 d, a time by which significant cholinergic sprouting has occurred (Fagan and Gage, 1994). Then the animals were killed, and the magnitude of lesion-induced sprouting was assessed. The anti-FGF-2 antibody has been shown to be neutralizing and specific for FGF-2 (Baird and Ling, 1987; A. Baird, unpublished observations). In all, 12 of 14 animals exhibited complete and discrete PP lesions. Of these animals, those receiving antibody infusions (i.e., anti-FGF-2 or normal serum) exhibited evidence of antibody penetration into the hippocampus, as determined by the presence of antibody immunoreactivity within tissue sections (Fig. 5). Sprouting of the commissural/associational projection was defined as an increase in width of the inner ML in the denervated hippocampus as compared with the contralateral side (Lynch et al., 1974). Cholinergic sprouting was defined as a greater AChE-positive fiber density in the denervated ML as compared with that in the contralateral, unlesioned ML within each animal (i.e., percentage of increase).
Fig. 5.
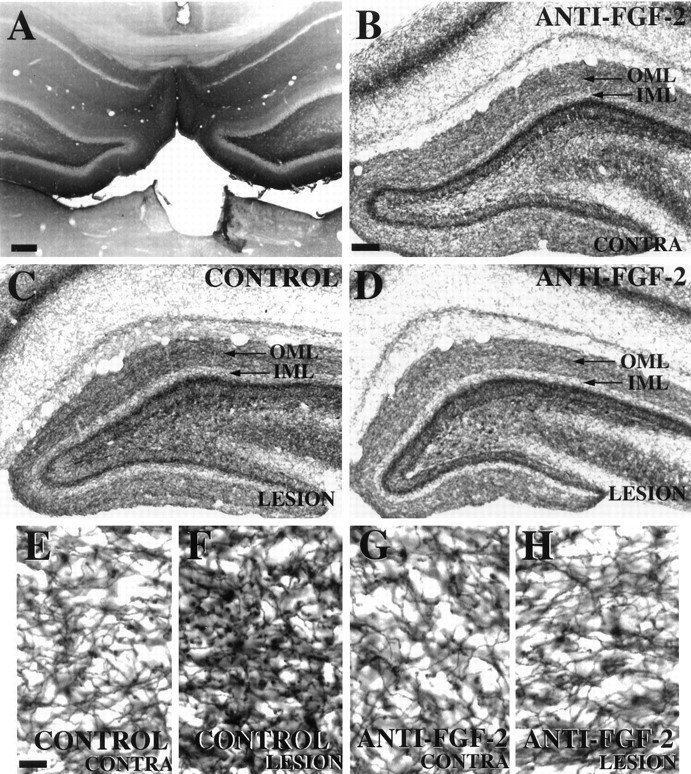
Photomicrographs of hippocampal tissue sections processed for (A) immunocytochemistry for normal serum and (B–H) histochemistry for AChE 14 d after unilateral PP transection. A, Immunoreactivity is observed within hippocampal parenchyma, demonstrating successful antibody penetration into this region after chronic ICV infusion.B, Contralateral (unlesioned) hippocampus from an animal receiving chronic anti-FGF-2 infusions displays the normal pattern of AChE-positive fibers within the outer molecular layer (OML, arrow) and inner molecular layer (IML) of the dentate gyrus. This pattern on the contralateral side is consistent between animals regardless of infusate (CSF, normal serum, or anti-FGF-2). C, Lesioned hippocampus from an animal receiving control infusate exhibits an increase in density of AChE-positive fibers in the OMLof the dentate gyrus. D, Lesioned hippocampus from an animal receiving neutralizing antibodies to FGF-2 displays no obvious increase in AChE-positive fiber density within the OMLof the dentate gyrus as compared with the contralateral side (B). E–H are high-power views of the OMLs from these same animals. Subsequent quantification of fiber density (see Fig. 6) was performed on tissue viewed at this high-power magnification. Scale bars: 500 μm in A; 50 μm inB–D; 12.5 μm in E–H.
Antibodies to FGF-2 did not seem to affect the sprouting of the commissural/association projection, because there was no statistically significant difference between the magnitude of expansion of the inner ML between the two groups (control = 14.0% ± 2.53 increase,n = 5; anti-FGF-2 = 8.1% ± 4.49 increase,n = 7; t = 1.013, p > 0.05). In regard to the cholinergic sprouting, because lesion-induced shrinkage of the outer ML artifactually could contribute to an observed increase in fiber density within the denervated region, the width of the outer ML on the lesioned and contralateral sides was measured as described in Materials and Methods, and the percentage of shrinkage was calculated for each animal. Then the mean shrinkage in each group was compared to assess the effect of antibody infusion specifically on this parameter. Statistical analysis revealed no statistically significant difference between groups in the magnitude of lesion-induced shrinkage of the denervated outer ML (control = 17.8% ± 4.24,n = 5; anti-FGF-2 = 11.3% ± 2.76,n = 7; t = 1.351, p > 0.05). Qualitative assessment of AChE-positive fiber density within the denervated ML suggested a clear attenuation of cholinergic sprouting with anti-FGF-2 treatment as compared with the control groups (Fig. 5). In fact, many animals receiving the FGF-2 antibodies seemed to exhibit little or no sprouting at all, despite complete PP lesions. Quantitative analysis confirmed that the percentage of increase in density of AChE-positive fibers within the denervated ML of animals receiving anti-FGF-2 was statistically less than the percentage of increase in animals given artificial CSF or normal serum (control = 64% increase ± 5.9, n = 5; anti-FGF-2 = 33% increase ± 12.4, n = 7; t = 1.985, p < 0.05) (Fig. 6). These results suggest a functional role for endogenous FGF-2 in the cholinergic sprouting that takes place in the outer ML of the DG in response to PP transection. The “normal” sprouting exhibited by two of the seven animals receiving anti-FGF-2 (see outlying points in Fig.6) may represent biological variability observed in in vivolesion paradigms or may reflect methodological variability (e.g., differences in antibody penetration into the tissue, in situantibody activity, etc.).
Fig. 6.

Histogram showing percentage of increase in AChE-positive fiber density in the denervated molecular layer (ML) of animals receiving ICV infusions of anti-FGF-2 or control infusates (i.e., CSF or normal serum) for 14 d after unilateral PP transection. Values for individual animals are presented as the percentage of increase in AChE-positive fiber density in the denervated ML as compared with the contralateral side. Student’s ttests demonstrated no statistical difference between mean values for animals receiving CSF or normal serum (t = −1.45,p > 0.05), so these groups were combined to make a single control group and subsequently were compared to values from animals receiving anti-FGF-2. Open triangles identify individual animals receiving control infusate, and open circles identify individual animals receiving neutralizing antibodies to FGF-2. Bold horizontal line identifies mean value for each group (CONTROL andANTI-FGF-2). Asterisk indicates difference from control values, p < 0.05.
Expression of FGF receptor (FGFR1) in the medial septum
To address the issue of whether endogenous FGF-2 could be acting directly on medial septal cholinergic neurons to induce their sprouting in this paradigm, we investigated the expression of its receptor, FGFR1, on these neurons. We performed double-labeling immunocytochemistry for ChAT and FGFR1, followed by confocal laser imaging, on tissue sections from adult rat brain. FGFR1 immunoreactivity (IR) was observed in many neurons in the medial septal region, a subset of which was also immunopositive for ChAT (Fig. 5), demonstrating that both cholinergic and noncholinergic neurons of the medial septum express the receptor for FGF-2. However, ChAT-IR always was colocalized with FGFR1-IR, indicating that most, if not all, ChAT-positive neurons in this region express the FGF-2 receptor (Fig.5). These results suggest that cholinergic neurons of the medial septum can respond directly to FGF-2.
DISCUSSION
Many investigators have identified changes in gene expression in the hippocampus after entorhinal denervation (Poirier et al., 1990,1991; Lampert-Etchells et al., 1991; Nichols et al., 1991; Beck et al., 1993; Morgan et al., 1993; Gwag et al., 1994; Guthrie et al., 1995;Jucker et al., 1995; Steward, 1995), but the relevance of such events for the subsequent neuronal sprouting has not been addressed directly. Our approach was to evaluate changes in trophic factor gene expression and then test the functional relevance of identified molecules by blocking bioactivity in vivo via administration of neutralizing antibodies during the course of lesion-induced synaptic reorganization. We used RT-PCR to investigate quantitative changes in mRNAs encoding trophic molecules known to be present in the hippocampus and hypothesized to be involved in neuronal plasticity. Among the changes we did observe of several trophic factor mRNAs was an increase in FGF-2. In addition, FGF-2 immunoreactivity was localized within GFAP-positive astrocytes throughout the hippocampus, including hypertrophic astrocytes distributed specifically within the denervated outer ML, consistent with a previous report (Gomez-Pinella et al., 1992).
To address the functional significance of this transcriptional increase in FGF-2, we assessed the magnitude of cholinergic sprouting in animals receiving chronic ICV infusions of neutralizing antibodies specific for FGF-2 and compared it with that observed in lesioned animals receiving infusate controls. Animals given FGF-2 antibodies displayed a clear reduction in cholinergic sprouting. In fact, many of these animals exhibited virtually no sprouting at all despite histological verification of complete PP lesions. To our knowledge, these results are the first to show that endogenous FGF-2 promotes axonal sprouting in the injured adult brain. Furthermore, immunocytochemical localization of receptors for FGF-2 (i.e., FGFR1) (Lee et al., 1989;Dionne et al., 1990) on these cholinergic neurons suggests that FGF-2 may act directly on these neurons to induce the lesion-induced sprouting response.
It is likely that the increase in AChE staining that was inhibited by antibodies to FGF-2 represents sprouting from septal cholinergic neurons for several reasons. The increase in AChE staining after PP lesions is abolished in the presence of a fimbria–fornix lesion (Lynch et al., 1972). This argues that a septohippocampal projection is required for the increase in AChE staining after PP lesions. Strongly arguing that the increase in AChE staining in the outer ML is attributable to sprouting of septal afferents are data from Stanfield and Cowan, who showed that, after injections of radiolabeled tracer in the medial septal region, anterograde labeling of hippocampal fibers is increased in the outer ML after PP lesions (Stanfield and Cowan, 1982). These two studies, however, do not rule out the possibility that noncholinergic septal afferents contribute to the increased AChE staining in this model. However, studies by Gomez-Pinella (1987) as well as our own argue that the cells in the septohippocampal pathway, which account for the AChE-positive sprouting in this model, are cholinergic. It was shown that the pattern of p75NGFR-immunoreactive fibers in the normal hippocampus is similar to that seen for AChE-positive fibers and that after PP lesioning there is a similar pattern and increase in density of p75NGFR-immunoreactive fibers in the outer ML as is seen for AChE staining. Using two independent antibodies to p75NGFR (MC-192, mouse monoclonal antibody, gift of E. Johnson (Washington University, St. Louis, MO); REX, a rabbit polyclonal antibody, gift of L. Reichardt (University of California, San Francisco, CA), we have confirmed this finding (D. Holtzman, data not shown). We also have observed that after PP lesions the density oftrkA immunoreactive hippocampal fibers (stained with a rabbit antibody specific to trkA) also is increased in the outer ML. Because (1) both trkA and p75NGFR are localized specifically to cholinergic neuronal cell bodies within the septum, the axons of which project to the hippocampus (Hefti et al., 1986; Holtzman et al., 1992; Holtzman et al., 1994), and (2) intrinsic hippocampal neurons do not express these markers (Hefti et al., 1986;Holtzman et al., 1992, 1994), the majority of the sprouting visualized by AChE staining seems to be attributable to sprouting from basal forebrain cholinergic neurons.
Because our assessment of cholinergic sprouting used a measure of the density of fibers per unit area of hippocampus (i.e., outer ML), significant tissue shrinkage artifactually could mimic an observed increase in fiber density. That we observed significantly less of an increase in fiber density in animals treated with an anti-FGF-2 antibody compared with controls, despite no statistical difference in shrinkage between the groups, suggests that anti-FGF-2 is, indeed, inhibiting cholinergic sprouting in this paradigm. There was, however, a trend for there to be less shrinkage of the outer ML in animals receiving anti-FGF-2. Although it is possible that this trend contributed in part to the difference in fiber density observed between the groups, we feel it is unlikely that differences in shrinkage alone can account for the majority of the observed effects of the anti-FGF-2 antibody infusions. Supporting this argument is the fact that we found no significant correlation between the percentage of shrinkage and percentage of increase in fiber density within each group (Spearman correlation; control, n = 5, p > 0.05; anti-FGF-2, n = 7, p > 0.05). Other studies using this lesion model system have not addressed this issue rigorously (Lynch et al., 1974; Van der Zee et al., 1992; Fagan and Gage, 1994).
Role of FGF-2 in cholinergic sprouting after PP transection
The attenuation of cholinergic sprouting observed in animals receiving infusions of anti-FGF-2 antibodies suggests that FGF-2 is involved functionally in eliciting, propagating, or maintaining these neuronal structural changes. The exact mechanism by which FGF-2 acts to influence these changes, however, remains unclear. Our demonstration of colocalization of FGFR1 and ChAT immunoreactivity within neurons of the medial septum suggests that FGF-2 can act directly on these sprouting cholinergic neurons. This observation is consistent with the known survival- and neurite-promoting effects of FGF-2 on these neurons (Anderson et al., 1988; Walicke, 1988; Otto et al., 1989; Matsuda et al., 1991). However, others have reported an absence of FGFR1 mRNA in this region (Wanaka et al., 1990), although lack of signal may reflect the different sensitivities of in situ hybridization versus immunocytochemistry. Although FGF-2 may act directly on basal forebrain cholinergic neurons to stimulate their sprouting, the effect may not require FGF-2 secretion, followed by subsequent binding to neuronal receptors and retrograde transport to the cell body. First, the mechanism of FGF-2 release from the cell is not well understood, given its lack of an identifiable signal sequence (Abraham et al., 1986). It may be, however, that FGF-2 release is not a prerequisite for biological action, because both secretory and cell-associated forms of the protein promote the survival of hippocampal neurons in vitro (Ray et al., 1995). Second, projecting basal forebrain cholinergic neurons have been reported not to transport125I-FGF-2 retrogradely that has been injected into the hippocampus in vivo (Ferguson and Johnson, 1991). Thus, the growth-promoting activity of FGF-2 in this model system may not require its uptake and retrograde transport by sprouting neurons but, rather, may be attributable to local interactions between receptors on growth cones and bound FGF-2 either on the cell surface or in the extracellular matrix.
Alternatively, FGF-2 may act to promote cholinergic sprouting indirectly by acting via glial cells. FGF-2 is an astrocyte mitogen (Pettman et al., 1985; Perraud et al., 1988) and can regulate NGF synthesis and secretion by astrocytes in culture (Spranger et al., 1990; Vige et al., 1991; Yoshida and Gage, 1991, 1992). The fact that astrocytes themselves produce FGF-2 (Ferrara et al., 1988; Hatten et al., 1988), including those within the denervated ML (Guthrie et al., 1995), suggests that astrocyte-derived FGF-2 could be acting in an autocrine and/or paracrine manner to induce putative production of additional trophic molecules in this model. Thus, antibodies to FGF-2 may block cholinergic sprouting indirectly by inhibiting astrocyte proliferation and subsequent production of trophic molecules, which themselves act directly on basal forebrain cholinergic neurons. This hypothesis generates two testable predictions. First, FGF-2 antibody infusions should block the astrocyte response after PP transection. Contrary to this prediction, we observed no difference in patterns of GFAP immunoreactivity in animals receiving anti-FGF-2 versus control infusates for 2 weeks (data not shown). However, the astrocyte response normally peaks within the first week after denervation, returning to normal patterns by 30 d (Fagan and Gage, 1994). Therefore, a more valid test of this hypothesis would be to assess astrocyte reactivity earlier than 14 d postlesion. The second prediction is that anti-FGF-2 infusions should block increases in NGF and/or other trophic molecules after the lesion. Because we did not observe any increase in NGF at the time points investigated, it is unlikely that the effect of anti-FGF-2 antibodies is via alterations in NGF levels. We cannot, however, rule out the possibility that FGF-2 in some way does alter NGF availability, bioactivity, or distribution, which in turn affects cholinergic sprouting. In total, our data are more consistent with the possibility that effects of FGF-2 on cholinergic sprouting are direct.
Role of neurotrophins in cholinergic sprouting after PP transection
Hippocampal BDNF mRNA levels have been reported not to change 2–10 d after EC lesion (Lapchak et al., 1993). In contrast, we observed a significant decrease of BDNF and NT-3 mRNA 16 hr after PP transection. This discrepancy may reflect differences in the sensitivity of the two methods used to quantify mRNA levels or may indicate a time-dependent change in neurotrophin transcription in this model. The decrease we observed may reflect a rapid transynaptic downregulation of neurotrophin synthesis by hippocampal neurons as a consequence of disrupting the major glutamatergic input to the hippocampus, because glutamate has been shown to modulate neurotrophin transcription in a variety of paradigms (Thoenen, 1995). The duration and functional significance of the early decrease we observe remain unclear.
Reports of increases in NGF-like bioactivity (Crutcher and Collins, 1986) and redistribution of NGF-like immunoreactivity within the denervated ML (Conner et al., 1994) after EC lesion provide support for the idea that NGF is involved in this lesion-induced cholinergic sprouting. In further support of this hypothesis, daily ICV injections of anti-NGF antibodies resulted in an attenuated cholinergic sprouting response (Van der Zee et al., 1992). Although results from this antibody delivery study suggest that NGF may play a role in the lesion-induced cholinergic sprouting, our data suggest that it is unlikely to be attributable to a general increase in NGF, because we did not detect increases in NGF mRNA or protein within the denervated hippocampus at any time point investigated. It does remain possible that there are small changes in NGF that are masked by the relatively high levels normally found in the hippocampus (Korsching et al., 1985;Large et al., 1986; Whittemore et al., 1986) or that NGF mRNA may be degraded rapidly (Layer and Shooter, 1983; Heumann et al., 1984;Whittemore et al., 1988). A lack of general increase in NGF protein also may merely reflect an increase in efficiency of retrograde transport of NGF out of the hippocampus by stimulated basal forebrain cholinergic neurons. It is also possible that baseline levels of endogenous NGF may in some way permit sprouting to occur and that its neutralization via chronic antibody delivery disrupts this process. Alternatively, this sprouting may be the result of a redistribution of available NGF specifically within a spatially restricted region (i.e., denervated outer ML) (Conner et al., 1994). This last scenario would provide a mechanism by which axons within a particular area would be exposed to elevated levels of trophic molecules without the need for a general increase in trophic factor transcription or translation.
In situ hybridization analyses demonstrate increases in a number of astrocytic mRNAs, including GFAP (Steward et al., 1990), apolipoprotein-E (Poirier et al., 1991), sulfated glycoprotein-2 (Day et al., 1990), and the noncatalytic form of the trkB receptor (Beck et al., 1993) after EC lesion. Although inconsistent with our data from an early time point (i.e., 16 hr), this last finding is especially interesting, given that this nonsignaling trkB receptor has been proposed to be a “presentation” receptor (Klein et al., 1990). This could provide a mechanism by which reactive astrocytes in the denervated ML serve as substrates for axonal growth by binding growth factors (produced by glia or neurons) on their membrane surface and presenting them to growing cholinergic axons (Kawaja and Gage, 1991) in much the same way as has been hypothesized for the p75NGFR receptor in Schwann cells in the injured PNS (Tanuichi et al., 1986). Alternatively, astrocytes may provide a substrate for growth that is independent of growth factors. For example, the cell adhesion molecule NCAM has been reported to be upregulated in reactive astrocytes within the denervated ML in this model (Jucker et al., 1995). Given the emerging recognition of complex interactions between trophic molecules in the regulation of their synthesis and activity, it is likely that the establishment of neuronal connectivity and reconnectivity is the result of the coordinate spatial and temporal action of multiple growth factors, substrates, and extracellular matrix molecules.
These authors contributed equally to this work.
This research was supported by Grants from National Institutes of Health (AG10435, P01AG10435, and P01NS28121 to F.H.G.) and the American Paralysis Association. D.M.H. was supported by a Paul Beeson Physician-Faculty Scholar Award from the American Federation for Aging Research. We thank S. Forbes, L. Randolph-Moore, and Dr. K. Yoshida for their technical assistance; Dr. S. Thode for her contribution to primer design; and Dr. A. Baird for the gift of the anti-FGF-2 antibody.
Correspondence should be addressed to Dr. Anne M. Fagan, Department of Neurology, Center for the Study of Nervous System Injury, Washington University School of Medicine, 660 South Euclid Avenue, Box 8111, St. Louis, MO 63110.
REFERENCES
- 1.Abraham JA, Mergia A, Whang JL, Tumolo A, Friedman J, Hjerrild KA, Gospodarowicz D, Fiddes JC. Nucleotide sequence of a bovine clone encoding the angiogenic protein, basic fibroblast growth factor. Science. 1986;233:545–548. doi: 10.1126/science.2425435. [DOI] [PubMed] [Google Scholar]
- 2.Amaral DG, Avendano C, Cowan WM. The effects of neonatal 6-hydroxydopamine treatment on morphological plasticity in the dentate gyrus of the rat following entorhinal lesions. J Comp Neurol. 1980;194:171–191. doi: 10.1002/cne.901940109. [DOI] [PubMed] [Google Scholar]
- 3.Anderson KJ, Dam D, Lee S, Cotman CW. Basic fibroblast growth factor prevents death of lesioned cholinergic neurons in vivo. Nature. 1988;332:360–361. doi: 10.1038/332360a0. [DOI] [PubMed] [Google Scholar]
- 4.Baird A, Ling N. Fibroblast growth factors are present in the extracellular matrix produced by endothelial cells in vitro: implications for a role of heparinase-like enzymes in the neovascular response. Biochem Biophys Res Commun. 1987;142:428–435. doi: 10.1016/0006-291x(87)90292-0. [DOI] [PubMed] [Google Scholar]
- 5.Barde YA. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- 6.Batchelor PE, Armstrong DM, Blaker SM, Gage FH. Nerve growth factor receptor and choline acetyltransferase colocalization in neurons within the rat forebrain: response to fimbria-fornix transection. J Comp Neurol. 1989;284:187–204. doi: 10.1002/cne.902840204. [DOI] [PubMed] [Google Scholar]
- 7.Beck KD, Lamballe F, Klein R, Barbacid M, Schauwecker PE, McNeill TH, Finch CE, Hefti F, Day JR. Induction of non-catalytic trkB neurotrophin receptors during axonal sprouting in the adult hippocampus. J Neurosci. 1993;13:4001–4014. doi: 10.1523/JNEUROSCI.13-09-04001.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchman VL, Davies AM. Different neurotrophins are expressed and act in developmental sequence to promote the survival of embryonic sensory neurons. Development (Camb) 1993;118:989–1001. doi: 10.1242/dev.118.3.989. [DOI] [PubMed] [Google Scholar]
- 9.Conner JM, Fass-Holmes B, Varon S. Changes in nerve growth factor immunoreactivity following entorhinal cortex lesions: possible molecular mechanism regulating cholinergic sprouting. J Comp Neurol. 1994;345:409–418. doi: 10.1002/cne.903450307. [DOI] [PubMed] [Google Scholar]
- 10.Crutcher KA, Collins F. Entorhinal lesions result in increased nerve growth factor-like growth-promoting activity in medium conditioned by hippocampal slices. Brain Res. 1986;399:383–389. doi: 10.1016/0006-8993(86)91533-7. [DOI] [PubMed] [Google Scholar]
- 11.Davies AM, Bandtlow C, Heumann R, Korsching S, Rohrer H, Thoenen H. Timing and site of nerve growth factor synthesis in developing skin in relation to innervation and expression of the receptor. Nature. 1987;326:353–358. doi: 10.1038/326353a0. [DOI] [PubMed] [Google Scholar]
- 12.Day JR, Laping NJ, McNeill TH, Schreiber SS, Pasinetti G, Finch CE. Castration enhances expression of glial fibrillary acidic protein and sulfated glycoprotein-2 in the intact and lesion-altered hippocampus of the adult male rat. Mol Endocrinol. 1990;4:1995–2002. doi: 10.1210/mend-4-12-1995. [DOI] [PubMed] [Google Scholar]
- 13.Deller T, Frotscher M, Nitsch R. Sprouting of crossed entorhinodentate fibers after unilateral entorhinal lesion: anterograde tracing of fiber reorganization with phaseolus vulgaris-leucoagglutinin (PHAL). J Comp Neurol. 1996;365:42–55. doi: 10.1002/(SICI)1096-9861(19960129)365:1<42::AID-CNE4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 14.Diamond J, Coughlin L, MacIntyre L, Holmes M, Visheau B. Evidence that endogenous β-nerve growth factor is responsible for the collateral sprouting, but not the regeneration, of nociceptive axons in adult rats. Proc Natl Acad Sci USA. 1987;84:6596–6600. doi: 10.1073/pnas.84.18.6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diamond J, Foerster A, Holmes M, Coughlin M. Sensory nerves in adult rats regenerate and restore sensory function to the skin independently of endogenous NGF. J Neurosci. 1992a;12:1467–1476. doi: 10.1523/JNEUROSCI.12-04-01467.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diamond J, Holmes M, Coughlin M. Endogenous NGF and nerve impulses regulate the collateral sprouting of sensory axons in the skin of the adult rat. J Neurosci. 1992b;12:1454–1466. doi: 10.1523/JNEUROSCI.12-04-01454.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dionne AD, Crumley G, Bellot F, Kaplow JM, Searfoss G, Ruta M, Burgess WH, Jaye M, Schlessinger J. Cloning and expression of two distinct high-affinity receptors cross-reacting with acidic and basic fibroblast growth factors. EMBO J. 1990;9:2685–2692. doi: 10.1002/j.1460-2075.1990.tb07454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ernfors P, Persson H. Developmentally regulated expression of BDNF/NT-3 mRNA in rat spinal cord motoneurons and detection of BDNF mRNA in embryonic dorsal root ganglion. Eur J Neurosci. 1991;3:953–961. doi: 10.1111/j.1460-9568.1991.tb00031.x. [DOI] [PubMed] [Google Scholar]
- 19.Ernfors P, Bengzon J, Kokaia A, Persson H, Lindvall O. Increased levels of messenger RNAs for neurotrophic factors in the brain following kindling epileptogenesis. Neuron. 1991;7:165–176. doi: 10.1016/0896-6273(91)90084-d. [DOI] [PubMed] [Google Scholar]
- 20.Fagan AM, Gage FH. Cholinergic sprouting in the hippocampus: a proposed role for IL-1. Exp Neurol. 1990;110:105–120. doi: 10.1016/0014-4886(90)90055-w. [DOI] [PubMed] [Google Scholar]
- 21.Fagan AM, Gage FH. Mechanisms of sprouting in the adult CNS: cellular responses in areas of terminal degeneration and reinnervation in the rat hippocampus. Neuroscience. 1994;58:705–725. doi: 10.1016/0306-4522(94)90449-9. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson IA, Johnson EM. Fibroblast growth factor receptor-bearing neurons in the CNS: identification by receptor-mediated retrograde transport. J Comp Neurol. 1991;313:693–706. doi: 10.1002/cne.903130412. [DOI] [PubMed] [Google Scholar]
- 23.Ferrara N, Ousley F, Gospodarowicz D. Bovine brain astrocytes express basic fibroblast growth factor, a neurotrophic and angiogenic mitogen. Brain Res. 1988;462:223–232. doi: 10.1016/0006-8993(88)90550-1. [DOI] [PubMed] [Google Scholar]
- 24.Friedman WJ, Olson L, Persson H. Cells that express brain-derived neurotrophic factor mRNA in the developing postnatal rat brain. Eur J Neurosci. 1991;3:688–697. doi: 10.1111/j.1460-9568.1991.tb00854.x. [DOI] [PubMed] [Google Scholar]
- 25.Gage FH, Olejniczak P, Armstrong DM. Astrocytes are important for sprouting in the septohippocampal circuit. Exp Neurol. 1988;102:2–13. doi: 10.1016/0014-4886(88)90073-8. [DOI] [PubMed] [Google Scholar]
- 26.Gallyas F, Wolff JR, Bottcher H, Zaborszky L. A reliable and sensitive method to localize terminal degeneration and lysosomes in the central nervous system. Stain Technol. 1980;55:299–306. doi: 10.3109/10520298009067258. [DOI] [PubMed] [Google Scholar]
- 27.Gasser UE, Weskamp G, Otten U, Dravid AR. Time course of the elevation of nerve growth factor (NGF) content in the hippocampus and septum following lesions of the septohippocampal pathway in rats. Brain Res. 1986;376:351–356. doi: 10.1016/0006-8993(86)90198-8. [DOI] [PubMed] [Google Scholar]
- 28.Gloster A, Diamond J. Sympathetic nerves in adult rats regenerate normally and restore pilomotor function during an anti-NGF treatment that prevents their collateral sprouting. J Comp Neurol. 1992;326:363–374. doi: 10.1002/cne.903260305. [DOI] [PubMed] [Google Scholar]
- 29.Gomez-Pinella F, Won-Kyun Lee J, Cotman CW. Basic FGF in adult rat brain: cellular distribution and response to entorhinal lesion and fimbria–fornix transection. J Neurosci. 1992;12:345–355. doi: 10.1523/JNEUROSCI.12-01-00345.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guthrie KM, Nguyen T, Gall CM. Insulin-like growth factor-1 mRNA is increased in deafferented hippocampus: spatiotemporal correspondence of a trophic event with axon sprouting. J Comp Neurol. 1995;352:147–160. doi: 10.1002/cne.903520111. [DOI] [PubMed] [Google Scholar]
- 31.Gwag BJ, Sessler F, Kimmerer K, Springer JE. Neurotrophic factor mRNA expression in dentate gyrus is increased following angular bundle transection. Brain Res. 1994;647:23–29. doi: 10.1016/0006-8993(94)91394-3. [DOI] [PubMed] [Google Scholar]
- 32.Hagg T, Vahlsing HL, Manthorpe M, Varon S. Nerve growth factor infusion into the denervated adult rat hippocampal formation promotes its cholinergic reinnervation. J Neurosci. 1990;10:3087–3092. doi: 10.1523/JNEUROSCI.10-09-03087.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatten ME, Lynch M, Rydel RE, Sanchez J, Joseph-Silverstein J, Moscatelli D, Rifkin D. In vitro neurite extension by granule neurons is dependent upon astroglial-derived fibroblast growth factor. Dev Biol. 1988;125:280–289. doi: 10.1016/0012-1606(88)90211-4. [DOI] [PubMed] [Google Scholar]
- 34.Hedreen JC, Bacon JC, Price DL. A modified histochemical method to visualize acetylcholinesterase-containing axons. J Histochem Cytochem. 1985;33:134–140. doi: 10.1177/33.2.2578498. [DOI] [PubMed] [Google Scholar]
- 35.Hefti F, Hartikka J, Salvatierra A, Weiner W, Mash D. Localization of nerve growth factor receptors in cholinergic neurons of the human basal forebrain. Neurosci Lett. 1986;69:37–41. doi: 10.1016/0304-3940(86)90410-6. [DOI] [PubMed] [Google Scholar]
- 36.Heisenberg CP, Cooper JD, Berke J, Sofroniew MV. NMDA potentiates NGF-induced sprouting of septal cholinergic fibers. Neuro- Report. 1994;5:413–416. doi: 10.1097/00001756-199401120-00010. [DOI] [PubMed] [Google Scholar]
- 37.Heumann R, Schwab M, Merkl R, Thoenen H. Nerve growth factor-mediated induction of choline acetyltransferase in PC12 cells: evaluation of the site of action of nerve growth factor and the involvement of lysosomal degradation products of nerve growth factor. J Neurosci. 1984;4:3039–3050. doi: 10.1523/JNEUROSCI.04-12-03039.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holtzman DM, Lowenstein DH. Selective inhibition of axon outgrowth by antibodies to NGF in a model of temporal lobe epilepsy. J Neurosci. 1995;15:7062–7070. doi: 10.1523/JNEUROSCI.15-11-07062.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holtzman DM, Li Y, Parada LF, Kinsman S, Chen CK, Valletta JS, Zhou J, Long J, Mobley WC. p140trk mRNA marks NGF-responsive forebrain neurons: evidence that trk gene expression is induced by NGF. Neuron. 1992;9:465–478. doi: 10.1016/0896-6273(92)90184-f. [DOI] [PubMed] [Google Scholar]
- 40.Holtzman DM, Kilbridge J, Bredt D, Black S, Li Y, Clary D, Reichardt L, Mobley WC. NOS induction by NGF in basal forebrain cholinergic neurons: evidence for regulation of brain NOS by a neurotrophin. Neurobiol Dis. 1994;1:51–60. doi: 10.1006/nbdi.1994.0007. [DOI] [PubMed] [Google Scholar]
- 41.Isaacson LG, Saffran BF, Crutcher KA. Nerve growth factor-induced sprouting of mature, uninjured sympathetic axons. J Comp Neurol. 1992;326:327–336. doi: 10.1002/cne.903260302. [DOI] [PubMed] [Google Scholar]
- 42.Jucker M, Mondadori C, Mohajeri H, Bartsch U, Schachner M. Transient upregulation of NCAM mRNA in astrocytes in response to entorhinal cortex lesions and ischemia. Mol Brain Res. 1995;28:149–156. doi: 10.1016/0169-328x(94)00206-t. [DOI] [PubMed] [Google Scholar]
- 43.Kawaja MD, Gage FH. Reactive astrocytes are substrates for the growth of adult CNS axons in the presence of elevated levels of nerve growth factor. Neuron. 1991;7:1019–1030. doi: 10.1016/0896-6273(91)90346-2. [DOI] [PubMed] [Google Scholar]
- 44.Kawaja MD, Rosenberg MB, Yoshida K, Gage FH. Somatic gene transfer of nerve growth factor promotes the survival of axotomized septal neurons and the regeneration of their axons in adult rats. J Neurosci. 1992;12:2849–2864. doi: 10.1523/JNEUROSCI.12-07-02849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- 46.Korsching S. The neurotrophic factor concept: a reexamination. J Neurosci. 1993;13:2739–2748. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korsching S, Auburger G, Heumann R, Scott J, Thoenen H. Levels of nerve growth factor and its mRNA in the central nervous system of the rat correlate with cholinergic innervation. EMBO J. 1985;4:1389–1393. doi: 10.1002/j.1460-2075.1985.tb03791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lampert-Etchells M, McNeill TH, Laping NJ, Zarow C, Finch CE, May PC. Sulfated glycoprotein-2 is increased in rat hippocampus following entorhinal cortex lesioning. Brain Res. 1991;563:101–106. doi: 10.1016/0006-8993(91)91520-b. [DOI] [PubMed] [Google Scholar]
- 49.Lapchak PA, Araujo DM, Hefti F. BDNF and trkB mRNA expression in the rat hippocampus following entorhinal cortex lesion. NeuroReport. 1993;4:191–194. doi: 10.1097/00001756-199302000-00019. [DOI] [PubMed] [Google Scholar]
- 50.Large TH, Bodary SC, Clegg DO, Weskamp G, Otten U, Reichardt LF. Nerve growth factor gene expression in the developing rat brain. Science. 1986;234:352–355. doi: 10.1126/science.3764415. [DOI] [PubMed] [Google Scholar]
- 51.Layer PG, Shooter EM. Binding and degradation of nerve growth factor by PC12 pheochromocytoma cells. J Biol Chem. 1983;258:3012–3018. [PubMed] [Google Scholar]
- 52.Lee PL, Johnson DE, Cousens LS, Fried VA, Williams LT. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science. 1989;245:57–60. doi: 10.1126/science.2544996. [DOI] [PubMed] [Google Scholar]
- 53.Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-β1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992;117:395–400. doi: 10.1083/jcb.117.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindvall O, Ernfors P, Bengzon J, Kokaia Z, Smith ML, Siesjo BK, Persson H. Differential regulation of mRNAs for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin 3 in the adult rat following cerebral ischemia and hypoglycemic coma. Proc Natl Acad Sci USA. 1992;89:648–652. doi: 10.1073/pnas.89.2.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lynch G, Matthews DA, Mosko S, Parks T, Cotman C. Induced acetylcholinesterase-rich layer in rat dentate gyrus following entorhinal cortex lesions. Brain Res. 1972;42:311–318. doi: 10.1016/0006-8993(72)90533-1. [DOI] [PubMed] [Google Scholar]
- 56.Lynch G, Stanfield B, Parks T, Cotman CW. Evidence for selective post-lesion axonal growth in the dentate gyrus of the rat. Brain Res. 1974;69:1–11. doi: 10.1016/0006-8993(74)90365-5. [DOI] [PubMed] [Google Scholar]
- 57.Maisonpierre PC, Belluscio L, Friedman B, Alderson R, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 58.Matsuda S, Saito H, Nishiyama N. Effect of fibroblast growth factor on neurons cultured from various regions of postnatal rat brain. Brain Res. 1991;520:310–316. doi: 10.1016/0006-8993(90)91720-2. [DOI] [PubMed] [Google Scholar]
- 59.Morgan TE, Nichols NR, Pasinetti GM, Finch CE. TGF-β1 mRNA increases in macrophage/microglial cells of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration. Exp Neurol. 1993;120:291–301. doi: 10.1006/exnr.1993.1063. [DOI] [PubMed] [Google Scholar]
- 60.Nichols NR, Laping NJ, Day JR, Finch CE. Increases in transforming growth factor-β mRNA in hippocampus during responses to entorhinal cortex lesions in intact and adrenalectomized rats. J Neurosci Res. 1991;28:134–139. doi: 10.1002/jnr.490280114. [DOI] [PubMed] [Google Scholar]
- 61.Otto D, Frotscher M, Unsicker K. Basic fibroblast growth factor and nerve growth factor administered in gelfoam rescue medial septal neurons after fimbria-fornix transection. J Neurosci Res. 1989;22:83–91. doi: 10.1002/jnr.490220111. [DOI] [PubMed] [Google Scholar]
- 62.Perraud F, Labourdette G, Miehe M, Loret C, Sensenbrenner M. Comparison of the morphological effects of acidic and basic fibroblast growth factors on rat astroblasts in culture. J Neurosci Res. 1988;20:1–11. doi: 10.1002/jnr.490200102. [DOI] [PubMed] [Google Scholar]
- 63.Peterson DA, Lucidi-Phillipi CA, Murphy DP, Ray J, Gage FH. Fibroblast growth factor-2 protects entorhinal layer II glutamatergic neurons from axotomy-induced death. J Neurosci. 1996;16:886–898. doi: 10.1523/JNEUROSCI.16-03-00886.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pettman B, Weibel M, Sensenbrenner M, Labourdette G. Purification of two astroglial growth factors from bovine brain. FEBS Lett. 1985;189:102–108. doi: 10.1016/0014-5793(85)80851-6. [DOI] [PubMed] [Google Scholar]
- 65.Poirier J, May PC, Osterburg HH, Geddes J, Cotman C, Finch CE. Selective alterations of RNA in rat hippocampus after entorhinal cortex lesioning. Proc Natl Acad Sci USA. 1990;87:303–307. doi: 10.1073/pnas.87.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poirier J, Hess M, May PC, Finch CE. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Mol Brain Res. 1991;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- 67.Ray J, Hogg J, Beutler AS, Takayama H, Baird A, Gage FH. Expression of biologically active basic fibroblast growth factor by genetically modified rat primary skin fibroblasts. J Neurochem. 1995;64:503–513. doi: 10.1046/j.1471-4159.1995.64020503.x. [DOI] [PubMed] [Google Scholar]
- 68.Rich KM, Yip HK, Osborne PA, Schmidt RE, Johnson EM. Role of nerve growth factor in the adult dorsal root ganglion neuron and its response to injury. J Comp Neurol. 1984;230:110–118. doi: 10.1002/cne.902300110. [DOI] [PubMed] [Google Scholar]
- 69.Rocamora N, Palacios JM, Mengod G. Limbic seizures induce a differential regulation of the expression of nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 in the rat brain. Mol Brain Res. 1992;13:27–33. doi: 10.1016/0169-328x(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 70.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual, 2nd Ed (Nolan C, ed). Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 71.Schnurch H, Risau W. Differentiating and mature neurons express the acidic fibroblast growth factor gene during chick neural development. Development (Camb) 1991;111:1143–1154. doi: 10.1242/dev.111.4.1143. [DOI] [PubMed] [Google Scholar]
- 72.Spranger M, Lindholm D, Bandtlow C, Gnahn H, Nahar-Noe M, Thoenen H. Regulation of nerve growth factor (NGF) synthesis in the rat central nervous system: comparison between the effects of interleukin-1 and various growth factors in astrocyte cultures and in vivo. Eur J Neurosci. 1990;2:69–76. doi: 10.1111/j.1460-9568.1990.tb00382.x. [DOI] [PubMed] [Google Scholar]
- 73.Springer JE, Gwag BJ, Sessler FM. Neurotrophic factor mRNA expression in dentate gyrus is increased following in vivo stimulation of the angular bundle. Mol Brain Res. 1994;23:135–143. doi: 10.1016/0169-328x(94)90219-4. [DOI] [PubMed] [Google Scholar]
- 74.Stanfield BB, Cowan WM. The sprouting of septal afferents to the dentate gyrus after lesions of the entorhinal cortex in adult rats. Brain Res. 1982;232:162–170. doi: 10.1016/0006-8993(82)90619-9. [DOI] [PubMed] [Google Scholar]
- 75.Steward O. Reinnervation of dentate gyrus by homologous afferents following entorhinal cortex lesions in adult rats. Science. 1976;194:426–428. doi: 10.1126/science.982024. [DOI] [PubMed] [Google Scholar]
- 76.Steward O. The process of reinnervation in the dentate gyrus of adult rats: gene expression by neurons during the period of lesion-induced growth. J Comp Neurol. 1995;359:391–411. doi: 10.1002/cne.903590304. [DOI] [PubMed] [Google Scholar]
- 77.Steward O, Cotman CW, Lynch GS. Growth of a new fiber projection in the brain of adult rats: reinnervation of the dentate gyrus by the contralateral entorhinal cortex following ipsilateral entorhinal lesions. Brain Res. 1974;114:181–200. doi: 10.1007/BF00239017. [DOI] [PubMed] [Google Scholar]
- 78.Steward O, Torre ER, Phillips LL, Trimmer PA. The process of reinnervation in the dentate gyrus of adult rats: time course of increases in mRNA for glial fibrillary acidic protein. J Neurosci. 1990;10:2373–2384. doi: 10.1523/JNEUROSCI.10-07-02373.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stockli KA, Lillien LE, Naher-Noe M, Breitfeld G, Hughes RA, Raff MC, Thoenen H, Sendtner M. Regional distribution, developmental changes, and cellular localization of CNTF mRNA and protein in the rat brain. J Cell Biol. 1991;115:447–459. doi: 10.1083/jcb.115.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Storm-Mathisen J. Choline acetyltransferase and acetylcholinesterase in fascia dentata following lesions of the entorhinal afferent. Brain Res. 1974;80:181–197. doi: 10.1016/0006-8993(74)90683-0. [DOI] [PubMed] [Google Scholar]
- 81.Takeda A, Onodera H, Sugimoto A, Kogure K, Obinata M, Shibahara S. Coordinated expression of messenger RNAs for NGF, BDNF, and NT-3 in the rat hippocampus following transient forebrain ischemia. Neuroscience. 1993;55:23–31. doi: 10.1016/0306-4522(93)90451-k. [DOI] [PubMed] [Google Scholar]
- 82.Tanuichi M, Clark HB, Johnson EM. Induction of nerve growth factor receptor in Schwann cells after axotomy. Proc Natl Acad Sci USA. 1986;83:4094–4098. doi: 10.1073/pnas.83.11.4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 84.Van der Zee CEEM, Fawcett J, Diamond J. Antibody to NGF inhibits collateral sprouting of septohippocampal fibers following entorhinal cortex lesion in adult rats. J Comp Neurol. 1992;326:91–100. doi: 10.1002/cne.903260108. [DOI] [PubMed] [Google Scholar]
- 85.Vige X, Costa E, Wise BC. Mechanism of nerve growth factor mRNA regulation by interleukin-1 and basic fibroblast growth factor in primary cultures of rat astrocytes. Mol Pharmacol. 1991;40:186–192. [PubMed] [Google Scholar]
- 86.Walicke PA. Basic and acidic fibroblast growth factors have trophic effects on neurons from multiple CNS regions. J Neurosci. 1988;8:2618–2627. doi: 10.1523/JNEUROSCI.08-07-02618.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wanaka A, Johnson EM, Milbrandt J. Localization of FGF receptor mRNA in the adult rat central nervous system by in situ hybridization. Neuron. 1990;5:267–281. doi: 10.1016/0896-6273(90)90164-b. [DOI] [PubMed] [Google Scholar]
- 88.Weskamp G, Gasser UE, Dravid AR, Otten U. Fimbria-fornix lesion increases nerve growth factor content in adult rat septum and hippocampus. Neurosci Lett. 1986;70:121–126. doi: 10.1016/0304-3940(86)90449-0. [DOI] [PubMed] [Google Scholar]
- 89.Whittemore SR, Ebendal T, Larkfors L, Olson L, Seiger A, Stromberg I, Persson H. Developmental and regional expression of beta-nerve growth factor messenger RNA and protein in the rat central nervous system. Proc Natl Acad Sci USA. 1986;83:817–821. doi: 10.1073/pnas.83.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Whittemore SR, Larkfors L, Ebendal T, Holets VR, Ericsson A, Persson H. Increased β-nerve growth factor messenger RNA and protein levels in neonatal rat hippocampus following specific cholinergic lesions. J Neurosci. 1987;7:244–251. doi: 10.1523/JNEUROSCI.07-01-00244.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Whittemore SR, Friedman PL, Larhammar D, Persson H, Gonzalez-Carvajarl M, Holets VR. Rat β-nerve growth factor sequence and site of synthesis in the adult hippocampus. J Neurosci Res. 1988;20:403–410. doi: 10.1002/jnr.490200402. [DOI] [PubMed] [Google Scholar]
- 92.Williams LR, Vahlsing HL, Lindamood T, Varon S, Gage FH, Manthorpe M. A small-gauge cannula device for continuous infusion of exogenous agents into the brain. Exp Neurol. 1987;95:743–754. doi: 10.1016/0014-4886(87)90313-x. [DOI] [PubMed] [Google Scholar]
- 93.Woodroofe MN, Sarna GS, Wadhwa M, Hayes GM, Loughlin AJ, Tinker A, Cuzner ML. Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: evidence of a role for microglia in cytokine production. J Neuroimmunol. 1991;33:227–236. doi: 10.1016/0165-5728(91)90110-s. [DOI] [PubMed] [Google Scholar]
- 94.Woodward WR, Nishi R, Meshul CK, Williams TE, Coulombe M, Eckenstein FP. Nuclear and cytoplasmic localization of basic fibroblast growth factor in astrocytes and CA2 hippocampal neurons. J Neurosci. 1992;12:142–152. doi: 10.1523/JNEUROSCI.12-01-00142.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wool IG, Chan YL, Paz V, Olvera J. The primary structure of rat ribosomal proteins: the amino acid sequences of L27a and L28 and corrections in the sequences of S4 and S12. Biochem Biophys Acta. 1990;1050:69–73. doi: 10.1016/0167-4781(90)90143-p. [DOI] [PubMed] [Google Scholar]
- 96.Yoshida K, Gage FH. Fibroblast growth factors stimulate nerve growth factor synthesis and secretion by astrocytes. Brain Res. 1991;538:118–126. doi: 10.1016/0006-8993(91)90385-9. [DOI] [PubMed] [Google Scholar]
- 97.Yoshida K, Gage FH. Cooperative regulation of nerve growth factor synthesis and secretion in fibroblasts and astrocytes by fibroblast growth factor and other cytokines. Brain Res. 1992;569:14–25. doi: 10.1016/0006-8993(92)90364-f. [DOI] [PubMed] [Google Scholar]