

Abstract
Reports that nitric oxide synthase (NOS) inhibition prevents the induction of long-term potentiation (LTP) have been controversial. Recent evidence suggests that NO may help to regulate the threshold for LTP induction. We have tested this hypothesis by examining the effects of stimulus frequency and train duration on synaptic plasticity in the presence of either NO donors or NOS inhibitors. Two different NO donors facilitated LTP induction by stimuli that normally produced only short-term potentiation, whereas NOS inhibitors blocked LTP to stimuli that normally produce small LTP. NO donors facilitated LTP induction even when NMDA receptors were blocked, indicating that NO need not act via NMDA receptors. NO donors and NOS inhibitors were without effect on long-term depression (LTD), suggesting that they act on a distinct potentiating mechanism. Thus, NO could contribute to the establishment of plasticity under physiologically relevant conditions by selectively increasing the probability of LTP induction.
Keywords: nitric oxide, synaptic plasticity, learning and memory, long-term potentiation, long-term depression, hippocampal slice
Since nitric oxide (NO) was first described as a membrane-soluble neuronal messenger, its role in neuronal plasticity has been disputed. Initial reports (Böhme et al., 1991; O’Dell et al., 1991; Schuman and Madison, 1991; Haley et al., 1992) indicated that inhibitors of nitric oxide synthase (NOS) completely blocked the induction of long-term potentiation (LTP) in the CA1 cell field of hippocampal slices. This finding helped kindle controversy about whether LTP expression results from a presynaptic mechanism despite the fact that early stages of induction are postsynaptic (Bliss et al., 1990). Perhaps more importantly, it provided a list of possible LTP expression mechanisms based on known biological targets for NO, including the enzymes guanylyl cyclase (Haley et al., 1992; Zhou et al., 1994) and ADP-ribosyltransferase (Schuman et al., 1994), as well as other important proteins, including NMDA receptors (Lei et al., 1992; Manzoni et al., 1992). Moreover, the discovery of a key link in a mechanism of use-dependent plasticity offered another tool for exploring the relationship between LTP and learning (Chapman et al., 1992; Böhme et al., 1993).
Subsequent investigations, however, have suggested that the role of NO in hippocampal LTP is limited (Gribkoff and Lum-Ragan, 1992; Chetkovich et al., 1993; Haley et al., 1993; Williams et al., 1993; Bannerman et al., 1994a–c) or nonexistent (Musleh et al., 1993; Cummings et al., 1994). In vitro recordings in CA1 at 30–32°C with standard tetanic stimulation indicated that LTP induction was unaffected even by high concentrations of NOS inhibitors. Williams et al. (1993) described a number of experimental factors (e.g., the temperature at which slice experiments were conducted and the age of the animals) that were not held constant across laboratories and argued that these could explain the apparent discrepancy in the reported effects of NOS inhibitors.
One factor that influences the effectiveness of NOS inhibitors in blocking LTP induction is the strength (i.e., intensity, frequency, or duration) of tetanic stimulation. Several groups have noted that LTP induced by weak tetanic stimulation is blocked by NOS inhibitors, whereas stronger tetanic stimulation leads to NO-independent potentiation (Chetkovich et al., 1993; Haley et al., 1993; O’Dell et al., 1994). The generation of NO, therefore, could be critical for LTP induction under the more physiologically relevant conditions in which a small number of synapses are activated briefly at moderately high frequency.
Although it is possible that NO affects plasticity by facilitating a potentiation mechanism (Zhuo et al., 1993, 1994), it is also possible that it contributes to LTP induction by inhibiting long-term depression (LTD). Several investigators (Dudek and Bear, 1993; Mulkey et al., 1993; Mayford et al., 1995) have suggested that LTP and LTD represent opponent processes of phosphorylation and dephosphorylation, respectively. If so, failure to induce LTP with moderate tetanic stimuli [e.g., 25 pulses delivered at 10 or 50 Hz in control normal artificial cerebrospinal fluid (ACSF)] might reveal a balance point at which the opponent mechanisms of LTP and LTD are in a state of equilibrium (Bear et al., 1987; Artola et al., 1990). If NO (or any other molecule) blocked LTD selectively, the net effect would be to lower the threshold for LTP induction by removing the opponent process. Two observations support a role for NO in this type of process. First, NO can reduce the activity of NMDA receptors (Manzoni et al., 1992; Lei et al., 1992) that are required for the induction of LTD. Second, Izumi and Zorumski (1993) have reported that NOS inhibitors can block the induction of LTD in hippocampal slices.
To test these hypotheses, we have examined the effects of NO donors and NOS inhibitors in the CA1 region of hippocampal slices across a range of stimuli designed to evoke LTP, LTD, or neither. Neither donors nor inhibitors affected homosynaptic LTD induced by prolonged low-frequency stimulation (LFS) at 1–3 Hz. On the other hand, 25-pulse trains of high-frequency stimulation (HFS) at 10–50 Hz produced only short-term potentiation in ACSF but produced LTP in the presence of the NO donors, even in slices in which NMDA receptors were blocked. NOS inhibitors also blocked LTP induced by 900 pulses delivered at 30 Hz, a protocol that produces small but significant potentiation in control slices. The results therefore favor a model in which NO adjusts the threshold for the induction of synaptic plasticity by selectively facilitating the induction of LTP.
MATERIALS AND METHODS
Slice preparation. All experiments were conducted in hippocampal slices prepared from 14- to 140-d-old male or female Sprague Dawley rats. Rats were anesthetized by intraperitoneal injection of sodium pentobarbital (75 mg/kg) and decapitated with a small animal guillotine. Their brains were removed rapidly to chilled (0–4°C) ACSF containing (in mm): 125 NaCl, 2 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 d-glucose, 10 MgSO4, and 0.5 CaCl2). Each hippocampus was dissected out and sectioned at 450 μm on a Vibratome (TPI, St. Louis, MO). After sectioning, slices were transferred to room temperature ACSF (of the same composition as the dissecting medium with 1.5 mmMg2+ and 2.5 mm Ca2+), where they were left to recover for at least 1 hr before they were transferred to a submersion-type recording chamber. Experiments were performed at a constant temperature of 32 ± 0.2°C with a flow rate for the ACSF of 1–2 ml/min.
Recording arrangement. Insulated stainless steel recording electrodes (Frederick Hare, Brunswick, ME) were placed in stratum radiatum of CA1, and monopolar insulated stainless steel stimulating electrodes were placed in stratum radiatum at the CA2–CA1 border. Constant current stimuli were delivered once every 15 sec, and responses were amplified (1000×), digitized (5–10 kHz), and analyzed on-line by custom-written software, using LabVIEW (National Instruments, Austin, TX). Analyses included measurement of the initial slope of the negative component of the field EPSP (fEPSP), as well as the maximum response amplitude. Baseline stimulus intensity was adjusted to elicit a fEPSP with slope 25–30% of the maximum evoked response, with no evidence of a population spike.
Experimental protocols. Baseline stimulation proceeded for at least 10 min before either tetanus or bath application of NO donors. For experiments in which tetanus was delivered in the presence of NO donors, the donors were added to the bathing medium after the establishment of a stable baseline. Donor application had no effect on baseline responses. Tetanic stimulation was delivered 20 min after donor wash-in, and the donors were washed out 5 min after tetanus. NOS inhibitors were added to the bathing medium as soon as each slice was transferred, and slices were incubated for at least 30 min before tetanic stimulation.
Each slice was given one of several different tetanus protocols, encompassing five stimulus frequencies. LFS consisted of 100, 600, or 900 pulses delivered at 1 or 3 Hz. HFS consisted of 25 pulses delivered at either 10 or 50 Hz, 900 pulses delivered at 10 or 30 Hz, or theta burst stimulation, consisting of four-pulse bursts of 100 Hz stimulation, separated by 200 msec (see Fig. 6 for summary). After either LFS or HFS, baseline stimulation was resumed for 30–60 min. LTP or LTD was defined as statistically significant increases or decreases (determined by t test) in the fEPSP slope 55–60 min post-tetanus, as compared with the pretetanus baseline.
Fig. 6.
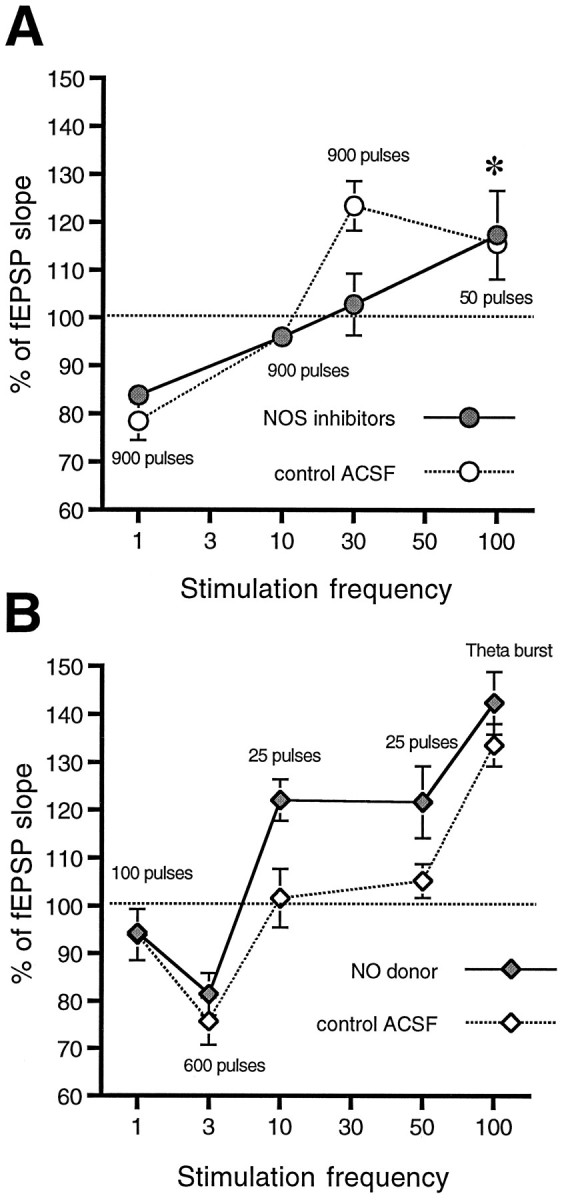
The effects of NO donors and NOS inhibitors are on potentiation, but not depression. A, NOS inhibitors have no effect on plasticity induced by 900 pulses at either 1 Hz (which produces large LTD both in control ACSF and in 100 μml-NAME) or 10 Hz (which produces no significant change in either control or inhibited slices). In contrast, the effect of 100 μm NOArg is pronounced and significant on the potentiation normally produced by 900 pulses at 30 Hz in control ACSF. The data for 100 Hz stimulation (asterisk) are taken from Haley et al. (1993) to demonstrate that, at higher frequency, LTP can be independent of NO. B, The effects of NO donors are complementary to those of NOS inhibitors; donors have no effect on stimuli that normally produce LTD but facilitate LTP induction to stimuli that are subthreshold in control ACSF. NO donors do not produce further facilitation when normally suprathreshold tetani (e.g., theta bursts) are delivered.
Drug preparation. NOS inhibitors (Sigma, St. Louis, MO) were prepared from stock solutions (100 mm); l-NAME was dissolved in dH2O, and NOArg was dissolved in 100 mm HCl. Stock solutions were made freshly every 2–3 d. Hydroxylamine (H2NOH) (Sigma) was dissolved in dH2O and was protected from exposure to light until used. Fresh SNOC was prepared daily as described (Lei et al., 1992) froml-cysteine, sodium nitrite, and hydrochloric acid. SNOC was prepared on ice and stored in a refrigerator for no more than 3 hr as a concentrated (100 mm) stock solution. SNOC was diluted to 100 μm in ACSF immediately before superfusion of the slices. When freshly prepared, SNOC is a clear red solution, which fades to clear colorless as NO dissipates spontaneously, leaving a solution containing only the disulphide cystine. For control experiments described below, SNOC was left at room temperature for 24 hr until all color had left the solution. The HCl used in the preparation of NOArg and SNOC had no effect on the pH of the ACSF when diluted to final concentration (1000-fold dilution).
RESULTS
Effects of NO donors on HFS-induced plasticity
A train of 25 pulses delivered at either 10 or 50 Hz did not produce LTP in the absence of NO donors (Fig. 1). Sixty minutes after tetanus, the fEPSP slope was not significantly different from control for either 10 Hz stimulation (103 ± 6%,n = 5, n.s.) or 50 Hz stimulation (105 ± 3%,n = 23, n.s.). When the NO donor hydroxylamine (H2NOH) was added to the ACSF (at 200 μm) 20 min before tetanus, however, both HFS protocols produced increases in fEPSP slope, which lasted at least 60 min (120 ± 5%,n = 8, p < 0.05 for 10 Hz; 123 ± 7%, n = 9, p < 0.02 for 50 Hz). Although these differences were reliable between slices, we also examined the effects of 200 μm H2NOH within slices (Fig. 1B). An HFS train consisting of 25 pulses at 50 Hz was delivered 10 min after a stable baseline was established in normal ACSF. Sixty minutes later, H2NOH was washed in for 20 min, followed by a repetition of the HFS. HFS did not produce LTP in the control medium (99 ± 2%, n = 9, n.s.) but did when the same tetanus was given to the same slices in the presence of H2NOH (112 ± 3%, n = 9, p < 0.0001).
Fig. 1.

H2NOH facilitates LTP induction. A 25-pulse stimulus train delivered at either 10 or 50 Hz (A) produced only short-term potentiation in slices bathed in normal ACSF (open symbols). In the presence of 200 μm H2NOH (filled symbols), both 10 and 50 Hz HFS produced potentiation that was still significant after 60 min. H2NOH was washed in 20 min before tetanus, and washed out 5 min after tetanus (horizontal line in A). A 25-pulse stimulus at 50 Hz was delivered to nine additional slices in control ACSF (B). As in A, responses returned to baseline after 60 min. H2NOH was washed onto these same slices for 20 min (horizontal line in B), followed by repetition of the 50 Hz HFS, which this time produced significant enhancement of the field EPSP slope, which lasted at least 60 min after the tetanus. Error bars in all figures represent SEM.
One potential source of artifact when NO donors are used is that NO itself is not causing the effect but the moieties that remain after NO has been released. We therefore replicated our H2NOH findings by delivering HFS in the presence of 100 μmS-nitrosocysteine (SNOC), which degrades spontaneously to NO and the relatively nonreactive disulphide cystine (Ignarro, 1990; Lei et al., 1992). The subthreshold tetanus of 25 pulses delivered at 50 Hz produced a significant increase in the fEPSP slope when delivered in the presence of 100 μm SNOC, as compared with 50 Hz HFS in control medium (117 ± 4%, n = 15,p < 0.05; Fig. 2). To provide further support that the source of the enhancement was NO, we administered subthreshold HFS in the presence of SNOC that had been left at room temperature for at least 24 hr and had changed from a clear red solution to a clear colorless one, indicating that the SNOC has degraded to cystine alone (Lei et al., 1992). In the presence of this degraded SNOC solution, the 50 Hz HFS did not produce a potentiation that was significantly different from the potentiation produced by control 50 Hz HFS (109 ± 3%, n = 7 n.s.), although the enhancements that followed SNOC and degraded SNOC were not significantly different from each other.
Fig. 2.

The specific NO donor SNOC facilitates LTP induction. The application of 100 μm SNOC has an effect identical to that of H2NOH. The 25 pulses delivered at 50 Hz produced significant potentiation in the presence of SNOC prepared freshly and stored on ice. SNOC that had been allowed to degrade to cystine by leaving the SNOC stock solution overnight at room temperature had a less pronounced effect on tetanus, producing an enhancement that was not significantly greater than control 50 Hz HFS 60 min after tetanus.
NO could facilitate LTP induction by several methods, including enhancing the sensitivity or activity of enzymes (e.g., guanylyl cyclase or ADP ribosyltransferase) or directly modulating NMDA receptors. To test the latter possibility, we first delivered high-frequency stimulation in the presence of 100 μmdl-AP5 (Fig. 3A). As expected (Collingridge et al., 1983), tetanus delivered in the presence of AP5 did not result in potentiation at 30 min (100 ± 3%,n = 7, n.s.). Ten minutes after tetanus, 100 μm SNOC was washed in, followed 20 min later by a second 25-pulse 50 Hz tetanus (Fig. 3A), which this time did produce significant potentiation at 30 min (113 ± 8%,n = 7, p < 0.05). When this protocol was repeated with SNOC that had been left at room temperature for at least 24 hr, the depleted SNOC proved unable to facilitate potentiation (Fig. 3B; 103 ± 5%, n = 4, n.s.).
Fig. 3.

AP5 does not block SNOC-facilitated LTP. Tetanic stimulation consisting of 25 pulses delivered at 50 Hz was delivered to slices bathed in 100 μmdl-AP5. Ten minutes after tetanus, AP5 plus either 100 μm SNOC (A) or SNOC depleted of NO by maintaining at room temperature for at least 24 hr (B) was washed in. Twenty minutes later (30 min after the first tetanus), slices were tetanized again. SNOC was washed out 5 min after the second tetanus. Significant potentiation was recorded only in the presence of SNOC.
Effects of NO donors on plasticity induced by low-frequency stimulation
We delivered LFS (600 pulses at 3 Hz) to hippocampal slices taken from young rats (14–35 d old) in the presence or absence of 200 μm H2NOH (Fig.4A) to test the hypothesis that NO’s apparent facilitation of LTP might result from its blocking LTD. The fEPSP slope was depressed 30 min after tetanus in both control (76 ± 5%, n = 6, p < 0.01) and H2NOH (81 ± 4%, n = 6,p < 0.01)-treated slices. Although the rather strongly depressing 3 Hz, 600 pulse tetanus revealed no effect of H2NOH, the effects of NO donors on LTD might be similar to their effects on LTP, i.e., only be detectable around the LTD induction threshold. We therefore delivered a weaker LFS of 100 pulses at 1 Hz to another group of slices taken from young animals in the presence or absence of 200 μm H2NOH (Fig.4B). Once again, we saw no effect of the donor on LTD (control, 94 ± 5%, n = 3; H2NOH, 94 ± 3%, n = 5).
Fig. 4.
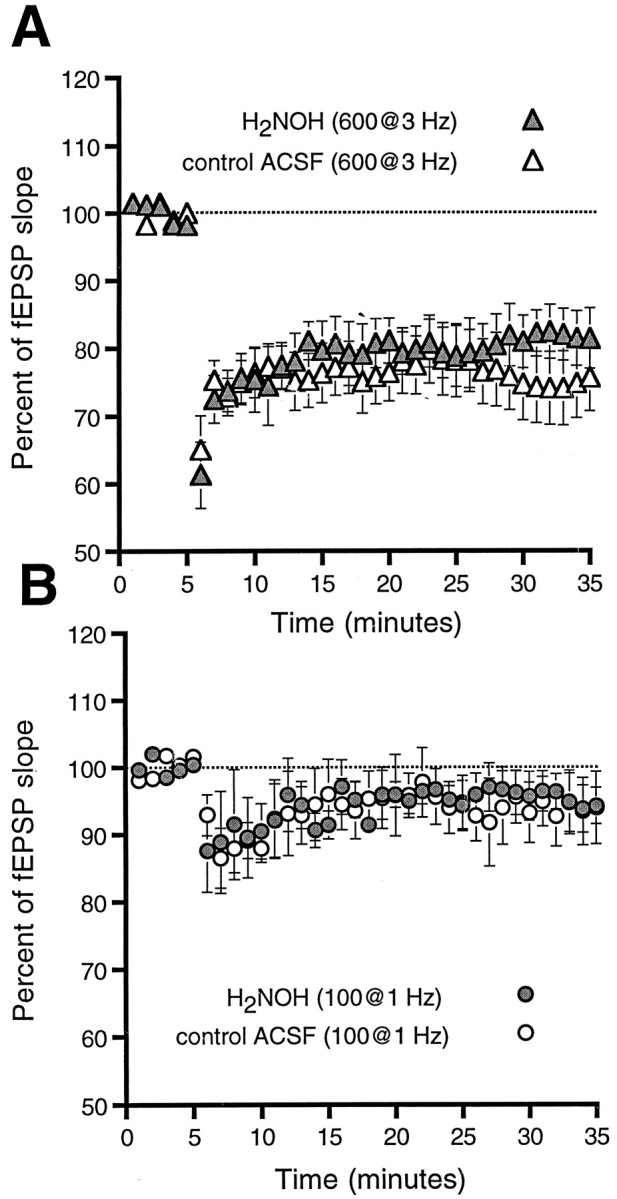
H2NOH has no effect on LTD. Slices taken from young rats and bathed in either control ACSF (open symbols) or ACSF containing 200 μmH2NOH (filled symbols) were given stimuli designed to produce either large LTD (600 pulses delivered at 3 Hz, A) or minimal LTD (100 pulses at 1 Hz,B). The addition of H2NOH to the bath had no effect on the plasticity induced by these stimuli.
Effects of NOS inhibitors on LFS- and HFS-induced plasticity
If the effect of NO on the plasticity threshold is selective for LTP, inhibitors of NO synthesis should block LTP without affecting LTD. We used 900-pulse stimulus trains delivered at three different frequencies (1, 10, and 30 Hz) in normal ACSF or in the presence of NOS inhibitors (1 mml-NAME or 100 μmNOArg). NOS inhibitors were without effect at 1 Hz, where both control and l-NAME-treated slices demonstrated LTD (l-NAME, 84 ± 3%, n = 7; control, 78 ± 4%, n = 6), and at 10 Hz, where neither control nor NOArg-treated slices showed any significant change from pretetanus (see Fig. 6A). Consistent with previous results (Chetkovich et al., 1993; Haley et al., 1993; O’Dell et al., 1994), 900 pulses at 30 Hz, which induced LTP in control slices (123 ± 5%, n = 6), did not produce significant changes in fEPSP slope in the presence of NOArg (103 ± 6%,n = 6; Fig. 5).
Fig. 5.

NOS inhibitors prevent LTP induction by prolonged stimulation at 30 Hz. Delivery of 900 pulses at 30 Hz in control ACSF (open circles) produces LTP of the field EPSP slope. The same tetanus delivered in the presence of 100 μm NOArg leads to only short-term potentiation. Slices were exposed to NOArg for at least 30 min before tetanus and throughout the post-tetanus measurement period.
Figure 6 summarizes the effects of both NO donors and NOS inhibitors on stimuli, which in control ACSF induces LTD (1, 3 Hz), LTP (30 Hz, 900 pulses; 100 Hz, 50 pulses), or no change (25 pulses delivered at either 10 or 50 Hz). The results demonstrate no effects on LTD and a consistent effect on LTP near the induction threshold; NO donors facilitate LTP induction, and NOS inhibitors prevent it.
DISCUSSION
The administration of NO donors to hippocampal slices facilitates the induction of LTP. Tetanic stimulation sufficient to produce only short-term potentiation in normal ACSF produced LTP in the presence of compounds that donate NO. Consistent with previous findings (Chetkovich et al., 1993; Haley et al., 1993; O’Dell et al., 1994), some tetanic stimuli that did produce LTP in normal ACSF could produce only short-term potentiation in the presence of NOS inhibitors. In contrast, neither NO donors nor NOS inhibitors had any detectable effect on homosynaptic LTD.
Although NO-facilitated LTP induction is activity-dependent, it is apparently not NMDA receptor-dependent (see also Zhou et al., 1993). Mild tetanic stimulation, which produced only short-term potentiation in control ACSF and no potentiation in dl-AP5, was sufficient to induce LTP in slices treated with AP5 plus SNOC. If NO modulates potentiation solely by affecting the NMDA receptor (Izumi et al., 1992; Lei et al., 1992; Manzoni et al., 1992), then blocking NMDA receptors should prevent NO-facilitated potentiation. Because it does not, we must assume that NO-facilitated LTP is NMDA receptor-independent. This does not, however, rule out the possibility that NO modulation of NMDA receptors could contribute to normal tetanus-induced plasticity.
The failure of AP5 to block NO-facilitated LTP indicates that NO-facilitated LTP is most likely independent of postsynaptic calcium influx as well; the mild tetanic stimulation that produces LTP in the presence of SNOC and AP5 is unlikely to activate significant voltage-dependent calcium currents postsynaptically. Three possible alternatives that could explain the facilitatory effect of NO on LTP induction are that (1) NO interacts with calcium released from intracellular stores in the postsynaptic neuron [e.g., by activation of postsynaptic metabotropic glutamate receptors or the cyclic ADP-ribose signaling pathway (Willmott et al., 1996)], (2) NO itself can initiate increases in postsynaptic calcium concentration by stimulating release from intracellular stores, or (3) exogenous NO is acting presynaptically in an activity-dependent manner. The third possibility suggests that NO could, indeed, act as a retrograde messenger, normally requiring postsynaptic activation only to generate activation of the Ca2+/calmodulin-dependent NOS.
Our data indicate that it is possible to facilitate LTP selectively, without affecting LTD. Recent evidence suggests that LTD and LTP may be complementary phenomena with a final common element in which phosphorylation of some substrate(s) by activity-dependent protein kinase(s) produces LTP, whereas LTD produces effects that are the opposite of LTP by activity-dependent phosphatase-mediated dephosphorylation of the same substrates (Bear and Malenka, 1994;Mayford et al., 1995). If this were true, it would not be possible to facilitate LTP without affecting LTD. If NO enhanced the sensitivity of a kinase to postsynaptic activity (e.g., by enhancing its calcium sensitivity), then stimuli that normally produce weak LTD would produce LTP, because the ratio of activated kinase to activated phosphatase would increase. The same would be true if the ability of the kinase to phosphorylate its substrate(s) were enhanced. Even if NO directly modified the substrate(s), there would have to be an effect of LTD if the same substrate were responsible for both potentiation and depression.
Artola, Bröcher, and Singer (1990) have suggested that there are distinct thresholds for the induction of LTD and LTP in the rat visual cortex. Although LTD was associated with small postsynaptic depolarization from rest and LTP was associated with large depolarization, there was a relatively broad range of postsynaptic potentials between the thresholds, which produced little or no lasting change in synaptic efficacy. Our data are consistent with these observations and suggest that the role of endogenous NO is in narrowing the range of stimulus conditions that produce no plasticity by selectively shifting the LTP induction threshold to the left. Thus, in slices stimulated in control ACSF, prolonged stimulation at 3 Hz produces LTD, and HFS of 25 pulses delivered at either 10 or 50 Hz produces no lasting change (Fig. 6). In contrast, in the presence of NO donors, 600 pulses delivered at 3 Hz still produce depression, but increasing the frequency to 10 Hz and delivering only 25 pulses produce LTP. In the presence of NOS inhibitors, stimuli that normally produce modest LTP [e.g., 900 pulses at 30 Hz (Fig. 6) or 25 pulses at 100 Hz (Haley et al., 1993)] are without effect, thus extending the range of activities that fall between the LTD and LTP induction thresholds.
Although other explanations exist, it is most likely that the results described here are attributable to effects of NO. First, there is agreement between the effects of NOS inhibitors and NO donors, with the former making LTP induction more difficult and the latter making it more likely. It is improbable that this specific pattern of results would have occurred had both the donor effect and the inhibitor effect been artifactual. Second, our use of two donors with different NO release mechanisms and different non-NO moieties reduces the risk that the facilitation of LTP was attributable to a molecule other than NO. In particular, SNOC, which releases only NO and cystine, entirely duplicated the effects of H2NOH on LTP induction threshold, whereas spent SNOC, containing only cystine, was without effect.
Controversy over the role of NO in activity-dependent plasticity is unlikely to abate in the near future. The present studies suggest that, although LTP can be induced in the CA1 region of hippocampus in the absence of NOS activity, NO can contribute to the induction of enhanced synaptic responses. Moreover, the conditions under which NO donors facilitate (and NOS inhibitors prevent) LTP induction are likely to be relevant to endogenously generated synaptic plasticity.
Footnotes
This work was supported by National Science Foundation Grant IBN 9410131 (P.F.C.) and a doctoral dissertation fellowship from the University of Minnesota (P.L.M.). We thank Drs. Kevin Fox and Alcino Silva for critical comments on this manuscript.
Correspondence should be addressed to Dr. Paul F. Chapman, University of Wales, Cardiff, Physiology Unit, School of Molecular and Medical Biosciences, P.O. Box 911, Cardiff, CF1 3US, United Kingdom
REFERENCES
- 1.Artola A, Bröcher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- 2.Bannerman DM, Butcher SP, Morris RGM. Intracerebroventricular injection of a nitric oxide synthase inhibitor does not affect long-term slope potentiation in vivo. Neuropharmacology. 1994a;33:1387–1397. doi: 10.1016/0028-3908(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 3.Bannerman DM, Chapman PF, Kelly PAT, Butcher SB, Morris RGM. Inhibition of nitric oxide synthase does not impair spatial learning. J Neurosci. 1994b;14:7404–7414. doi: 10.1523/JNEUROSCI.14-12-07404.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannerman DM, Chapman PF, Kelly PAT, Butcher SB, Morris RGM. Inhibition of nitric oxide synthase does not prevent the induction of LTP in vivo. J Neurosci. 1994c;14:7415–7425. doi: 10.1523/JNEUROSCI.14-12-07415.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 6.Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synapse modification. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- 7.Bliss TVP, Errington ML, Lynch MA, Williams JH. Presynaptic mechanisms in hippocampal long-term potentiation. Cold Spring Harb Symp Quant Biol. 1990;55:119–129. doi: 10.1101/sqb.1990.055.01.015. [DOI] [PubMed] [Google Scholar]
- 8.Böhme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. Possible involvement of nitric oxide in long-term potentiation. Eur J Pharmacol. 1991;199:379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- 9.Böhme GA, Bon C, Lemaire M, Reibaud M, Piot O, Stutzmann JM, Doble A, Blanchard JC. Altered synaptic plasticity and memory formation in nitric oxide synthase inhibitor-treated rats. Proc Natl Acad Sci USA. 1993;90:9191–9194. doi: 10.1073/pnas.90.19.9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapman PF, Atkins CM, Allen MT, Haley JE, Steinmetz JE. Inhibition of nitric oxide synthesis impairs two different forms of learning. NeuroReport. 1992;3:567–570. doi: 10.1097/00001756-199207000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Chetkovich DM, Klann E, Sweatt JD. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. NeuroReport. 1993;4:919–922. doi: 10.1097/00001756-199307000-00020. [DOI] [PubMed] [Google Scholar]
- 12.Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral commissural pathway of the rat hippocampus. J Physiol (Lond) 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cummings JA, Nicola SM, Malenka RC. Induction in the rat hippocampus of long-term potentiation (LTP) and long-term depression (LTD) in the presence of a nitric oxide synthase inhibitor. Neurosci Lett. 1994;176:110–114. doi: 10.1016/0304-3940(94)90883-4. [DOI] [PubMed] [Google Scholar]
- 14.Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gribkoff VK, Lum-Ragan JT. Evidence for nitric oxide synthase inhibitor-sensitive and insensitive hippocampal synaptic plasticity. J Neurophysiol. 1992;68:639–642. doi: 10.1152/jn.1992.68.2.639. [DOI] [PubMed] [Google Scholar]
- 16.Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- 17.Haley JE, Malen PL, Chapman PF. Nitric oxide synthase inhibitors block long-term potentiation induced by weak but not strong tetanic stimulation at physiological brain temperatures in rat hippocampal slices. Neurosci Lett. 1993;160:85–88. doi: 10.1016/0304-3940(93)90919-c. [DOI] [PubMed] [Google Scholar]
- 18.Ignarro LJ. Biosynthesis and metabolism of endothelium-derived nitric oxide. Annu Rev Pharmacol Toxicol. 1990;30:535–560. doi: 10.1146/annurev.pa.30.040190.002535. [DOI] [PubMed] [Google Scholar]
- 19.Izumi Y, Zorumski CF. Nitric oxide and long-term synaptic depression in the rat hippocampus. NeuroReport. 1993;4:1131–1134. [PubMed] [Google Scholar]
- 20.Izumi Y, Clifford DB, Zorumski CF. Inhibition of long-term potentiation by NMDA-mediated nitric oxide release. Science. 1992;257:1273–1276. doi: 10.1126/science.1519065. [DOI] [PubMed] [Google Scholar]
- 21.Lei SZ, Pan Z-H, Aggarwal SK, Chen H-SV, Hartman J, Sucher NJ, Lipton S. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- 22.Manzoni O, Prezeau L, Marin P, Deshager S, Bockaert J, Fagni L. Nitric oxide-induced blockade of NMDA receptors. Neuron. 1992;8:653–662. doi: 10.1016/0896-6273(92)90087-t. [DOI] [PubMed] [Google Scholar]
- 23.Mayford M, Wang J, Kandel ER, O’Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 24.Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- 25.Musleh WY, Shahi K, Baudry M. Further studies concerning the role of nitric oxide in LTP induction and maintenance. Synapse. 1993;13:370–375. doi: 10.1002/syn.890130409. [DOI] [PubMed] [Google Scholar]
- 26.O’Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation—evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci USA. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- 28.Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- 29.Schuman EM, Meffert MK, Schulman H, Madison DV. An ADP-ribosyltransferase as a potential target for nitric oxide action in hippocampal long-term potentiation. Proc Natl Acad Sci USA. 1994;91:11958–11962. doi: 10.1073/pnas.91.25.11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams JH, Li YG, Nayak A, Errington ML, Murphy KPSJ, Bliss TVP. The suppression of long-term potentiation in rat hippocampus by inhibitors of nitric oxide synthase is temperature and age dependent. Neuron. 1993;11:877–884. doi: 10.1016/0896-6273(93)90117-a. [DOI] [PubMed] [Google Scholar]
- 31.Willmott N, Sethi JK, Walseth TF, Lee HC, White AM, Galione A. Nitric oxide-induced mobilization of intracellular calcium via the cyclic ADP-ribose signaling pathway. J Biol Chem. 1996;271:3699–3705. doi: 10.1074/jbc.271.7.3699. [DOI] [PubMed] [Google Scholar]
- 32.Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260:1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]
- 33.Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]