

Abstract
Alzheimer’s disease (AD) is a devastating neurological disorder characterized by loss of cognitive skills and progressive dementia. The pathological hallmark of AD is the presence of numerous senile plaques throughout the hippocampus and cerebral cortex associated with degenerating axons, neurofibrillary tangles, and gliosis. The core of the senile plaque primarily is composed of the 39–43 amino acid β-amyloid peptide (Aβ), which forms fibrils of β-pleated sheets. Although considerable genetic evidence implicates Aβ in the pathogenesis of AD, a direct causal link remains to be established.
Senile plaques are foci of local inflammatory processes, as evidenced by the presence of numerous activated microglia and acute phase proteins. Aβ has been shown to elicit inflammatory responses in microglia; however, the intracellular events mediating these effects are largely unknown. We report that exposure of microglia and THP1 monocytes to fibrillar Aβ led to time- and dose-dependent increases in protein tyrosine phosphorylation of a population of proteins similar to that elicited by classical immune stimuli such as immune complexes. The tyrosine kinases Lyn, Syk, and FAK were activated on exposure of microglia and THP1 monocytes to Aβ, resulting in the tyrosine kinase-dependent generation of superoxide radicals. The present data support a role for oxidative damage in the pathogenesis of AD, provide an important mechanistic link between Aβ and the generation of reactive oxygen intermediates, and identify molecular targets for therapeutic intervention in AD.
Keywords: Alzheimer’s disease, β-amyloid, microglia, THP1 monocytes, signal transduction, tyrosine kinase, inflammatory, superoxide, piceatannol, RAGE, scavenger receptor
Dementia of the Alzheimer type is the most prevalent form of dementia of the aged. The pathological hallmark of Alzheimer’s disease (AD) is the presence of numerous senile plaques associated with degenerating neurons, neurofibrillary tangles (Selkoe, 1991), and marked gliosis throughout the hippocampus and cerebral cortex (Itagaki et al., 1989). The senile plaque is composed primarily of the β-amyloid protein (Aβ; Glenner and Wong, 1984; Masters et al., 1985). Aβ is a 39–43 amino acid peptide derived from the larger amyloid precursor protein (APP) as a result of proteolytic processing (Cole et al., 1989; Golde et al., 1992). Aβ forms fibrils that aggregate and form deposits comprising the core of senile plaques. Considerable genetic evidence has implicated Aβ in AD pathogenesis (Selkoe, 1996); however, the relationship between Aβ and neuronal death and gliosis is incompletely understood.
It has been postulated that the progressive pathology associated with AD is a consequence of local inflammatory reactions. This view has been supported by clinical studies demonstrating the efficacy of anti-inflammatory drug treatments in reducing the incidence of dementia (McGeer and McGeer, 1996). Microglia, the main immune effector cells within the brain (Leong and Ling, 1992), are the predominant glial cell type present within senile plaques (Itagaki et al., 1989). Microglia that are in direct contact with senile plaques exhibit an activated phenotype, as evidenced by elevated expression of HLA-DR, complement receptors, and immunoglobulin receptors (McGeer et al., 1989, 1993). In addition, acute phase proteins (Abraham et al., 1988; Griffin et al., 1989; McGeer et al., 1989; Cataldo and Nixon, 1990; Bauer et al., 1991) are present in AD-afflicted brain tissue at significantly elevated levels and are known to be secreted by reactive microglia (Araujo and Cotman, 1992). A critical question concerning the pathogenesis of AD is whether Aβ is directly capable of eliciting a local inflammatory response that is damaging to neurons.
The presence of reactive microglia and their secretory products within senile plaques suggest that microglia respond to constituents of the plaques, leading to the acquisition of an activated phenotype. The signal transduction pathways subserving the phenotypic changes mainly are unknown. Importantly, microglia that are in direct contact with senile plaques exhibit high levels of tyrosine-phosphorylated proteins (Wood and Zinsmeister, 1991), suggesting sustained activation of intracellular signaling processes. The activation of tyrosine kinases is the initial step in regulating a variety of cellular processes, including proliferation, differentiation, and inflammatory responses. Numerous inflammatory stimuli are known to activate tyrosine kinases such as Lyn and Syk in monocytes and macrophages, resulting in the release of cytokines and superoxide (Pfefferkorn and Fanger, 1989;Agarwal et al., 1993; Ghazizadeh et al., 1994; Crowley et al., 1996).
We report here that exposure of microglia and THP1 monocytes to fibrillar forms of Aβ resulted in the activation of tyrosine kinase-dependent intracellular signaling systems and the generation of superoxide radicals. These responses, however, are not linked to scavenger receptors or the receptor for advanced glycation end products, which recently have been shown to bind Aβ. These findings support a role for oxidative damage in the pathophysiology of AD and provide a mechanistic link between Aβ and the acquisition of an activated phenotype in microglia and the generation of local inflammatory responses. Moreover, identifying signal transduction pathways that are activated on exposure of the cells to Aβ provides molecular targets for therapeutic interventions in AD.
MATERIALS AND METHODS
Materials. The anti-phosphotyrosine antibody PY20 and the anti-paxillin mAb were obtained from Transduction Labs (Lexington, KY). The anti-phosphotyrosine antibody 4G10 was obtained from Upstate Biotechnology (Lake Placid, NY). Anti-FcγRI (mAb 32.2) and anti-FcγRII (mAb IV.3) were obtained from Medarex (Annendale, NJ). Affinity-purified polyclonal antisera to Lyn, Syk, and FAK were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Affinity-purified horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit antibody was obtained from Boehringer Mannheim (Indianapolis, IN). Goat anti-mouse F(ab)2 was obtained from Cappel (West Chester, PA) Peptides corresponding to amino acids 25–35 of Aβ (Aβ25–35), amino acids 1–28 of Aβ (Aβ1–28), and Substance P were obtained from American Peptide (Sunnyvale, CA). Nonfibrillar Aβ1–40 (Bachem, Philadelphia, PA) was prepared by dissociating fibrils in hexafluoroisopropanol, followed by lyophilization and reconstitution in sterile distilled water, and then used immediately. Fibrillar Aβ1–40 was prepared by reconstitution of the lyophilized peptide in sterile distilled water, followed by incubation for 1 week at 37°C. Aβ1–42 and scrambled Aβ25–35 (SC25–35; NAMGKILSGIG) were synthesized at Gliatech (Cleveland, OH). All peptides were solubilized in sterile distilled H2O. Serum amyloid A, human LDL, and acetylated human LDL were kind gifts of Dr. Frederick DeBeer (University of Kentucky). LPS, ferricytochrome C type III, nitroblue tetrazolium (NBT), and superoxide dismutase were obtained from Sigma (St. Louis, MO). Protein A-agarose, fatty acid-free BSA, and piceatannol were obtained from Boehringer Mannheim. BSA was maleylated as previously described (Haberland and Fogelman, 1985).
Cell culture. THP-1 cells were maintained in RPMI-1640 (Whittaker Bioproducts, Walkersville, MD) supplemented with 10% heat-inactivated fetal calf serum (FCS), 5 × 10−5m 2-mercaptoethanol, 5 mm HEPES, and 1.5 μg/ml gentamicin in an atmosphere of 5% CO2. Jurkat cells were maintained in the the same medium but without 2-mercaptoethanol. Microglia and astrocytes were derived from the brains of neonatal rats as previously described but with some modifications (Giulian and Baker, 1986). Cerebral cortices were isolated from postnatal day 0 (P0) Sprague Dawley rats, and meninges and blood vessels were removed completely. Cortices were minced with a sterile razor blade, and cells were dissociated in PBS containing 0.25% trypsin and 1 mm EDTA for 30 min at 25°C. Digestion was terminated by adding an equal volume of DMEM/F12 medium (Life Technologies, Gaithersburg, MD) containing 20% FCS, and cells were triturated to obtain a single-cell suspension. Cells were plated in 75 mm flasks coated with poly-l-lysine (0.1 mg/ml; Sigma) at a density of 5 × 107 cells per flask. Media were replaced the next day with DMEM/F12 containing 20% FCS. Cells were grown for 7 d without changing the medium to allow microglial proliferation. Microglia were harvested by shaking for 30 min on a rotary shaker at 120 rpm. Purity of cultures was determined by staining with the microglial marker Griffonia simplicifoliaIsolectin B4 (Sigma). Astrocytes were recovered after removal of microglia and passaged three times, generating highly enriched cultures of astrocytes.
Cell stimulation. Tissue culture dishes were coated with nitrocellulose (Lagenaur and Lemmon, 1987) and derivatized by adding 48 pmol/mm2 Aβ peptides in distilled water and allowed to dry. Microglia (5 × 106 cells), astrocytes (5 × 106 cells), THP-1 cells (1 × 107), and Jurkat cells (1 × 107 cells) were resuspended in 1 ml of HBSS and added to underivatized dishes or dishes derivatized with immobilized peptide for 5 min. In some cases the cells were stimulated by adding peptides or stimulants in solution. High-affinity (FcγRI) and low-affinity (FcγRII) immunoglobulin receptors were cross-linked by incubation of 1 × 107 THP1 monocytes with 2 μg of anti-FcγRI or anti-FcγRII in ice-cold RPMI for 30 min. Cells were pelleted and resuspended in HBSS (37°C) in the absence or presence of 10 μg of goat anti-mouse F(ab)2 for 5 min. Then cells were lysed in 0.5 ml of ice-cold Triton buffer [1% Triton X-100 and (in mm): 10 Tris, pH 7.4, 140 NaCl, 1 Na3VO4, 10 NaF, 1 EDTA, 1 EGTA, and 2 PMSF].
Immunoprecipitation and Western blotting. Protein content of lysates was quantitated by the method of Bradford, using BSA as a standard (Bradford, 1976). Triton buffer lysates were precleared by incubation with 10 μl of protein A-agarose for 30 min and then incubated with 1 μg of antibody per milligram of lysate protein for 2 hr, followed by adding 10 μl of 50% (v/v) protein A-agarose for 1 hr at 4°C. The immunoprecipitates were washed three times in lysis buffer. In some instances the phosphotyrosine-containing proteins were eluted from the immune complex by adding 40 mmp-nitrophenylphosphate (PNP). Otherwise, the immune complexes were solubilized directly in Laemmli sample buffer, boiled for 5 min, and then resolved by SDS/PAGE under reducing conditions. For Western blot analysis of Lyn, cells were lysed in 300 μl of ice-cold RIPA buffer ]1% Triton, 0.1% SDS, 0.5% deoxycholate, and (in mm): 20 Tris, pH 7.4, 150 NaCl, 10 NaF, 1 Na3VO4, 1 EDTA, 1 EGTA, and 2 PMSF]. Insoluble material was removed by centrifugation at 10,000 × gat 4°C for 10 min. Laemmli sample buffer was added to RIPA lysates, which were resolved by SDS/PAGE under reducing conditions. The proteins were transferred to PolyScreen membranes (DuPont NEN, Boston, MA) and then blocked in TBS-T (10 mm Tris, pH 7.5, 100 mm NaCl, and 0.05% Tween 20) containing 3% BSA overnight at 4°C. The blots were incubated with the appropriate primary antibody for 1 hr, washed three times in TBS-T, incubated for 1 hr with goat anti-mouse or goat anti-rabbit antibodies conjugated to HRP in TBS-T plus 5% nonfat dried milk, and washed three times in TBS-T, followed by detection with an enhanced chemiluminescence (ECL) detection system (DuPont NEN). In some instances blots were stripped by incubation for 30 min at 50°C in stripping buffer (62.5 mm Tris, pH 6.8, 100 mm β-mercaptoethanol, and 2% SDS) and reprobed with other antibodies.
Tyrosine kinase assays. THP1 monocytes were stimulated with Aβ25–35, and tyrosine-phosphorylated proteins were immunoprecipitated with PY20 as described above. Tyrosine kinase activity then was assayed by phosphorylation of polyGluTyr (PGT; 4:1 Glu, Tyr; Sigma) (Guan and Shalloway, 1992). PGT assays were performed by adding 10 μl (0.1 μg) aliquots of phosphotyrosine-containing proteins eluted with 100 μl of Triton buffer containing 40 mm PNP, which were added to 40 μl of PGT kinase buffer (50 mm Tris, pH 7.4, 10 mm MnCl2, 10 μm ATP, and 30 cpm/fmol [32P]ATP) and incubated at 25°C for 30 min. Reactions were terminated by adding Laemmli sample buffer and were boiled for 5 min. Samples were resolved by SDS/PAGE under reducing conditions, and radioactivity was quantitated by Cerenkov counting.
In vitro kinase assays were performed with THP1 monocytes, which were grown for 2 d in media alone or supplemented with 50 ng/ml lipopolysaccharide (LPS; Sigma), and then stimulated as described above and lysed in Triton buffer (Ghazizadeh et al., 1994). Phosphotyrosine-containing proteins were immunoprecipitated, and the immunoprecipitates were washed three times in Triton buffer and once in HEPES buffer containing (in mm) 25 HEPES, pH 7.4, 150 NaCl, and 1 Na3VO4, followed by incubation in kinase buffer (25 mm HEPES, pH 7.4, 10 mmMnCl2, 1 μm ATP, and 150 cpm/fmol [32P]ATP) for 5 min at 25°C in a final volume of 40 μl. Reactions were terminated by adding Laemmli buffer and were boiled for 5 min. Incorporated radioactivity was quantitated by Cerenkov counting of the excised gel lane.
The enzymatic activity of Lyn was analyzed from unstimulated THP1 monocytes or THP1 monocytes stimulated on 60 pmol/mm2surface-bound Aβ25–35 (Ghazizadeh et al., 1994).32P-labeled anti-phosphotyrosine immune complexes were obtained as described for in vitro kinase assays. Reactions were terminated, and immune complexes were dissociated by adding 70 μl of Triton buffer containing 3% SDS and were boiled for 3 min. Eluted proteins were diluted 10-fold with Triton buffer and reimmunoprecipitated with 1 μg of anti-Lyn antisera and 10 μl of protein A-agarose for 3 hr. Proteins were resolved by SDS/PAGE under reducing conditions, and 32P-labeled proteins were detected by autoradiography.
Measurement of superoxide production.O2− production was measured by the reduction of ferricytochrome C, as previously described (Pick, 1986). Microglia (2 × 105 cells) were added to 48-well tissue culture dishes and allowed to adhere overnight, whereas THP1 monocytes (5 × 105 cells) were added to dishes in 0.5 ml HBSS containing 80 μm ferricytochrome C type III immediately before use. Media were removed from microglia, and stimulants were added in 0. 5 ml of HBSS containing 80 μm ferricytochrome C type III (Sigma) in duplicate and incubated for 90 min. Stimulants were added directly to the THP1 monocytes in duplicate and incubated for 90 min. The specificity of the reaction was verified by adding 40 μg of superoxide dismutase (Sigma) to one set of samples. In some instances THP1 monocytes and microglia were pretreated for 1 hr with 25 μg/ml piceatannol, followed by stimulation of the cells with peptides in the presence of piceatannol. The medium was collected, and production of superoxide was determined spectrophotometrically by measurement of the reduction of ferricytochrome C at 550 nm and converted to moles of O2−, using an extinction coefficient of 21 × 103m−1cm−1. Production of O2− is expressed as nanomole O2− per 90 min per 2 × 105cells. Intracellular production of O2− was assayed by the reduction of NBT, as previously described (Pick, 1986). THP1 monocytes (1 × 106) were suspended in 0.5 ml of HBSS containing 1 mg/ml NBT. Stimulants were added to cells, followed by incubation at 37°C for 10 min. Cells were pelleted rapidly, supernatants were removed, and cells were lysed by sonication in RIPA buffer to release reduced NBT precipitates. Reduction of NBT was measured by the change in absorbance at 550 nm. Cells treated with vehicle only served as blanks. Assays were performed in duplicate.
RESULTS
Aβ stimulates tyrosine phosphorylation in microglia and THP1 monocytes
We tested whether Aβ could initiate intracellular signaling events via the stimulation of the activity of tyrosine kinases by exposing primary cultures of rat microglia and astrocytes, as well as human THP1 monocytes and Jurkat cells, to Aβ peptides that had been immobilized on the surface of tissue culture dishes. The interaction of microglia and THP1 monocytes with Aβ1–40 or a peptide derived from the C terminus of Aβ, Aβ25–35, elicited a rapid and dramatic increase in tyrosine phosphorylation of a similar population of proteins (Fig. 1A,B). Astrocytes and Jurkat cells were unresponsive, demonstrating that Aβ-stimulated protein tyrosine phosphorylation was specific to cells within the microglial lineage (Fig. 1A).
Fig. 1.
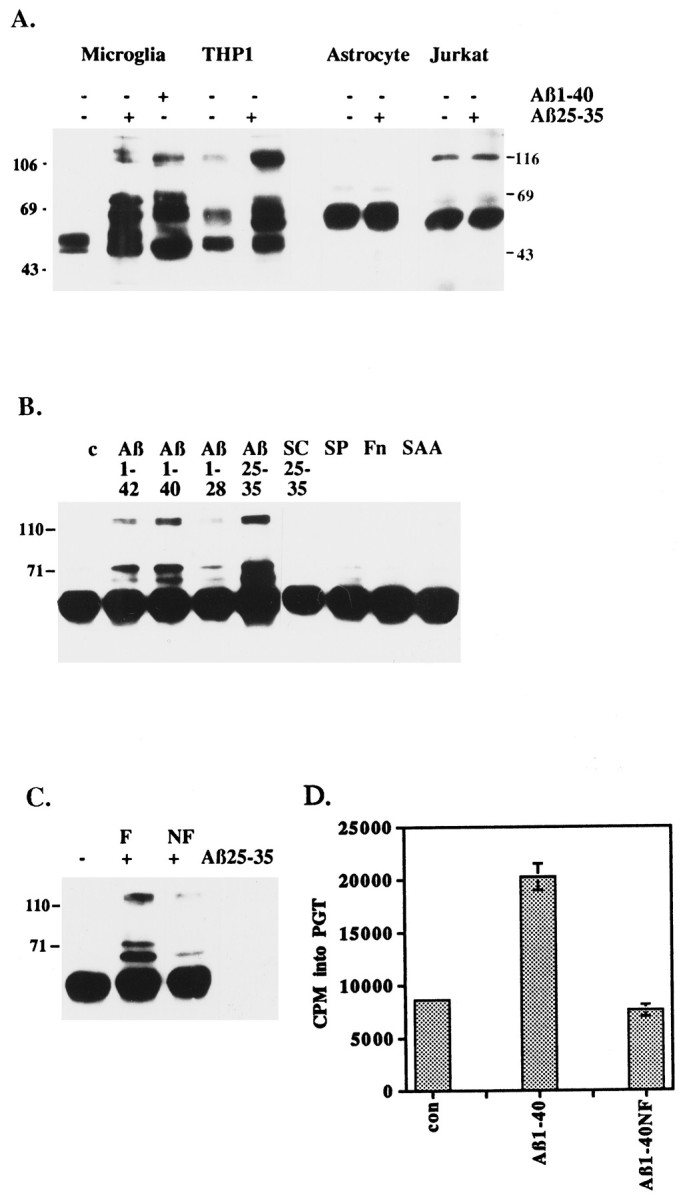
Fibrillar Aβ peptides stimulate increased tyrosine phosphorylation and tyrosine kinase activity in microglia and THP1 monocytes. A, Exposure of microglia and THP1 monocytes, but not astrocytes and Jurkat cells, to Aβ peptides leads to increased protein tyrosine phosphorylation. Western blot analysis of tyrosine phosphoproteins was performed on phosphotyrosine immunoprecipitates from primary rat microglia and astrocytes, human THP1 monocytes, and Jurkat T-lymphocytes. The cells were exposed for 5 min to Aβ peptides (48 pmol/mm2) immobilized on the surface of tissue culture dishes or to an underivatized surface. The broad band at 55 kDa is IgG heavy chain. B, Ligand specificity of Aβ-stimulated tyrosine phosphorylation in THP1 monocytes. Tissue culture dishes were underivatized (c) or derivatized with 48 pmol/mm2 Aβ1–42, Aβ1–40, Aβ1–28, Aβ25–35, scrambled Aβ25–35 (SC25–35; NAMGKILSGIG), substance P (SP), serum amyloid A (SAA), or 20 μg/ml fibronectin (Fn). THP1 monocytes were exposed to the immobilized substrates or an underivatized surface for 5 min, followed by Western blot analysis with anti-phosphotyrosine antibodies. The broad band at 55 kDa is IgG heavy chain. C, Stimulation of protein tyrosine phosphorylation in THP1 monocytes by fibrillar Aβ25–35, but not nonfibrillar Aβ25–35. THP1 monocytes were incubated for 5 min in the absence (−) or presence (+) of 20 μm fibrillar (F) Aβ25–35 or nonfibrillar (NF) Aβ25–35 in suspension. Western blot analysis of tyrosine phosphoproteins was performed on phosphotyrosine immunoprecipitates. The broad band at 55 kDa is IgG heavy chain. D, Fibrillar Aβ1–40 stimulates tyrosine kinase activity in THP1 monocytes. Tyrosine kinase activity in THP1 monocytes was measured after a 5 min exposure of the cells to fibrillarAβ1–40 or nonfibrillar Aβ1–40 (Aβ1–40NF) immobilized on a dish. Tyrosine kinase activity was measured from phosphotyrosine immunoprecipitates, with polyGluTyr as a substrate. Proteins were resolved by SDS-PAGE, the gel was dried, and the lanes were cut and subjected to Cerenkov counting. The positions of the molecular weight standards (kDa) are indicated.
Aβ contained within senile plaques primarily consists of Aβ1–42 and Aβ1–40 in a fibrillar β-pleated sheet conformation (Masters et al., 1985). THP1 monocytes were exposed to fibrillar Aβ1–42 and Aβ1–40, which had been immobilized on tissue culture dishes. Aβ induced markedly elevated levels of protein tyrosine phosphorylation (Fig. 1B). Then the biologically active domain within Aβ was defined with peptides derived from the N terminus (Aβ1–28) and C terminus (Aβ25–35) of Aβ. The active domain of Aβ was found to be restricted to amino acids 25–35, a region capable of forming β-pleated sheets (Terzi et al., 1994). Control peptides included scrambled Aβ25–35 (SC25–35), substance P (SP), fibronectin, and serum amyloid A (SAA). These peptides elicited no alteration in protein tyrosine phosphorylation.
The dependence of the conformation of Aβ on its ability to induce tyrosine phosphorylation in THP1 monocytes was examined. A fibrillar conformation of Aβ25–35 was far more effective in stimulating tyrosine phosphorylation than nonfibrillar Aβ25–35 (Fig.1C). Similarly, the activation of tyrosine kinase activity was dependent on exposure of the cells to fibrillar Aβ1–40 (Fig.1D). Exposure of the cells to nonfibrillar Aβ1–40 had no effect on the enzymatic activity of tyrosine kinases. These observations are consistent with previous studies, which have shown that a fibrillar confirmation of Aβ is critical for biological responses such as neurotoxicity (Pike et al., 1990) and stimulation of IL-1β release from THP1 monocytes (Lorton et al., 1996).
Time course and dose–response of Aβ-stimulated tyrosine phosphorylation
Exposure of THP1 monocytes to surface-bound Aβ rapidly stimulated protein tyrosine phosphorylation, reaching maximal levels in 5 min and returning to basal levels by 30 min, as measured by Western blot analysis with anti-phosphotyrosine antibodies. Aβ-induced tyrosine kinase activity was measured in parallel assays by an enzymatic assay of the tyrosine kinases by phosphorylation of the substrate PGT. Aβ stimulated rapid activation of tyrosine kinase activity with similar kinetics, reaching peak levels in 5 min (Fig.2A). We did not consistently observe significant increases in tyrosine kinase activity from 30 to 60 min.
Fig. 2.

Time course and dose–response of Aβ25–35-stimulated tyrosine phosphorylation in THP1 monocytes.A, THP1 monocytes were stimulated for the indicated times on 60 pmol/mm2 Aβ25–35 bound to tissue culture dishes. Tyrosine-phosphorylated proteins were immunoprecipitated with anti-phosphotyrosine mAb (PY20), followed by elution with 40 mmp-nitrophenylphosphate. Proteins (0.6 μg) were resolved by SDS-PAGE, transferred to polyvinylidene fluoride (PVDF), and subjected to Western blot with anti-phosphotyrosine mAb (PY20). In parallel assays, proteins (0.1 μg) were analyzed in a kinase assay by phosphorylation of the tyrosine kinase substrate polyGluTyr (PGT). B, THP1 monocytes were stimulated with the indicated quantities of Aβ25–35, surface-bound or in solution, for 5 min. Tyrosine-phosphorylated proteins were immunoprecipitated with anti-phosphotyrosine mAb (PY20), resolved by SDS-PAGE, transferred to PVDF, and subjected to Western blot with anti-phosphotyrosine mAb (4G10). The broad band at 55 kDa is IgG heavy chain.
The dose–response of Aβ-stimulated tyrosine phosphorylation was examined. The response of THP1 monocytes to surface-bound fibrillar Aβ or fibrillar Aβ in solution was compared. Fibrillar Aβ25–35 added in solution to THP1 monocytes stimulated maximal tyrosine phosphorylation between 80–100 μm Aβ25–35, and surface-bound Aβ25–35 stimulated maximal levels of tyrosine phosphorylation in THP1 monocytes at a peptide density between 80–100 pmol/mm2 (Fig. 2B). However, fibrillar Aβ added in solution consistently stimulated greater increases in tyrosine phosphorylation at lower quantities of peptide than surface-bound Aβ. This observation may suggest that Aβ in solution is more potent than surface-bound Aβ or that similar quantities of Aβ may be more accessible to cells in solution than bound to a surface. We did not consistently detect any further stimulation of tyrosine phosphorylation at higher levels of peptides (data not shown). The maximal response was elicited by similar quantities of Aβ25–35 presented to THP1 monocytes in suspension (1 ml, 80 μm = 80 nmol) or surface-bound (80 pmol/mm2 = 77 nmol). Repeated examination of Aβ-stimulated tyrosine phosphorylation did not reveal significant qualitative or quantitative differences in the populations of tyrosine-phosphorylated proteins observed in THP1 monocytes when stimulated with maximal quantities of Aβ in solution or bound to a surface.
Aβ and activation of immunoglobulin G receptors elicits tyrosine phosphorylation of a common population of proteins
Monocytes respond to a variety of immune stimuli by activation of tyrosine kinases. A direct comparison of the response of THP1 monocytes to Aβ and to activation of the high- and low-affinity immunoglobulin receptors (FcγRI and FcγRII) revealed that these stimuli induced phosphorylation of a similar population of proteins, suggesting that Aβ activates common elements within an inflammatory response pathway (Fig. 3). Therefore, we initiated studies to establish the identities of several of the tyrosine kinases activated on exposure of microglia and THP1 monocytes to Aβ.
Fig. 3.

Comparison of tyrosine phosphorylation in THP1 monocytes stimulated with Aβ, FcγRI (RI), or FcγRII (RII). THP1 monocytes were stimulated for 5 min with 60 pmol/mm2Aβ25–35 bound to a tissue culture dish, mAb 32.2 (anti-FcγRI), mAb 32.2 (anti-FcγRI) cross-linked with goat anti-mouse F(ab)2, mAb IV.3 (anti-FcγRII), or mAb IV.3 (anti-FcγRII) cross-linked with goat anti-mouse F(ab)2. Tyrosine phosphoproteins were analyzed by immunoprecipitation, followed by Western blot with anti-phosphotyrosine mAb (4G10).
Identification of Aβ-stimulated tyrosine-phosphorylated proteins
Activation of monocytes by immune stimuli, such as FcγRI and FcγRII cross-linking, results in the activation of Src family tyrosine kinases such as Lyn (Kiener et al., 1993; Ghazizadeh et al., 1994). To determine whether Aβ activated Lyn, we exposed microglia and THP1 monocytes to Aβ25–35. Src family tyrosine kinases have been shown to associate with the Triton-insoluble cytoskeleton (Clark and Brugge, 1993), so Lyn was extracted from the cytoskeleton by incubation in RIPA buffer. RIPA lysates were analyzed by Western blot with anti-phosphotyrosine mAb, followed by stripping the blot and reprobing with anti-Lyn antisera. In parallel assays, the effect of Aβ25–35 on the enzymatic activity of Lyn was examined from Triton lysates of THP1 monocytes that were sonicated briefly to disrupt cytoskeleton. Lysates were immunoprecipitated with anti-phosphotyrosine mAb, and immunoprecipitates were incubated with [32P]ATP.32P-labeled proteins were eluted from immune complexes by boiling 3 min in Triton buffer containing 3% SDS and were diluted 10-fold in Triton buffer. The proteins were reimmunoprecipitated with anti-Lyn antisera, and immunoprecipitates were resolved by SDS-PAGE and analyzed by autoradiography. Exposure of microglia and THP1 monocytes to Aβ25–35 stimulated both the tyrosine phosphorylation and enzymatic activity of Lyn (Fig.4A,B).
Fig. 4.
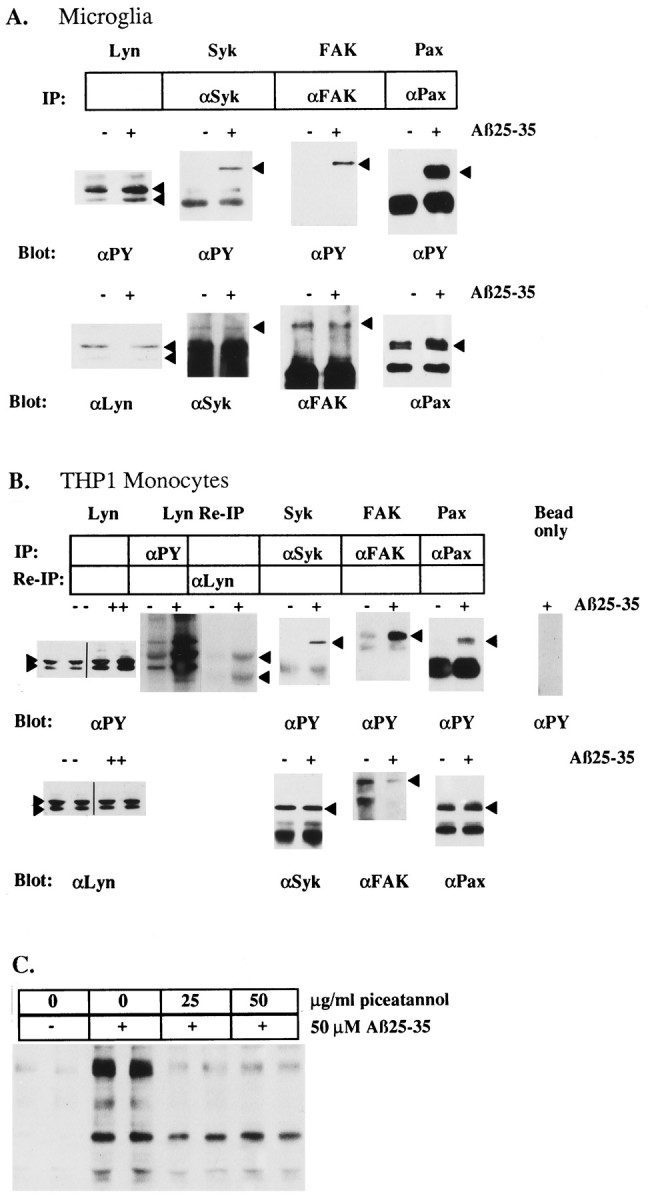
Identification of Aβ25–35-stimulated tyrosine-phosphorylated proteins in microglia and THP1 monocytes and the effect of the tyrosine kinase inhibitor piceatannol.A, Aβ25–35-stimulated tyrosine phosphorylation ofLyn, Syk, FAK, and paxillin (Pax) in primary cultures of rat microglia. B, THP1 monocytes were evaluated by immune precipitation and Western blot analysis with the indicated antibodies. Lyn identification and enzymatic activation in THP1 monocytes also was evaluated by immunoprecipitation with an anti-phosphotyrosine antibody, followed by incubation of the immune complex with [32P]ATP. The radiolabeled Lyn was released from the immune complex and then reprecipitated with an anti-Lyn antibody and visualized by autoradiography. Arrowheadsdenote migration of the respective proteins. C, THP1 monocytes were pretreated for 1 hr with the indicated amounts of piceatannol. Then cells were exposed to 50 μm Aβ25–35 for 5 min. Cellular lysates were resolved by SDS-PAGE, transferred to PVDF, and subjected to Western blot with anti-phosphotyrosine mAb (4G10).
The cytosolic tyrosine kinase Syk is an important signaling component that is tyrosine-phosphorylated and enzymatically activated by a variety of inflammatory stimuli in monocytes and macrophages (Greenburg et al., 1994; Crowley et al., 1996). Aβ stimulation of microglia and THP1 monocytes led to increased tyrosine phosphorylation of Syk (Fig.4A,B). Pretreatment of THP1 monocytes with piceatannol, a tyrosine kinase inhibitor that preferentially inhibits Syk (Oliver et al., 1994), significantly reduced Aβ-stimulated tyrosine phosphorylation of a subset of cellular proteins, which included p72Syk (Fig. 4C). Thus, Aβ activated the tyrosine kinases, Lyn and Syk, which are the most proximal catalytic elements comprising a signal transduction pathway mediating the activation of these cells.
Microglia are the predominant glial cell type found within the senile plaques (Itagaki et al., 1989), suggesting that microglia are likely to migrate to and interact with these structures. The cytosolic tyrosine kinase, FAK, has been shown to be important in cellular adhesion, migration, and inflammatory responses (Furuta et al., 1995;Hamawy et al., 1995; Ilic et al., 1995). Examination of phosphotyrosine-labeled proteins from Aβ-stimulated microglia and THP1 monocytes revealed a prominent 125 kDa phosphoprotein, the tyrosine phosphorylation of which was stimulated markedly by exposure to Aβ bound to surfaces or added in solution (Fig.2B). This protein was identified as p125FAK by immunoprecipitation with anti-FAK antisera, followed by anti-phosphotyrosine blot (Fig.4A,B).
One of the primary targets of FAK and the src family tyrosine kinases such as Lyn is the cytoskeletal-associated protein, paxillin (Minoguchi et al., 1994; Bellis et al., 1995). Paxillin has been shown to be involved in linking membrane proteins and signaling molecules to the actin cytoskeleton and colocalizes at focal adhesions and phagolysosomes in macrophages (Greenburg et al., 1994). Exposure of microglia and THP1 monocytes to Aβ25–35 resulted in a dramatic stimulation of the tyrosine phosphorylation of paxillin in both cell types (Fig. 4A,B).
Stimulation of respiratory burst in microglia and THP1 monocytes by Aβ
To establish whether Aβ is capable of directly activating microglia and stimulating the release of potentially harmful inflammatory products, we studied the effects of Aβ on the production of superoxide radicals in microglia and THP1 monocytes. Adding Aβ peptides to microglia and THP1 monocytes resulted in the production of O2−, which was blocked by the presence of superoxide dismutase (Fig. 5A). The stimulation of O2− release was dependent on addition of fibrillar Aβ1–40 and Aβ25–35. Adding nonfibrillar Aβ25–35 to microglia stimulated 50% less O2− production than fibrillar Aβ25–35, and this modest response is most likely a consequence of aggregation of Aβ25–35 during the 90 min time course of the assay (Fig. 5B; Terzi et al., 1994). Importantly, pretreatment of microglia and THP1 monocytes with piceatannol, the Syk-selective tyrosine kinase inhibitor, blocked the production of superoxide, demonstrating that Aβ-stimulated superoxide production is linked to the activation of Syk or kinases downstream of Syk (Fig.5C).
Fig. 5.

Fibrillar Aβ peptide stimulates respiratory burst in microglia and THP1 monocytes. A, Rat microglia were incubated in the absence (c) or presence of 60 μm fibrillar Aβ1–40, Aβ25–35, or scrambled Aβ25–35 (SC25–35). The peptides were added alone or in the presence of 40 μg of superoxide dismutase (SOD), and O2− production was measured. THP1 monocytes were incubated in absence (c) or presence of 60 μmAβ25–35. B, Fibrillar, but not nonfibrillar, Aβ25–35 stimulated release of O2−from rat microglia. Microglia were incubated with 40 μmfibrillar Aβ25–35 or 40 μm nonfibrillar Aβ25–35 (Aβ25–35NF), 1 μg/ml phorbol myristate acetate (PMA), or vehicle only (c) in duplicate wells for 90 min. C, Aβ25–35-stimulated release of O2− from THP1 monocytes and microglia is blocked by piceatannol. Microglia and THP1 monocytes were pretreated for 1 hr with ± 25 μg/ml piceatannol (P). Cells were unstimulated (con) or stimulated for 90 min with 60 μm Aβ25–35 ± 25 μg/ml piceatannol. Supernatants were collected, and production of superoxide was determined spectrophotometrically by measurement of the reduction of ferricytochrome C at 550 nm and converted to moles of O2−, using an extinction coefficient of 21 × 103m−1 cm−1. Production of O2− is expressed as nanomole O2− per 90 min per 2 × 105cells.
Effect of scavenger receptor and advanced glycation end product receptor ligands on tyrosine phosphorylation and respiratory burst
Recently, Aβ was shown to interact at the cell surface with class A scavenger receptors and the receptor for advanced glycation end products (RAGE; El Khoury et al., 1996; Yan et al., 1996), both of which have been linked to the production of reactive oxygen species. RAGE is postulated to immobilize Aβ at the cell surface, where Aβ then generates reactive oxygen species extracellularly (Hensley et al., 1994). The scavenger receptor, which binds both Aβ and advanced glycation end products, has been shown to mediate adhesion of microglia to Aβ, resulting in the generation of reactive oxygen species. The intracellular signal transduction pathways activated by scavenger receptors are not well described, and RAGE has not been shown to be linked to signaling pathways or to elicit cellular effects. We tested whether binding of Aβ to these receptors was likely to be responsible for activation of tyrosine kinases and subsequent generation of reactive oxygen species. THP1 monocytes were exposed to the scavenger receptor ligands, maleylated-BSA and acetylated-LDL, and the RAGE ligand, glycated BSA in combination with iron-saturated lactoferrin. These ligands failed to elicit de novo increases in protein tyrosine phosphorylation. Conversely, Aβ stimulated dramatic increases in protein tyrosine phosphorylation of multiple proteins (Fig. 6A). Because Aβ and glycated proteins have been postulated to generate oxygen radicals (Sakurai and Tsuchiya, 1988; Hensley et al., 1994) spontaneously, the intracellular reduction of NBT was used to assay cell-dependent generation of superoxide (Pick, 1986). Aβ was a potent stimulus of generation of intracellular superoxide radicals. We were unable to detect scavenger receptor ligand and RAGE ligand-stimulated intracellular superoxide production (Fig. 6B). We were, however, able to measure a modest level of scavenger receptor-mediated superoxide production from phorbol ester-differentiated THP1 monocytes (data not shown). These data indicate that Aβ is likely to activate different signaling pathways in monocytic cells than classical scavenger receptor and RAGE ligands. Also, we have been unable to implicate FcγRI, FcγRII, and tachykinin receptors directly in this response (data not shown). The identity of the microglial receptor(s) subserving these rapid effects of fibrillar Aβ remains unknown.
Fig. 6.

Effect of scavenger receptor and RAGE ligands on protein tyrosine phosphorylation and intracellular respiratory burst.A, THP1 monocytes were stimulated with 50 μm Aβ25–35 (Aβ) or 20 μg/ml ofLDL, acetylated LDL (Ac-LDL), maleylated BSA (m-BSA), BSA, BSA plus lactoferrin (BSA Lac), glycated BSA (AGE), or glycated BSA plus lactoferrin (AGE Lac) for 2 min in HBSS. Cells were lysed in RIPA buffer, equal quantities of protein (50 μg) were resolved by SDS-PAGE, and proteins were transferred to PVDF. Tyrosine-phosphorylated proteins were detected by Western blot with 4G10. B, THP1 monocytes were stimulated with 50 μm Aβ25–35 (Aβ) acetylated LDL (Ac-LDL), maleylated BSA (m-BSA), or glycated BSA plus lactoferrin (AGE-L) for 10 min in HBSS containing nitroblue tetrazolium. Cells were pelleted, supernatants were removed, and cells were lysed by sonication in RIPA buffer. Generation of superoxide was measured by the change in absorbance of reduced NBT at 550 nm.
DISCUSSION
We report that microglia are able selectively to detect fibrillar forms of Aβ, resulting in the activation of intracellular signaling cascades. These observations provide an important mechanistic link in understanding how these cells acquire an activated phenotype in the AD brain. It is of particular interest that the signaling pathways activated in response to Aβ also are used by these cells (and other cells of this lineage) to respond to conventional immune stimuli and result in common biological effects such as cytokine secretion (Debets et al., 1988) and superoxide production (Pfefferkorn and Fanger, 1989). Macrophages and microglia express a complex set of cell-surface proteins that mediate inflammatory responses to a wide variety of immune stimuli. Scavenger receptors or RAGE does not seem to mediate the rapid effects of Aβ on the activation of tyrosine kinases and downstream signaling events leading to the generation of superoxide. These data provide evidence that Aβ may interact with other membrane proteins linked to intracellular signal transduction pathways. These findings also suggest that scavenger receptor and RAGE occupancy elicit the production of reactive oxygen species through mechanistically distinct pathways that have not been defined.
Activation of tyrosine kinases of the src family, such as Lyn and Lck, is an initial step in signal transduction cascades of several immune responses in monocytes, macrophages, and lymphocytes. Stimulation of microglia and THP1 monocytes with Aβ also has led to activation of Lyn. Lyn is associated constitutively with the cytoplasmic domain of the FcγRII (Ghazizadeh et al., 1994) receptor as well as the gamma subunit of FcγRI and FcεRI (Wang et al., 1994) and becomes activated on clustering of the receptors by immune complexes. Lyn then phosphorylates these receptors on two tyrosine residues within the tyrosine activation motif (TAM), allowing the interaction and activation of downstream effector molecules such as members of Syk/ZAP70 tyrosine kinase family (Burkhardt et al., 1994; Shiue et al., 1995).
The tyrosine kinases of the Syk/ZAP70 family are also critical components for several immune responses in monocytes, macrophages, and lymphocytes and have been shown to be essential to B-cell maturation (Cheng et al., 1995; Turner et al., 1995; Crowley et al., 1996). Syk is activated by binding to phosphotyrosine residues within TAMs present on the cytoplasmic domain of the receptor or receptor subunits (Shiue et al., 1995). It is also possible that Syk may be activated by a direct interaction with Lyn, because Syk has been shown to be coimmunoprecipitated with Lyn (Sidorenko et al., 1995).
Syk has been shown to be essential for phagocytosis of immune complexes (Indik et al., 1995). Exposure of microglia and THP1 monocytes to Aβ led to tyrosine phosphorylation of p72Syk. It is not known, however, whether the Aβ-stimulated activation of Syk represents an early step in the phagocytosis of Aβ by microglia. Microglia in culture have been shown to be capable of phagocytosis of Aβ (Frackowiak et al., 1992); however, it remains unclear whether microglia are capable of phagocytosing Aβ-comprising senile plaques (Frautschy et al., 1992;el-Hachimi and Fonchin, 1994).
The tyrosine kinase inhibitor, piceatannol, has been shown to inhibit Syk (Oliver et al., 1994) selectively. The specific action of this drug has established the requirement for Syk in the IgE-stimulated release of serotonin from RBL-2H3 rat mast cells. We have observed that one of the biological consequences of microglial interactions with Aβ is the activation of an inflammatory response, evidenced by the generation of superoxide radicals. Superoxide radicals are produced via the action of NADPH oxidase, a multisubunit complex, the assembly of which is stimulated by extracellular stimuli. Significantly, protein tyrosine phosphorylation has been shown to be critical for the activation of NADPH oxidase, presumably via activation of downstream signaling cascades, including the MAP kinases (Dusi et al., 1994). Phosphosphorylation of p47phox is essential for the assembly of the NADPH oxidase complex (Curnutte et al., 1994). The Aβ-stimulated production of superoxide radicals was arrested by pretreatment of microglia and THP1 monocytes with piceatannol. Therefore, inhibition of the activity of Syk or other elements within this signaling pathway represents molecular targets for blockade of microglial inflammatory responses to Aβ.
FAK has been shown to be involved in regulating integrin-mediated cellular adhesion and migration. Activation of FAK is known to occur at focal adhesions and mediates the assembly of large protein complexes linking the cytoskeleton to membrane signal transduction machinery in numerous cell types (Clark and Brugge, 1995), including monocytes (Lin et al., 1995). Microglia are the predominant cell type found within the core of senile plaques, suggesting that microglia must migrate to and interact with components of the plaques. We have demonstrated that the interaction of microglia and THP1 monocytes with either surface-bound fibrillar Aβ or fibrillar Aβ in solution stimulated the tyrosine phosphorylation of FAK. Thus, it is likely that activation of FAK is involved in microglial adhesion to and migration on Aβ within senile plaques. In addition, Aβ-stimulated tyrosine phosphorylation of FAK may affect its interactions with other components of the Aβ-stimulated signal transduction pathway by serving as a site for the formation of signaling complexes (Clark and Brugge, 1995).
A consequence of microglial Aβ interactions is the activation of a complex signal transduction cascade resulting in the production of superoxide, which was found to be dependent on interaction of the cells with fibrillar conformations of Aβ. This finding is significant, because AD pathology is associated with mature senile plaques possessing Aβ in a fibrillar or β-pleated sheet conformation. In addition, other investigators have found that the biological effects of Aβ, such as neurotoxicity (Pike et al., 1990) and stimulation of IL-1β release from THP1 monocytes (Lorton et al., 1996), are dependent on a fibrillar conformation of the peptide. It is unlikely that the responses to fibrillar Aβ described here are mediated by RAGE, because it was identified on the basis of its ability to bind Aβ monomers.
Importantly, previous activation of microglia and THP1 monocytes was not necessary for Aβ to induce release of superoxide. The ability of Aβ to elicit the production of reactive oxygen species is consistent with a previous report showing that Aβ stimulated the release of NO2− from primary cultures of rodent microglia (Meda et al., 1995). However, the latter response was entirely dependent on priming microglia with INFγ. In addition, the role of NO2− in human macrophage responses to inflammatory stimuli remains controversial (Albina, 1995), and recent studies have failed to detect iNOS mRNA in cultures of activated human microglia (Walker et al., 1995). The present data demonstrate that Aβ alone is sufficient to activate complex intracellular signaling processes in microglia, resulting in the release of toxic inflammatory products, and support a role for oxidative damage in the pathogenesis of AD.
Recently, RAGE and scavenger receptors have been shown to interact with Aβ at the cell surface, where the peptide may elicit potentially harmful responses in microglia and neurons via generation of reactive oxygen species (El Khoury et al., 1996; Yan et al., 1996). However, occupancy of these receptors with saturating quantities of their specific ligands does not stimulate changes in protein tyrosine phosphorylation or intracellular O2− production, in contrast to the effect of Aβ. RAGE is believed to act as a tether that binds Aβ to cell surfaces, where reactive oxygen species are generated extracellularly by an undefined mechanism. In contrast, the data presented here demonstrate that fibrillar Aβ activates signal transduction pathways in microglia, leading to intracellular generation of superoxide as well as its release in the extracellular space (Figs.5, 6). Although Aβ has been shown to interact with scavenger receptors and RAGE, the present data demonstrate that these receptors do not mediate the activation of tyrosine kinases and generation of superoxide detected here. Moreover, these data provide evidence for the existence of other Aβ-interactive species that are linked to the signal transduction pathways in these cells. However, it remains possible that fibrillar Aβ may bind scavenger receptors or RAGE but activate intracellular processes distinct from natural ligands, including acetylated LDL and glycated BSA, respectively.
These data provide support for the view that the pathogenesis of AD comprises a series of events initially characterized by the production of Aβ, aggregation of Aβ into fibrils, and deposition of Aβ fibrils as extracellular plaques within the brain. Microglia initially interact with Aβ fibrils and initiate a rapid, complex cellular response, resulting in the elaboration of potentially toxic products, including reactive oxygen intermediates. The elaboration of cytokines and other mediators of inflammation, such as complement components, proteases, and protease inhibitors, then may generate a feed-forward inflammatory process. The progressive neuropathological changes in the AD brain and the accompanying deterioration in cognitive ability are likely to be, in part, a consequence of ongoing local inflammatory responses in the brain mediated by microglia. Inflammation, generation of reactive oxygen intermediates, and cytokine release are recurring mechanisms in the pathophysiology of many neurodegenerative diseases (Halliwell, 1992; Brown et al., 1996; Lorton et al., 1996). Indeed, the hypothesis that AD pathogenesis involves an ongoing inflammatory response is supported by recent epidemiological analysis demonstrating that long-term anti-inflammatory drug treatment is correlated with a lower incidence of dementia (McGeer and McGeer, 1996).
Footnotes
Support for this work was provided by grants from the National Institute on Aging (AG08012) and American Health Assistance Foundation to G.E.L. We thank Drs. Karp Herrup, Patrick McGeer, Bruce Trapp, and Andre Nel for their comments on this manuscript.
Correspondence should be addressed to Dr. Gary Landreth, Alzheimer Research Laboratory, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106-4928.
REFERENCES
- 1.Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor α1-antichymotrypsin in the brain amyloid deposits of Alzheimer disease. Cell. 1988;52:487–501. doi: 10.1016/0092-8674(88)90462-x. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal A, Salem P, Robbins KC. Involvement of p72syk, a protein tyrosine kinase, in Fcγ receptor signaling. J Biol Chem. 1993;268:15900–15905. [PubMed] [Google Scholar]
- 3.Albina JE. On the expression of nitric oxide synthase by human macrophages. Why no NO? J Leukoc Biol. 1995;58:643–649. doi: 10.1002/jlb.58.6.643. [DOI] [PubMed] [Google Scholar]
- 4.Araujo DM, Cotman CW. β-Amyloid stimulates glial cells in vitro to produce growth factors that accumulate in senile plaques in Alzheimer’s disease. Brain Res. 1992;569:141–145. doi: 10.1016/0006-8993(92)90380-r. [DOI] [PubMed] [Google Scholar]
- 5.Bauer J, Strauss S, Scheiter-Gasser U, Ganter U, Shlegel P, Witt I, Yolk B, Berger M. Interleukin-6 and alpha-2-macroglobulin indicate an acute phase in Alzheimer’s disease cortices. FEBS Lett. 1991;285:111–114. doi: 10.1016/0014-5793(91)80737-n. [DOI] [PubMed] [Google Scholar]
- 6.Bellis SL, Miller JT, Turner CE. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J Biol Chem. 1995;270:17437–17441. doi: 10.1074/jbc.270.29.17437. [DOI] [PubMed] [Google Scholar]
- 7.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 8.Brown DR, Schmidt B, Kretzschmar HA. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature. 1996;380:345–347. doi: 10.1038/380345a0. [DOI] [PubMed] [Google Scholar]
- 9.Burkhardt AL, Saouaf SJ, Mahajan S, Bolen JB. Involvement of nonreceptor protein tyrosine kinases in multichain immune recognition receptor signal transduction. Adv Exp Med Biol. 1994;365:131–141. doi: 10.1007/978-1-4899-0987-9_14. [DOI] [PubMed] [Google Scholar]
- 10.Cataldo AM, Nixon RA. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc Natl Acad Sci USA. 1990;87:3861–3865. doi: 10.1073/pnas.87.10.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature. 1995;378:303–306. doi: 10.1038/378303a0. [DOI] [PubMed] [Google Scholar]
- 12.Clark EA, Brugge JS. Redistribution of activated pp60src to integrin-dependent cytoskeletal complexes in thrombin-stimulated platelets. Mol Cell Biol. 1993;13:1863–1871. doi: 10.1128/mcb.13.3.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 14.Cole GM, Huynh TV, Saitoh T. Evidence for lysosomal processing of amyloid beta-protein precursor in cultured cells. Neurochem Res. 1989;10:933–939. doi: 10.1007/BF00965926. [DOI] [PubMed] [Google Scholar]
- 15.Crowley MT, Harmer SL, DeFranco AL. Activation-induced association of a 145 kDa tyrosine-phosphorylated protein with Shc and Syk in B-lymphocytes and macrophages. J Biol Chem. 1996;271:1145–1152. doi: 10.1074/jbc.271.2.1145. [DOI] [PubMed] [Google Scholar]
- 16.Curnutte JT, Erickson RW, Ding J, Badwey JA. Reciprocal interactions between protein kinase C and components of the NADPH oxidase complex may regulate superoxide production by neutrophils stimulated with phorbol ester. J Biol Chem. 1994;269:10813–10819. [PubMed] [Google Scholar]
- 17.Debets JM, Van der Linden CJ, Dieteren IE, Leeuwenberg JF, Buurman WA. Fc-receptor cross-linking induces rapid secretion of tumor necrosis factor (cachectin) by human peripheral blood monocytes. J Immunol. 1988;141:1197–1201. [PubMed] [Google Scholar]
- 18.Dusi S, Donini M, Rossi F. Tyrosine phosphorylation and activation of neutrophils: a possible role for MAP kinases and for a 75 kDa protein. Biochem J. 1994;304:243–250. doi: 10.1042/bj3040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.el-Hachimi KH, Fonchin JF. Do microglial cells phagocytose the beta/A4-amyloid senile plaque core of Alzheimer disease? C R Acad Sci III. 1994;317:445–451. [PubMed] [Google Scholar]
- 20.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 21.Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol (Berl) 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- 22.Frautschy SA, Cole GM, Baird A. Phagocytosis and deposition of vascular beta-amyloid in rat brains injected with Alzheimer beta amyloid. Am J Pathol. 1992;140:1389–1399. [PMC free article] [PubMed] [Google Scholar]
- 23.Furuta Y, Ilic D, Kanazawa S, Takeda N, Yamamoto T, Aizawa S. Mesodermal defect in late phase of gastrulation by a targeted mutation of focal adhesion kinase, FAK. Oncogene. 1995;11:1989–1995. [PubMed] [Google Scholar]
- 24.Ghazizadeh S, Bolen JB, Fleit HB. Physical and functional association of Src-related protein tyrosine kinases with Fc gamma RII in monocytic THP-1 cells. J Biol Chem. 1994;269:8878–8884. [PubMed] [Google Scholar]
- 25.Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glenner GC, Wong CW. Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 27.Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- 28.Greenburg S, Chang P, Silverstein SC. Tyrosine phosphorylation of the γ subunit of Fcγ receptors, p72syk, and paxillin during Fc receptor-mediated phagocytosis in macrophages. J Biol Chem. 1994;269:3897–3902. [PubMed] [Google Scholar]
- 29.Griffin W, Stanley L, Ling C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guan JL, Shalloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature. 1992;358:690–692. doi: 10.1038/358690a0. [DOI] [PubMed] [Google Scholar]
- 31.Haberland ME, Fogelman AM. Scavenger receptor-mediated recognition of maleyl bovine plasma albumin and the demaleylated protein in human monocyte macrophages. Proc Natl Acad Sci USA. 1985;82:2693–2697. doi: 10.1073/pnas.82.9.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 33.Hamawy MM, Minoguchi K, Swaim WD, Mergenhagen SE, Siraganian RP. A 77 kDa protein associates with pp125FAK in mast cells and becomes tyrosine-phosphorylated by high affinity IgE receptor aggregation. J Biol Chem. 1995;270:12305–12309. doi: 10.1074/jbc.270.20.12305. [DOI] [PubMed] [Google Scholar]
- 34.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 36.Indik ZK, Park JG, Pan ZQ, Schreiber AD. Induction of phagocytosis by a protein tyrosine kinase. Blood. 1995;85:1175–1180. [PubMed] [Google Scholar]
- 37.Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 38.Kiener PA, Rankin BM, Burkhardt AL, Schieven GL, Gilliland LK, Rowley RB, Bolen JB, Ledbetter JA. Cross-linking of Fcγ receptor I (FcγRI) and receptor II (FcγRII) on monocytic cells activates a signal transduction pathway common to both Fc receptors that involves the stimulation of p72Syk protein tyrosine kinase. J Biol Chem. 1993;268:24442–24448. [PubMed] [Google Scholar]
- 39.Lagenaur C, Lemmon V. An L1-like molecule, the 8D9 antigen, is a potent substrate for neurite extension. Proc Natl Acad Sci USA. 1987;84:7753–7757. doi: 10.1073/pnas.84.21.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leong SK, Ling E-A. Ameboid and ramified microglia: their interrelationship and response to brain injury. Glia. 1992;7:39–47. doi: 10.1002/glia.440060106. [DOI] [PubMed] [Google Scholar]
- 41.Lin TH, Rosales C, Mondal K, Bolen JB, Haskill S, Juliano RL. Integrin-mediated tyrosine phosphorylation and cytokine message induction in monocytic cells. J Biol Chem. 1995;270:16189–16197. doi: 10.1074/jbc.270.27.16189. [DOI] [PubMed] [Google Scholar]
- 42.Lorton D, Kocsis J-M, King L, Madden K, Brunden KR. β-Amyloid induces increased release of interleukin 1β from lipopolysaccharide-activated human monocytes. J Neuroimmunol. 1996;67:21–29. doi: 10.1016/0165-5728(96)00030-6. [DOI] [PubMed] [Google Scholar]
- 43.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid core protein in Alzheimer’s disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGeer PL, McGeer EG. Anti-inflammatory drugs in the fight against Alzheimer’s disease. Ann NY Acad Sci. 1996;777:213–220. doi: 10.1111/j.1749-6632.1996.tb34421.x. [DOI] [PubMed] [Google Scholar]
- 45.McGeer PL, Akiyama H, Itagaki S, McGeer EG. Immune system response in Alzheimer’s disease. Can J Neurol Sci. 1989;16:516–527. doi: 10.1017/s0317167100029863. [DOI] [PubMed] [Google Scholar]
- 46.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in degenerative neurological diseases. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- 47.Meda L, Cassatella MA, Szendrei GI, Otovos L, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by β-amyloid protein and interferon γ. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 48.Minoguchi K, Kihara H, Nishikata H, Hamawy MM, Siraganian RP. Src family tyrosine kinase Lyn binds several proteins including paxillin in rat basophilic leukemia cells. Mol Immunol. 1994;31:519–529. doi: 10.1016/0161-5890(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 49.Oliver JM, Burg DL, Wilson BS, McLaughlin JM, Geahlen RL. Inhibition of mast cell Fc epsilon R1-mediated signaling and effector function by the Syk selective inhibitor, piceatannol. J Biol Chem. 1994;269:29697–29703. [PubMed] [Google Scholar]
- 50.Pfefferkorn LC, Fanger MW. Cross-linking of the high affinity Fc receptor for human immunoglobulin G1 triggers transient activation of NADPH oxidase activity. J Biol Chem. 1989;264:14112–14120. [PubMed] [Google Scholar]
- 51.Pick E. Microassays for superoxide and hydrogen peroxide production and nitroblue tetrazolium reduction using an enzyme immunoassay microplate reader. Methods Enzymol. 1986;132:407–421. doi: 10.1016/s0076-6879(86)32026-3. [DOI] [PubMed] [Google Scholar]
- 52.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1990;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakurai T, Tsuchiya S. Superoxide production from nonenzymatically glycated protein. FEBS Lett. 1988;236:406–410. doi: 10.1016/0014-5793(88)80066-8. [DOI] [PubMed] [Google Scholar]
- 54.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:915–922. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 55.Selkoe DJ. Amyloid β-protein and the genetics of Alzheimer’s disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 56.Shiue L, Zoller MJ, Brugge JS. Syk is activated by phosphotyrosine-containing peptides representing the tyrosine-based activation motifs of the high affinity receptor for IgE. J Biol Chem. 1995;270:10498–10502. doi: 10.1074/jbc.270.18.10498. [DOI] [PubMed] [Google Scholar]
- 57.Sidorenko SP, Law C-L, Chandran KA, Clark EA. Human spleen tyrosine kinase p72Syk associates with the Src-family kinase p53/p56Lyn and a 120 kDa phosphoprotein. Proc Natl Acad Sci USA. 1995;92:359–363. doi: 10.1073/pnas.92.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Terzi E, Holzemann G, Seelig J. Reversible random coil-β-sheet transition of the Alzheimer β-amyloid fragment (25–35). Biochemistry. 1994;33:1345–1350. doi: 10.1021/bi00172a009. [DOI] [PubMed] [Google Scholar]
- 59.Turner M, Mee PJ, Costello PS, Williams O, Price AA, Duddy LP, Furlong MT, Geahlen RL, Tybulewicz VL. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature. 1995;378:298–302. doi: 10.1038/378298a0. [DOI] [PubMed] [Google Scholar]
- 60.Walker DG, Kim SU, McGeer PL. Complement and cytokine expression in cultured microglia derived from postmortem human brains. J Neurosci Res. 1995;40:478–493. doi: 10.1002/jnr.490400407. [DOI] [PubMed] [Google Scholar]
- 61.Wang AV, Scholl PR, Geha RS. Physical and functional association of the high affinity immunoglobulin G receptor (Fc gamma RI) with the kinases Hck and Lyn. J Exp Med. 1994;180:1165–1170. doi: 10.1084/jem.180.3.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wood J, Zinsmeister P. Tyrosine phosphorylation systems in Alzheimer’s disease pathology. Neurosci Lett. 1991;121:12–16. doi: 10.1016/0304-3940(91)90637-9. [DOI] [PubMed] [Google Scholar]
- 63.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmitt AM. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]